
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated fabrication of antibacterial and osteoinductive electrospun fibrous scaffolds via electrochemical deposition†
Yingbo Wang‡
*a,
Ya Gao‡a,
Guoqiang Xu‡b,
Han Liuc,
Yi Xiangc and
Wenguo Cui *c
*c
aCollege of Chemical Engineering, Xinjiang Normal University, 102 Xinyi Road, Urumqi 830054, China. E-mail: ybwang20002575@163.com; Fax: (+86) 09914333279; Tel: (+86) 09914333279
bDepartment of Prosthodontics, The First Affiliated Hospital of Xinjiang Medical University, 393 Xinyi Road, Urumqi 830054, China
cShanghai Institute of Traumatology and Orthopaedics, Shanghai Key Laboratory for Prevention and Treatment of Bone and Joint Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, 197 Ruijin 2nd Road, Shanghai 200025, P. R. China. E-mail: wgcui80@hotmail.com; Fax: +86-21-64370045; Tel: +86-21-64370045 ext. 663332
First published on 6th March 2018
Abstract
Electrospun fibrous scaffolds have attracted much research interest due to their many applications in orthopedics and other relevant fields. However, poor surface bioactivity of the polymer scaffold body significantly limits the implementation of many potential applications, and an effective solution remains a great challenge for researchers. Herein, a highly efficient method, namely pulsed electrochemical deposition (ED) with co-electrospinning nano-Ag dopant, to fabricate poly(L-lactic acid) (PLLA)/nano-Ag composite fibers is presented. The resulting product demonstrated excellent antibacterial properties, as well as strong capabilities in facilitating the precipitation of calcium phosphate crystals at fiber surfaces and in promoting osteogenic differentiation. In the process of ED, the conductivity of the fibers was observed to increase due to the nano-Ag dopant. Upon applying pulse signals when charging, water electrolysis occurred in micro-reactive regions of anodic fibers, forming OH−, an alkaline environment that allowed the supersaturation of calcium phosphate. When discharging, the calcium phosphate in the solution diffused rapidly and reduced the concentration polarization, reforming a homogeneous electrolyte. The realization of efficient bioactive coatings at fiber surfaces was achieved in a highly efficient manner by repeating the above charging and discharging processes. Therefore, ED can be adopted to simplify and accelerate the fabrication process of an osteogenetic and antibacterial electrospun fibrous scaffold.
1. Introduction
Electrospun fibers resemble the natural extracellular matrix (ECM); their structural similarities allow the engineering of an ideal bionic environment for cell adhesion and reproduction, thus promoting the growth of new tissues. At present, electrospun fibrous scaffolds have many applications in orthopedics and other relevant fields.1 However, because the main body of electrospun fibers is predominantly polymer based, orthopedic applications are limited by poor surface bioactivity, which remains a great challenge for researchers. On the other hand, hydroxyapatite (HA), the main inorganic component of most naturally occurring calcified tissues in humans, is currently considered an optimal material for osteoinduction due to its high bioactivity.2 He et al.3 showed that an HA coating on the surface of poly(L-lactic acid) (PLLA) fibers promoted the deposition of calcium and phosphorus ions in a physiological environment; thus, it facilitated the adhesion and reproduction of osteocytes, as well as osteoblastic differentiation. As a result, HA was able to accelerate the integration between the implant and its surrounding tissues, forming a uniform identity.3 Therefore, the fabrication of electrospun fibrous scaffolds with HA coatings have the potential to significantly increase the bioactivity and biocompatibility of implant materials.At present, the most commonly used method for coating HA on electrospun fibers is liquid immersion.4 Hu et al. used an electrospinning method to make HA micro-nanoparticle composite PLLA fibrous scaffolds.5 The HA composite material was able to provide nucleation sites for calcium phosphate in an in vitro immersion experiment; thus, it increased the bioactivity of the fibrous scaffold, forming an enriched layer of Ca–P at the fiber surfaces. However, in the process of integrating HA micro-nanoparticles into the fibrous scaffold, HA was often observed to agglomerate due to the separation of organic and inorganic phases. As a result, the mechanical properties of the material were significantly compromised. In vivo, the addition of regulating agents, such as cell growth factors, amino acids etc., can increase the deposition and growth efficiencies of calcium phosphate.6 Guo et al. bio-mineralized PLLA/gelatin nanofibers in an amino-acid-rich artificial body fluid and found that the presence of amino acids increased the preferential growth of HA crystals along the c axis, promoting the formation of HA acicular crystals.7 However, in vitro bio-mineralization in simulated body fluid (SBF) needs at least several weeks for the desirable outcomes, and long immersion times can lead to drug release and material degradation.8–10 Therefore, an efficient deposition method for calcified HA bioactive coatings on fibrous scaffold surfaces remains a challenge and an urgent issue that needs to be solved.
Pulsed electrodeposition (ED) is an important technique used to control the rate of a reaction. When the pulsed signals are applied, the electrolyte inside the electro-bath alternates the charging and discharging processes. The intermittent nature of the reaction at the electrode favors the diffusion process and it mitigates the risk of concentration polarization. In this way, the efficiency of deposition can be greatly improved.11 Hence, the pairing of pulsed ED and electrospinning techniques has the potential to construct fibrous scaffolds on a calcium phosphate composite surface at a relatively fast speed. Due to the electric field inside the pulsed ED solution, fibrous membranes are able to maintain a state of supersaturation of calcium and phosphate ions at micro-reactional regions of the cathode. The ions and electrospun fibers have high specific surface areas; the resulting molecular interactions give rise to the fast nucleation and growth processes of calcium phosphate crystals.12 In addition, because ED takes place in a solution, it is possible to achieve a porous fibrous scaffold or matrix with a uniformly distributed calcium phosphate coating; it is also possible to precisely control the morphology and size of the calcium phosphate crystals by adjusting the electrochemical parameters. Therefore, ED is a promising method for increasing the degree of mineralization while at the same time decreasing the mineralization time at surfaces of the material; it thereby accelerates the HA coating process at fibrous surfaces. However, an electrospun scaffold made of degradable PLLA has poor conductivity; consequently, it is difficult for rapid calcium phosphate deposition to take place directly in the electrolyte during ED.
To further improve the ED efficiency at composite surfaces, metals with high conductivity, such as Cu, Ni and nano-Ag are used as co-electrospinning dopants.13–15 Surfaces of Cu and Ni nanoparticles are prone to oxidation as their elemental forms are unstable.16 In comparison, nano-Ag has high specific conductance and is chemically stable; hence it has received much research interest as an antibacterial agent in medically-related fields.17 Nano-Ag in PLLA fibers will enhance the conductive properties of the composite scaffold, thereby accelerating the mineralization process of calcium phosphate coatings and significantly increasing the efficiency of scaffold fabrication; the composite scaffold product will also inherit the antibacterial and bioactivity properties from nano-Ag.
The structure of our PLLA/nano-Ag composite fibrous scaffold and the ED mineralization process taking place at the surfaces are illustrated in Scheme 1. This study reports a composite fibrous scaffold fabricated by electrospinning which has the majority of highly conductive nano-Ag distributed uniformly at the fiber surfaces. In the process of ED, fiber conductivity was observed to increase due to the nano-Ag dopant; upon applying the pulse signals when charging, water electrolysis occurred at micro-reactive regions of anodic fibers, forming OH−, an alkaline environment that supported the supersaturation of calcium phosphate. When discharging, calcium phosphate in the solution diffused rapidly and reduced the concentration polarization, reforming a homogeneous electrolyte. The realization of efficient bioactive coatings at fiber surfaces was quickly achieved by repeating the charging and discharging processes. The paper systematically investigated the morphologies of the calcium phosphate particles deposited at fiber surfaces at various electrolyte concentrations, 42 mmol L−1, 16.7 mmol L−1 and 5 mmol L−1. Using SEM, it was observed that calcium phosphate deposition at fiber surfaces appeared to be well-dispersed and the particles appeared to be finer when the ED electrolyte concentration was 16.7 mmol L−1 (Fig. S1†).
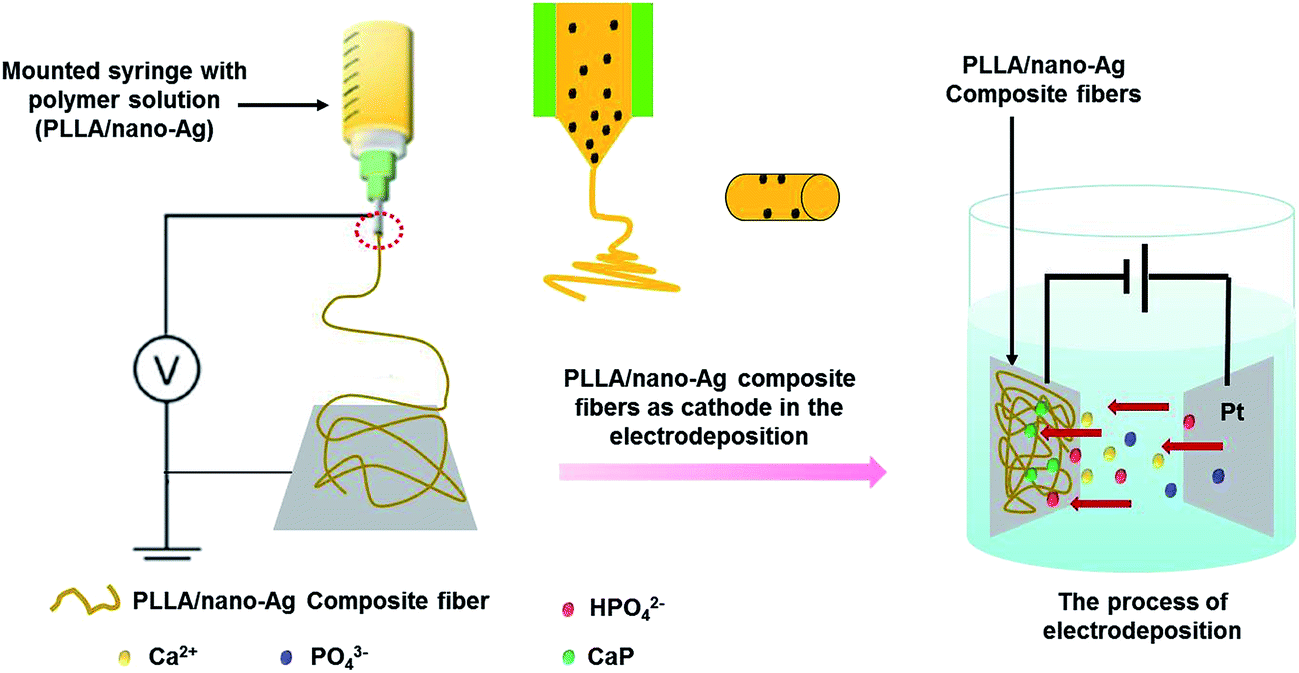 | ||
| Scheme 1 A schematic diagram of the experimental setup for fabricating mineralized nanofibers by combining electrospinning and electrodeposition methods. | ||
2. Materials and methods
2.1. Materials
Chemicals were obtained from the following suppliers: poly(L-lactic acid) (PLLA) from Jinan Daigang Biomaterial Co., Ltd; nano-Ag (diameter < 100 nm) from Sigma-Aldrich; dichloromethane (DCM) from Tianjin Yongsheng Fine Chemicals Co., Ltd; N,N-dimethylformamide (DMF) from Tianjin no. 3 Chemical Reagent Factory; calcium nitrate tetrahydrate (Ca(NO3)2·4H2O) from Tianjin Zhiyuan Chemical Reagent Co., Ltd; and ammonium dihydrogen phosphate (NH4H2PO4) from Tianjin Shengao Chemical Reagent Co., Ltd.2.2. Instruments
A thin layer of gold was sprayed on top of each fiber composite sample to increase its surface conductivity, and scanning electron microscopy (SEM; SU8010, Japan) was used to capture its morphology. Software Image was used for dimensional analyses such as the average size and distribution of fibers. Transmission electron microscopy (TEM; JEM-2100F, Japan) was used to capture the morphology of nano-Ag alone and in PLLA. Thermogravimetric (TG) analysis (TGA-DSA 2960 TA instruments) was conducted to determine the nano-Ag content in the fiber composite samples. A heating rate of 10 °C min−1 from 10 to 800 °C was used to measure the heat properties of the fiber samples in a nitrogen environment.2.3. Electrospinning solution
Nano-Ag solutions (3 wt%, 5 wt% and 7 wt%) were prepared in DMF; the mixtures were stirred thoroughly, and sonicated. PLLA solutions (10 wt%, 12 wt%, 14 wt% and 16 wt%) were prepared in DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF solution (2:1, wt%) and stirred thoroughly. Nano-Ag and PLLA mixtures were blended together and the resulting solutions were used for electrospinning.
:DMF solution (2:1, wt%) and stirred thoroughly. Nano-Ag and PLLA mixtures were blended together and the resulting solutions were used for electrospinning.
2.4. Experimental procedures
A 20 mL plastic syringe with a no. 6 medical stainless-steel needle head was filled with electrospinning solution. The electrospun fibers were collected 15 cm from the needle tip, with the sample release rate set at a constant 3 mL h−1 and an electric field of 18 kV cm−1 from a DC source. At the end of the process, electrospun fibers were collected on an electrode collector or a tin foil (20 cm × 13 cm × 0.2 cm) wrapped around the spinning core. During electrolytic deposition, the nanofibers were collected on a 200–300 μm-thick polar plate. When simulating the artificial body fluid environment, the nanofibers collected on the tin foil were modified to have the same thickness as that obtained from ED.2.5. Electrodeposition
Under constant voltage, the platinum (20 mm × 20 mm × 0.2 mm) and stainless-steel fiber plates were set to opposite polarities, with the platinum plate functioning as the counter electrode and the stainless-steel fiber plates functioning as the working electrodes. The two electrodes were set 2.5 cm apart and the electrochemical beaker (250 mL) was bathed to maintain a specific temperature. The electrolyte was a mixture of 16.7 mmol L−1 Ca(NO3)2·4H2O and 10 mmol L−1 NH4H2PO4. Based on results from ED, the mineralized PLLA was retrieved from the stainless-steel electrode, dried naturally and characterized.2.6. Humoral mineralization stimulation
The fiber composite from supersaturated calcium phosphate solution (SCPS) was mineralized in vitro. The sample product was tailored into a 20 mm × 20 mm square and left immersed in 40 mL SCPS at 37 °C, with the solution changed every 24 h. The ionic concentration of the solution was set to match that in blood (Table S1†) and Tris–HCl was used as a buffer with the pH set to 6.2–6.3 at room temperature. When the sample was retrieved, it was washed with deionized (DI) water and kept at 37 °C in a vacuum drying oven.2.7. Ion release tests
The composite coating was soaked in a 40 mL phosphate solution (PBS, pH = 7.4) at 37 °C for 10 days and the release profiles of Ca2+ and Ag+ were recorded. Atomic absorption spectrophotometry was used to determine the absorbance values of Ca2+ and Ag+; their respective concentrations were determined from standard curves, and values were used to investigate the physiological instability of the composite coating layer (Table S2†).2.8. Antibacterial capability
Escherichia coli and Staphylococcus aureus were selected for both qualitative and quantitative analyses, and experiments were performed with three groups: PLLA/nano-Ag, ED, and SCPS, each with three parallel samples. The spread plate method was used for qualitative analysis and the film adhering method was used for quantitative analysis.2.9. Qualitative analysis
Escherichia coli and Staphylococcus aureus were cultured on LB medium at 37 °C for 12 h, and then a moderate amount of each was picked by inoculation loop and cultured in a fluid nutrient medium for 12 h to obtain a bacterial suspension solution. Next, 10 mL 1 × 108 cell per mL suspension solutions were made from each of Escherichia coli and Staphylococcus aureus using PBS. The samples were left at 37 °C in a 200 rpm swing bed for 12 h. In the end, 100 μL of each was obtained for the corresponding spread plate, which was kept in an incubator at 37 °C for 12 h; colonies were recorded and photographed.2.10. Quantitative analysis
The antibacterial rate was investigated with the film adhering method. Both bacterial strains were cultured in LB medium at 37 °C for 12 h and the process was repeated three times to obtain purer colonies. A colony was inoculated to a fluid nutrient medium kept at 37 °C, in a 200 rpm swing bed for 12 h. Solutions (3.0 × 107 cell per mL) were made using PBS, and diluted 103-fold. Each sample to be measured was placed on a slide and sterile water was added to the bottom of the Petri dish to prevent evaporation. A 50 μL droplet was added to the sample surface and cultured at 37 °C for 24 h; then the bacterial solution was blended with the PBS solution, shaken well, smeared onto the spread plate, kept for 24 h and counted for the number of colonies. The antibacterial capabilities of the samples were calculated using the formula shown below. PLLA/nano-Ag was used as the control group; experiments for three parallel samples were conducted in each case and an average bacterial ratio was calculated.where CCCG is the colony count of the control group and CCEG is the colony count of the experimental group.
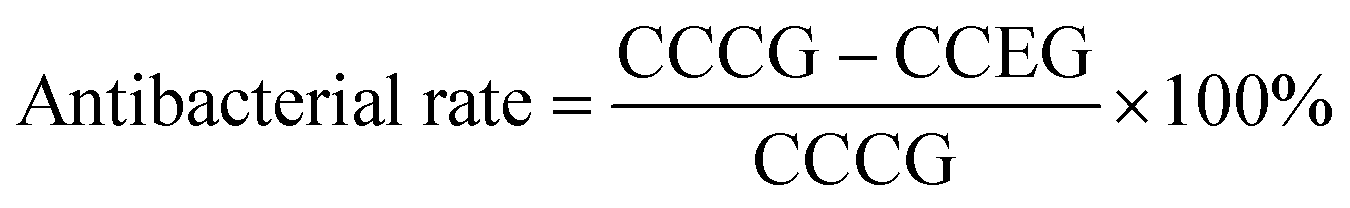
2.11. Osteoblast culture
MC3T3-E1 cells (osteoblastic cell line, extraction of primary osteoblasts from the skull of a 2–3 day old Sprague Dawley (SD) rat) were selected to investigate the biocompatibility of the electrospun fibers. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the First Affiliated Hospital of Xinjiang Medical University, and approved by the Animal Ethics Committee of the First Affiliated Hospital of Xinjiang Medical University.There were three experimental groups (PLLA/nano-Ag, ED and SCPS) and for each of them there were seven parallel sample groups. The samples were placed into six-well culture dishes and MC3T3-E1 cells were inoculated onto the sample surfaces with an inoculum density of 7.5 × 103·mL−1. L-DMEME nutrient medium (3.0 mL) was added to each well and samples were kept in a CO2 incubator at 37 °C and treated as follows: (1) after 1, 3, 5, 7 days, samples from each of the three groups were washed three times using PBS and sealed twice by serum, each for 30 min; then 2.5% glutaraldehyde solution was used for immobilization, and gradient ethanol solution was used for dealcoholization. After the dried samples were plated with thin layers of gold for conductivity, SEM was used to capture the morphologies of the cells. (2) After 1, 3, 5, 7 days, cell proliferation and viability were determined by CCK-8 assay. Cell suspensions were inoculated onto a 96-well culture dish with 100 μL in each well and three parallel samples for each case. Upon adding 10 μL of CCK-8, the samples were kept in a CO2 incubator at 37 °C for 3 h and photometric absorption values at 450 nm were measured.2.12. Statistical analysis
All data are presented as means ± standard deviation. Statistical analysis was carried out using a One Sample t test (assuming unequal variance). The difference between two sets of data was considered statistically significant when P < 0.05.3. Results and discussion
As shown in Fig. 1, PLLA fibers had a smooth surface (Fig. 1a and b) while PLLA/nano-Ag composite fibers were evenly distributed and their surfaces were covered by nano-Ag particles (Fig. 1d and e). The diameters of the nano-Ag particles were smaller than 100 nm and the fibers completely encapsulated the particles (Fig. 1c). The PLLA and PLLA/nano-Ag water contact angles were found to be 132.2° and 139.1° respectively through contact angle measurements (Fig. 1a and d). Because nano-Ag dopant increased the roughness of the fiber surfaces, PLLA/nano-Ag fibers displayed more hydrophobicity than PLLA fibers.18 The aforementioned structures also allowed the composite to slowly release the nano-Ag dopant, achieving a long-term antibacterial effect. An experiment was conducted to investigate the quantity and consistency of Ag+ release; it found that the Ag+ level stayed constant at 1.0 ppm in the composite fibers for the first 10 days (Fig. 1f), indicating a desirable trait of steady release and showing that nano-Ag can be used as a continuous antibacterial dopant in PLLA. | ||
| Fig. 1 Electrospun PLLA/nano-Ag composite fiber characterization. (a) SEM and water contact angle of the PLLA fibers; (b) TEM of the PLLA fibers; (c) SEM of the nano-Ag; (d) SEM and water contact angle of the PLLA/nano-Ag; (e) TEM of the PLLA/nano-Ag; (f) release profile of Ag+ from PLLA/nano-Ag. | ||
To optimize the physical properties of the composite fibrous scaffold, the processing parameters of the electrospun PLLA/nano-Ag composite fibrous scaffold were systematically explored. Shown in Fig. S2† are the diameter profiles and fiber morphologies of PLLA fabricated at different concentrations. When the concentration was relatively low (10%, 12%), the average diameters of the fibers were 300 nm and 370 nm; the fibers were unevenly distributed and there were observable beads. The presence of beads could be attributed to the non-uniformity of the fibers as a result of low surface tension due to low PLLA concentration. As the concentration increased (14%, 16%), the average diameters were observed to increase to 930 nm and 1040 nm. The fibers were evenly distributed because the solution viscosity increased the surface tension, which became too great for the electric field forces to overcome; thus, the average diameters increased accordingly.19 Therefore, concentrations of PLLA of between 14% and 16% should be selected for superior performances.
Fig. S3† shows the morphology, diameter distribution and TG graph of fibers fabricated at various nano-Ag concentrations. When the concentration of nano-Ag was lower than 3%, no adhesion was observed among fibers and the overall fiber thickness was even; the average diameter of the fibers was 724 nm. When the concentration of nano-Ag was 5%, the composite fibers were uniform and the average diameter was 893 nm (Fig. 1d). When the concentration of nano-Ag was 7%, a small proportion of fibers started to agglomerate and the average diameter was 768 nm. The aggregation could be attributed to the fact that when the concentration was high, nano-Ag introduced discontinuities into the fiber chains by occupying spaces among nanoparticles, thus lowering the tenacity of the fibers.20 As the concentration of nano-Ag increased, the amount of residual nano-Ag increased as well. For concentrations of 3%, 5% and 7% discussed above, the corresponding residuals were 2.03%, 4.64% and 4.89%. Therefore, composite fibers should be optimal when the concentration of nano-Ag is from 3% to 5%.
Bioactivity is a main criterion for the effectiveness of integration at the interface of bone repairing materials and human bones. Mineralization can enhance the bioactivity of a material, promote osteoblast activity and facilitate osteogenesis.21 Fig. S4† shows the mineralization conditions at PLLA fiber surfaces. Fig. S4a† illustrates that, in the ED mineralization process, when the average fiber diameter was 930 nm, the quantity of calcium phosphate calcification was proportional to deposition time. Because pores existed among the PLLA fibers that covered the stainless-steel surfaces, the calcium ions in the solution migrated towards the cathode under the effect of an electric field. When the average fiber diameter increased to 1040 nm, the quantity of calcium phosphate calcification was initially proportional but then inversely proportional to deposition time. One plausible explanation could be that the thicker fibers had decreased pore sizes, which in turn reduced field stress. In the process of deposition (35 min), as the fiber diameter increased, the increasing amount of calcified calcium phosphate offered a greater surface area. After 35 min, potentially due to the lengthy electrolysis time, the calcium phosphate coating thickened, which in turn lowered the fiber porosity, As a consequence, the hydrogen produced from the electrolysis reaction could not vent properly, causing the shedding of some calcium phosphate salt. The most commonly used way to determine the bioactivity of a material is by in vivo immersion in an SBF environment; the product of this method, apatite, integrates easily with bioactive bones.22 Our research compares ED to in vivo immersion. Fig. S4b† presents pictures of calcified SCPS and shows that quantities of calcium phosphate calcification were inversely proportional to fiber diameters. Since the nucleation sites for calcium and phosphate ions were the same, fibers with smaller diameters offered greater specific surface areas which resulted in greater quantities of calcium phosphate calcification.3 To conclude, calcium phosphate crystals grow faster and calcify more easily when the calcium phosphate is in a hyper-saturated state sustained by electric fields in ED.
The quantity of calcium phosphate calcification is a function of the nano-Ag content in composite fibers. Fig. S5a† illustrates the mineralization by ED: when the nano-Ag content was low (≤4.64%), the quantity of calcium phosphate mineralization at fiber surfaces was proportional to the deposition time. As the nano-Ag content increased, the tendency for calcium phosphate to calcify increased as well. This was the case because nano-Ag increased the conductivity of the material, which was beneficial for the deposition of calcium phosphate.23 When the nano-Ag content increased to 4.89%, the calcification quantity initially increased but then dropped. This was because the thickening of the mineralization layer resulted in a decrease in the degree of porosity, which caused the conductivity to drop, making hydrogen gases produced in the electrolysis hard to vent.24 In the meantime, fibers with greater diameters (DAg 2.03% < DAg 4.89% < DAg 4.64%) provided greater surface areas, which promoted the deposition of calcium phosphate. Fig. S5b† illustrates the mineralization of SCPS and shows that there is no correlation between the quantity of calcium phosphate calcification at fiber surfaces and the nano-Ag content added. As previously discussed, fiber diameters were closely related to the quantity of calcium phosphate mineralization at fiber surfaces. The smaller the diameter, the greater the specific surface area, which resulted in a greater quantity of calcium phosphate deposition at fiber surfaces. Both mineralization methods, ED and SCPS, can therefore improve the bioactivity but the mineralization time needed differs significantly. The ED mineralization method took 1 hour or less. Furthermore, the morphology, size and crystallinity could be modified by changing the processing parameters such as the temperature of the electrolyte, the electrical potential and deposition time.25 In comparison, SCPS mineralization took longer to finish and it is worth noting that prolonged immersion could trigger drug release and lead to material degradation.26
Fig. S6† displays a collection of fiber topographies resulting from both ED and SCPS calcification methods. Fig. S6a and b† show topographies of calcium phosphate depositions at PLLA fiber surfaces before and after the addition of nano-Ag using ED. Before adding nano-Ag, the calcium phosphate coating at the PLLA fiber surface appeared to be acicular with low crystallinity. After adding nano-Ag, the calcium phosphate coating at the PLLA fiber surface appeared to be flake-like and well-distributed with high crystallinity. The changes in topography could be explained by the fact that the nano-Ag distributed at the fiber surface increased the electric current density at the fiber surface.27 Consequently, calcium phosphate was able to deposit at fiber surfaces at electrochemically higher reaction rates. Fig. S6c and d† show topographies of calcium phosphate depositions at PLLA fiber surfaces before and after the addition of nano-Ag using SCPS. After the addition of nano-Ag, which increased the bioactivity of the PPLA fibers, calcium phosphate deposition sites at the PLLA fiber surfaces appeared to be flake-like and globular; this could be the result of increasing nucleation sites at the PLLA surfaces due to the nano-Ag. Before the addition of nano-Ag, the calcium phosphate deposition at the PLLA fiber surface was negligible. Thus, it is concluded that nano-Ag can promote in vitro bio-mineralization of PLLA fiber surfaces.
To analyze the influence of ED and SCPS bio-mineralization methods on the growth and evolution process of calcium phosphate at PLLA/nano-Ag composite fiber surfaces, the conditions of calcium phosphate at various deposition times were recorded. Fig. 2 provides observations of the morphologies at 15, 30 and 60 minutes during the ED process. The calcium phosphate depositions at the composite fiber surfaces appeared to be nanoparticles at 15 min (Fig. 2a). The nano-structures are closely related to deposition time; Eliaz et al. reported 3D growth of calcium monophosphide on titanium after instantaneous nucleation, 2D continuous nucleation and ED (approximately 12 min).28 Zhang et al. reported on a coating that initially formed an octacalcium phosphate layer, which also functioned as the precursor for HA.29 The high pH value near the cathode was the driving force for transformation. As shown in Fig. 2b and a uniform layer of flaky calcium phosphate first formed at the fiber surfaces. Then the calcium phosphate at the fiber surfaces showed an inhomogeneous tendency at 60 min (Fig. 2c) because lengthy electrolysis times produce an excessively thick coating, blocking hydrogen gases from the electrolysis reaction from venting promptly.24 According to the bio-mineralization mechanism outlined in Fig. 2d, pores existed among fibers and positive ions migrated towards the cathode under the electric field; water electrolysis took place at the cathode, producing hydrogen gases and hydroxyl groups; and the calcium phosphate near the composite fibers reached a hyper-saturated state in the alkaline environment at the cathode. In addition, micro-reactions of fibers took place in the alkaline environment; some PLLA reacted with water during ED and formed activated carboxyl groups. Activated carboxyl groups were susceptible to nucleation reactions with calcium ions and calcium phosphate, realizing the rapid growth of the calcium phosphate crystals.30
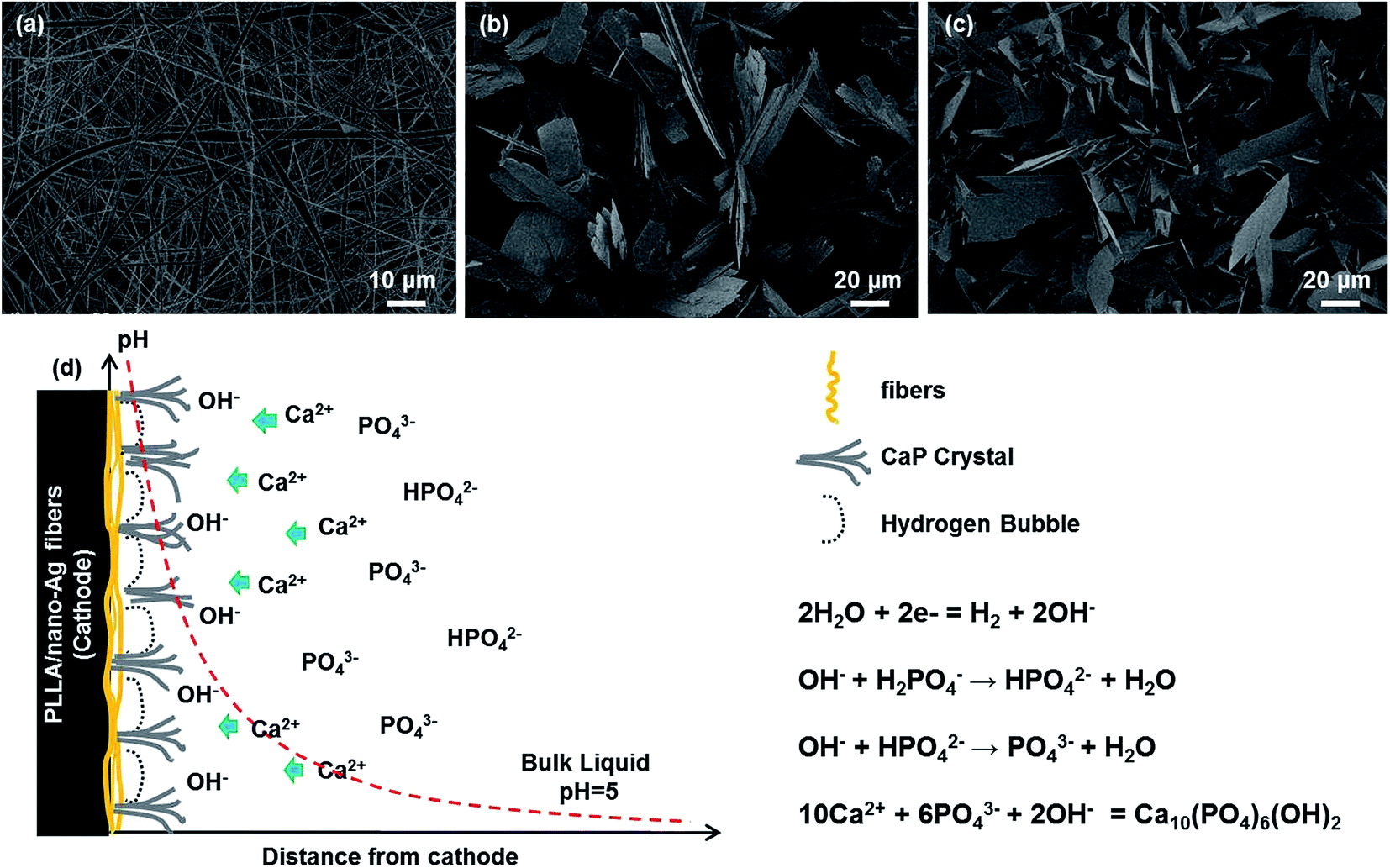 | ||
| Fig. 2 SEM of composite fiber calcification during the ED process: (a) 15 min; (b) 30 min; (c) 60 min. (d) Deposition mechanism. | ||
Fig. S7† shows the bio-mineralized calcium phosphate growth process for fibers immersed in SCPS. No marked change was observed at the fiber surfaces on day 3, but nanoparticles started to be scattered on the fiber surfaces on day 7 and the structure of the nanoparticles was believed to be closely related to the deposition time.31 On day 12, flakes of bio-mineralized sediments began to form at the fiber surfaces.32 Based on the observations above, the deposition mechanism can be divided into three stages: (1) during the dissolution phase, the fiber surfaces interact with SCPS; some fibers are hydrolyzed, producing active carboxyl functional groups. (2) During the deposition phase, negative ions at the fiber surfaces attract and form an enriched layer of calcium ions from SCPS. (3) During the salt formation phase, the enriched layer of calcium ions attracts phosphate groups, resulting in the calcium phosphate coating.33 Studies have shown that the bio-mineralization period of SCPS is longer than that of ED, for an equal amount of bio-mineralization. Further, the morphology of calcium phosphate can be changed to accelerate the bio-mineralization process of the calcium phosphate coating by simply adjusting the reaction parameters in ED. In this article, the experiments below were carried out with an ED bio-mineralization period set to 60 minutes.
As shown in Fig. 3, after 12 hours of incubation at constant temperature, Escherichia coli colonies piled up to create a lawn of bacteria at the fiber surface of the PLLA/nano-Ag control group, and on top of it there was a layer of viscous and pasty substances, visible to the naked eye, indicating strong metabolism of the bacteria. Only a small number of Staphylococcus aureus colonies were observed on the control fiber surface while no distinct colonies were marked on the ED or SCPS surfaces signifying their strong antibacterial abilities. Results from a quantitative analysis regarding antibacterial capabilities showed the antibacterial ratio of the PLLA/nano-Ag fibers to Escherichia coli to be 10% and the antibacterial ratio of the PLLA/nano-Ag fibers to Staphylococcus aureus to be 95%; this agreed with data from Zhang et al.34 ED and SCPS surfaces showed an antibacterial ratio of 99.5% and above against Escherichia coli and Staphylococcus aureus. These high ratios could be explained by the fact that the space between flakes of calcium phosphate facilitated nano-Ag release; in addition, the sharp edges of the calcium phosphate flakes could damage the bacterial cell membranes.35 Experimental outcomes from both quantitative and qualitative analyses showed that ED and SCPS had strong antibacterial activities.
 | ||
| Fig. 3 Antibacterial effects of the composite fibers. | ||
Nano-Ag is able to enhance the antibacterial activity of composite fibers significantly. The primary mechanisms behind the antibacterial activity are metal-ion effects and photo-catalysis.36,37 The chemical structure of Ag bestows strong catalytic ability; in addition, Ag has a high reduction potential, thus it can produce atomic oxygen in its surrounding environment. Atomic oxygen is a strong oxidizing agent, which is ideal for sterilization purposes. Further, an Ag+ ion can pull itself towards the thiol groups on bacterial proteases, and then deactivate the proteases, causing the eventual death of the bacteria. Finally, the Ag+ ion can migrate away from the dead bacteria and repeat the cycle stated above again and again. That is the reason for the remarkable sustainability of the antibacterial activity of Ag.
Fig. 4 shows different morphologies of osteocytes cultured at composite fiber surfaces (ED 60 min, SCPS 12 days) during a seven-day period. From day 1 to day 3, adhesion started to take place at the fiber surfaces; on day 5, osteocytes began to spread on the fiber surfaces; and on day 7, osteocytes became well dispersed at the fiber surfaces. The distribution at ED fiber surfaces was particularly good, likely due to its higher degree of roughness.38 Studies showed that ED fibers facilitate cell adhesion and adequately maintain cellular morphology. Furthermore, alkaline phosphatase (ALP) activity assays found no marked difference in the differentiation ability of osteocytes at the different surfaces on day 5. However, the differentiation ability of osteocytes at ED fiber surfaces was significantly higher than that at SCPS fiber surfaces on day 7; and the differentiation abilities of both ED and SCPS fiber surfaces were higher than that of a surface without calcium phosphate. In addition, 5% Ag had no impact on osteoblasts, while the ED fiber surface was shown to have strong capability to induce osteogenic differentiation. In conclusion, this paper has reported a pulsed ED method that can accelerate the bio-mineralization process of Ag-doped fiber surfaces, forming an antibacterial fibrous scaffold with high bioactivity and strong osteogenesis capability.
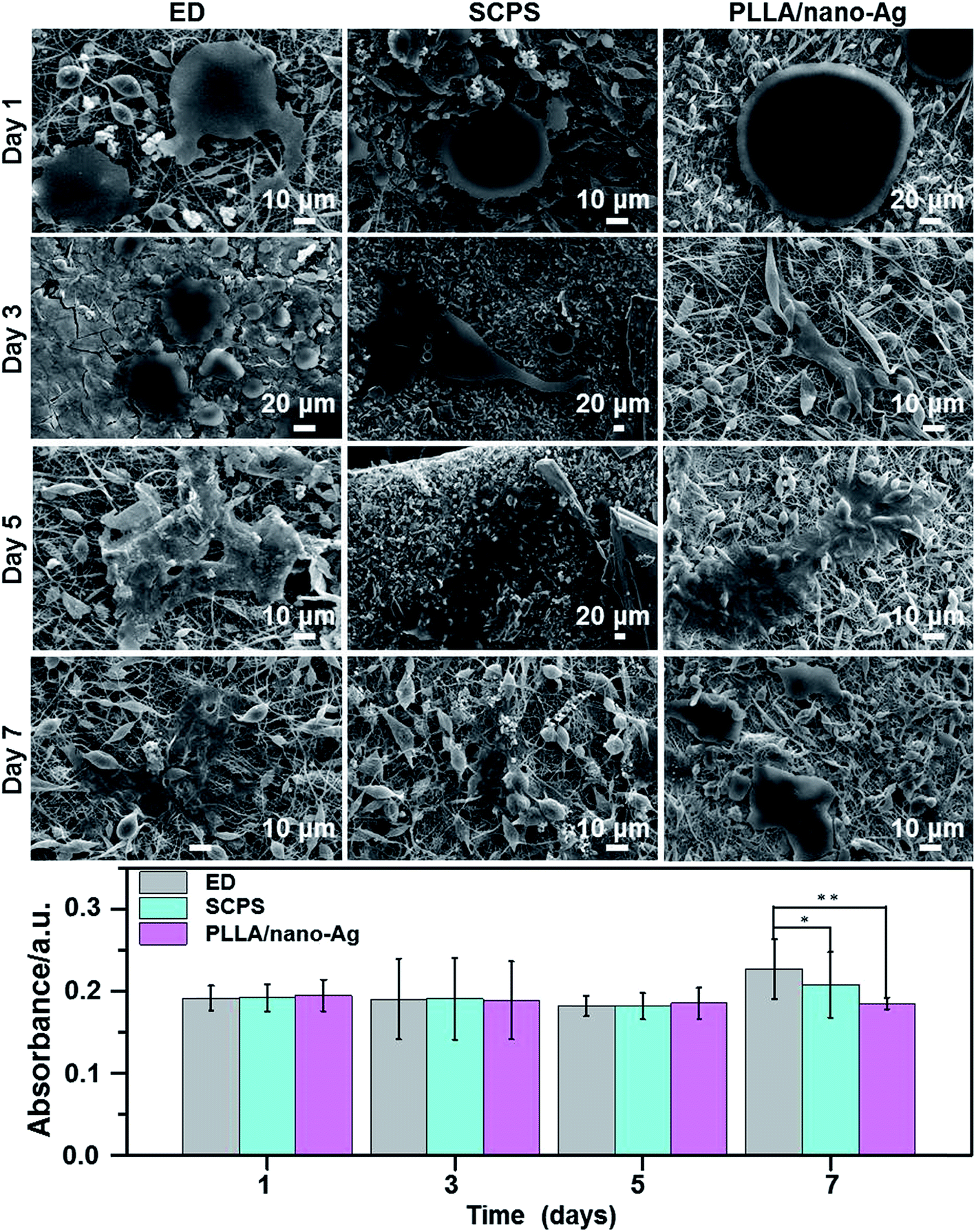 | ||
| Fig. 4 ALP activity of osteoblasts cultured on different nano-fibrous matrices after 1, 3, 5 and 7 days in culture. Data are expressed as mean ± SD (n = 3). A significant difference between groups is indicated by *(P < 0.05 compared with the neat PLLA/nano-Ag matrix) and **(the value of P for ED, SCPS and neat PLLA/nano-Ag matrix). | ||
4. Conclusions
Herein we have presented a method for fabricating PLLA/nano-Ag composite fibers that facilitates the growth of calcium phosphate crystals at the fiber surfaces and promotes osteogenic differentiation, thus significantly improving the fabrication efficiency of osteoinductive electrospun fibers. The study found the fiber diameter to be proportional to the PLLA concentration and the optimized concentration to be 14%. The fiber diameters were observed to be even when the concentration of nano-Ag was relatively low and the morphology showed the highest degree of homogeneity when the concentration of Ag was between 3% and 5%. The release rate of Ag+ ions in composite fibers was highly stable and remained at 1.0 ppm per day for 10 days. ED was utilized to accelerate the bio-mineralization process of calcium phosphate, and comparisons to traditional SCPS were made and analyzed. The quantity of bio-mineralized calcium phosphate at fiber surfaces was directly proportion to the deposition time and closely related to the fiber diameter as well as to the nano-Ag concentration; in an ED process, when the concentration of nano-Ag was 5%, the larger the fiber diameter, the greater the amount of calcium phosphate deposited, and the shorter the bio-mineralization process. Experimentally, the ED composite also demonstrated strong antibacterial properties and the capability to induce osteoblast differentiation at fiber surfaces. Therefore, ED can be adopted to simplify and accelerate the fabrication process of an osteogenetic and antibacterial electrospun fibrous scaffold.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by The Natural Science Foundation of Xinjiang Uygur Autonomous Region (2015211A037).Notes and references
- P. Bhattacharjee, D. Naskar, H. W. Kim, T. K. Maiti, D. Bhattacharya and S. C. Kundu, Eur. Polym. J., 2015, 71, 490–509 CrossRef CAS.
- I. Y. M. Baba, I. Wimpenny, O. Bretcanu, K. Dalgarno and A. J. E. Haj, J. Biomed. Mater. Res., 2017, 105, 1775–1785 CrossRef PubMed.
- C. He, X. Jin and P. X. Ma, Acta Biomater., 2013, 10, 419–427 CrossRef PubMed.
- K. Shin, T. Acri, S. Geary and A. K. Salem, Tissue Eng., Part A, 2017, 23, 1169–1180 CrossRef CAS PubMed.
- Y. Hu, S. Zou, W. Chen, Z. Tong and C. Y. Wang, Colloids Surf., B, 2014, 122, 559–565 CrossRef CAS PubMed.
- Y. H. Hsiao, S. R. Yang, E. M. Brey, I. M. Chu and M. H. Cheng, Plast. Reconstr. Surg., 2016, 4, 1–7 Search PubMed.
- Y. Guo, J. Lan, C. Zhang, C. Man, Q. Cai and X. Yang, Appl. Surf. Sci., 2015, 349, 538–548 CrossRef CAS.
- G. Wei and P. X. Ma, J. Biomed. Mater. Res., 2006, 78, 306–315 CrossRef PubMed.
- J. Chen, B. Chu and B. S. Hsiao, J. Biomed. Mater. Res., 2006, 79, 307–317 CrossRef PubMed.
- R. Zhang and P. X. Ma, Macromol. Biosci., 2004, 4, 100–111 CrossRef CAS PubMed.
- S. Liu, H. Li, L. Zhang, X. Yin and Y. Guo, Mater. Sci. Eng., C, 2017, 79, 100–107 CrossRef CAS PubMed.
- Y. Ling, X. Yi, Y. Jia, Y. Wang and W. Cui, ACS Appl. Mater. Interfaces, 2017, 9, 5023–5030 Search PubMed.
- G. A. Gelves, U. Sundararaj and J. A. Haber, Macromol. Rapid Commun., 2010, 26, 1677–1681 CrossRef.
- A. Lonjon, P. Demont, E. Dantras and C. J. Lacabanne, Non-Cryst. Solids., 2012, 358, 236–240 CrossRef CAS.
- G. Gelves, B. Lin, U. Sundararaj and J. Haber, Adv. Funct. Mater., 2010, 16, 2423–2430 CrossRef.
- Y. L. Huang, S. M. Yuen, C. C. M. Ma, C. Y. Chuang, K. C. Yu and C. C. Teng, Compos. Sci., Technol, 2009, 69, 1991–1996 CrossRef CAS.
- L. Li, Y. Li, J. Li, L. Yao, A. F. T. Mak, F. Ko and L. Qin, J. Nanomater., 2009, 4, 9–14 Search PubMed.
- C. Hu, S. Liu, B. Li, H. Yang, C. Fan and W. Cui, Adv. Healthcare Mater., 2013, 2, 1314–1321 CrossRef CAS PubMed.
- Y. Gao, Y. B. Wang, Y. M. Wang and W. Cui, Mar. Drugs, 2016, 14, 192–206 CrossRef PubMed.
- I. Cacciotti, E. Fortunati, D. Puglia, J. M. Kenny and F. Nanni, Carbohydr. Polym., 2014, 103, 22–31 CrossRef CAS PubMed.
- P. Huang, X. Bi, J. Gao, L. Sun, S. Wang and S. Chen, J. Mater. Chem. B, 2016, 4, 2090–2101 RSC.
- H. Park, D. J. Lim, S. H. Lee and H. Park, J. Biomed. Nanotechnol., 2016, 12, 2076–2082 CrossRef CAS PubMed.
- C. T. Pan, T. L. Yang, Y. C. Chen, C. Y. Su, S. P. Ju and K. H. Hung, J. Nanomater., 2015, 16, 155–160 Search PubMed.
- Y. C. Chai, S. Truscello, S. V. Bael, F. P. Luyten, J. Vleugels and J. Schrooten, Acta Biomater., 2011, 7, 2310–2319 CrossRef CAS PubMed.
- H. Wang, N. Eliaz, Z. Xiang, H. P. Hsu, M. Spector and L. W. Hobbs, Biomaterials, 2006, 27, 4192–4203 CrossRef CAS PubMed.
- J. Chen, B. Chu and B. S. Hsiao, J. Biomed. Mater. Res., 2010, 79, 307–317 Search PubMed.
- P. V. Mayuri and P. Ramesh, J. Mater. Sci., 2016, 51, 1–8 CrossRef.
- N. Eliaz, O. Ritman-Hertz, D. Aronov, E. Weinberg, Y. Shenhar and G. Rosenman, J. Mater. Sci. Mater. Med., 2011, 22, 1741–1752 CrossRef CAS PubMed.
- R. Zhang, Z. Song, L. Yin, N. Zheng, H. Tang, H. Lu, N. P. Gabrielson, Y. Lin, K. Kim and J. Cheng, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 98–110 CrossRef CAS PubMed.
- C. He, G. Xiao, X. Jin, C. Sun and P. X. Ma, Adv. Funct. Mater., 2010, 20, 3568–3576 CrossRef CAS PubMed.
- R. Zhang, N. Metoki, O. Sharabani-Yosef, H. Zhu and N. Eliaz, Adv. Funct. Mater., 2016, 26, 7965–7974 CrossRef CAS.
- Q. Cai, Q. Xu, Q. Feng, X. Cao, X. Yang and X. Deng, Appl. Surf. Sci., 2011, 257, 10109–10118 CrossRef CAS.
- S. Liu, H. Li, L. Zhang, X. Yin and Y. Guo, Mater. Sci. Eng. C Mater. Biol. Appl., 2017, 79, 100–107 CrossRef CAS PubMed.
- Z. Zhang, Y. Wu, Z. Wang, X. Zhang, Y. Zhao and L. Sun, Mater. Sci. Eng. C Mater. Bio. Appl., 2017, 78, 706–735 CrossRef CAS PubMed.
- V. Uskoković, S. S. Batarni, J. Schweicher, A. King and T. A. Desai, ACS Appl. Mater. Interfaces, 2013, 5, 2422–2431 Search PubMed.
- B. S. Atiyeh, M. Costagliola, S. N. Hayek and S. A. Dibo, Burns, 2007, 33, 139–148 CrossRef PubMed.
- M. Goudouri, E. Kontonasaki, U. Lohbauer and A. R. Boccaccini, Acta Biomater., 2014, 10, 3795–3810 CrossRef PubMed.
- A. Abdal-Hay, L. D. Tijing and J. K. Lim, Chem. Eng. J., 2013, 215, 57–64 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01011k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |