
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mechanistic investigations of photochemical carbonyl elimination and oxidative addition reactions of (η5-C5H5)M(CO)3, (M = Mn and Re) complexes†
Zheng-Feng Zhanga and
Ming-Der Su *ab
*ab
aDepartment of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan. E-mail: midesu@mail.ncyu.edu.tw
bDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
First published on 20th March 2018
Abstract
We used computational methods to explore the mechanisms of the photochemical decarbonylation and the Si–H bond activation reaction of the group 7 organometallic compounds, η5-CpM(CO)3 (M = Mn and Re). The energies of both conical intersections and the intersystem crossings, which play a decisive role in these photo-activation reactions, are determined. Both intermediates and transition states in either the singlet or triplet states are also computed to furnish a mechanistic interpretation of the whole reaction paths. In the case of Mn, four types of reaction pathways (path I–path IV) that lead to the final insertion product are examined. The theoretical findings suggest that at the higher-energy band (295 nm) the singlet-state channel is predominant. As a result, the conical intersection mechanism (i.e., path I) prevails. However, at the lower-energy band (325 nm) the triplet-state channel occurs. In such a situation, the intersystem crossing mechanism (i.e., path IV) can successfully explain its CO-photodissociation mechanism. In the case of Re, on the other hand, the theoretical evidence reveals that only the singlet state-channel is superior. In consequence, the conical intersection mechanism (i.e., path V) can more effectively explain its photochemical decarbonylation mechanism. These theoretical analyses agree well with the available experimental observations.
1. Introduction
The photochemical oxidative addition of hydrosilanes to vacant coordination sites of transition-metal centers has attracted intense interest since its discovery by Graham and Jetz in 1971.1 Indeed, the study of Si–H bond activation by coordinatively unsaturated organometallic complexes represents a decisive step in the catalytic hydrosilylation of unsaturated substrates to form Si–C bonds and is of outstanding significance in organometallic chemistry, as well as for industrial processes.2–15The chemistry of 18-electron d6 CpM(CO)3 (Cp = η5-C5H5; M = Mn and Re) complexes, whose molecular structures have been described as a “three-legged-piano-stool”, have been extraordinarily studied and applied in various fields by several generations of chemists since their discovery.16–24 In particular, the photochemistry of CpMn(CO)3 has been widely investigated by many experimental laboratories because this manganese tricarbonyl complex can provide significant photochemical advantages. For instance, CpMn(CO)3 can serve as an important model system for the evolution and application of sophisticated techniques for examining excited-state dynamics.25–30 Moreover, CpMn(CO)3 can be used as a readily available precursor for a variety of substituted manganese cyclopentadienyl complexes.31–33 In addition, this manganese complex has recently been used as a photocatalyst for H2 production.34
Through the elegant researches accomplished by Harris and colleagues,35–37 the photochemical Si–H bond activation reactions by the Group VIIB, d6 η5-CpM(CO)3 (M = Mn and Re) have been experimentally explored (Scheme 1). From their study, it was found that the photolysis of η5-CpMn(CO)3, following the extrusion of one CO ligand, leads to the production of η5-CpMn(CO)2 in its singlet or triplet electronic states. On the other hand, the photolysis of one CO group from the η5-CpRe(CO)3 complex results only in the construction of η5-CpRe(CO)2 in the singlet electronic state. The subsequent reactions for both η5-CpMn(CO)2 and η5-CpRe(CO)2 intermediates can be partitioned into two routes, including the initial solvation of the dicarbonyls through either the Si–H bond or an alkyl group of the solvent species. Although both photodecarbonylation and oxidative addition reactions of (η5-C5H5)M(CO)3, (M = Mn and Re) complexes have been experimentally investigated,35–37 their photochemical mechanisms are still not understood in detail. For instance, on the basis of the available experimental results,35–37 it appears that a direct crossover from the excited state to the ground state hyper-surface should take place. However, until now, neither experimental nor theoretical study have confirmed on such photochemical reaction mechanisms of the 18-electron d6 CpM(CO)3 transition metal complexes.
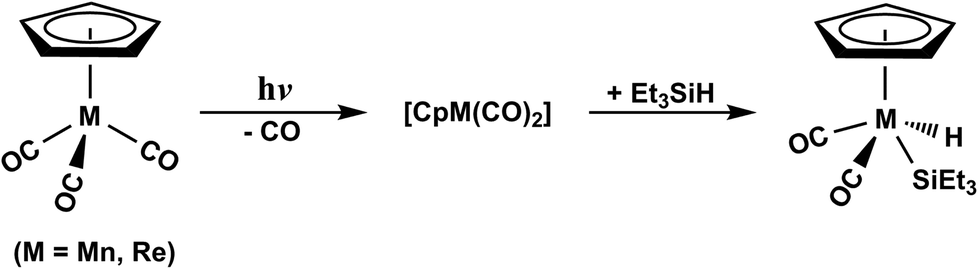 | ||
| Scheme 1 Experimental results. See ref. 35–37. | ||
It is these unexplored problems that motivate our examination of the mechanisms involved in the CO-photoelimination as well as the Si–H bond activation reaction by the d6 η5-CpM(CO)3 complexes. The reason for not studying them theoretically could be due to the fact that the sophisticated computational techniques necessary for these photochemical reactions were not yet available during the 1990s.38–41 In this work, therefore, the mechanisms for the photochemical carbonyl elimination and the Si–H bond insertion reaction in trimethylsilane using the Group 7 d6 η5-CpM(CO)3 (M = Mn and Re) molecules, eqn (A), have been theoretically undertaken.42
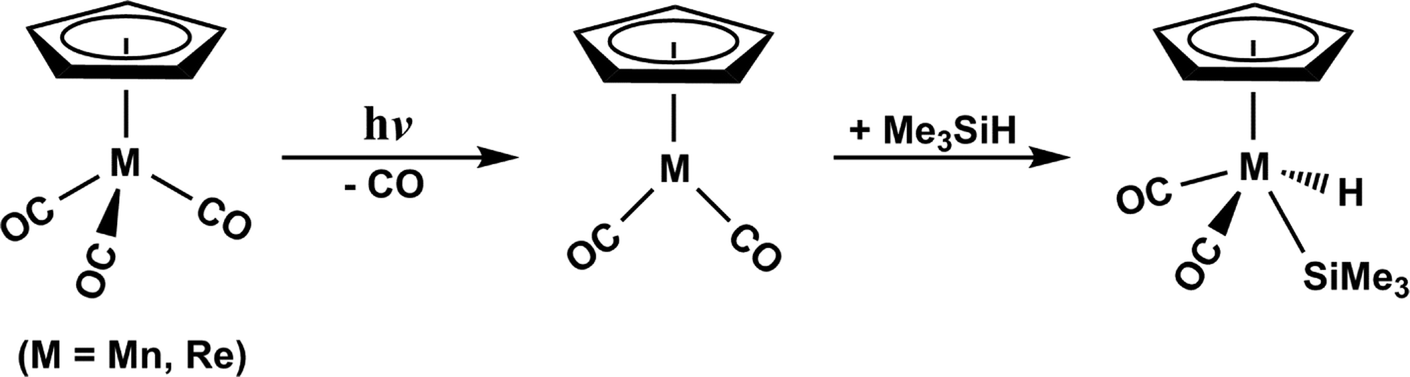 | (A) |
The aim of this work is to provide a better comprehension for the photochemical reactions of the 18-electron d6 η5-CpM(CO)3 compounds. This understanding may help to anticipate the overall reaction course of various known and/or as yet unknown η5-CpM(CO)3 systems in order to gain a superior control over them.
