
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effects of boron-doped silicone resin and a layered double hydroxide modified with sodium dodecyl benzenesulfonate for enhancing the flame retardancy of polycarbonate
Yi Jiang a,
Zhifeng Hao*a,
Hongsheng Luo*a,
Zehui Shaoa,
Qian Yua,
Ming Suna,
Yong Keb and
Yilong Chenb
a,
Zhifeng Hao*a,
Hongsheng Luo*a,
Zehui Shaoa,
Qian Yua,
Ming Suna,
Yong Keb and
Yilong Chenb
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, P. R. China. E-mail: haozhifeng3377@163.com
bHeyuan Jingwang Electronic Technology (Longchuan) Co., Ltd, Heyuan 517300, P. R. China
First published on 20th March 2018
Abstract
To improve the flame retardancy of polycarbonate (PC), a novel and environmentally friendly flame retardant was synthesized by combining boron-doped silicone resin (BSR) with a layered double hydroxide (LDH) modified with sodium dodecyl benzenesulfonate (SDBS) which was denoted as DBS-LDH/BSR. The structure of the hybrid was characterized by Fourier transform infrared (FTIR) spectroscopy and X-ray photoelectron spectroscopy (XPS), which indicated that BSR was successfully combined with DBS-LDH. X-ray diffraction (XRD) studies showed that the reaction of BSR occurred only on the surface of DBS-LDH. In addition, scanning electron microscopy (SEM) was used to further verify the combination of DBS-LDH with BSR. PC exhibited the optimum flame retardancy following the incorporation of 10 wt% DBS-LDH/BSR (5 wt% DBS-LDH and 5 wt% BSR). Based on thermogravimetric analysis, the char residue of this PC composite in air at 750 °C increased to 3.60 wt%. Mechanical test showed that the DBS-LDH/BSR could affect the mechanical properties after incorporation into PC. According to the UL-94 vertical burning test, the flame retardant rating of the PC composite improved to V-0. Furthermore, the limiting oxygen index (LOI) value of the PC composite increased to 34%. According to the cone calorimeter test, the peak heat release rate (PHRR) dramatically decreased by 44%. The morphology of the PC composite after combustion was characterized by SEM, which revealed that the pores of the composite were smaller than those of pure PC. This result was attributed to the limited spread of oxygen and heat permeation. Thus, both DBS-LDH and BSR contributed to the synergistic effects of reducing the fire hazard of PC.
1. Introduction
Polycarbonate (PC), a tough thermoplastic resin with carbonate groups, has been widely used in many fields, including glass assembly and the automotive and electronics industries.1,2 However, due to its high flammability and the production of large amounts of smoke and toxic gases during combustion, which is accompanied by severe dripping, researchers have improved the flame retardancy of this material.3–5Recently, it has made some progress in the fire safety of PC.6 For example, halogen flame retardants form some of the most widely applied flame retardant materials, but they do have clear disadvantages: generation of toxic and corrosive gases during combustion.7 Therefore, non-halogen flame retardants8–10 have been paid more attention today. Some environmental flame retardants based on phosphorus, nitrogen, silicon, boron and other elements have gradually substituted halogen flame retardants. Even though the flame retardancy of PC has dramatically improved, one type flame retardant may affect other properties of the material, such as the mechanical properties, dispersion and thermal stability. Greater flame-retardant loadings are frequently added in polymers to improve flame retardancy, but this approach is detrimental to the intrinsic properties of the polymers. To improve the flame retardancy of PC, we examined the synergistic effects of two types of flame retardants.
Layered double hydroxides (LDHs) have shown to offer good flame retardancy because of their unique chemical composition. These are potentially eco-friendly flame retardants for improving the fire-retardant properties of polymers.11,12 The flame retardant mechanism of LDHs can be attributed to the combination of the following three functions: (1) heat absorption, (2) gas dilution, and (3) char formation. The high polarity of these materials greatly affects their dispersion in the polymer matrix. Additionally, to achieve the desired flame retardancy, high LDH loadings are necessary. Thus, researchers have applied organic anions to modify LDHs, such as fatty acid salts,13 sulfonates,14 and phosphates,15 which improves the dispersion of LDHs in polymers.16–18 In addition, some synergistic effects of LDHs with other fire retardants have become the focus in the field of flame retardant technology. For example, ammonium polyphosphate,19 red phosphorus,20 zinc borate,21 graphene oxide,22 and intumescent flame retardant (IFR)23 play roles as synergists to improve the flame retardancy of polymers. It is reported that interlayer gallery distance of LDHs can be enlarged after modified with sodium dodecyl benzenesulfonate (SDBS), which enhances the compatibility of LDHs with polymer matrix.17 High contents of single inorganic flame retardants are known to affect the mechanical properties of bulk polymers. Thus, the combination of inorganic and organic flame retardants is a good strategy for maintaining the properties of polymers.
Organic silicone,24,25 as organic flame retardant, has been applied to many polymers and turns into a promising class of cost-effective, minimally toxic, drip-proof, and eco-friendly flame retardants. During combustion, the organic silicone flame retardant can transfer to the surface of the polymer, forming inorganic oxygen and heat-insulating protective layers containing Si–O and Si–C bonds. These layers can inhibit the release of decomposed product and protect the polymer from further burning. Furthermore, boron (B)-containing flame retardants have been reported to show good flame retardancy.26,27 They are often used as synergists in flame-retardant systems. B-containing compounds can promote the formation of char during combustion,28,29 which can inhibit the effusion of volatile combustibles and protect the char layer from further oxidation. Therefore, to obtain highly effective flame retardants, we introduce boron into pure silicone resins, marked as B-doped silicone resin (BSR). In our previous work, we found that BSR possessed better thermal stability than pure silicon resins.30 Thus, we apply BSR as a flame retardant in this study.
In this paper, we focus on the synergistic effects of combining BSR with DBS-LDH as flame retardants on PC composites. The intercalation of DBS ions in LDH increases the distance between layers, which reduces the polarity of LDH in PC. In addition, the siloxane monomers used for preparing the silicone resin can hydrolyze via hydrolysis. After that, the hydrolyzed siloxanes can dehydrate with the hydroxyls of boric acid, hence B element can combine with these siloxanes. Subsequently, we can prepare BSR through polycondensation. Furthermore, BSR with hydroxyls can dehydrate with the hydroxyls of DBS-LDH (Scheme 1). The combination of organic BSR with DBS-LDH can improve the compatibility of inorganic particles in PC. Therefore, the newly synthetic DBS-LDH/BSR is anticipated to reduce the fire hazard of the PC composites.
