
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAluminium based binary catalytic system for the solvent free conversion of CO2 to carbonates with high activity and selectivity†
Ran Ma *,
Haojie Sun and
Yuanzhi Cui
*,
Haojie Sun and
Yuanzhi Cui
College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, 6 Xuefu Road, Weiyang District, Xi’an, Shaanxi 710021, China. E-mail: maran@sust.edu.cn
First published on 21st March 2018
Abstract
An easily prepared and low-cost aluminium based metal complex catalyst was prepared using kojic acid (Hkoj) as a ligand, and this developed oxo-coordinated Al(koj)3 complex showed high activity and selectivity for the CO2 fixation reaction with epoxides under mild conditions without any organic solvents. Various cyclic carbonates were obtained in excellent yields (up to 99%). This stable catalytically active Al(koj)3 has strong Lewis acidity for the activation of epoxides, and meanwhile the hydroxy group in Al(koj)3 may play a role in boosting the catalytic activity through possible hydrogen bonding interactions with the epoxide.
Introduction
Recent daily and weekly values of CO2 concentration measured at Mauna Loa Observatory have remained above 400 parts per million.1 This is a significant symbolic event in the global warming process, because part of global warming is attributed to increasing carbon dioxide levels in atmosphere, which is mainly caused by the combustion of huge amounts of fossil fuels (such as coal, petroleum, and natural gas). The rising CO2 concentration may cause a pronounced change to our ecosystem because plant species are very sensitive to the increasing CO2 concentration, as this will influence the behavior of stomata, the microscopic holes in leaves that allow leaves to take up CO2, and as a result will cause changes to land plant photosynthesis. The existing fossil fuel reserves are limited and are being gradually depleted, and alternative resources such as CO2 and biomass have received extensive attention.2 In this context, the utilization of CO2 as an inexpensive, ubiquitous, nontoxic, non-flammable, renewable, and green basic raw material has drawn wide attention in academia and industry.3 Many useful fine chemical products, which have previously been derived from the limited fossil fuel resources, are currently produced using CO2 as an alternative C1 building block, such as methanol, urea, carbonate, formamide and carboxylic acid.4 However, just a fraction of the huge amount of CO2 has been fixed into value-added products and the thermodynamic stability and kinetic inertness of CO2 limits its conversion under mild conditions. A large energy input is required, such as higher CO2 pressure and higher reaction temperatures, thus resulting in huge production costs. Therefore, development of high-efficiency, inexpensive, eco-friendly processes for CO2 conversion and utilization are most urgently needed.5One of the methods used to overcome the obstacles in CO2 utilization is the use of high-energy small-membered ring compounds, e.g. epoxides, as the reactants with a negative Gibbs free energy for the reaction. Also, the addition of CO2 to epoxides is a 100% atom-economical reaction, which is in accordance with the principles of green chemistry, and thus this method represents one of the most attractive routes for CO2 utilization.6 Cyclic carbonates and polycarbonate products have been studied with particular interest due to their widespread application in industry, e.g. as aprotic high-boiling polar solvents for organic synthesis (replacing DMF, DMSO, NMP and acetonitrile), electrolytes for lithium-ion batteries, monomers for polymeric materials (polycarbonates, and isocyanate-free polyurethanes), and intermediates in the synthesis of fine chemicals.7 In the past few years, plenty of catalysts have been exploited for the synthesis of carbonates from CO2 and epoxides, including metal-based catalysts (such as salen–metal compounds, porphyrin–metal compounds, metal oxides, metal–organic frameworks and so on), miscellaneous catalytic systems, ionic liquids, onium salts, and mono or binary metal-free catalysts.8 Catalysts are used to activate the epoxide or/and CO2 and reduce the activation energy of the transformation. The epoxide, activated by the catalyst, is liable to go through ring cleavage-addition with CO2 upon nucleophilic attack by Br− or I−, to form an alkoxide anion, ultimately affording the corresponding cyclic carbonate (Scheme 1). Thus, electron-deficient metal catalysts are privileged for this reaction. In this respect, metal complexes based on porphyrins and metallosalen complexes have been widely investigated due to their unique Lewis acidities.9 In addition, oxo-coordinated metal complexes also displayed high catalytic activity in the cycloaddition reaction of CO2 with epoxides.10 In our previous work, a tetraoxo-coordinated zinc complex, i.e. 2-hydroxypyridine N-oxide zinc(II), Zn(OPO)2, was used and demonstrated remarkable turnover frequencies (up to 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1).11 Therefore, we continued to consider highly active oxo-coordinated metal complexes for the synthesis of cyclic carbonates.