2. Methodology
In the current study, the GAUSSIAN 09 package of programs43 have been used to investigated the key points on the potential energy surfaces of excited-state and singlet ground states for both η5-CpMn(CO)3 and η5-CpRe(CO)3 complexes. The traditional molecular orbital energy diagram for the d6 η5-CpM(CO)3 complex with the “three-legged-piano-stool” conformation can be found elsewhere.44 According to the previous theoretical studies,38,44 the singlet ground state of the η5-CpMn(CO)3 complex with the CS symmetry should have a closed shell occupation: …(46a′)2(47a′)2(48a′′)2(49a′)2(50a′′)2(51a′)2, which corresponds to the …(πCp/x2 − y2)2(πCp/3dyz)2(πCp)2(3dxy)2(3dxz)2(3dz2 + σsp)2 configuration. See Fig. 1. In addition, five low-lying virtual orbitals conform to π*Cp/3dxz(52a′′), π*Cp/3dyz(53a′), π*Cp/3dxy(54a′), π*Cp(55a′′), π*Cp/σsp(56a′). Accordingly, an active space confined to twelve electrons correlated in eleven active orbitals has been applied. That is to say, the ground state CASSCF (complete active space self-consistent field) with an optimized geometry (12 electrons, 11 orbitals) using the active space has thus been utilized to compute energies of the critical points on both singlet and triplet potential energy surfaces.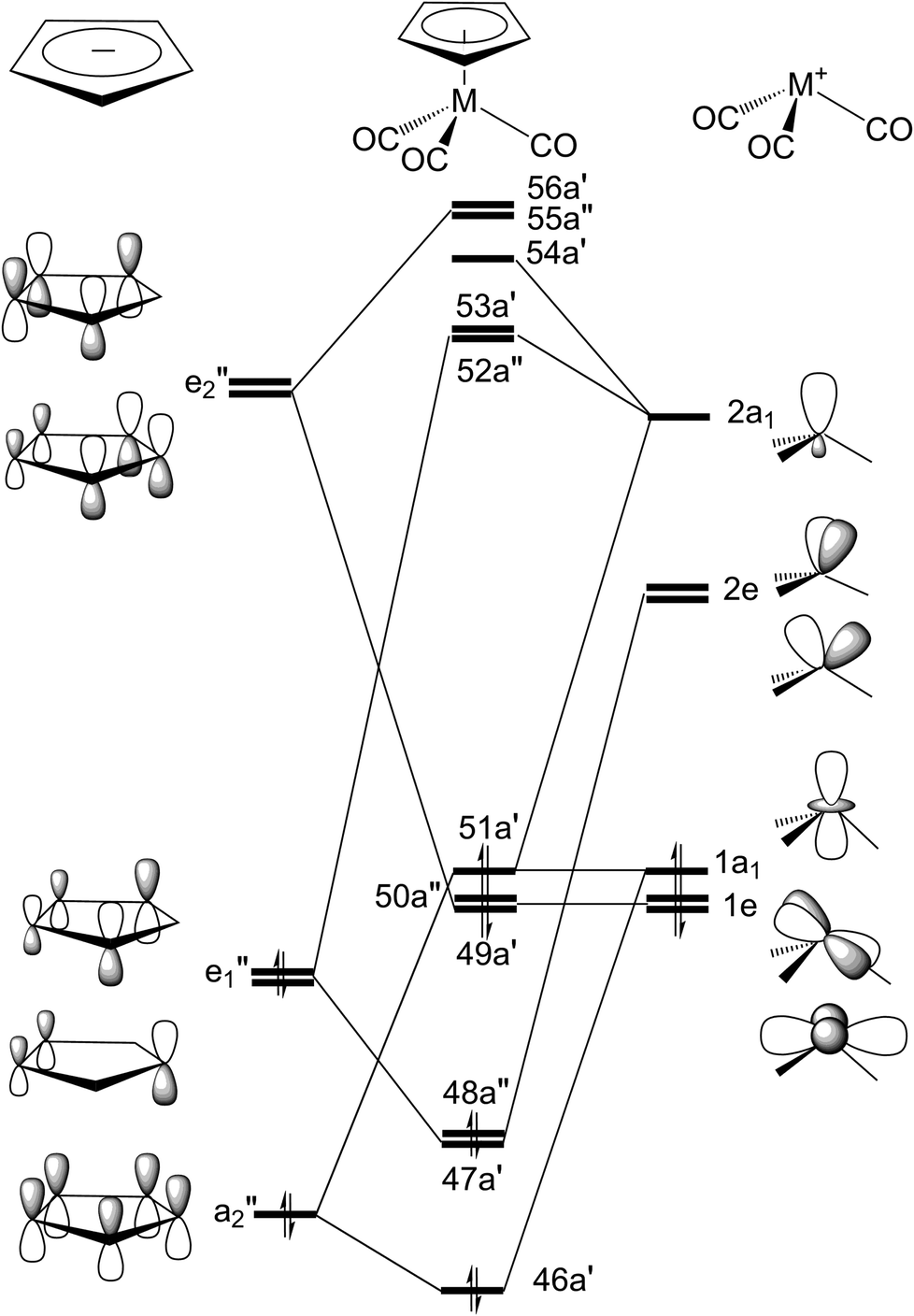 | ||
| Fig. 1 Valence molecular orbitals of the η5-CpM(CO)3 (M = Mn and Re) complex. | ||
Two kinds of sophisticated calculations were employed here. One is the calculation of the conical intersection,45–49 which is achieved in the (f − 2)-dimensional intersection space, based on the method of Bearpark et al.50 The other type of calculation is intersystem crossing optimization.45,46 Using the computational method previously mentioned with state-average orbitals,51 the optimized lowest energy points of T1/S0 surface crossings can be obtained. For this, a weighting of 50%/50% for the T1/S0 crossing was chosen to ensure the triplet and singlet states in the state-averaging procedure chosen to make sure the triplet and singlet states in the state-averaging procedure.52 Both computational methods are already implemented in the Gaussian 09 program.43
The CASSCF/Def2-SVPD53 method was initially carried out to optimize the critical points on the potential energy surfaces. After the optimizations, the CASSCF wave function was taken as the reference function and then based on this, the dynamic correlation contributions can be considered by means of a second-order perturbation procedure (MP2-CAS),54,55 in conjunction with a larger basis set (Def2-TZVPD).56 As a result, the MP2-CAS(14,13)/Def2-TZVPD//CAS(14,13)/Def2-SVPD (energies) levels of theory have been applied for all the key points on the potential energy surfaces of either the singlet or the triplet excited and the singlet ground states. Hereafter, the MP2-CAS(14,13)/Def2-TZVPD//CAS(14,13)/Def2-SVPD method will be abbreviated as MP2-CAS.