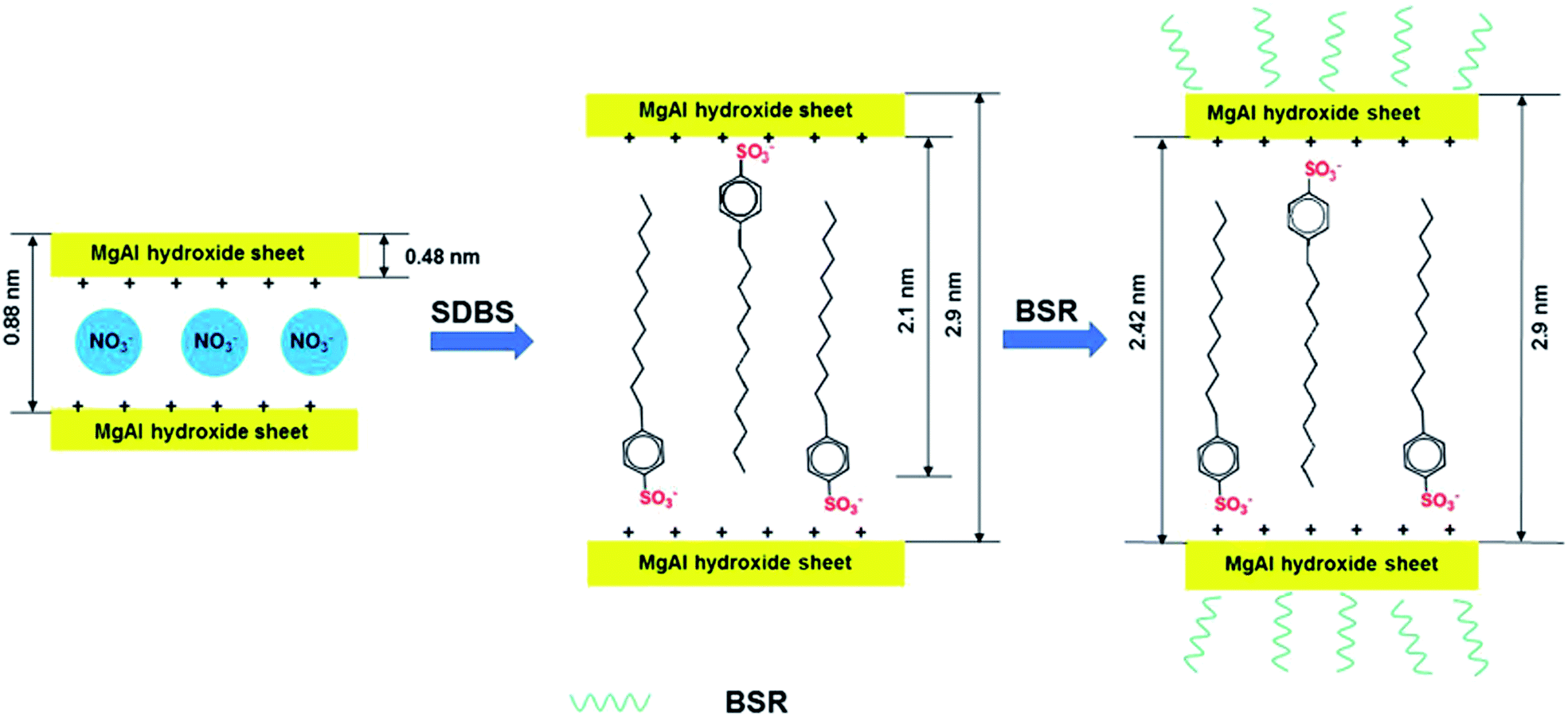 | ||
| Scheme 1 Schematic illustration of the synthetic route to DBS-LDH/BSR. | ||
2. Experiment
2.1 Materials
Magnesium nitrate hexahydrate (Mg(NO3)2·6H2O), aluminium nitrate nonahydrate (Al(NO3)3·9H2O), SDBS, sodium hydroxide (NaOH), boric acid, concentrated hydrochloric acid (HCl, 36.5%) and ethanol (99.7%) were supplied by Tianjin Damao Chemical Reagent Factory (Tianjin, China) and were used without further purification. Diethoxydimethylsilane and phenyltriethoxysilane were provided by Suzhou Chuangjia Plastic Co., Ltd. PC was purchased from Suzhou Chuangjia Plastic Co., Ltd.2.2 Synthesis of DBS-LDH/BSR
DBS-LDH was prepared using a co-precipitation method.31 SDBS solution (0.1 M) was introduced into a 250 ml three-neck round flask that was equipped with constant pressure liquid funnels, reflux-condenser and pH meter and a magnetic stirrer. A mixture solution containing Mg(NO3)2·6H2O (0.13 M) and Al(NO3)3·9H2O (0.06 M) was then slowly added to the above flask with vigorous stirring at a constant pH = 10.0 ± 0.5, adjusted by the synchronous dropwise addition of 1 M NaOH solution. During the reaction, the resultant slurry was continuously stirred for 30 min at 50 °C. Subsequently, we aged the slurry for 24 h at a temperature of 70 °C. Finally, the white product was filtered and washed thoroughly with distilled water until the pH of the solution was about 7, and then dried in a freeze dryer for 24 h, which was marked as DBS-LDH.BSR was synthesized using a hydrolysis-polycondensation method.30 Diethoxydimethylsilane (6.34 g), phenyltriethoxysilane (24.0 g), 40 ml absolute alcohol, and 1.73 g boric acid were added into a 250 ml four-necked which was equipped with a magnetic stirrer, a water condenser, and a constant pressure liquid funnel. The resulting mixture was vigorously stirred. At the temperature of 65 °C, 3.5 ml deionized water and 0.9 ml hydrochloric acid (1 M) were added into the above flask dropwise. Then the mixture was kept refluxing for 2 h, obtaining the hydrolysate. Subsequently, the hydrolysate was heated to 135 °C under vacuum (0.06 MPa) to remove the ethanol and other small molecules. Finally, the resulting product was transparent viscous liquid and recorded as BSR.
Here was the typical procedure to combine DBS-LDH with BSR. BSR (6 g) was first added into an aqueous alcohol solution containing 200 ml of alcohol and 20 ml of water under stirring for 30 min. After that, 6 g of DBS-LDH was added, following with stirring the mixture for 3 h. The pH of the system was adjusted to 9–10 with sodium hydroxide. And the reaction continued for another 24 h. Finally, the resulting product was filtered and thoroughly washed with deionized water until the pH was 7. Finally, the product was dried in a freeze dryer for 24 h, and was marked as DBS-LDH/BSR.
2.3 Preparation of PC/DBS-LDH/BSR
PC and its composites were prepared by adding different contents of flame retardants (DBS-LDH, BSR or DBS-LDH/BSR), and the components are listed in Table 1. Briefly, PC composite with 10 wt% DBS-LDH was prepared as follows: 90 wt% PC and 10 wt% DBS-LDH were mixed in a co-rotating twin-screw extruder with a screw temperature profile of 180, 178, 178, 178, 178, and 183 °C from the hopper to the die. The resulting mixture was injected and moulded into various sheets with a temperature profile of 215, 215, and 215 °C from the hopper to the nozzle. PC/BSR10 and PC/DBS-LDH/BRS composites were prepared using the same method.| Sample | Component (wt%) | ||
|---|---|---|---|
| PC | DBS-LDH | BSR | |
| PC | 100 | 0 | 0 |
| PC/DBS-LDH10 | 90 | 10 | 0 |
| PC/BSR10 | 90 | 0 | 10 |
| PC/DBS-LDH7/BSR3 | 90 | 7 | 3 |
| PC/DBS-LDH5/BSR5 | 90 | 5 | 5 |
2.4 Characterization
Fourier transform infrared (FTIR) spectroscopy was used to identify the chemical structures of the prepared samples, and the spectra were recorded on a Thermo Scientific Nicolet 6700 FTIR spectrometer.X-ray diffraction (XRD) patterns of the prepared samples were recorded with an XPERT-PRO X-ray diffractometer. The Cu Kα (λ = 0.1542 nm) radiation source was operated at 40 kV and 30 mA with a scan rate of 2° min−1 from 2.5° to 30°.