000 h−1).11 Therefore, we continued to consider highly active oxo-coordinated metal complexes for the synthesis of cyclic carbonates.
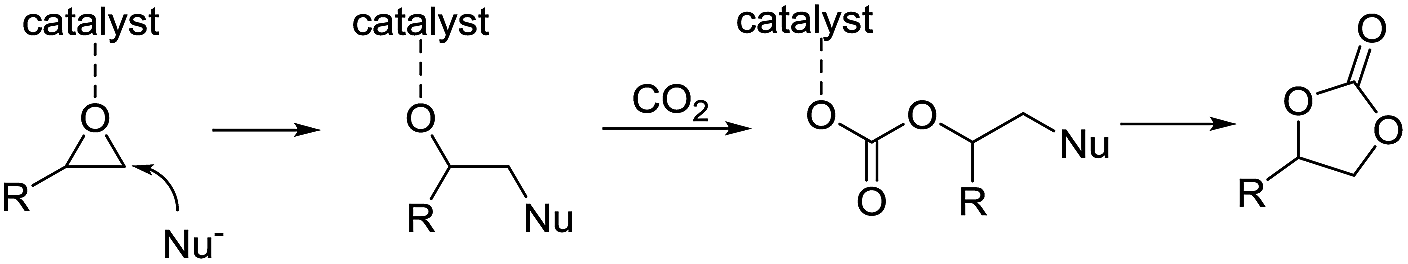 | ||
| Scheme 1 Possible mechanism for the catalytic formation of cyclic carbonates. | ||
Kojic acid (5-hydroxy-2-(hydroxymethyl)-4-pyrone, Hkoj) is considered to be one of the safest food products and is widely used as a food additive to prevent enzymatic browning.12 Hkoj has chelating activity and its metal complexes have reasonable hydrolytic stability, neutral charge and significant lipophilicity. Thus these kojic acid based metal complexes, e.g. Al(koj)3, may have strong Lewis acidities for the activation of epoxides, and meanwhile the hydroxy group in Al(koj)3 may play a role in boosting the catalytic activity through the possible hydrogen bonding interactions with the epoxide (Scheme 2). Hence, in this manuscript, we conducted a thorough investigation to explore the catalytic activity of these kojic acid based metal complexes, which are easy to prepare, low cost, multifunctional for the cycloaddition of CO2 with epoxides.