3. Results and discussion
3.1 Mechanism for the photoactivation reaction of η5-CpMn(CO)3 in the singlet state channel
The mechanism of the CO-photoextrusion reaction for η5-CpMn(CO)3 (Mn-S0-Rea) is first examined and discussed in this section. On the basis of the available experimental reports,35–37 it was found that the photolysis of the Mn tricarbonyl complex under the excitations at both 295 nm (=96.9 kcal mol−1) and 325 nm (=88.0 kcal mol−1) yields η5-CpMn(CO)2 in its singlet or triplet electronic states. The CAS(12,11)/Def2-SVPD method has thus been utilized to compute the vertical excitation energies of the Franck–Condon (FC) zone. It should be noted that the FC zone maintains the geometry of Mn-S0-Rea in its ground state (S0) even in its singlet or triplet excited states. The CASSCF computational results given on the left-hand side of Fig. 2 indicate that the energy levels of the excited states increase in the order: Mn-S0-Rea (0.0 kcal mol−1) < Mn-T1-FC (69.0 kcal mol−1) < Mn-T2-FC (75.6 kcal mol−1) < Mn-S1-FC (83.8 kcal mol−1) < Mn-S2-FC (92.8 kcal mol−1) < Mn-T3-FC (104.8 kcal mol−1) < Mn-S3-FC (107.4 kcal mol−1). It should be emphasized here that the energy splittings of the organometallic compounds are usually much smaller than those of the organic molecules, since the transition-metal atom has five d orbitals, while the carbon atom has 3 p orbitals. As a consequence, once light is absorbed by the organometallic species, it may jump to the low-lying unoccupied orbitals, from which it would produce several nearly degenerate states, such as the metal-centered (MC) or metal-to-ligand charge-transfer (MLCT) states. Subsequently, this excited molecule relaxes through the conical intersection (CI) or the intersystem crossing (ISC) adjacent to the Frank–Condon zone.52–55 Fig. 2 demonstrates that when Mn-S0-Rea is photo-irradiated with 295 nm light into an excited electronic state, Mn-S0-Rea may arrive at a singlet excited electronic state (Mn-S2-FC). It then relaxes, branching between the S1 and S0 or T1 and S0 states. The same phenomenon also occurs in the case of Mn-S0-Rea under irradiation with 325 nm light. On the basis of the present computational results, it is therefore concluded that the photochemical carbonylation reaction of Mn-S0-Rea would take place from either a singlet excited state (Mn-S1-FC) or a triplet excited state (Mn-T1-FC) within the FC zone, which agrees well with available experimental findings.35–37 The CAS(14,13)/Def2-SVPD optimized structures for path I of η5-CpMn(CO)3 (Mn-S0-Rea) are collected in Fig. 3 and 4.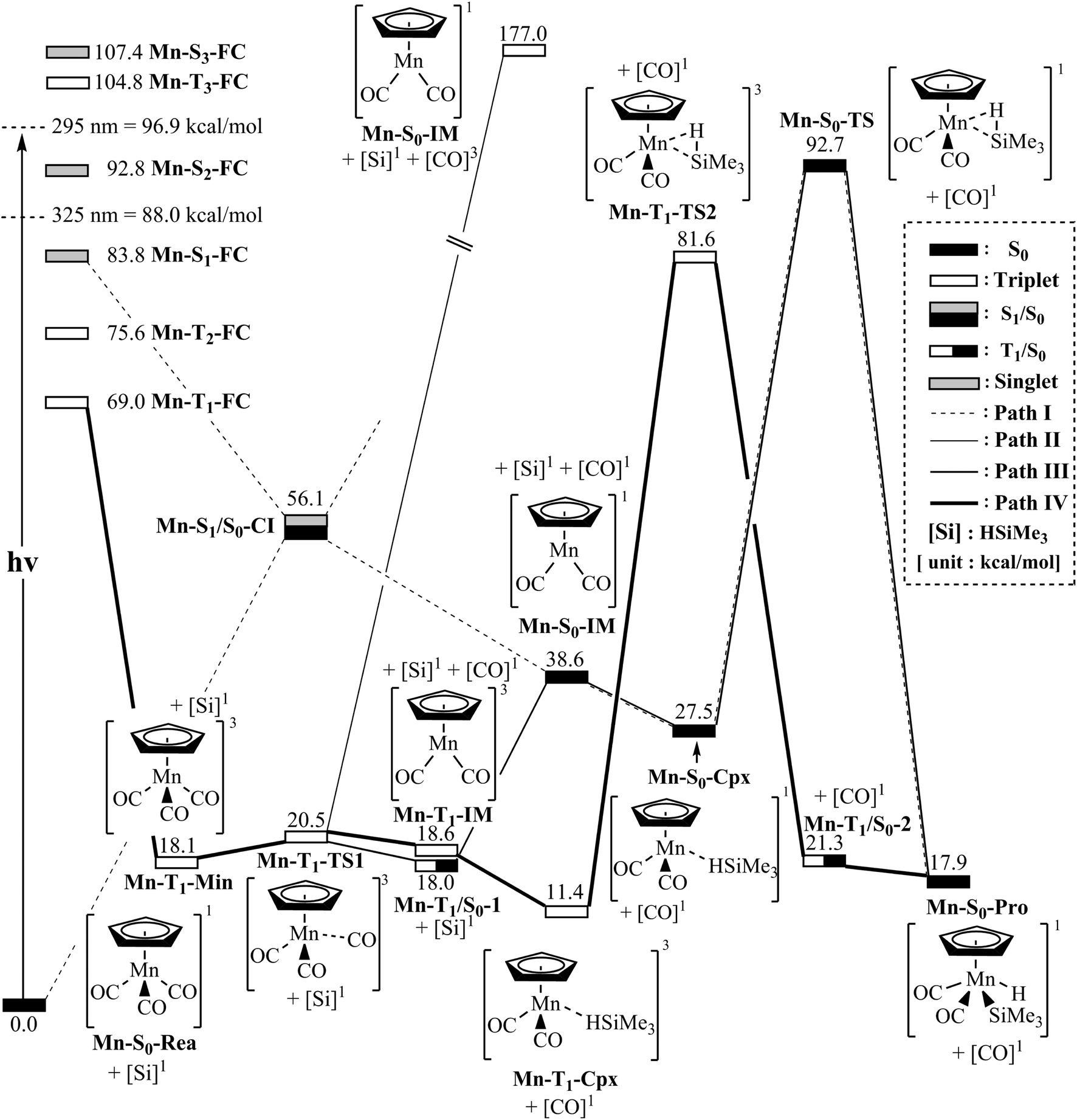 | ||
| Fig. 2 Energy profiles for the photo-extrusion reactions for η5-CpMn(CO)3 (Mn-S0-Rea). The abbreviations, FC and CI, respectively represent the Frank–Condon and conical intersection. The relative energies are given at the MP2-CAS-(14,13)/Def2- TZVPD//CAS(14,13)/Def2-SVPD level of theory. All energies (in kcal mol−1) are given with respect to the reactant (Mn-S0-Rea). For the crucial points of the CASSCF optimized structures, see Fig. 3. For more information, see the text. | ||
 | ||
| Fig. 3 The CAS(14,13)/Def2-SVPD geometries (in Å and deg) for path I (the singlet state) of η5-CpMn(CO)3 (Mn-S0-Rea), the conical intersection (CI) and the final product (Pro). The derivative coupling and the gradient difference vectors are computed using CASSCF at the conical intersection, Mn-S1/S0-CI. Some hydrogen atoms are omitted for clarity. | ||
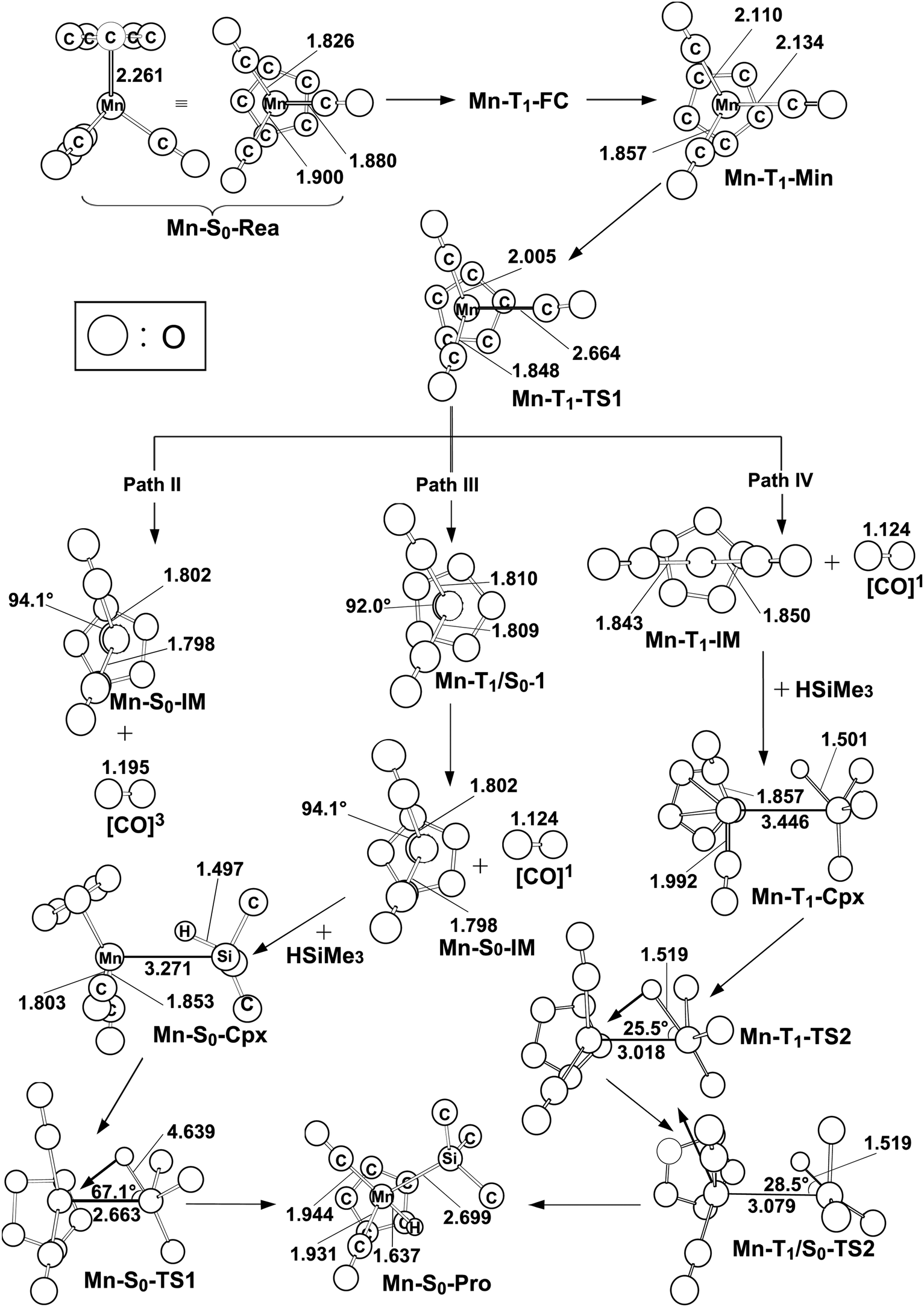 | ||
| Fig. 4 The CAS(14,13)/Def2-SVPD geometries (in Å and deg) for paths II, III, and IV (triplet states) of the critical points of reactant η5-CpMn(CO)3 (Mn-S0-Rea) on the potential energy surfaces. The bold arrows indicate the principal atomic motions in the transition state eigenvector. The relative energies for each species see Fig. 1. Some hydrogen atoms are omitted for clarity. | ||
From the above analysis, we initially investigate the photochemical oxidative addition reaction of Mn-S0-Rea through the singlet electronic state channel. As Fig. 2 shows, after being photo-irradiated by 295 nm light, Mn-S0-Rea may finally move to the photo-excited Mn-S1-FC. Then, this excited species undergoes a radiation-less decay to a singlet [η5-CpMn(CO)2]1 intermediate (Mn-S0-IM) via a conical intersection point (Mn-S1/S0-CI). Our MP2-CAS calculations indicate that Mn-S1/S0-CI and Mn-S0-IM are about 56 and 39 kcal mol−1 above the ground-state reactant (Mn-S0-Rea). Subsequently, this singlet dicarbonyl intermediate would interact with trimethylsilane to yield a singlet precursor complex (Mn-S0-Cpx), which is computed to be about 28 kcal mol−1 above the starting material. In other words, the complexation energy of the Mn-S0-IM is about 11 kcal mol−1. It is noteworthy that since the complexation energy of Mn-S0-Cpx is quite low, compared to the further barrier height (65 kcal mol−1) from Mn-S0-Cpx to Mn-S0-TS, the Mn-S0-Cpx complex should be easily detected by experiments. Indeed, this theoretical finding is confirmed by the available experimental observations.35–37 This singlet dicarbonyl species next undergoes intermolecular oxidative addition to a Si–H bond of (CH3)3Si–H by way of a transition state (Mn-S0-TS),57 whose energy is computed to be about 93 kcal mol−1 above Mn-S0-Rea. Since the initial photo-excitation energy is experimentally reported to be 96.9 kcal mol−1 (=295 nm),35–37 the barrier height (65 kcal mol−1) of this oxidative addition reaction should be easily overcome to achieve the final product (Mn-S0-Pro), whose energy is estimated to be about 18 kcal mol−1 above the initial reactants (Mn-S0-Rea and (CH3)3SiH) as shown in Fig. 2. Accordingly, the present theoretical findings reveal that the mechanism for the photochemical singlet reaction channel (path I) of the η5-CpMn(CO)3 complex should be represented as follows: ([Si]1 acts for (CH3)3SiH in the singlet ground state)