X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos AXIS His spectrometer using a monochromatized Al Kα X-ray source (1486.6 eV photons).
Thermal stabilities of the flame retardants, pure PC and its composites were studied by thermogravimetric analysis (TGA) (NETZSCH), which was performed with a heating rate of 10 °C min−1 under air and nitrogen at a flow rate of 40 ml min−1 from 30 °C to 800 °C.
Mechanical tests were performed using an AI-7000M servo control system universal testing machine according to the ASTM D3039-08 method, at a speed of 10 mm min−1.
Limiting oxygen index (LOI) values were measured using an oxygen index instrument (Beijing Xinsheng Zhuo Rui Technology Co., Ltd, China). The samples' dimensions were 120 × 10 × 4 mm3, and the tests were performed according to standard ASTM D2863.
Vertical burning tests were performed in a burning chamber (Beijing Xinsheng Zhuo Rui Technology Co., Ltd, China) on samples with dimensions of 125 × 13 × 3 mm3 according to standard ASTM D3801.
Morphological features of the flame retardants, the surfaces and cross-sections of the char residues were investigated by scanning electron microscopy (SEM).
Combustion tests were performed on a cone calorimeter FTT007 (Fire Testing Technology, UK) according to ISO 5600 standard procedures with 100 × 100 × 3 mm3 specimens. Each specimen was exposed horizontally to a 35 kW m−2 external heat flux.
3. Results and discussion
3.1 Characterization of DBS-LDH/BSR
Fig. 1 shows the FTIR spectra of NO3-LDH, DBS-LDH, BSR and DBS-LDH/BSR. As can be seen in the spectrum of NO3-LDH, the peak at 3430 cm−1 is ascribed to the O–H group. The absorption of NO3− appears at 1350 cm−1. In the spectrum of BSR, the peak at 3460 cm−1 is attributed to the stretching vibrations of O–H groups. The absorption bands at 2965–2870 cm−1 are ascribed to the stretching vibrations of C–H units (including –CH3 or –CH2CH3). The characteristic peaks at 1110 cm−1 and 827 cm−1 are due to the stretching modes and bending vibrations of Si–O–Si,32 respectively. The absorption by O–C bonds occurs at 1050 cm−1. The peaks at 1410 cm−1, 881 cm−1 and 700 cm−1 belong to the stretching and in-plane shear vibrations of B–O units.33 In the spectrum of DBS-LDH, the characteristic peak at 3490 cm−1 is assigned to the stretching vibrations of the –OH groups attached to Al and Mg ions in the layers.30 The sulfonate stretching bands occur at 1180 cm−1 and 1040 cm−1,34 confirming the exchange of DBS ions in the interlayer space of LDH (consistent with the results of XRD), as shown in Scheme 1. In the spectrum of DBS-LDH/BSR, the peak at 3490 cm−1 (O–H) is broader than that of DBS-LDH, indicating a slight reduction in the number of hydroxyl groups. This may be due to the dehydration of hydroxyls between DBS-LDH and BSR (in accordance with the results shown in Fig. 3). The broad band at 1250–1000 cm−1 is due to the overlap of O–C, Si–O and sulfonate vibrations.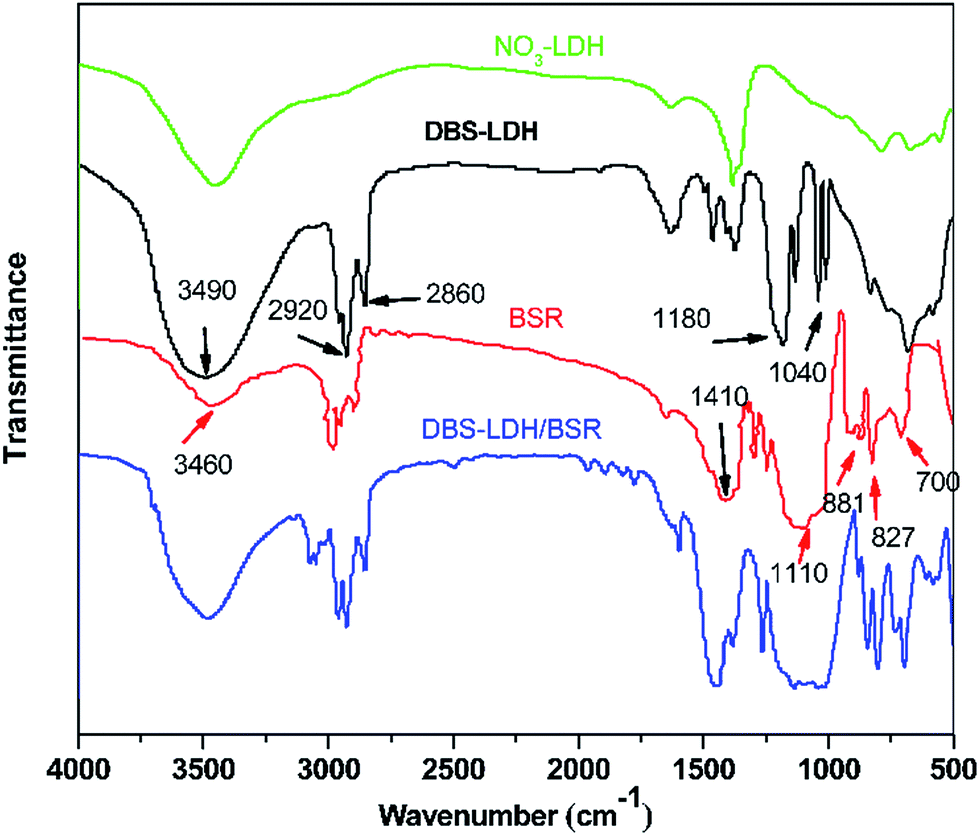 | ||
| Fig. 1 FTIR spectra of DBS-LDH, BSR and DBS-LDH/BSR. | ||
Fig. 2 shows the XPS wide-scan spectra of DBS-LDH/BSR. The peaks at 74 eV, 1305 eV, 532.5 eV and 284.8 eV are assigned to Al 2p, Mg 1s, O 1s and C 1s, respectively,35 verifying the presence of DBS-LDH in the hybrid composite. Furthermore, the peaks at binding energies of 101.23 eV and 193.33 eV are ascribed to Si 2p36 and B 1s37,38, which further confirms that BSR has successfully combined with DBS-LDH.