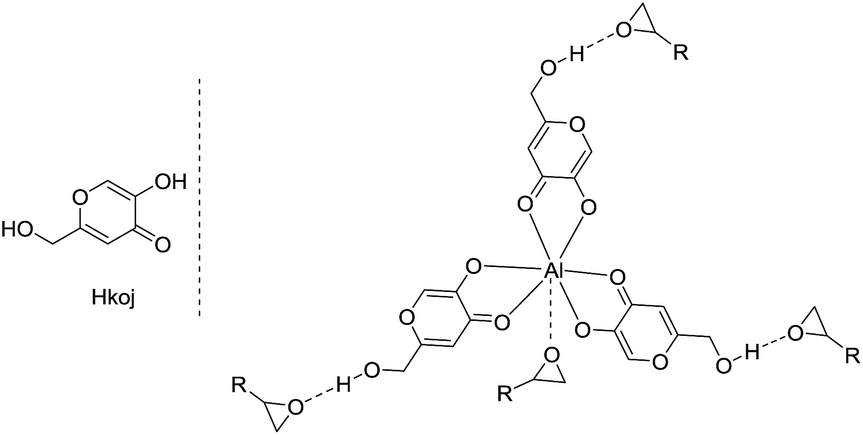 | ||
| Scheme 2 Multifunctional metal complexes used for the activation of epoxides in this work. | ||
Results and discussion
First, different kojic acid based metal complexes were prepared according to methods reported in the literatures13 and the structures were confirmed using FT-IR and elemental analysis. The cycloaddition of styrene oxide with CO2 was chosen as a benchmark reaction (Table 1). TBAI (tetrabutylammonium iodide) was used as a co-catalyst for this transformation due to its excellent nucleophilicity. The initial screening experiments were conducted at 100 °C, with 1 MPa CO2 for 10 h with a catalyst loading of 0.25 mol%. The effect of different metal centers in the complexes was evaluated for the given reaction. Zn(koj)2 and Mn(koj)2, which have been reported as potential radioprotective agents,12a moderately promoted the reaction (40% and 52% yields, respectively, entries 1–2). An uncommon iron complex, Fe(koj)3, also showed comparable reactivity (entry 3). Note that the reaction using Al(koj)3 as a catalyst gave a good yield of 69% with the assistance of TBAI (entry 4). In the absence of Al(koj)3, the yield dropped from 69% to 21% (entry 4 vs. 5). To clarify the role of the hydroxy group in Al(koj)3, we also examined the reaction using Hkoj and a slightly higher yield of carbonate was obtained (entry 5 vs. 6). This result may indicate that the hydroxy group in Al(koj)3 plays a role in boosting the catalytic activity through the possible hydrogen bonding interactions with the epoxide.8d No reaction occurred without any catalyst (entry 7).
|
|||
|---|---|---|---|
| Entry | Cata./mol% | TBAI/mol% | Yieldb/% |
| a The reaction was carried out on a 5.0 mmol scale of styrene oxide at 100 °C under 1 MPa CO2 (initial).b NMR yield using 1,1,2,2-tetrachloroethane as the internal standard. | |||
| 1 | Zn(koj)2/0.25 | 0.25 | 40 |
| 2 | Mn(koj)2/0.25 | 0.25 | 52 |
| 3 | Fe(koj)3/0.25 | 0.25 | 50 |
| 4 | Al(koj)3/0.25 | 0.25 | 69 |
| 5 | — | 0.25 | 21 |
| 6 | Hkoj/0.75 | 0.25 | 43 |
| 7 | — | — | 0 |
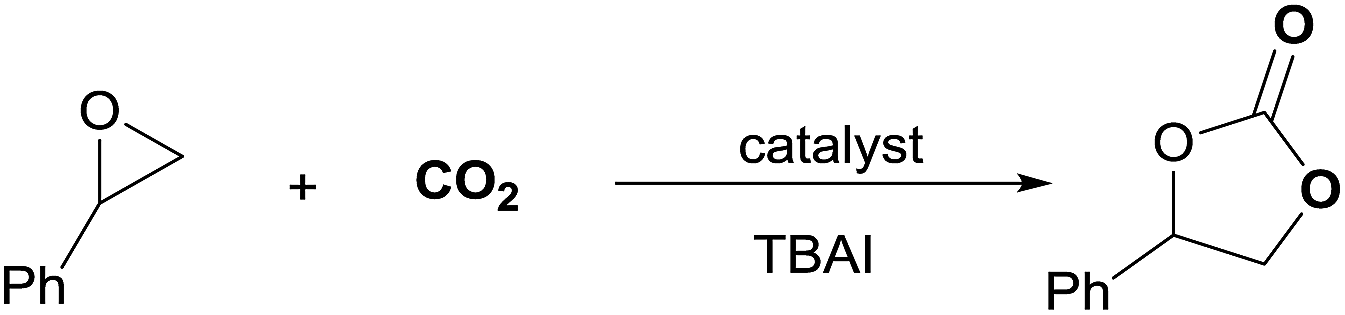
We continued our investigations by optimizing the reaction parameters using Al(koj)3 as the catalyst (Table 2). Remarkably, with increasing temperature from 100 °C to 120 °C, the yield of styrene carbonate increased significantly from 69% to 93% (entry 1 vs. 2). The yield decreased sharply when the reaction was conducted at 80 °C, which thus suggested that 120 °C was the appropriate temperature for the cycloaddition reaction (entry 2 vs. 3). From a thermodynamics point of view, the cycloaddition of epoxides with CO2 is an exothermic reaction and 120 °C is an easy operation temperature for industrial production. Then, the effect of catalyst loading on the reaction was studied. A quantitative yield was obtained even after decreasing the catalyst loading from 0.25 mol% to 0.15 mol% (entry 4). Even at a loading of 0.05 mol%, a yield of 66% was achieved (entries 5–6). Thus, we defined the optimal reaction conditions for the cycloaddition as 0.15 mol% Al(koj)3 at 120 °C with 1 MPa CO2 under solvent-free conditions.
| Entry | Cata./mol% | T/°C | Yieldb/% |
|---|---|---|---|
| a Reaction conditions: styrene oxide (5.0 mmol), TBAI (0.25 mol%), and 1 MPa CO2 (initial).b NMR yield with 1,1,2,2-tetrachloroethane as the internal standard. | |||
| 1 | Al(koj)3/0.25 | 120 | 95 |
| 2 | Al(koj)3/0.25 | 100 | 69 |
| 3 | Al(koj)3/0.25 | 80 | 13 |
| 4 | Al(koj)3/0.15 | 120 | 94 |
| 5 | Al(koj)3/0.1 | 120 | 72 |
| 6 | Al(koj)3/0.05 | 120 | 66 |
In order to evaluate the efficiency and general applicability of this oxo-coordinated Al(koj)3 complex, a series of epoxides (terminal and internal) was examined for the synthesis of carbonates using CO2 in the presence of Al(koj)3 (Table 3). Propylene carbonate is widely used as an electrolyte and alternative green solvent in organic syntheses. An excellent yield of up to 99% was obtained using this developed catalyst system (entry 1). Using TBAB (tetrabutylammonium bromide) instead of TBAI also gave a quantitative yield of propylene carbonate. Furthermore, the reusability of the Al(koj)3/TBAI system was examined for the synthesis of propylene carbonate, and no significant loss in its catalytic activity was observed after three successive runs (entry 1). The results achieved mark this protocol as an attractive method for potential future industrial applications. Other terminal epoxides smoothly converted to the corresponding cyclic carbonates in high yields and with excellent selectivities (entries 2–6). As for disubstituted epoxides, e.g. isobutylene oxide and cyclohexene oxide, these showed inferior activity even under harsh conditions (longer reaction times and increased amount of catalyst) compared with terminal counterparts, due to the increased steric hindrance (entries 7–8).8c
|
||
|---|---|---|
| Entry | Substrate | Yieldb/% |
| a Conditions: epoxide (5 mmol), Al(koj)3 (0.15 mol%), TBAI (0.25 mol%), 1 MPa CO2, 120 °C, and 10 h.b NMR yield with 1,1,2,2-tetrachloroethane as the internal standard. The yield in parentheses is the isolated yield.c TBAB instead of TBAI.d After three cycles using Al(koj)3/TBAI.e Al(koj)3 (0.25 mol%), 2 MPa CO2, and 15 h. | ||
| 1 | 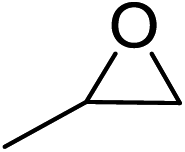 |
99(94), 94(91)c, 96(95)d |
| 2 | 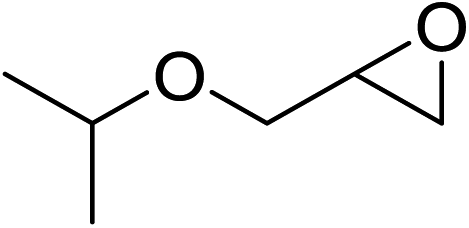 |
95(91) |
| 3 | 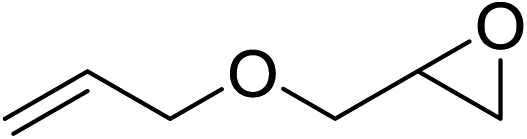 |
91(89) |
| 4 | 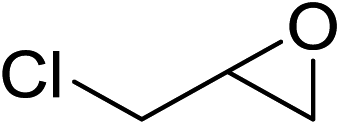 |
90(88) |
| 5 | 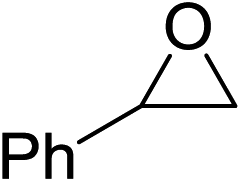 |
94(91) |
| 6 | 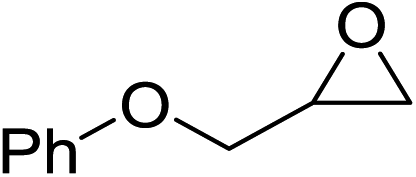 |
95(90) |
| 7e | 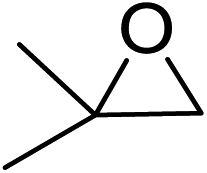 |
42(40) |
| 8e | 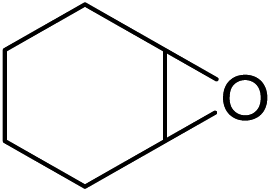 |
37(34) |
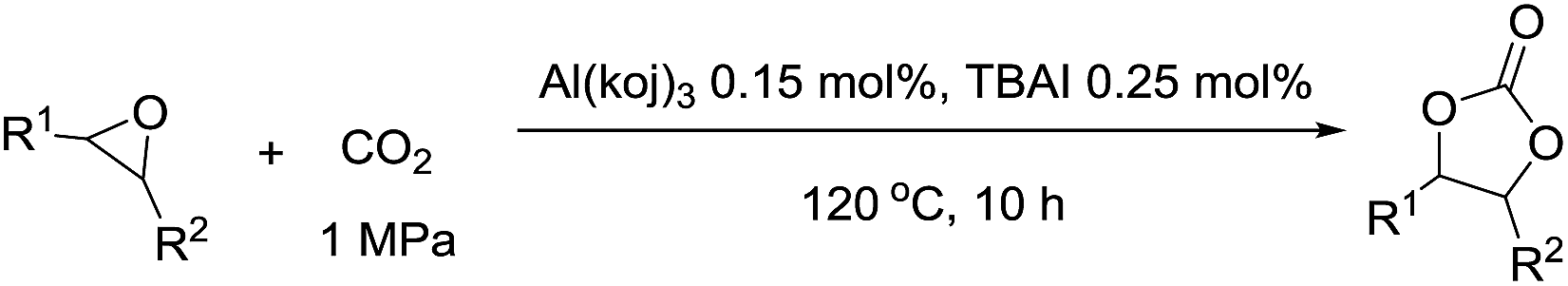
Based on previous studies14 and the above experiments, a plausible mechanism for this Al(koj)3 catalyzed coupling of CO2 with epoxides was proposed and depicted in Scheme 3. Lewis acid metal catalyst activates the epoxide, followed by nucleophilic attack of the iodine anion from TBAI, to form the metal-coordinated iodo alkoxide. Then, the addition of CO2 leads to the formation of the cyclic carbonate and regeneration of the catalyst. Notably, the hydroxy group in Al(koj)3 may also play a role in boosting the catalytic activity through possible hydrogen bonding interactions with the epoxide.
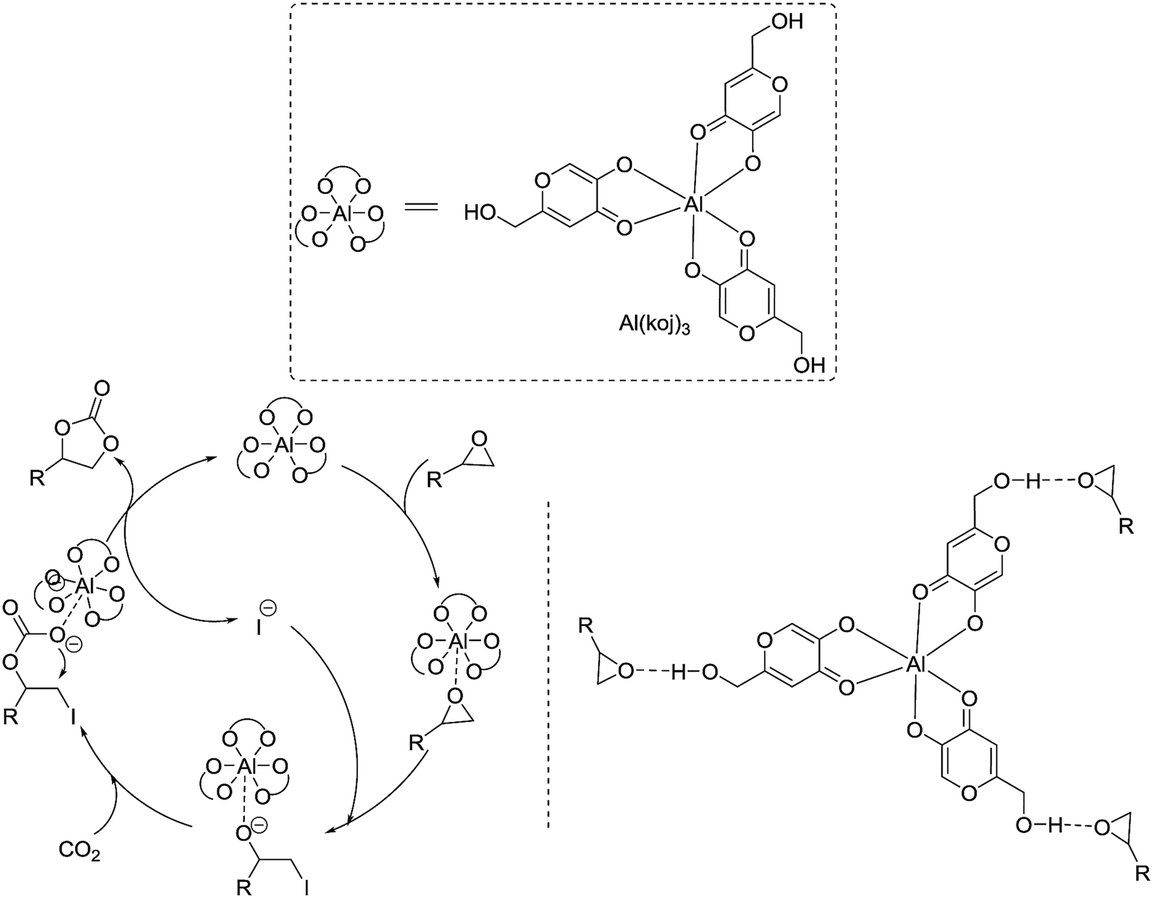 | ||
| Scheme 3 Plausible reaction pathway for the coupling of epoxides with CO2 catalyzed by Al(koj)3/TBAI. | ||
Conclusions
In summary, oxo-coordinated Al(koj)3 was developed as an efficient catalyst for the synthesis of carbonates from epoxides with CO2 under mild conditions without any solvent. A series of terminal epoxides were converted to the corresponding carbonates in yields of up to 99%. Moreover, this developed catalyst system can be reused at least three times with no significant decrease in the propylene carbonate yield. This developed catalyst system shows great potential for industrialization due to the fact that it is easily prepared, inexpensive, has stable catalytic activity and requires reaction conditions that are easily realized.Experimental
General information
TBAI and epoxides were purchased from Alfa Aesar, Aladdin, and J&K Chemical and were used without further treatment. Kojic acid, Zn(OAc)2, Mn(OAc)2·2H2O, Fe(NO3)3·9H2O, and Al(NO3)3 were from Energy Chemical. CO2 with a purity of 99.99% was commercially available. The products were characterized in CDCl3 using a Varian Bruker 400 spectrometer.Synthesis and characterization of the kojic acid metal complexes13
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1517 (CC); 1277, 1219 (C–O). Elemental analysis: [calcd for C12H10O8Zn·1/2H2O], calc.: C, 41.47; H, 2.90. Found: C, 41.2; H, 2.98.O); 1527 (CC); 1307, 1249 (C–O). Elemental analysis: [calcd for C18H15O10Al], calc.: C, 48.01; H, 3.36. Found: C, 48.09; H, 3.30.O); 1528 (CC); 1292, 1246 (C–O). Elemental analysis: [calcd for C12H10O8Mn], calc.: C, 42.75; H, 2.99. Found: C, 42.48; H, 3.05.O); 1513 (CC); 1276, 1249 (C–O). Elemental analysis: [C18H15O10Fe·1/2H2O], calc.: C, 45.12; H, 3.16. Found: C, 45.19; H, 3.12.
O); 1517 (CC); 1277, 1219 (C–O). Elemental analysis: [calcd for C12H10O8Zn·1/2H2O], calc.: C, 41.47; H, 2.90. Found: C, 41.2; H, 2.98.O); 1527 (CC); 1307, 1249 (C–O). Elemental analysis: [calcd for C18H15O10Al], calc.: C, 48.01; H, 3.36. Found: C, 48.09; H, 3.30.O); 1528 (CC); 1292, 1246 (C–O). Elemental analysis: [calcd for C12H10O8Mn], calc.: C, 42.75; H, 2.99. Found: C, 42.48; H, 3.05.O); 1513 (CC); 1276, 1249 (C–O). Elemental analysis: [C18H15O10Fe·1/2H2O], calc.: C, 45.12; H, 3.16. Found: C, 45.19; H, 3.12.General procedure for the cycloaddition reaction of epoxides with CO2
A mixture of the epoxide (5 mmol), metal catalyst, and TBAI was placed into a glass tube, and then the tube was placed into a 25 ml stainless steel autoclave. The reactor was charged with CO2 to the desired pressure. Then, the autoclave was placed into an oil-bath and heated. After the reaction, the reactor was cooled in an ice-water bath, and the CO2 was carefully vented. Diethyl ether was added to extract the carbonate products and unreacted epoxide. The yield of cyclic carbonate was determined using NMR analysis with 1,1,2,2-tetrachloroethane as the internal standard. The products were purified using column chromatography with ethyl acetate–petroleum ether as the eluent. The precipitated catalyst was washed with diethyl ether five times and dried using a pressure blowing concentrator ready to be used directly for the next reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Shaanxi University of Science and Technology for the financial support of this research (2016GBJ-20).References
- Mauna Loa Observatory (Hawaii), Scripps Institution of Oceanography at UC San Dieg, https://scripps.ucsd.edu/programs/keelingcurve/, Accessed 23 Dec 2017.
- M. Y. He, Y. H. Sun and B. X. Han, Angew. Chem., Int. Ed., 2013, 52, 2 Search PubMed.
- (a) K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524 RSC; (b) C. Chauvier and T. Cantat, ACS Catal., 2017, 7, 2107 CrossRef CAS.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (b) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS; (c) Y. N. Li, R. Ma, L. N. He and Z. F. Diao, Catal. Sci. Technol., 2014, 4, 1498 RSC; (d) B. Yu and L. N. He, ChemSusChem, 2015, 8, 52 CrossRef CAS PubMed; (e) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (f) Y. Y. Gui, W. J. Zhou, J. H. Ye and D. G. Yu, ChemSusChem, 2017, 10, 1337 CrossRef CAS PubMed.
- (a) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (b) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, 2010 Search PubMed.
- (a) M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651 RSC; (b) H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- (a) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554 CrossRef PubMed; (b) B. Yu, Z. F. Diao, C. X. Guo, C. L. Zhong, L. N. He, Y. N. Zhao, Q. W. Song, A. H. Liu and J. Q. Wang, Green Chem., 2013, 15, 2401 RSC.
- (a) J. Steinbauer, A. Spannenberg and T. Werner, Green Chem., 2017, 19, 3769 RSC; (b) S. Yue, P. P. Wang, X. J. Hao and S. L. Zang, J. CO2 Util., 2017, 21, 238 CrossRef CAS; (c) Q. N. Zhao, Q. W. Song, P. Liu, Q. X. Zhang, J. H. Gao and K. Zhang, Chin. J. Chem., 2018, 36, 187 CrossRef CAS; (d) S. Kaneko and S. Shirakawa, ACS Sustainable Chem. Eng., 2017, 5, 2836 CrossRef CAS; (e) C. Maeda, J. Shimonishi, R. Miyazaki, J. Hasegawa and T. Ema, Chem.–Eur. J., 2016, 22, 6556 CrossRef CAS PubMed; (f) S. He, F. Wang, W. L. Tong, S. M. Yiu and M. C. W. Chan, Chem. Commun., 2016, 52, 1017 RSC; (g) H. Zhou, G. X. Wang, W. Z. Zhang and X. B. Lu, ACS Catal., 2015, 5, 6773 CrossRef CAS; (h) M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, ACS Catal., 2015, 5, 3398 CrossRef CAS; (i) S. Sopena, G. Fiorani, C. Martin and A. W. Kleij, ChemSusChem, 2015, 8, 3248 CrossRef CAS PubMed; (j) R. Ma, L. N. He, X. F. Liu, X. Liu and M. Y. Wang, J. CO2 Util., 2017, 19, 28 CrossRef CAS.
- (a) C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134 CrossRef CAS PubMed; (b) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adan, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228 CrossRef CAS PubMed.
- (a) Y. Yang, Y. Hayashi, Y. Fujii, T. Nagano, Y. Kita, T. Ohshima, J. Okuda and K. Mashima, Catal. Sci. Technol., 2012, 2, 509 RSC; (b) M. V. Escárcega-Bobadilla, M. M. Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem.–Eur. J., 2013, 19, 2641 CrossRef PubMed.
- R. Ma, L. N. He and Y. B. Zhou, Green Chem., 2016, 18, 226 RSC.
- (a) S. Emami, S. J. Hosseinimehr, S. M. Taghdisia and S. Akhlaghpoorb, Bioorg. Med. Chem. Lett., 2007, 17, 45 CrossRef CAS PubMed; (b) H. S. Rho, S. M. Ahn, D. S. Yoo, M. K. Kim, D. H. Cho and J. Y. Cho, Bioorg. Med. Chem. Lett., 2010, 20, 6569 CrossRef CAS PubMed; (c) V. M. Nurchi, G. Crisponi, J. I. Lachowicz, S. Murgia, T. Pivetta, M. Remelli, A. Rescigno, J. Niclós-Gutíerrez, J. M. González-Pérez, A. Domínguez-Martín, A. Castiñeiras and Z. Szewczuk, J. Inorg. Biochem., 2010, 104, 560 CrossRef CAS PubMed.
- (a) M. C. Barret, M. F. Mahon, K. C. Molloy, J. W. Steed and P. Wright, Inorg. Chem., 2001, 40, 4384 CrossRef CAS PubMed; (b) A. F. Sowinski and K. P. Dockery, US Pat., U.S.20120105553, 2012; (c) O. Vajragupta, P. Boonchoong, Y. Sumanont, H. Watanabe, Y. Wongkrajang and N. Kammasu, Bioorg. Med. Chem., 2003, 11, 2329 CrossRef CAS PubMed.
- (a) J. Martínez, J. A. Castro-Osma, A. Earlam, C. Alonso-Moreno, A. Otero, A. Lara-Sánchez, M. North and A. Rodríguez-Diéguez, Chem.–Eur. J., 2015, 21, 9850 CrossRef PubMed; (b) J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323 CrossRef; (c) C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gómez, E. Martin, E. C. Escudero-Adán, C. Bo and A. W. Kleij, Chem.–Eur. J., 2014, 20, 2264 CrossRef CAS PubMed; (d) M. E. Wilhelm, M. H. Anthofer, M. Cokoja, I. I. E. Markovits, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2014, 7, 1357 CrossRef CAS PubMed; (e) S. Gennen, M. Alves, R. Méreau, T. Tassaing, B. Gilbert, C. Detrembleur, C. Jerome and B. Grignard, ChemSusChem, 2015, 8, 1845 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00835c |
| This journal is © The Royal Society of Chemistry 2018 |