| Path I: Mn-S0-Rea + [Si]1 + hν → Mn-S1-FC + [Si]1 → Mn-S1/S0-CI + [Si]1 → Mn-S0-IM + [Si]1 + [CO]1 → Mn-S0-Cpx + [CO]1 → Mn-S0-TS + [CO]1 → Mn-S0-Pro + [CO]1 |
In brief, we proceed on the basis of the experimental works published by Harris and colleagues,35–37 who found that irradiation of η5-CpMn(CO)3 with the 295 nm light can lead to the higher-energy band, which associates with the formation of singlet [η5-CpMn(CO)2] (Mn-S0-IM) and then interact with the hydrosilanes to result in the oxidative addition products. The present computational results (path I) given in Fig. 2 are in accordance with the previous experimental findings.35–37
3.2 Mechanisms for the photoactivation reaction of η5-CpMn(CO)3 in the triplet state channel
On the other hand, when Mn-S0-Rea is photo-irradiated to its highly excited electronic states, then it may relax to the first triplet excited FC (Mn-T1-FC) point. Starting from Mn-T1-FC, this triplet excited FC species would decay to the lowest triplet state minimum (Mn-T1-Min). It may then undergo three possible reaction paths (i.e., path II, path III, and path IV), as shown in Fig. 2, to encounter an oxidative addition reaction with trimethylsilane and thereby generate the final insertion product (Mn-S0-Pro).In path II, the triplet intermediate (Mn-T1-Min) dissociates one CO group through a transition state (Mn-T1-TS1) to obtain two final products: one triplet CO molecule and one singlet manganese dicarbonyl complex (Mn-S0-IM). Nevertheless, our MP2-CAS computations suggest that the relative energy of the final points ([CO]3 + Mn-S0-IM) is estimated to be 177 kcal mol−1, which is much higher than the initial photoirradiation energy (295 nm = 96.9 kcal mol−1). As a result, the theoretical evidence indicates that the formation of one triplet CO and one singlet Mn-S0-IM molecule by way of the photoirradiation of Mn-S0-Rea with 295 nm light is very unlikely. Indeed, as far as we are aware, no such photoproducts have been experimentally detected in the photo-activation reaction for η5-CpMn(CO)3.35–37 The mechanism for path II of Mn-S0-Rea is shown as follows:
| Path II: Mn-S0-Rea + [Si]1 + hν → Mn-T1-FC + [Si]1 → Mn-T1-Min + [Si]1 → Mn-T1-TS1 + [Si]1 → [CO]3 + Mn-S0-IM + [Si]1 |
In path III, when the triplet manganese species (Mn-T1-Min) encounters one CO ligand dissociation via a triplet transition state (Mn-T1-TS1), it may subsequently proceed an intersystem crossing from the triplet state to the singlet state in the region of the T1/S0 intersection (Mn-T1/S0-1), as demonstrated in Fig. 2. From this intersection point, the manganese species produces one singlet CO molecule and one singlet Mn-S0-IM intermediate. Then, Mn-S0-IM would interact with trimethylsilane, following a reaction pathway similar to that shown above for path I (conical intersection path), to approach an oxidative addition η5-CpMn(CO)2(H) (SiMe3) molecule (Mn-S0-Pro). The mechanism for path III can thus be represented as follows:
| Path III: Mn-S0-Rea + [Si]1 + hν → Mn-T1-FC + [Si]1 → Mn-T1-Min + [Si]1 → Mn-T1-TS1 + [Si]1 → Mn-T1/S0-1 + [Si]1 → Mn-S0-IM + [Si]1 + [CO]1 → Mn-S0-Cpx + [CO]1 → Mn-S0-TS + [CO]1 → Mn-S0-Pro + [CO]1 |
As mentioned in previous research,35–37 there are two possibilities for the mechanisms of triplet reaction channels involved in the experimentally accessible region under irradiation of the parent η5-CpMn(CO)3 with 295 nm and 325 nm light. Two points are noteworthy as follows:
(i) At the higher-energy band (using the 295 nm light): It is well known that spin-allowed absorption cross-sections are basically larger than those for spin-forbidden excitations.58–63 Moreover, as seen in the left-hand side of Fig. 2, once Mn-S0-Rea is absorbed by light with 295 nm, the possibility for the Mn complex relaxing from the singlet FC zone to the triplet region is quite slim, since the excited Mn complex would initially jump to the nearby singlet excited state. Therefore, in principle, this 295 nm process should follow path I (the conical intersection mechanism; from the singlet excited state to the singlet ground state) to yield the final oxidative addition product (Mn-S0-Pro).
(ii) At the lower-energy band (using the 325 nm light): The theoretical results shown in Fig. 2 reveal that once the Mn complex starts from the Mn-T1-FC point, it may follow path III to proceed the Si–H activation reaction. However, the relative energy of Mn-S0-TS (93 kcal mol−1) with respect to the initial reactants (Mn-S0-Rea and (CH3)3SiH) is higher than the vertical photoexcitation energy of the 325 nm (=88.0 kcal mol−1) light. This strongly implies that if Mn-S0-Rea relies upon absorption of light by 325 nm, path III (the intersystem crossing mechanism) must be energetically unfeasible for the production of an oxidative addition Mn-S0-Pro molecule.
In short, based on the above analyses, it is expected that under UV photoirradiation, neither 295 nm nor 325 nm, Mn-S0-Rea proceeds along path III to form the final photoproduct, Mn-S0-Pro.
In the forth pathway, path IV, Mn-T1-Min can form one singlet CO molecule and one triplet [η5-CpMn(CO)2]3 complex (Mn-T1-IM) through a triplet transition state, Mn-T1-TS1. The MP2-CAS results represented in Fig. 2 suggest that the energy of this photo-induced point ([CO]1 + Mn-T1-IM) lies only about 19 kcal mol−1 above that of the starting point (Mn-S0-Rea). Subsequently, the Mn-T1-IM interacts with (CH3)3Si–H to form the precursor complex (Mn-T1-Cpx) in the triplet state. Then this triplet Mn-T1-Cpx complex undergoes a triplet transition state (Mn-T1-TS2) to reach the final singlet insertion (Mn-S0-Pro) through the Mn-T1/S0-1 intersystem crossing point. In other words, if the starting point (Mn-S0-Rea) absorbs light of 325 nm (=88.0 kcal mol−1) wavelength, the manganese complex has more than enough energy to overcome the barrier height (70.2 kcal mol−1) from Mn-T1-Cpx to Mn-T1-TS2. This manganese tricarbonyl molecule can then eventually reach the final oxidative addition compound (Mn-S0-Pro). Consequently, the mechanism for path IV of the η5-CpMn(CO)3 complex can be described as follows:
| Path IV: Mn-S0-Rea + [Si]1 + hν → Mn-T1-FC + [Si]1 → Mn-T1-Min + [Si]1 → Mn-T1-TS1 + [Si]1 → Mn-T1-IM + [Si]1 + [CO]1 → Mn-T1-Cpx + [CO]1 → Mn-T1-TS2 + [CO]1 → Mn-T1/S0-2 + [CO]1 →Mn-S0-Pro + [CO]1 |
In short, the present theoretical observations strongly demonstrate that the process of the irradiation on η5-CpMn(CO)3 with 325 nm light could follow either path I (the conical intersection mechanism, from singlet excited state to singlet ground state) or path IV (the intersystem crossing mechanism, from triplet excited state to singlet ground state) rather than path II or path III. Indeed, this theoretical conclusion agrees well with the available experimental findings, in which the irradiation of the 325 nm light would produce the spin-crossover product (Mn-S0-Pro) through the intersystem crossing channel.35–37
3.3 Mechanism for the photoactivation reaction of η5-CpRe(CO)3 in the singlet state channel
In this section, we discuss the mechanisms of the photochemical decarbonylation reaction and the oxidative addition reaction of the 18-electron η5-CpRe(CO)3 (Re-S0-Rea) molecule. The vertical excitation energies of the FC zone for the η5-CpRe(CO)3 complex, together with the relative energies of the key points with respect to the starting materials (Re-S0-Rea + (CH3)3SiH), were all calculated using the MP2-CAS method as mentioned above in the methodology section. Our computational results are summarized in Fig. 5 and the optimized geometries of the critical points on the Re potential energy surfaces are collected in Fig. 6.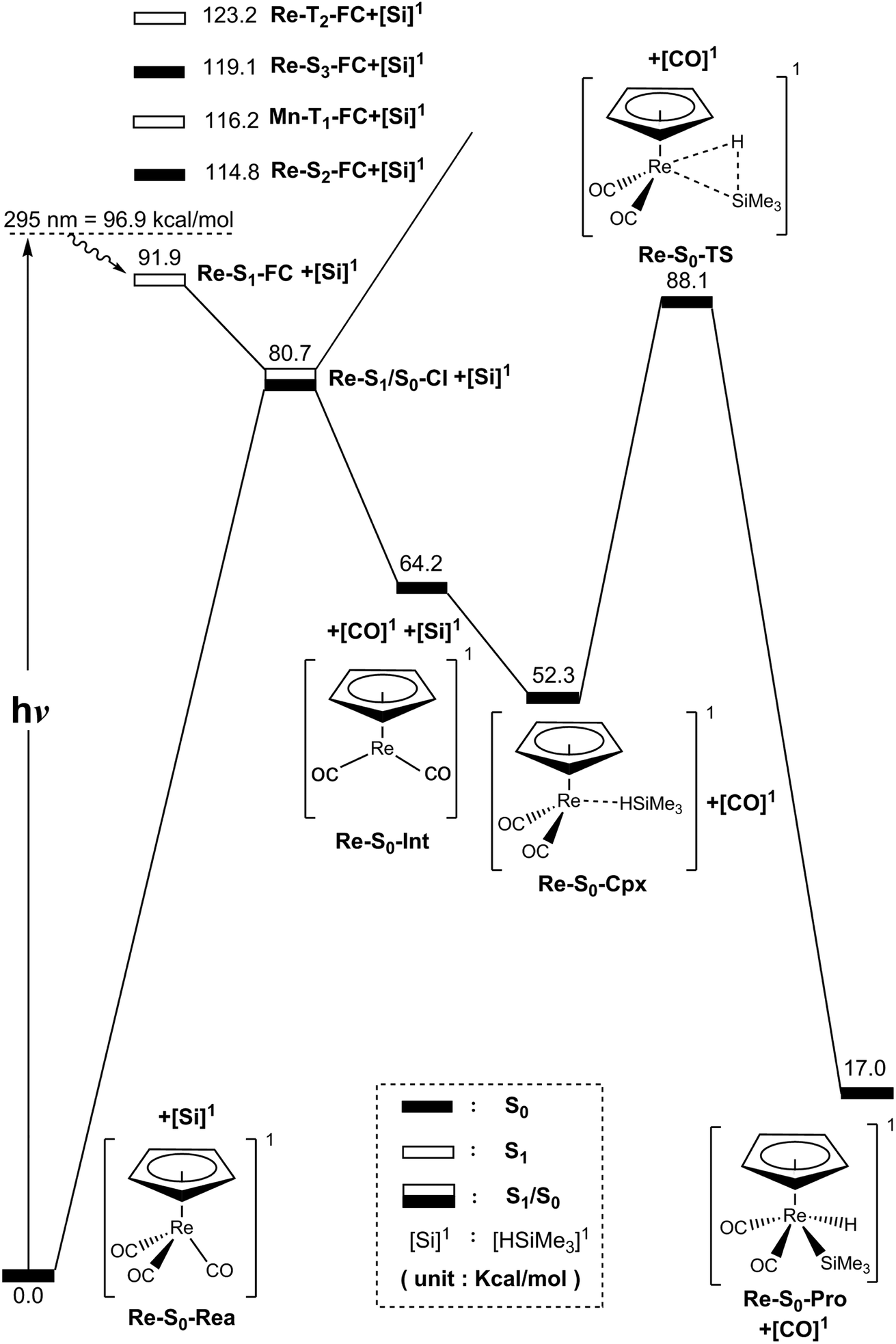 | ||
| Fig. 5 Energy profiles for the photo-extrusion reactions for η5-CpRe(CO)3 (Re-S0-Rea). The abbreviations, FC and CI, respectively, represent the Frank–Condon and conical intersection. The relative energies are given at the MP2-CAS-(14,13)/Def2- TZVPD//CAS(14,13)/Def2-SVPD level of theory. All energies (in kcal mol−1) are given with respect to the reactant (Re-S0-Rea). For the crucial points of the CASSCF optimized structures, see Fig. 6. For more information, see the text. | ||
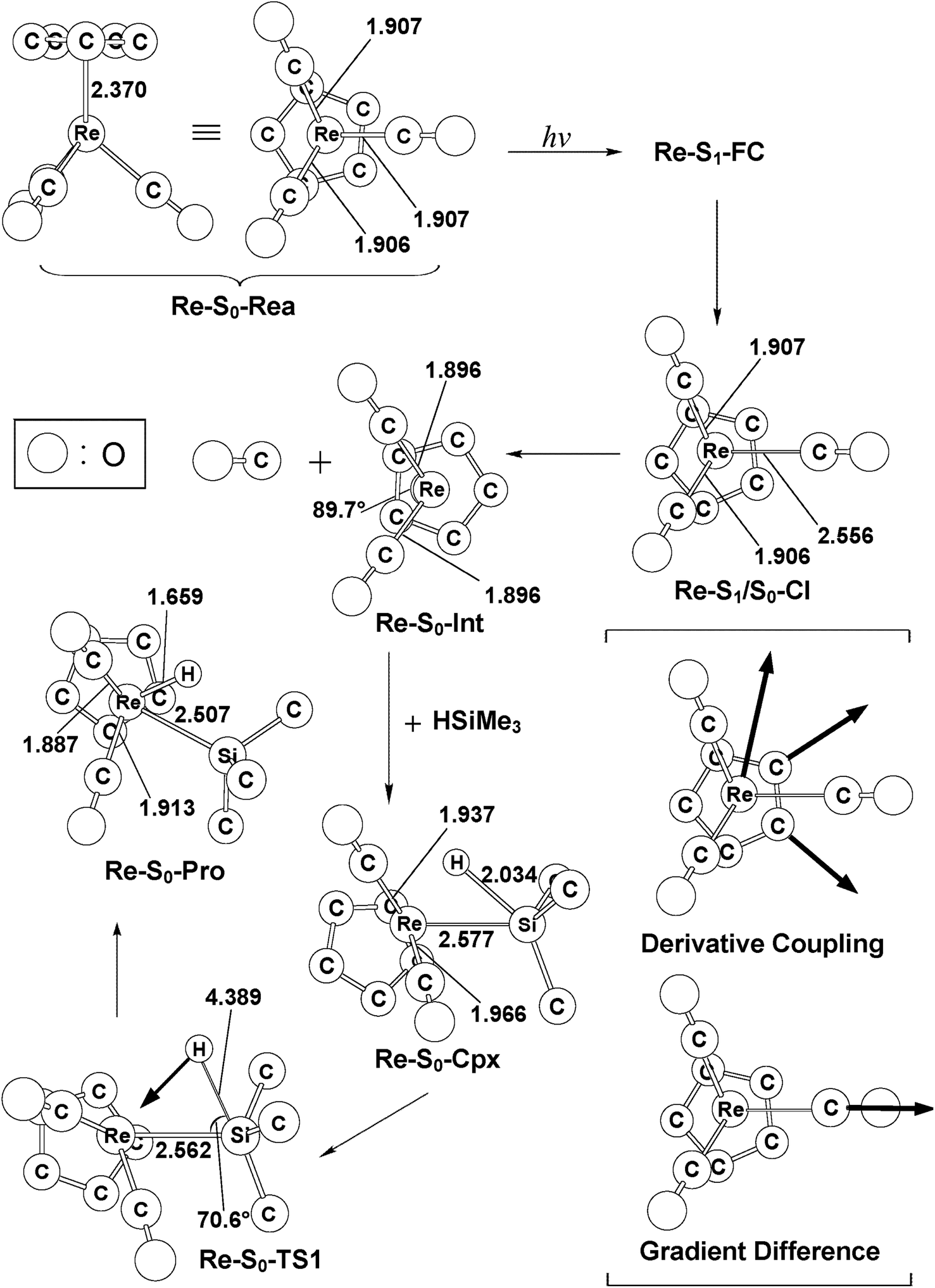 | ||
| Fig. 6 The CAS(14,13)/Def2-SVPD geometries (in Å and deg) for η5-CpRe(CO)3 (Re-S0-Rea), the conical intersection (CI) and the final product (Pro). The derivative coupling and the gradient difference vectors are computed using CASSCF at the conical intersection, Re-S1/S0-CI. The corresponding CASSCF vectors are shown in the inset. | ||