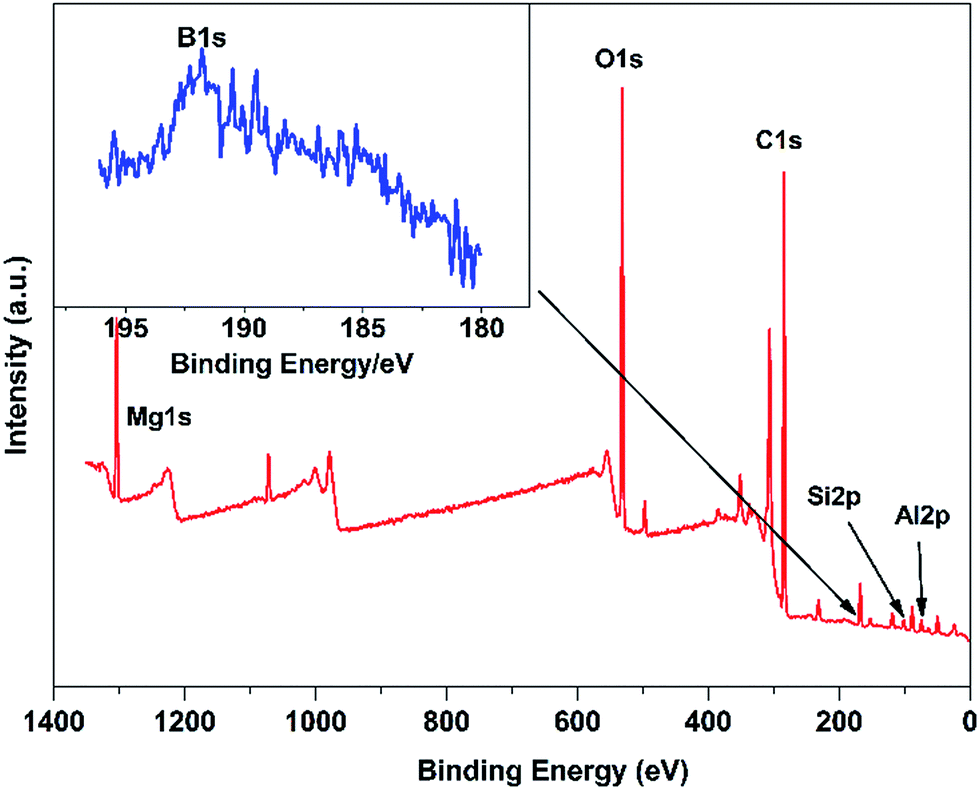 | ||
| Fig. 2 XPS wide-scan spectra of DBS-LDH/BSR. | ||
Fig. 3 shows the XRD patterns of NO3-LDH, DBS-LDH and DBS-LDH/BSR. The (003) reflection of NO3-LDH is located at 9.97°, giving a d003 distance of 0.88 nm. Following modification of NO3-LDH with SDBS, the (003) reflection shifts to a lower angle (2.95°). Thus, it can provide evidence for the intercalation of DBS ions into the interlayer galleries to increase the interlayer spacing (2.9 nm).30,39 Furthermore, the dispersion of DBS-LDH in the polymer has greatly improved over that of the unmodified LDH.11 The thickness of NO3-LDH has been reported to be 0.48 nm, and the distance between layers is 2.42 nm, which is nearly identical to the length of the DBS molecular chain (approximately 2.1 nm).40 Therefore, we can conclude that DBS vertically intercalates into NO3-LDH (Scheme 1). As shown in Fig. 3, DBS-LDH/BSR exhibits the same diffraction peaks as DBS-LDH, showing that the distance between the layers of DBS-LDH does not change upon combination with BSR. This result shows that the reaction between DBS-LDH and BSR occurs only on the surfaces of the DBS-LDH nanocrystals and does not change the bulk properties of DBS-LDH.
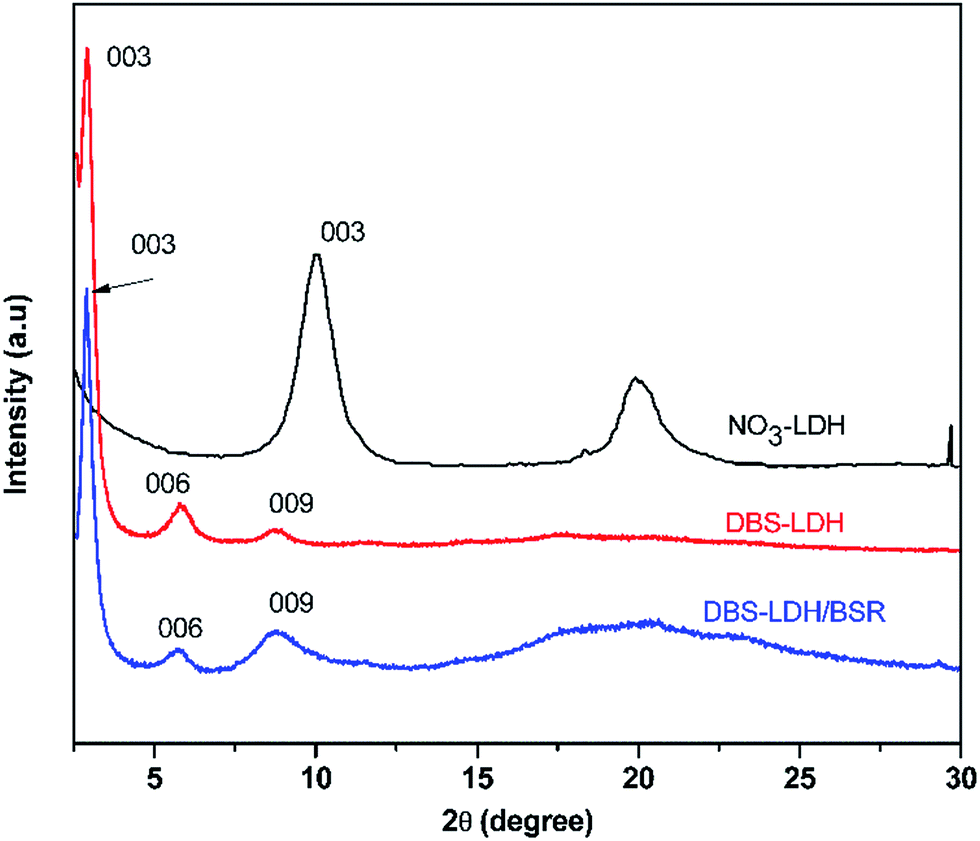 | ||
| Fig. 3 XRD patterns of NO3-LDH, DBS-LDH and DBS-LDH/BSR. | ||
Panels a, b and c in Fig. 4 display the SEM images of NO3-LDH, DBS-LDH and DBS-LDH/BSR. As can be seen from the images, all exhibit lamellar plates. Compared with NO3-LDH and DBS-LDH, the layer structure of DBS-LDH/BSR is destroyed to some extent, which is due to the BSR grafting onto the surface of DBS-LDH.
 | ||
| Fig. 4 SEM images of NO3-LDH, DBS-LDH and DBS-LDH/BSR. | ||
3.2 Thermal properties
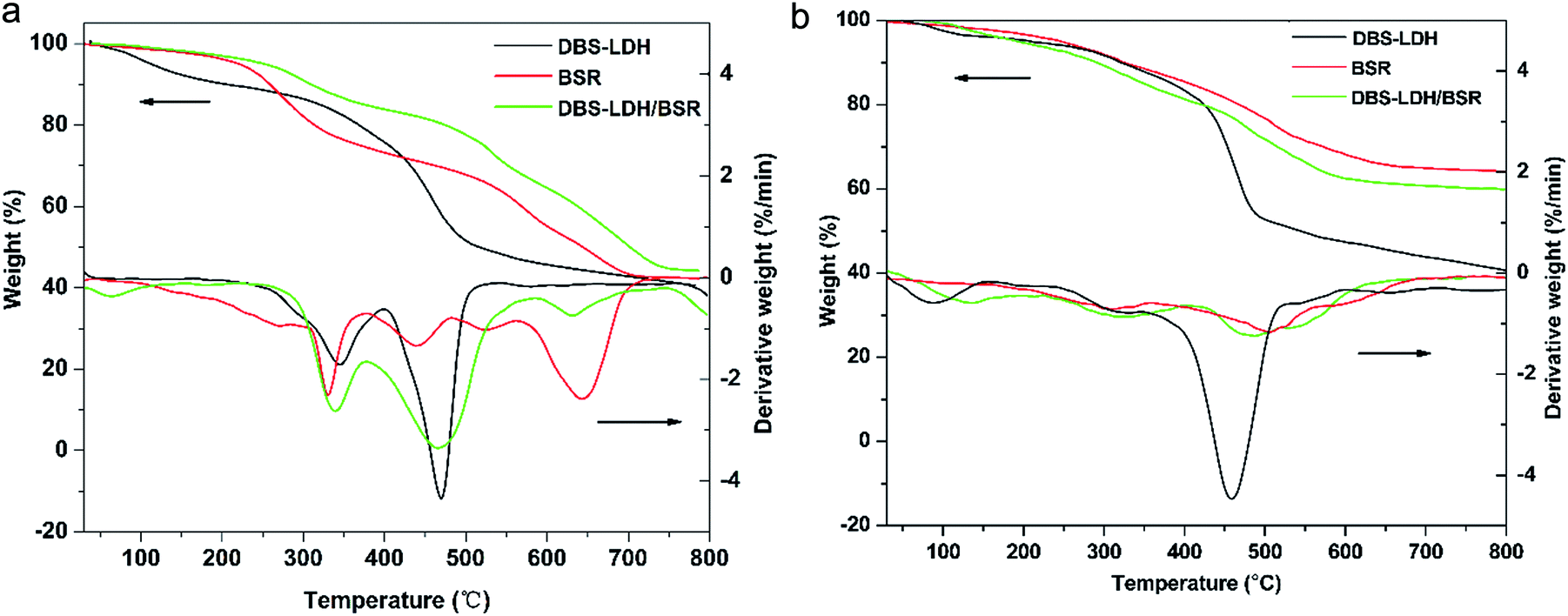 | ||
| Fig. 5 TG/DTG profiles for the flame retardants under (a) air atmosphere and (b) nitrogen with 10 °C min−1 heating rate, 40 ml min−1 gas flow rate and temperature range of 30–800 °C. | ||
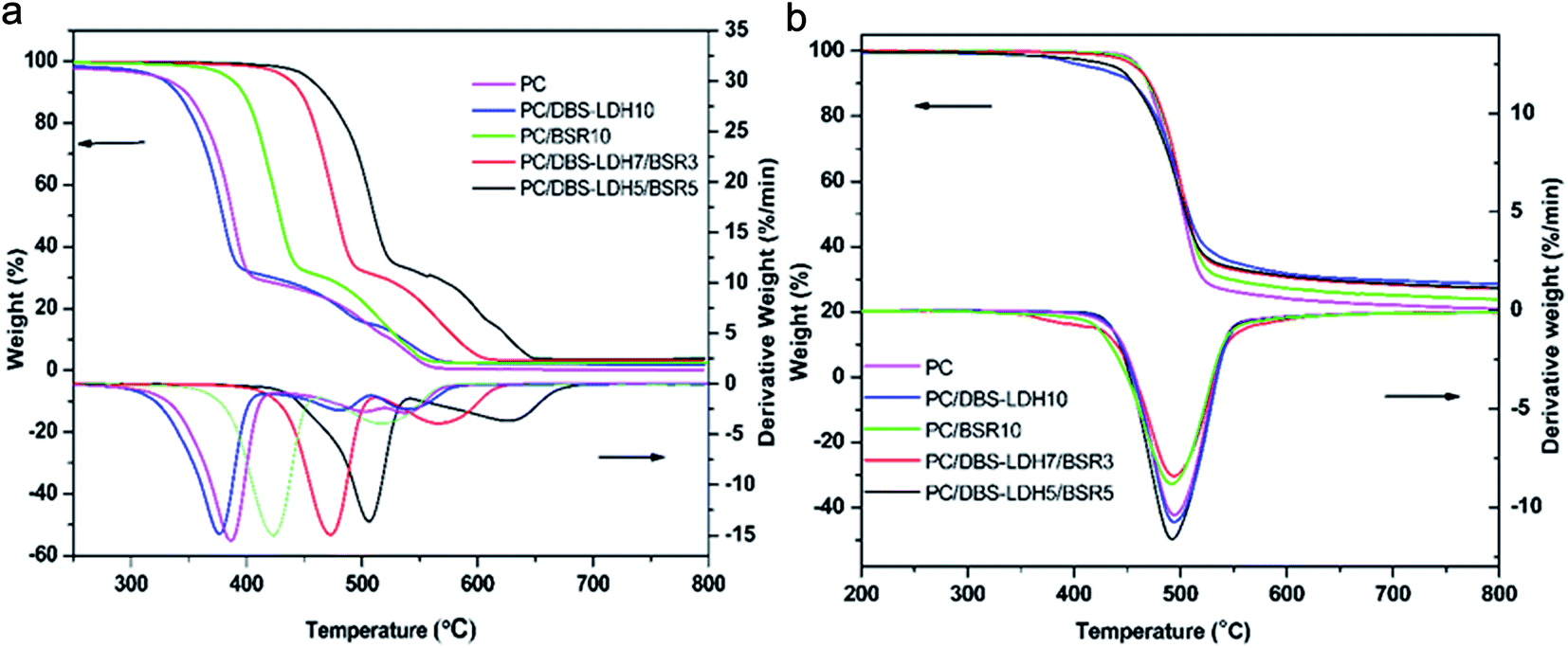 | ||
| Fig. 6 TG/DTG curves of the different filler loadings of the PC composites under (a) air and (b) nitrogen atmosphere with 10 °C min−1 heating rate, 40 ml min−1 gas flow rate and temperature range of 30–800 °C. | ||
| Sample | T5% (°C) | Tmax1 (°C) | Tmax2 (°C) | Char yield at 750 °C | ||||
|---|---|---|---|---|---|---|---|---|
| Air | Nitrogen | Air | Nitrogen | Air | Nitrogen | Air | Nitrogen | |
| PC | 324 | 462 | 388 | 495 | 493 | — | 0.20% | 20.90% |
| PC/DBS-LDH10 | 322 | 408 | 374 | 493 | 479 | — | 1.80% | 28.90% |
| PC/BSR10 | 386 | 457 | 422 | 494 | 521 | — | 2.58% | 24.00% |
| PC/DBS-LDH7/BSR3 | 432 | 453 | 474 | 492 | 568 | — | 3.20% | 27.20% |
| PC/DBS-LDH5/BSR5 | 452 | 437 | 507 | 491 | 632 | — | 3.60% | 27.40% |
3.3 Mechanical properties
Table 3 shows the mechanical properties of pure PC and its composites. As can be observed, the neat PC exhibits higher breaking elongation (4.49%), ultimate tensile strength (38.743 MPa), maximum strength (140.370 kgf) and fracture strength (3.866 MPa) comparing with its composites. In general, the flame-retardant fillers can lead to the reduction of mechanical properties of PC, which is mainly because of not much better additive-matrix compatibility. Even though the BSR is an organic flame retardant, its structure that includes stiff benzene rings may also affect the mechanical properties of PC. Additionally, the DBS-LDH is likely to form agglomerates, resulting in poorer interfacial interactions. Therefore, to obtain flame-retardant PC while to retain the mechanical properties, proper loadings of DBS-LDH and BSR seem to be essential.| Sample | Breaking elongation (%) | Ultimate tensile strength (MPa) | Maximum strength (kgf) | Fracture strength (MPa) |
|---|---|---|---|---|
| PC | 4.490 | 38.743 | 140.370 | 3.866 |
| PC/DBS-LDH10 | 2.851 | 22.088 | 80.028 | 2.156 |
| PC/BSR10 | 3.379 | 28.222 | 102.250 | 2.762 |
| PC/DBS-LDH7/BSR3 | 3.038 | 22.392 | 81.127 | 2.238 |
| PC/DBS-LDH5/BSR5 | 4.148 | 25.716 | 93.172 | 2.564 |
3.4 Flame retardant properties