As seen in the left-hand side of Fig. 5, the MP2-CAS calculations represent the relative FC energies increase in the order: Re-S0-Rea (0.0 kcal mol−1) < Re-S1-FC (91.90 kcal mol−1) < Re-S2-FC (114.8 kcal mol−1) < Re-T1-FC (116.2 kcal mol−1) < Re-S3-FC (119.1 kcal mol−1) < Re-T2-FC (123.2 kcal mol−1). These computational values clearly reveal that only the energy of the singlet Re-S1-FC point is below that of the reported irradiation light with 295 nm (=96.9 kcal mol−1).35–37 In other words, the MP2-CAS computations provide strong theoretical evidence that the CO-photoextrusion mechanism of the η5-CpRe(CO)3 complex should advance on the singlet potential energy surface. Indeed, it was experimentally found that photolysis of the Re-S0-Rea complex led only to a rhenium dicarbonyl η5-CpRe(CO)2 species in its singlet electronic ground state.35–37 We therefore focus on the singlet state through the whole reaction mechanism in the case of the Re complex (eqn (A)). That is to say, the conical intersection mechanism will be applied here to interpret the CO-photoextrusion process of the Re-S0-Rea compound.
Starting from the Re-S1-FC point, as seen in Fig. 5, the Re tricarbonyl funnels through S1/S0 conical intersection point (i.e., Re-S1/S0-CI), leading to one singlet [η5-CpRe(CO)2]1 intermediate (i.e., Re-S0-Int) and one singlet CO molecule. This theoretical finding has been confirmed by the experimental observations mentioned above.35–37 Subsequently, Re-S0-Int interacts with (CH3)3SiH to generate a singlet precursor complex, Re-S0-Cpx. This Re system then undergoes a Si–H bond activation reaction by way of a transition state (Re-S0-TS) to generate the final oxidative addition product (Re-S0-Pro). As given in Fig. 5, the present theoretical computations estimate that with respect to the energy of the starting materials (Re-S0-Rea + [Si]1), the Re-S1/S0-CI + [Si]1, Re-S0-Int + [Si]1, Re-S0-Cpx, Re-S0-TS, and Re-S0-Pro points are predicted to be 81, 64, 52, 88, and 17 kcal mol−1, based on the MP2-CAS computations. All of these energy values are smaller than that of the photoirradiation promotion energy with 295 nm light (=96.9 kcal mol−1). As a consequence, path V is anticipated to be energetically accessible since the reactant (Re-S0-Rea) has more than enough energy (96.9 kcal mol−1) to overcome the barrier height (35.8 kcal mol−1) from Re-S0-Cpx to Re-S0-TS when Re-S0-Rea absorbs light of the 295 nm wavelength. The mechanism for path V is thus described as follows:
| Path V: Re-S0-Rea + [Si]1 + hν → Re-S1-FC + [Si]1 → Re-S1/S0-CI + [Si]1 → Re-S0-Int + [Si]1 + [CO]1 → Re-S0-Cpx + [CO]1 → Re-S0-TS + [CO]1 → Re-S0-Pro + [CO]1 |
4. Conclusion
In this work, we used both CASSCF and MP2-CAS levels of theory to investigate the mechanisms of the CO-photoextrusion and the oxidative addition of a Si–H bond to the Group 7 transition metal complexes η5-CpM(CO)3 (M = Mn and Re). To our knowledge, this study represents the first theoretical examinations on the photochemical mechanisms of the cyclopentadiene tricarbonyl systems. Fig. 2 (Mn) and Fig. 5 (Re) demonstrate the comprehensive reaction mechanisms for the two kinds of organometallic molecules.42 As a result, this theoretical study allows four noteworthy conclusions to be drawn, as follows:(1) Although manganese and rhenium belong to the same family in the periodic table, our theoretical investigations strongly demonstrate that their photochemical reaction mechanisms are quite different from each other. This difference could be owing to the fact that their photochemical activities strongly depend on the nature of the electronic structures of such cyclopentadiene tricarbonyl complexes.
(2) The MP2-CAS computational results presented in this work strongly support the experimental evidence,35–37 in which the CO-photolysis of the η5-CpMn(CO)3 (Mn-S0-Rea) complex can generate a singlet dicarbonyl (Mn-S0-IM) and a triplet dicarbonyl (Mn-T1-IM) intermediate, whereas the CO-photoextrusion of the η5-CpRe(CO)3 (Mn-S0-Rea) molecule can generate only the singlet dicarbonyl transient complex (Re-S0-Int).
(3) In the case of η5-CpMn(CO)3 (Mn-S0-Rea), as shown in Fig. 2, the MP2-CAS results anticipate that photolysis of Mn-S0-Rea with 295 nm (the higher-energy band) can lead to the singlet state channel to be predominant. As a result, the Mn complex would follow path I (the conical intersection mechanism)39 to produce the final oxidative addition product (Mn-S0-Pro). On the other hand, the MP2-CAS calculations also predict that CO-photoelimination of Mn-S0-Rea with 325 nm (the lower-energy band) can result in the triplet state process to be prevailing. Therefore, this Mn molecule would pursue path IV (the intersystem crossing mechanism)39 to yield the final insertion product (Mn-S0-Pro).
(4) In the case of η5-CpRe(CO)3 (Re-S0-Rea), as shown in Fig. 5, our theoretical computations strongly predict that CO-photoextrusion of Re-S0-Rea with 295 nm can generate only the singlet state channel. As a consequence, using the conical intersection mechanism39 can successfully explain the photochemical decarbonylation of the Re complex.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Center for High-Performance Computing of Taiwan for generous amounts of computing time, and the Ministry of Science and Technology of Taiwan for the financial support. In particular, one of the authors (M.-D. Su) also wishes to thank Professor Michael A. Robb, Dr S. Wilsey, Dr Michael J. Bearpark, (University of London, UK) and Professor Massimo Olivucci (Universita degli Studi di Siena, Italy), for their encouragement and support during his stay in London. Special thanks are also due to reviewers 1 and 2 for very helpful suggestions and comments.References
- W. A. G. Graham and W. Jetz, Silicon-transition Metal Chemistry. I. Photochemical Preparation of Silyl(transition metal) Hydrides, Inorg. Chem., 1971, 10, 4–9 CrossRef CAS.
- B. Marciniec, Comprehensive Handbook on Hydrosilation, Pergamon Press, Oxford, UK, 1992 Search PubMed.
- B. Marciniec, Hydrosilation: A Comprehensive Review on Recent Advances, Springer, 2008 Search PubMed.
- A. Bande and J. Michl, Conformational Dependence of Sigma-electron Delocalization in Linear Chains: Permethylated Oligosilanes, Chem.–Eur. J., 2009, 15, 8504–8517 CrossRef CAS PubMed.
- R. D. Miller and J. Michl, Polysilane High Polymers, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS.
- G. I. Nikonov, Recent Advances in Nonclassical Interligand Si⋯H Interactions, Adv. Organomet. Chem., 2005, 53, 217–309 CrossRef CAS.
- R. Malacea, R. Poli and E. Manouri, Asymmetric hydrosilylation, Transfer Hydrogenation and Hydrogenation of Ketones Catalyzed by Iridium Complexes, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS.
- R. H. Morris, Asymmetric hydrogenation, transfer hydrogenation and hydrosilylation of ketones catalyzed by iron complexes, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- S. Diaz-Gonzaĺez and S. P. Nolan, Copper, Silver, and Gold Complexes in Hydrosilylation Reactions, Acc. Chem. Res., 2008, 41, 349–358 CrossRef PubMed.
- A. K. A. Roy, Review of Recent Progress in Catalyzed Homogeneous Hydrosilation (Hydrosilylation), Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef.
- D. Julienne, O. Delacroix and A. C. Gaumont, An Overview of the Synthesis of Alkenylphosphines, Curr. Org. Chem., 2010, 14, 457–482 CrossRef CAS.
- S. Greenberg and D. W. Stephan, Stoichiometric and Catalytic Activation of P–H and P–P Bonds, Chem. Soc. Rev., 2008, 37, 1482–1489 RSC.
- L. Coudray and J. L. Montchamp, You have full text access to this content Recent Developments in the Addition of Phosphinylidene-Containing Compounds to Unactivated Unsaturated Hydrocarbons: Phosphorus–Carbon Bond Formation by Hydrophosphinylation and Related Processes, Eur. J. Org. Chem., 2008, 21, 3601–3613 CrossRef PubMed.
- G. Alcaraz, M. Grellier and S. Sabo-Etienne, Bis σ-Bond Dihydrogen and Borane Ruthenium Complexes: Bonding Nature, Catalytic Applications, and Reversible Hydrogen Release, Acc. Chem. Res., 2009, 42, 1640–1649 CrossRef CAS PubMed.
- C. Deutsch, N. Krause and B. H. Lipshutz, CuH-Catalyzed Reactions, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed.
- M. S. Wrighton, Photochemistry of Metal Carbonyls, Chem. Rev., 1974, 74, 401–430 CrossRef CAS.
- T. M. Barnhart, R. F. Fenske and R. J. McMahon, Isomerism in Coordinatively Unsaturated Tricarbonyl-.eta.2-etheneiron Complexes, Inorg. Chem., 1992, 31, 2679–2681 CrossRef CAS.
- T. M. Barnhart and R. J. McMahon, Spectroscopic Observation of a Thermal Carbon-hydrogen Bond Insertion Reaction at 5 K: intramolecular Rearrangement of Fe(CO)3(.eta.2-C3H6) to Produce HFe(CO)3(.eta.3-C3H5), J. Am. Chem. Soc., 1992, 114, 5434–5435 CrossRef CAS.
- T. M. Barnhart, J. De Felippis and R. J. McMahon, Structure and Reactivity of [HFe(CO)3(η3
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3H5)], Angew. Chem., Int. Ed. Engl., 1993, 32, 1073–1074 CrossRef.
C3H5)], Angew. Chem., Int. Ed. Engl., 1993, 32, 1073–1074 CrossRef. - P. J. Giordano and M. S. Wrighton, Photosubstitution Behavior of Dicarbonyl(.eta.5-cyclopentadienyl)pyridinomanganese and -Rhenium and Related Complexes, Inorg. Chem., 1977, 16, 160–166 CrossRef CAS.
- M. P. Thornberry, C. Slebodnick, P. A. Deck and F. R. Fronczek, Synthesis and Structure of Piano Stool Complexes Derived from the Tetrakis(pentafluorophenyl)cyclopentadienyl Ligand, Organometallics, 2001, 20, 920–926 CrossRef CAS.
- (a) R. J. Batchelor, F. W. B. Einstein, R. H. Jones, J.-M. Zhuang and D. Sutton, Photochemical Carbon-hydrogen Bond Activation of Coordinated Propene in (.Eta.5-C5Me5)Re(CO)2(.eta.2-C3H6). X-ray Structure Determination of Exo and Endo Isomers of the Resulting.Eta.3-allyl(hydrido) Complex (.eta.5-C5Me5)Re(H)(CO)(.eta.3-C3H5), J. Am. Chem. Soc., 1989, 111, 3468–3469 CrossRef CAS; (b) J.-M. Zhuang and D. Sutton, Photolysis of (.Eta.5-pentamethylcyclopentadienyl)rhenium Tricarbonyl with Alkenes. Synthesis of (.eta.5-C5Me5)Re(CO)2(alkene) Complexes and Carbon-hydrogen Activation to Give Allyl Hydrido Derivatives, Organometallics, 1991, 10, 1516–1527 CrossRef CAS.
- J. Chetwynd-Talbot, P. Grebenik and R. Perutz, [(η-C5H5)2ReH] and [(η-C5H5)(η2-C5H6)Re(CO)2] in Low Temperature Matrices: Hydrogen Loss and Hydrogen Migration, J. Chem. Soc., Chem. Commun., 1981, 452–454 RSC.
- J. Chetwynd-Talbot, P. Grebenik and R. N. Perutz, Photochemical Studies of Rhenium.Eta.-cyclopentadienyl Complexes in Matrixes and in Solution: Detection of Rhenocene and Mechanisms of Hydrogen Transfer, Inorg. Chem., 1983, 22, 1675–1684 CrossRef CAS.
- J. Full, L. Gonzaĺez and C. Daniel, Photochemistry of A CASSCF/CASPT2 and TD-DFT Study of the Low-Lying Excited States of η5-CpMn(CO)3, J. Phys. Chem. A, 2001, 105, 184–189 CrossRef CAS.
- C. Daniel, J. Full, L. Gonzaĺez, C. Lupulescu, J. Manz, A. Merli, S. Vajda and L. Wöste, Deciphering the Reaction Dynamics Underlying Optimal Control Laser Fields, Science, 2003, 299, 536–539 CrossRef CAS PubMed.
- L. Gonzaĺez and J. Full, Special Issue on New Perspectives in Theoretical Chemistry, Theor. Chem. Acc., 2006, 116, 148–159 CrossRef.
- J. Full, L. Gonzaĺez and J. Manz, Quantum Chemistry Based Inversion of Experimental Pump-probe Spectra: Model Simulations for CpMn(CO)3, Chem. Phys., 2006, 329, 126–138 CrossRef CAS.
- T. Jiao, Z. Pang, T. J. Burkey, R. F. Johnston, T. A. Heimer, V. D. Kleiman and E. J. Heilweil, Ultrafast Ring Closure Energetics and Dynamics of Cyclopentadienyl Manganese Tricarbonyl Derivatives, J. Am. Chem. Soc., 1999, 121, 4618–4624 CrossRef CAS.
- Z. Pang, T. J. Burkey and R. F. Johnston, Design of Transition Metal Complexes with High Quantum Yields for Ligand Substitution: Efficient Photochemical Chelate Ring Closure in Cyclopentadienylmanganese Tricarbonyl Derivatives, Organometallics, 1997, 16, 120–123 CrossRef CAS.
- P. M. Treichel, in Comprehensive Organometallic Chemistry II, ed. AbelE. W., StoneF. G. A and WilkinsonG., Elsevier, Oxford, UK, 1995, ch. 5, vol. 6 Search PubMed.
- P. M. Treichel, in Comprehensive Organometallic Chemistry, ed. WilkinsonG., StoneF. G. A. and AbelE. W., Pergamon, Oxford, UK, 1982, ch. 29, vol. 4 Search PubMed.
- K. B. Caulton, Coordination Chemistry of the Manganese and Rhenium Fragments (C5H5)M(CO)2, Coord. Chem. Rev., 1981, 38, 1–43 CrossRef CAS.
- J. W. Kee, Y. Y. Tan, B. H. G. Swennenhuis, A. A. Bengali and W. Y. Fan, Hydrogen Generation from Water Upon CpMn (CO) 3 Irradiation in a Hexane/water Biphasic System, Organometallics, 2011, 30, 2154–2159 CrossRef CAS.
- H. Yang, K. T. Kotz, M. C. Asplund and C. B. Harris, Femtosecond Infrared Studies of Silane Silicon−Hydrogen Bond Activation, J. Am. Chem. Soc., 1997, 119, 9564–9565 CrossRef CAS.
- H. Yang, M. C. Asplund, K. T. Kotz, M. J. Wilkens, H. Frei and C. B. Harris, Reaction Mechanism of Silicon−Hydrogen Bond Activation Studied Using Femtosecond to Nanosecond IR Spectroscopy and Ab Initio Methods, J. Am. Chem. Soc., 1998, 120, 10154–10165 CrossRef CAS.
- H. Yang, K. T. Kotz, M. C. Asplund, M. J. Wilkens and C. B. Harris, Ultrafast Infrared Studies of Bond Activation in Organometallic Complexes, Acc. Chem. Res., 1999, 32, 551–560 CrossRef CAS.
- J. Full, L. González and C. Daniel, A CASSCF/CASPT2 and TD-DFT Study of the Low-Lying Excited States of η5-CpMn(CO)3, J. Phys. Chem. A, 2001, 105, 184–189 CrossRef CAS.
- There are two kinds of photochemical reaction paths to bring molecules from the high-energy excited energy states to the ground state in a radiationless way. One is conical intersection, which corresponds to crossings between states of the same multiplicity (mostly singlet/singlet). The other is intersystem crossing, which corresponds to crossings between states of the different multiplicity (mostly triplet/singlet). These two crossings can exhibit very effective channels for chemical deactivations of molecules without any light irradiations. For details, see: ref. 37, 38, 40 and 41.
- M. Klessinger and J. Michl, Conical intersection, in Excited States and Photochemistry of Organic Molecules, VCH Publishers, New York, 1995 Search PubMed.
- B. Valeur, Intersystem crossing, in Molecular Fluorescence: Principles and Applications, Wiley, New York, 2001 Search PubMed.
- In the VIIB group of the periodic table, there exist three elements, i.e., manganese (Mn), technetium (Tc), and rehenium (Re). However, Tc is the only synthetic element that does not exist on earth. In other words, Tc is a man-made element produced in a particle accelerator. This could be the reason that so far no photochemical reactions of the η5-CpTc(CO)3 complex have been experimentally reported yet. We thus did not consider its photochemical reaction mechanisms in this work.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al, Gaussian, Inc., Wallingford, CT, 2013.
- T. A. Albright, J. K. Burdett and M. H. Whangbo, Orbital Interaction in Chemistry, Wiley, New York, 1985, p. 381 Search PubMed.
- W. Domcke, D. R. Yarkony and H. Koppel, Conical Intersections, Adv. Ser. Phys. Chem., World Scientific, Singapore, 2004, vol 15 Search PubMed.
- W. Domcke, D. R. Yarkony and H. Koppel, in Conical Intersections: Theory, Computation and Experiment, World Scientific, Singapore, 2011 Search PubMed.
- B. G. Levine and T. J. Martinez, Isomerization Through Conical Intersections, Annu. Rev. Phys. Chem., 2007, 58, 613–634 CrossRef CAS PubMed.
- S. Gozem, H. L. Luk, I. Schapiro and M. Olivucci, Theory and Simulation of the Ultrafast Double-Bond Isomerization of Biological Chromophores, Chem. Rev., 2017, 117, 13502–13565 CrossRef CAS PubMed.
- I. Schapiro, F. Melaccio, E. N. Laricheva and M. Olivucci, Using the Computer to Understand the Chemistry of Conical Intersections, Photochem. Photobiol. Sci., 2011, 10, 867–886 CAS.
- M. J. Paterson, P. A. Hunt and M. A. Robb, Non-Adiabatic Direct Dynamics Study of Chromium Hexacarbonyl Photodissociation, J. Phys. Chem. A, 2002, 106, 10494–10504 CrossRef CAS , and references therein.
- D. R. Yarkony, Conical Intersections: Diabolical and Often Misunderstood, Acc. Chem. Res., 1998, 31, 511–518 CrossRef CAS.
- M. A. Robb, M. Garavelli, M. Olivucci and F. Bernardi, A Computational Strategy for Organic Photochemistry, Rev. Comput. Chem., 2000, 33, 87–146 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Bearpark, M. A. Robb and H. B. Schlegel, A Direct Method for the Location of the Lowest Energy Point on a Potential Surface Crossing, Chem. Phys. Lett., 1994, 223, 269–274 CrossRef CAS.
- J. J. W. McDouall, K. Peasley and M. A. Robb, A Simple MCSCF Perturbation Theory: Orthogonal Valence Bond Møller-Plesset 2 (OVB MP2), Chem. Phys. Lett., 1988, 148, 183–189 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Energy-adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- Since the correct isolation of transition state is paramount for constructing reaction paths, one reviewer recommend a brief discussion on a number of efficient transition state locating strategies like nudged elastic band. Several groups, however, have already intensively explored these studies. For intense, see ref. 44.
- G. A. Worth, G. Welch and M. J. Paterson, Wavepacket Dynamics Study of Cr(CO)5 After Formation by Photodissociation: Relaxation Through an (E ⊕ A) ⊗ e John-Teller Conical Intersection, Mol. Phys., 2006, 104, 1095–1105 CrossRef CAS.
- R. G. McKinlay, J. M. Zurek and M. J. Paterson, Vibronic Coupling in Inorganic Systems: Photochemistry, Conical Intersections, and The John-Teller and Pseudo-John-Teller Effects, Adv. Inorg. Chem., 2010, 62, 351–390 CrossRef CAS.
- N. M. S. Almeida, R. G. McKinlay and M. J. Paterson, Computation of Excited States of Transition Metal Complexes, Struct. Bonding, 2016, 167, 107–138 CrossRef CAS.
- S. A. Trushin, K. Kosma, W. Fuβ and W. E. Schmid, Wavelength-independent Ultrafast Dynamics and Coherent Oscillation of a Metal–Carbon Stretch Vibration in Photodissociation of Cr(CO)6 in the Region of 270–345 nm, Chem. Phys., 2008, 347, 309–323 CrossRef CAS.
- O. Buchardt, in Photochemistry of Heterocyclic Compounds, Wiley, New York, 1976 Search PubMed.
- J. Kopecky, in Organic Photochemistry: A Visual Approach, VCH Publishers, New York, 1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01118d |
| This journal is © The Royal Society of Chemistry 2018 |