The flame retardancy of PC and its composites is evaluated by the LOI and UL-94 test (Fig. 7). Pure PC has a low LOI value of 26% and a V-2 rating in the UL-94 vertical burning test. Upon adding 10 wt% DBS-LDH, the LOI value increases to 29%, and the material obtains a V-1 rating. With the incorporation of 10 wt% BSR, the LOI reaches 28%, and the UL-94 has a V-1 rating. When BSR is introduced (DBS-LDH/BSR hybrid composites), the flame retardancy of the PC composites further improves over that of pure PC. The LOI value increases to 34%, and a V-0 rating is obtained for PC/DBS-LDH5/BSR5. Thus, DBS-LDH and BSR together enhance the fire retardancy. Pure PC exhibits obvious flame drips in the UL-94 vertical burning test (Fig. 8). The occurrence of flame drips decreases substantially, but not completely, when 10 wt% DBS-LDH is added to PC. After the incorporation of 10 wt% BSR, the flame drips still exist. In contrast, when both DBS-LDH and BSR are incorporated, the flame drips disappear completely. As for PC/DBS-LDH5/BSR5 composite, DBS-LDH acts as a barrier to maximize the heat insulation properties and minimize the permeability of volatile degradation products, while BSR promotes char formation during the burning process. Hence, the composite reduces the fire hazard during combustion, and the flame retardancy of PC improves greatly.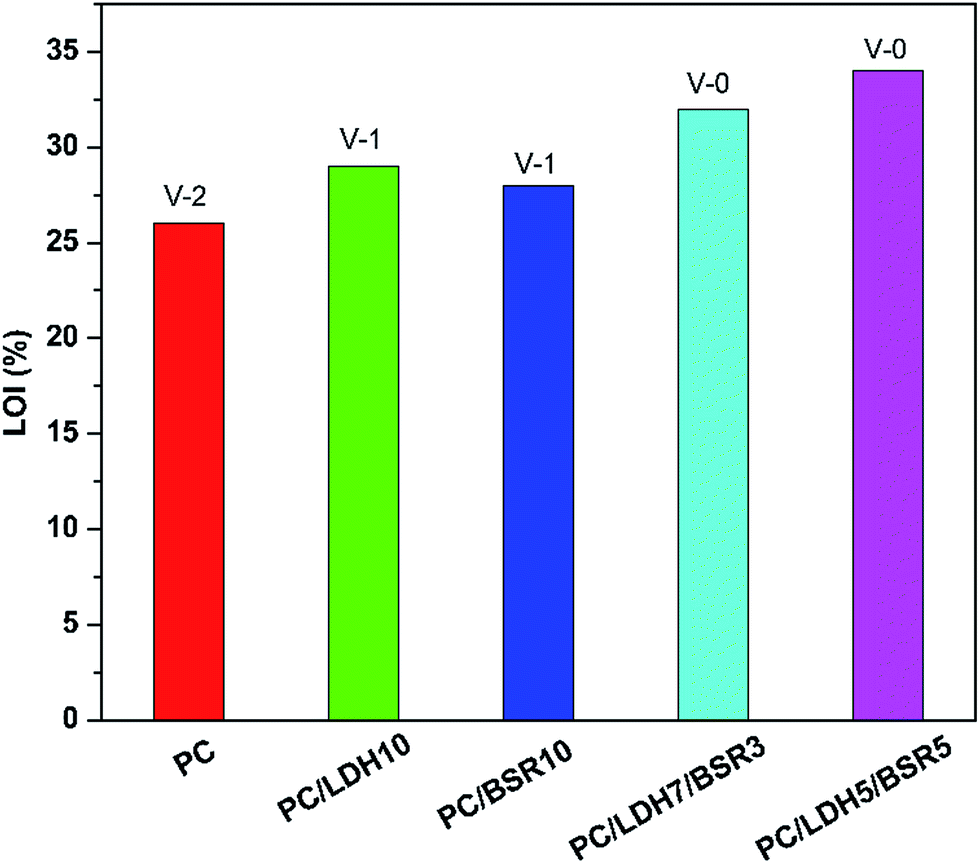 | ||
| Fig. 7 LOI values and UL-94 results of PC and its composites. | ||
 | ||
| Fig. 8 Digital photos of the char residues of the PC composites containing different amounts of flame retardants (a) PC, (b) PC/DBS-LDH10, (c) PC/BSR10, (d) PC/DBS-LDH7/BSR3 and (e) PC/DBS-LDH5/BSR5. | ||
The morphologies of the char residues of the PC composites are further examined after the LOI tests by SEM. Fig. 9 shows the SEM photographs obtained for the exterior and interior of the chars from pure PC and a PC composite. The exterior char of pure PC consists of a porous residue, and its interior char shows much larger pores after combustion, indicative of the high flammability and poor thermostability of the pure PC char. In the case of the PC/DBS-LDH5/BSR5, the interior char shows smaller pores than those of the PC char and a continuous structure. Furthermore, the exterior char layer also shows a compact structure with small pores. The char layer protects PC from the radiant heat and direct contact with the flame, effectively insulating the interior from gas and heat transfer. Therefore, the char layer plays an important role in improving the flame retardancy of PC.
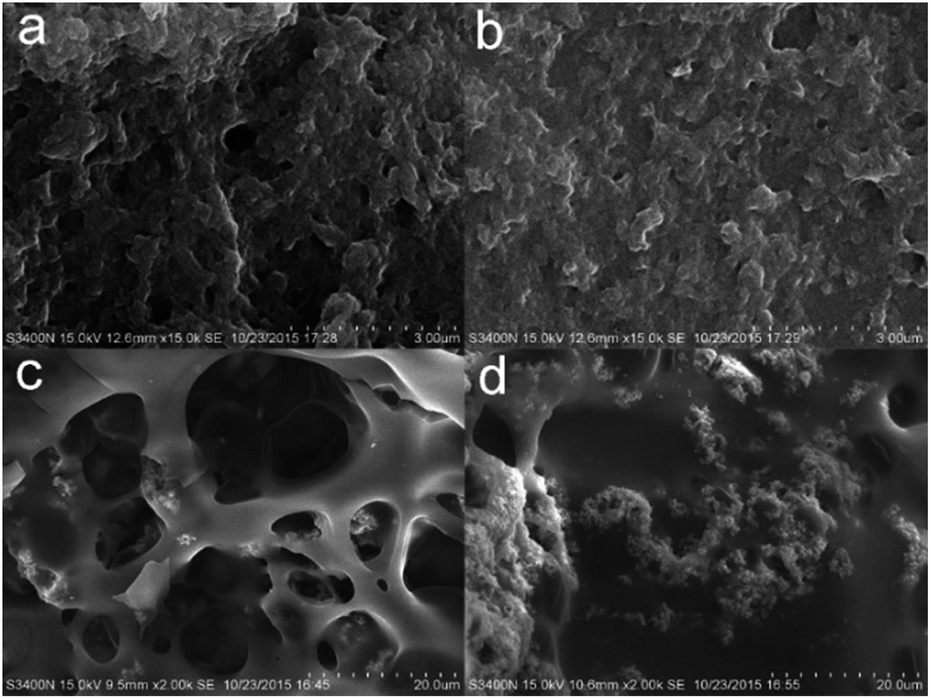 | ||
| Fig. 9 SEM images of the exterior char residues of (a) PC and (b) PC/DBS-LDH5/BSR5 and the interior residual chars of (c) PC and (d) PC/DBS-LDH5/BSR5. | ||
Fig. 10 shows the heat release rate (HRR) versus time curves of pure PC and its composites. The peak heat release rate (PHRR) decreases from 294 kW m−2 for pure PC to 239 kW m−2 (−19%), 236 kW m−2 (−19.7%), 210 kW m−2 (−29%) and 165 kW m−2 (−44%) for PC/DBS-LDH10, PC/BSR10, PC/DBS-LDH7/BSR3 and PC/DBS-LDH5/BSR5, respectively. For pure PC, the PHRR occurs at approximately 190 s, whereas those of PC/DBS-LDH10, PC/BSR10, PC/DBS-LDH7/BSR3 and PC/DBS-LDH5/BSR5 appear at 71 s, 136 s, 110 s and 149 s, respectively. For PC/BSR10, the PHRR decreases comparing with pure PC, which is attributed to the BSR forming char residue to protect the PC composite. The PHRR reaches earlier in the PC composites (except for PC/BSR10) than in pure PC due to the intercalation of the unstable DBS in LDH. However, the PHRR decreases dramatically upon introducing DBS-LDH and DBS-LDH/BSR into the PC. Specifically, the PC/DBS-LDH5/BSR5 composite shows a much better flame retardancy than all the other samples. As demonstrated by the first stage of the TGA curve obtained for the PC composites, the presence of DBS-LDH/BSR contributes to charring. The char residues, which can prevent heat transfer and flame spread during combustion, are very important for the flame retardancy of the PC composites. Due to the synergistic effects of DBS-LDH and BSR, the char yields of the PC composites after burning increase (Table 2), which can remarkably enhance flame retardancy of PC composites.
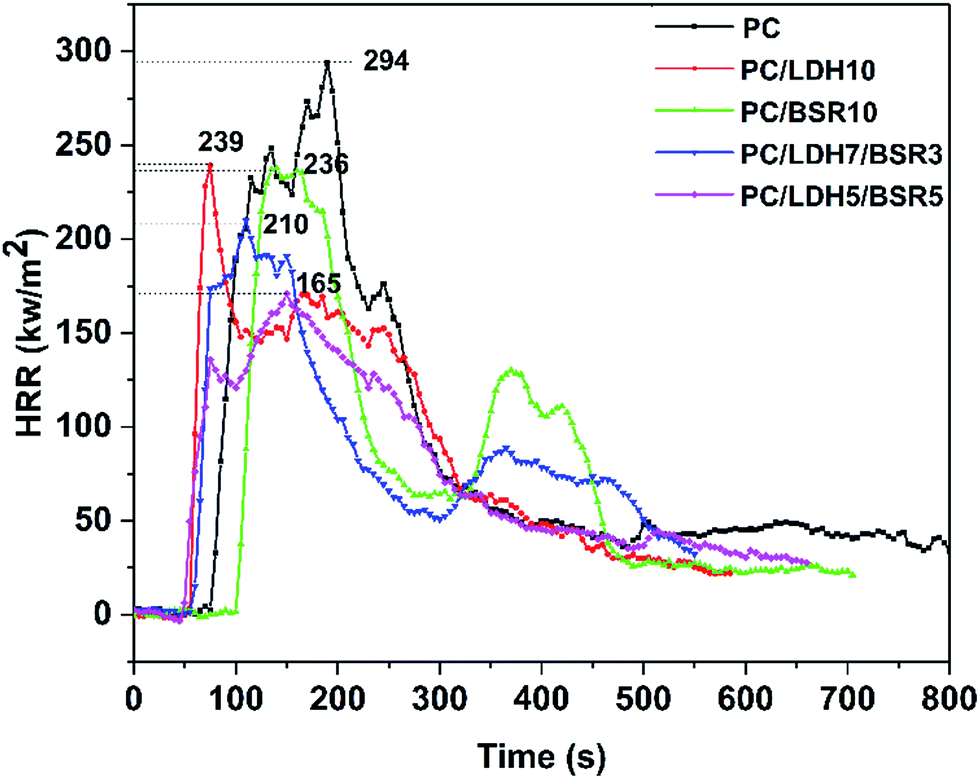 | ||
| Fig. 10 HRR versus time curves of PC and its composites determined from cone calorimeter tests. | ||
4. Conclusion
In this study, the flame retardancy of PC has dramatically improved through the synergistic effects of DBS-LDH and BSR. The results show that DBS-LDH/BSR improves the thermal stability of PC and that the TGA char yield of PC/DBS-LDH5/BSR5 at 800 °C increases to 3.75 wt% due to the synergistic effects of DBS-LDH and BSR. The results of the UL-94 vertical burning tests and the LOI tests reveal excellent flame retardancy following the addition of DBS-LDH5/BSR5 to PC. The LOI value increases to 34%, and this formulation passes the UL-94 test with a V-0 rating. The improved fire safety of the composite is mainly due to the physical barrier created by the DBS-LDH/BSR, which prevents oxygen from penetrating into the material and confines heat transmission. In addition, the formation of a better char layer protects the PC composites from further burning, which reduces the fire hazard of PC. However, with the incorporation of DBS-LDH5/BSR5, mechanical properties of PC/DBS-LDH5/BSR5 has decreased compared with pure PC. Therefore, it is a challenge for us to deal with.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
This work was supported by the Applied Science and Technology Research and Development Special Foundation of Guangdong Province (Grant No.: 2016B090930004).References
- J. Wang, H. Li, G. Li, Z. Liu, Q. Zhang, N. Wang and X. Qu, J. Appl. Polym. Sci., 2017, 134, 44978 Search PubMed.
- S. Stassi, V. Cauda, C. Ottone, A. Chiodoni, C. F. Pirri and G. Canavese, Nano Energy, 2015, 13, 474–481 CrossRef CAS.
- W. Zhang, X. Li and R. Yang, Polym. Adv. Technol., 2012, 23, 588–595 CrossRef CAS.
- H. Cai, X. Zhang, K. Xu, H. Liu, J. Su, X. Liu, Z. Fu, Y. Guo and M. Chen, Polym. Adv. Technol., 2012, 23, 765–775 CrossRef CAS.
- I. Taraghi, A. Fereidoon, S. Paszkiewicz, A. Szymczyk, R. Chylinska, A. Kochmanska and Z. Roslaniec, Polym. Adv. Technol., 2017, 28, 1794–1803 CrossRef CAS.
- S.-Q. Fu, J.-W. Guo, D.-Y. Zhu, Z. Yang, C.-F. Yang, J.-X. Xian and X. Li, RSC Adv., 2015, 5, 67054–67065 RSC.
- W. Yang, L. Song, Y. Hu, H. Lu and R. K. K. Yuen, Composites, Part B, 2011, 42, 1057–1065 CrossRef.
- W. Zhang, G. Camino and R. Yang, Prog. Polym. Sci., 2017, 67, 77–125 CrossRef CAS.
- X. Chen, W. Wang, S. Li and C. Jiao, J. Hazard. Mater., 2017, 324, 789–796 CrossRef CAS PubMed.
- X. Chen, W. Wang and C. Jiao, J. Hazard. Mater., 2017, 331, 257–264 CrossRef CAS PubMed.
- N. J. Kang, D. Y. Wang, B. Kutlu, P. C. Zhao, A. Leuteritz, U. Wagenknecht and G. Heinrich, ACS Appl. Mater. Interfaces, 2013, 5, 8991–8997 CAS.
- Z. Matusinovic and C. A. Wilkie, J. Mater. Chem., 2012, 22, 18701 RSC.
- N. Nhlapo, T. Motumi, E. Landman, S. M. C. Verryn and W. W. Focke, J. Mater. Sci., 2007, 43, 1033–1043 CrossRef.
- N. Iyi, Y. Ebina and T. Sasaki, J. Mater. Chem., 2011, 21, 8085 RSC.
- G. R. Williams, N. H. Rees and D. O'Hare, Solid State Sci., 2009, 11, 1229–1238 CrossRef CAS.
- X. Wang, E. N. Kalali and D.-Y. Wang, ACS Sustainable Chem. Eng., 2015, 3, 3281–3290 CrossRef CAS.
- E. N. Kalali, X. Wang and D.-Y. Wang, J. Mater. Chem. A, 2016, 4, 2147–2157 CAS.
- Q. Wang, J. P. Undrell, Y. Gao, G. Cai, J.-C. Buffet, C. A. Wilkie and D. O'Hare, Macromolecules, 2013, 46, 6145–6150 CrossRef CAS.
- C. Zhao, G. Peng, B. Liu and Z. Jiang, J. Polym. Res., 2011, 18, 1971–1981 CrossRef CAS.
- D. Zhou, W. He, N. Wang, X. Chen, J. Guo and S. Ci, Polym. Compos., 2016 DOI:10.1002/pc.24253.
- L. Wang, X. He, H. Lu, J. Feng, X. Xie, S. Su and C. A. Wilkie, Polym. Adv. Technol., 2011, 22, 1131–1138 CrossRef CAS.
- A. Edenharter, P. Feicht, B. Diar-Bakerly, G. Beyer and J. Breu, Polymer, 2016, 91, 41–49 CrossRef CAS.
- F. Luo, K. Wu, D. Li, J. Zheng, H. Guo, Q. Zhao and M. Lu, Polym. Compos., 2017, 38, 2762–2770 CrossRef CAS.
- D. J. McGoldrick, R. J. Letcher, E. Barresi, M. J. Keir, J. Small, M. G. Clark, E. Sverko and S. M. Backus, Environ. Pollut., 2014, 193, 254–261 CrossRef CAS PubMed.
- Y. Jiang, X. Li and R. Yang, J. Appl. Polym. Sci., 2012, 124, 4381–4388 CrossRef CAS.
- M. Doğan and E. Bayramlı, Polym. Adv. Technol., 2011, 22, 1628–1632 CrossRef.
- Q. Zhang, W. Zhang, J. Huang, Y. Lai, T. Xing, G. Chen, W. Jin, H. Liu and B. Sun, Mater. Des., 2015, 85, 796–799 CrossRef CAS.
- E. Mansour, J. Mol. Struct., 2012, 1014, 1–6 CrossRef CAS.
- K. Xie, A. Gao and Y. Zhang, Carbohydr. Polym., 2013, 98, 706–710 CrossRef CAS PubMed.
- Z. Hao, J. Zhang, Y. Wu, J. Yu and L. Yu, J. Appl. Polym. Sci., 2014, 131 DOI:10.1002/APP.40934.
- X. Wang, Y. Spörer, A. Leuteritz, I. Kuehnert, U. Wagenknecht, G. Heinrich and D.-Y. Wang, RSC Adv., 2015, 5, 78979–78985 RSC.
- M. D. Yuniati, T. Hirajima, H. Miki and K. Sasaki, Mater. Trans., 2015, 56, 1733–1741 CrossRef CAS.
- M. Środa and C. Paluszkiewicz, J. Mol. Struct., 2007, 834–836, 302–307 CrossRef.
- J. Stejskal, I. Sapurina, M. Trchová and E. N. Konyushenko, Macromolecules, 2008, 41, 3530–3536 CrossRef CAS.
- X. Yuan, Y. Wang, J. Wang, C. Zhou, Q. Tang and X. Rao, Chem. Eng. J., 2013, 221, 204–213 CrossRef CAS.
- Z. Jiang, S. Fang, C. Wang, H. Wang and C. Ji, Appl. Surf. Sci., 2016, 390, 993–1001 CrossRef CAS.
- X. Yang, J. Li, L. Xing, Y. Liao, M. Xu, Q. Huang and W. Li, Electrochim. Acta, 2017, 227, 24–32 CrossRef CAS.
- S. Wang, X. Wang, B. Jia and X. Jing, Appl. Surf. Sci., 2017, 392, 481–491 CrossRef CAS.
- M. Shabanian, N. Basaki, H. A. Khonakdar, S. H. Jafari, K. Hedayati and U. Wagenknecht, Appl. Clay Sci., 2014, 90, 101–108 CrossRef CAS.
- G. H. Lu, M. He, H. Lu, X. L. Xie and L. J. Wang, Mater. Rev. B: Res., 2015, 29, 62–67 CAS.
- B. N. Jang and C. A. Wilkie, Thermochim. Acta, 2005, 426, 73–84 CrossRef CAS.
- P. Deng, Y. Shi, Y. Liu, Y. Liu and Q. Wang, Appl. Surf. Sci., 2018, 427, 894–904 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |