
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecularly imprinted ‘traps’ for sulfonylureas prepared using polymerisable ion pairs†
Federica Pessagnoa,
Aliya Nur Hasanahb and
Panagiotis Manesiotis *a
*a
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, BT9 5AG Belfast, Northern Ireland, UK. E-mail: p.manesiotis@qub.ac.uk
bPharmaceutical Analysis and Medicinal Chemistry Department, Faculty of Pharmacy, Universitas Padjadjaran, Jl Raya Bandung Sumedang KM 21,5, Jatinangor, Indonesia
First published on 17th April 2018
Abstract
A novel approach towards recognition of sulfonylureas based on a polymerisable ion pair is presented. A solution association constant >105 M−1 between the model target glibenclamide and 4-vinylbenzyltrimethylammonium methacrylate is measured, and the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes verified. Subsequently prepared stoichiometrically imprinted polymers exhibit exceptionally high affinity and binding capacity for glibenclamide, owing to synergistic binding of both the neutral and deprotonated form of the drug by the ion pair monomer. The polymers are applied to the selective extraction of glibenclamide from blood serum samples, achieving recoveries of up to 98% and demonstrating excellent long-term stability, negating the need for regular sorbent regeneration.
:1 complexes verified. Subsequently prepared stoichiometrically imprinted polymers exhibit exceptionally high affinity and binding capacity for glibenclamide, owing to synergistic binding of both the neutral and deprotonated form of the drug by the ion pair monomer. The polymers are applied to the selective extraction of glibenclamide from blood serum samples, achieving recoveries of up to 98% and demonstrating excellent long-term stability, negating the need for regular sorbent regeneration.
Introduction
Sulfonylureas are a family of organic compounds with applications in medicine, mainly in the treatment of diabetes mellitus (type II), and agriculture, as herbicides. In both cases, such compounds are found in complex matrices and often at low concentrations, which makes their analytical determination a lengthy and laborious process, usually involving liquid–liquid or solid phase extraction, prior to analysis by HPLC-UV, HPLC-MS or capillary electrophoresis.1–4 Molecularly Imprinted Polymers (MIPs) have been previously used for the selective capture of glibenclamide (GLIB), a sulfonylurea drug. Wu et al. prepared MIP-coated micro-stir bars for the extraction of the sulfonylurea from herbal dietary supplements with recoveries of 81.9–101.4%,5 while Wang et al. achieved recoveries of 81.5–93.5% from health foods using dendritic grafting of MIPs onto magnetic nanoparticles.6 More recently, Ostovan et al. prepared hollow MIP nanoparticles for extraction of glibenclamide from urine with recoveries of 89.5%.7 In all these cases methacrylic acid was used as the functional monomer for recognition of the sulfonylurea.We have previously reported on our study of the interaction of glibenclamide (Fig. 1) with neutral and anionic receptors, and introduced tetrabutylammonium methacrylate (TBAM) as a novel recognition element for use in molecular imprinting,8 reversing the previously established polymerisable urea-carboxylate motif, studied by our group and others.9–11 We demonstrated that not only was the methacrylate anion capable of very strong association with the sulfonylurea moiety in solution, but under certain conditions it can deprotonate the acidic NH adjacent to the sulfonyl group, resulting in the formation of ‘narcissistic’ dimers12 between the neutral and anionic forms of GLIB, stabilised by the associated tetrabutylammonium cation. Consequently, when TBAM was used in the stoichiometric molecular imprinting of GLIB, the resulting polymers outperformed polymers prepared using acrylamide or methacrylic acid as the functional monomers. However, it was found that these polymers were ‘deactivated’ upon GLIB binding, by transfer of a proton from the template to the methacrylate moieties residing within the binding sites, thus negating the functional group complementarity between the two counterparts. This limitation was overcome by addition of a polymer regeneration step after each extraction cycle, using a dilute tetrabutylammonium hydroxide solution. Nonetheless, while no adverse effects on the stability or performance of the polymers were observed, it was decided to investigate alternative, more robust binding motifs.
 | ||
| Fig. 1 Chemical structures of VBTMA ion pair monomer, showing proposed primary interaction with GLIB, and structures of analogous sulfonylurea substances (top), and synthesis of VBTMA (bottom). | ||
Here, we wish to report, for the first time, the development of a novel polymerisable ion pair, whereby both anionic and cationic counterparts are permanently incorporated in the polymer matrix, and its application in the molecular imprinting of sulfonylureas. This approach is complementary to the field of ion-pair receptors, expertly reviewed in literature,13 as instead of employing a single receptor with heterotopic binding sites for both co-existing cationic and anionic partners of an ion pair, we employ a polymerisable ion pair to recognise both the neutral and anionic form of a sulfonylurea that do not co-exist but are different forms of the same molecule. Thus, even if the target molecule switches between the two forms by a change in the chemical environment, e.g. pH, the new polymer-bound receptor will be able to capture it, maximising the efficiency and application range of the material and revealing the true potential of the imprinted material. Furthermore, in a step change compared to previous reports of mixed ionic polymers14 and poly(ionic liquids) as molecular recognition elements,15–18 where one of the counterparts is mobile and can be exchanged during application of the material,19 the present design yields robust imprinted polymers that can be repeatedly used without loss in performance due to ion exchange, and without the need for regular regeneration. Furthermore, the novel materials exhibit exceptionally high affinity, as well as enhanced binding capacity and selectivity for the model sulfonylurea template, vastly outperforming previously reported sorbents.
Experimental
Materials and methods
Glibenclamide (GLIB), sodium methacrylate (SMA), 4-vinylbenzyl chloride (VBC), tetrabutylammonium hydroxide (TBAOH), tetrabutylammonium chloride (TBACl), ethyleneglycol dimethacrylate (EGDMA), 1-hydroxycyclohexyl phenyl ketone (UV initiator), acetic acid, trifluoroacetic acid (TFA), trimethylamine (TMA, 4.2 mol L−1 solution in ethanol), triethylamine (TEA), HPLC grade solvents, deuterated solvents, empty polypropylene solid-phase extraction (SPE) cartridges (3 mL) and 20 μm porous polyethylene frits were purchased from Sigma Aldrich (Gillingham, UK). Polymerisation inhibitors were removed from all monomers by filtration through a basic alumina column. Lithium bis(trifluoromethyl-sulfonyl)imide (LiNTf2) was purchased from Alfa Aesar (Heysham, UK). Glipizide (GLIP) was purchased from the Indonesian National Agency of Drug and Food Control. Gliclazide (GLIC) was provided by Dexa Medica Pharmaceuticals Industry (Tangerang, Indonesia). NMR spectra were collected on a Bruker Avance 600 MHz NMR spectrometer, and 1H NMR titrations and Job plots on a Bruker ECX 400 MHz NMR spectrometer (Coventry, UK). FT-IR spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR spectrometer equipped with an ATR attachment (Seer Green, UK). An Agilent 1100 HPLC instrument equipped with photodiode array detector was used in for all chromatographic separations. Analyses were performed by isocratic elution using a 40:60 water/acetonitrile mixture containing 0.01% TFA as the mobile phase and a Phenomenex Kinetex™ C18 column (5 μm, 150 mm × 4.6 mm i.d.) (Macclesfield, UK). The flow rate was 1 mL min−1 and the detection wavelength was set at 230 nm. A 12-port Phenomenex vacuum manifold was used for SPE experiments (Macclesfield, UK). Blood samples were provided by the Indonesian Red Cross.
Synthesis of 4-vinylbenzyltrimethylammonium methacrylate
4-vinylbenzyltrimethylammonium methacrylate (VBTMA) was prepared in two steps as follows: 1.53 g of inhibitor free VBC (10 mmol) and 20 mL of ethanol were transferred to a round-bottom flask and 4.75 mL (20 mmol) of a 4.2 M solution of TMA in ethanol were added. The reaction was allowed to proceed at room temperature for 18 h and the solvent was subsequently evaporated under reduced pressure. The product, 4-vinylbenzyltrimethylammonium chloride (VBTAC), was obtained as a white solid in quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 7.67–7.47 (m, 4H), 6.81 (dd, J = 17.7, 11.0 Hz, 1H), 5.96 (dd, J = 17.7, 0.8 Hz, 1H), 5.39 (dd, J = 10.9, 0.8 Hz, 1H), 4.56 (s, 2H), 3.35 (s, 9H); 13C NMR (151 MHz, DMSO-d6) δ 139.34, 136.32, 133.58, 128.29, 126.99, 116.68, 67.86, 52.17. HRMS: C12H18N+ calculated 176.1434, found 176.1358.In order to obtain the final monomer, 2.12 g (10 mmol) of VBTAC were dissolved in 50 mL of ethanol and 1.08 g (10 mmol) of sodium methacrylate were added. The mixture was stirred for 4 h at room temperature, then cooled at 0 °C for 1 h and finally centrifuged at 3000 rpm to remove the NaCl formed. The supernatant was evaporated to dryness under reduced pressure to yield the product, VBTMA, as a white solid with 88% yield. 1H NMR (600 MHz, DMSO-d6) δ 7.68–7.46 (m, 4H), 6.81 (dd, J = 17.7, 11.0 Hz, 1H), 5.96 (dd, J = 17.7, 0.8 Hz, 1H), 5.52 (d, J = 3.4 Hz, 1H), 5.39 (dd, J = 10.9, 0.8 Hz, 1H), 4.99–4.89 (m, 1H), 4.56 (s, 2H), 3.05 (s, 9H), 1.76 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 171.45, 146.24, 139.32, 136.33, 133.59, 128.33, 126.98, 116.66, 116.44, 67.86, 52.16, 20.88. A melting point of 160 °C was measured, however, the compound polymerised immediately upon melting.
Synthesis of glibenclamide tetrabutylammonium salt
The tetrabutylammonium salt of GLIB (GLIB-TBA) was prepared by mixing of equimolar amounts of GLIB and TBAOH in methanol, followed by evaporation of the solvent under reduced pressure to yield GLIB-TBA as a white solid in quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 8.23 (s, br, 1H), 7.69 (dd, J = 2.8, 1.8 Hz, 1H), 7.67–7.63 (m, 2H), 7.50 (dd, J = 8.9, 2.8 Hz, 1H), 7.21 (d, J = 8.3 Hz, 2H), 7.15 (d, J = 8.9 Hz, 1H), 5.54 (s, 1H), 3.80 (s, 3H), 3.51 (t, J = 7.2 Hz, 2H), 3.20–3.12 (m, 8H), 2.83 (t, J = 7.2 Hz, 2H), 1.63–1.52 (m, 8H), 1.36–1.25 (m, 8H), 0.94 (t, J = 7.4 Hz, 12H); 13C NMR (151 MHz, DMSO-d6) δ 163.89, 156.24, 146.79, 140.53, 131.99, 130.05, 128.08, 126.99, 125.16, 124.78, 114.64, 58.00, 56.75, 49.00, 41.07, 40.54, 35.10, 33.95, 25.96, 25.28, 23.54, 19.68, 13.96.Synthesis of 4-vinylbenzyltrimethylammonium bis(trifluoro-methylsulfonyl) imide
4-vinylbenzyltrimethylammonium bis(trifluoromethylsulfonyl) imide (VBTANTf2) was prepared by addition of two-fold excess of LiNTf2 to an aqueous solution of 4-vinylbenzyltrimethyl ammonium chloride (VBTAC), followed by solvent extraction of the aqueous phase with chloroform, drying of the organic layer with MgSO4, and solvent evaporation under reduced pressure, to finally yield a white solid. 1H NMR (600 MHz, CDCl3) δ 7.44 (dd, J = 41.3, 8.2 Hz, 4H), 6.71 (dd, J = 17.6, 10.9 Hz, 1H), 5.83 (d, J = 17.6 Hz, 1H), 5.38 (d, J = 11.1 Hz, 1H), 4.41 (s, 2H), 3.08 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 139.44 (s), 134.38 (s), 131.90 (s), 126.09 (s), 124.67 (s), 121.98–115.60 (q), 115.64 (s), 68.84 (s), 51.78–51.50 (t); 19F NMR (565 MHz, CDCl3) δ −78.94 (s).1H NMR titration experiments
The solution interactions of GLIB and GLIB-TBA with VBTMA, VBTAC and VBTANTf2, as well as the complexation of GLIB with TBACl, were studied by 1H NMR titrations in DMSO-d6. Thus, to a 1.0 mmol L−1 solution of the host (GLIB or GLIB-TBA), increasing amounts of each guest were added, until at least a 10-fold excess was reached. The complexation-induced shift (CIS) of several protons was followed and titration isotherms were constructed. The stoichiometry of the selected monomer–template complexes was confirmed using Job's method of continuous variation. Hence, equimolar solutions (10.0 mmol L−1) of the host and each guest were mixed in different ratios and a plot of Δδ against the molar fraction of monomer multiplied by the CIS (Xi × Δδ) was constructed.Preparation of imprinted polymers
Stoichiometrically imprinted and corresponding non-imprinted polymers, PGLIB, PGLIBTBA and NP respectively, were prepared by photochemically initiated free radical polymerisation. The compositions of all prepared polymers are presented in Table 2. Briefly, the template and the selected functional monomer were transferred into to a glass vial and mixed with the porogen. Upon complete dissolution, the cross-linker was added followed by the initiator. The resulting pre-polymerisation solutions were degassed by ultra-sonication for 5 min, purged with argon and then hermetically sealed. The vials were then placed in the chamber of a UVP CX-2000 UV curing reactor (UVP, Jena, Germany) and irradiated at 360 nm for 3 hours at room temperature. The resulting rigid monoliths were coarsely ground and washed with methanol in a Soxhlet apparatus for 24 h, in order to remove the template and any unreacted monomers. The coarse polymer particles were further ground using a mortar and pestle, wet-sieved with acetone, and the 25–50 μm fraction was collected, dried and stored at room temperature. The corresponding non-imprinted polymers were prepared in a similar fashion, omitting addition of the template to the pre-polymerisation mixture.| Guest | Host | |
|---|---|---|
| GLIB | GLIB-TBA | |
| VBTMA | >105 | 53 ± 6 |
| VBTAC | 34 ± 5 | No binding |
| VBTANTf2 | No binding | 22 ± 3 |
| TBACl | 3452 ± 230 | No binding |
| Polymer ID | Functional monomer | Template | Cross-linker | Porogen |
|---|---|---|---|---|
| PGLIB | VBTMA | GLIB | EDMA | CHCl3 |
| NP | VBTMA | — | EDMA | CHCl3 |
| PGLIB2 | TBAM/MAA 1:1 |
GLIB | EDMA | CHCl3 |
| NP2 | TBAM/MAA 1:1 |
— | EDMA | CHCl3 |
| PGLIBTBA | VBTAC | GLIB-TBA | EDMA | DMSO |
| PGLIBTBA(NTf2) | VBTANTf2 (exchanged) | GLIB-TBA | EDMA | DMSO |
| PGLIBTBA(XL) | — | GLIB-TBA | EDMA | DMSO/CHCl3 |
PGLIBTBA(NTf2) was prepared from PGLIBTBA by exchange of chloride counter anions with bis-triflimide. Briefly, 0.5 g of PGLIBTBA were suspended in 10 mL of distilled water containing 0.5 g of LiNTf2. The suspension was stirred at room temperature overnight and then polymer particles were filtered, washed with distilled water and dried under reduced pressure prior to use. Polymers PGLIBTBA2 and NP2 were prepared as described in our previous publication.8 An additional control polymer, PGLIBTBA(XL), was prepared in a similar fashion to PGLIBTBA, but without the addition of a functional monomer.
Rebinding experiments
Polymer affinity and capacity for each analyte were measured using equilibrium rebinding experiments performed in acetonitrile. Thus, 10 mg of each polymer were transferred in 2 mL glass vials and incubated with 1.5 mL of analyte solution of increasing concentrations (0.0–3.0 mmol L−1) for 24 hours. The supernatants were then analysed by HPLC using the method described above. The amount of analyte bound to the polymer was calculated by subtracting the amount determined after the rebinding experiment from the starting amount of the drug. The results were plotted as concentration of free analyte in solution (mol L−1) vs. the amount of analyte bound on the polymers (μmol g−1) to produce binding isotherms that were fitted using the appropriate binding model.Solid phase extractions
50 mg of imprinted or non-imprinted polymer particles (25–50 μm) were dry packed in 3 mL SPE cartridges using 20 μm porous polyethylene frits. Blood serum samples were prepared by centrifugation of the collected blood at 8000 rpm for 5 minutes at 14 °C and careful collection of the clear top layer. Blood serum samples were spiked with 5 mg L−1 of GLIB in 5% acetonitrile in water. Following an extensive optimisation process, the final extraction protocol consisted of an initial conditioning step with 1 mL of 5% acetonitrile in water, loading 2 mL of the spiked blood sample, followed by an aqueous wash (1 mL), a wash with 2 mL of 0.01% TEA in CHCl3, and a final elution with 0.5 mL of 1% acetic acid in methanol. Full vacuum was applied to the cartridges between each step for 2 minutes. In order to test the specificity of the prepared polymers, an equimolar mixture of GLIB, GLIC and GLIP (5 mg L−1 each) in 5% acetonitrile in water was spiked into blood serum samples and applied onto the SPE cartridges. The collected fractions were analysed by HPLC using the method described above.Results and discussion
Host-guest interactions in solution
The solution association of GLIB with the novel polymerisable ion pair system VBTMA was studied by a series of 1H NMR titrations in DMSO-d6. Several additional equilibria were also investigated in order to explain the behaviour observed by subsequently prepared imprinted polymers. The obtained association constants are outlined in Table 1. As seen in Fig. 2, VBTMA interacts strongly with GLIB, forming 1:1 complexes with an estimated stability constant Ka > 105 M−1, too strong to accurately determine by 1H NMR titration. Further insights into the nature of the formed complexes were offered by closer inspection of the collected 1H NMR spectra (Fig. 3), in particular the chemical shifts of the methacrylate protons and the methyl and methylene protons of the counter-cation. Thus, during the early stages of the titration experiment, where GLIB (host) was in excess compared to VBTMA (guest), and up to a ratio GLIB:VBTMA of 1:1, the signals corresponding to the two methacrylate protons were poorly defined and showed almost no change (Fig. 4). The chemical shift of those protons was also down-field from their position in the spectrum of the free monomer. Furthermore, the disappearance of the signal corresponding to the acidic sulfonylurea NH (initially at 10.31 ppm), and the concurrent up-field shift of the second sulfonylurea NH (initially at 6.32 ppm), are consistent with deprotonation of GLIB and protonation of methacrylate. Once an excess of VBTMA was added, the signals attributed to the methacrylate group moved towards their corresponding positions in the spectrum of the free monomer (4.99 ppm and 5.54 ppm), while the second sulfonylurea proton nearly disappeared under the methacrylate signal (5.59 ppm). The deprotonation event was also evidenced by the movements of the signals corresponding to the aromatic protons adjacent to the sulfonylurea group, whereby up-field shifts from 7.84 ppm to 7.86 ppm, and 7.48 ppm to 7.22 ppm were observed. It is noteworthy that the positions of the signals corresponding to GLIB protons at the end of the titration, are identical to the signals of GLIB-TBA, which further supports the deprotonation mechanism. Observation of the peaks corresponding to the methyl and methylene groups of the positively charged counterpart, show a gradual down-field movement of the signals throughout the titration, however, upon closer inspection of the plotted curves, the chemical shift change is sharper up to the 1:1 point of the experiment and becomes shallower when excess VBTMA has been added (see ESI†). This behaviour hints at an interaction of the quaternary ammonium cation with GLIB− that is possibly stronger that the force between the two partners of the polymerisable ion pair. This is a crucial attribute of the ion pair monomer as it means that when exposed to the sulfonylurea, both counterparts will favourably bind to the neutral or deprotonated form of the latter, and the interaction between them will not hinder the association to the third party. In an attempt to quantify this interaction, a titration experiment between GLIB-TBA and VBTMA revealed a weak yet significant association (Ka = 53 M−1) of the deprotonated host with the quaternary ammonium functionality of the ion pair monomer. Interestingly, movement was only detected for the signals corresponding to either the methyl or the methylene groups of VBTMA, and not the methacrylate protons.
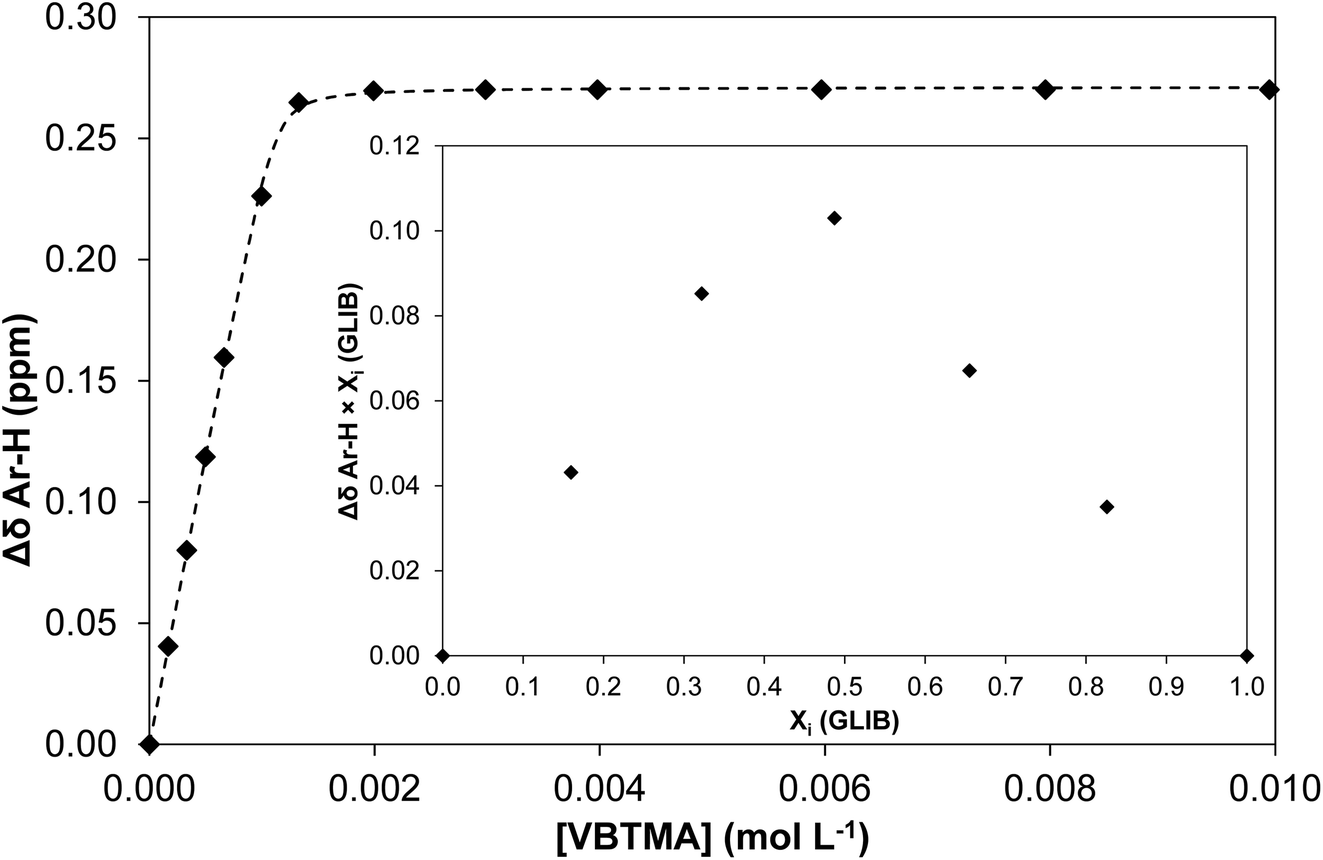 | ||
| Fig. 2 Binding isotherm obtained during 1H NMR titration of GLIB with VBTMA in DMSO-d6. Inset: Job plot for the association of GLIB with VBTMA in DMSO-d6, where the formation of 1:1 complexes is verified. | ||
 | ||
| Fig. 3 Overlay of characteristic 1H NMR spectra (DMSO-d6) obtained during the titration of GLIB vs. VBTMA, showing the disappearance of the acidic sulfonylurea proton and the movement of the second sulfonylurea proton, as well as the movement of the methacrylate vinyl protons. From top to bottom, 0 eq., 2 eq., 6 eq. and 10 eq. of VBTMA added to GLIB. | ||
 | ||
| Fig. 4 Change in chemical shift of the two vinyl protons (diamonds, left axis) and methyl protons (triangles, right axis) of methacrylate during the titration of GLIB vs. VBTMA in DMSO-d6. | ||
The binding mechanism was further investigated by titration of GLIB and GLIB-TBA vs. VBTAC. In the former case, weak association, again accompanied by movement of the methyl and methylene peaks of VBTAC, as well as the two sulfonylurea protons, was observed, which was attributed to interaction of the chloride ion with the sulfonylurea (see ESI†). This was supported by the strong association of GLIB with TBACl (Ka = 3452 M−1). The bulky tetrabutylammonium cation associates weakly with chloride, thus not hindering the interaction of the halide anion with the sulfonylurea. No binding was observed between GLIB-TBA and either VBTAC or TBACl, due to the electrostatic repulsion between the negative charged sulfonylurea and chloride anions. Lastly, when chloride was exchanged with the larger, non-coordinating, bis-triflimide anion, weak binding with the positive quaternary ammonium monomer was observed (Ka = 22 M−1), while no interaction with GLIB could be detected. It was thus concluded that the binding between GLIB and VBTAC is mediated by the formation of chloride bridges, while the GLIB⋯Cl− complexes electrostatically repel GLIB-TBA.
Evaluation of polymer performance
Physico-chemical characterisation of the prepared materials was conducted by surface area analysis and FT-IR. The specific surface area of PGLIB was 67.2 m2 g−1 with a pore diameter of 23.1 Å, and the corresponding values for NP were 73.1 m2 g−1 and 37.5 Å. Both sets of values are in the same range, so the porous structure of the two polymers should not influence the results of the subsequent rebinding experiments. FT-IR analysis revealed all the characteristic peaks for the incorporated functional groups (carboxylate: 1574 cm−1, 1388 cm−1; C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ester): 1722 cm−1; methylene bend: 1451 cm−1; C–C skeletal stretch: 1138 cm−1), while the spectra of both polymers were nearly identical, suggesting that the presence of the template did not impact the progress of the polymerisation reaction or the relative reactivity of the ion pair and cross-linking monomer, and that it had been fully removed during the polymer washing procedure (see ESI†).
O (ester): 1722 cm−1; methylene bend: 1451 cm−1; C–C skeletal stretch: 1138 cm−1), while the spectra of both polymers were nearly identical, suggesting that the presence of the template did not impact the progress of the polymerisation reaction or the relative reactivity of the ion pair and cross-linking monomer, and that it had been fully removed during the polymer washing procedure (see ESI†).
Evaluation of the polymer binding performance was conducted by means of equilibrium rebinding experiments, whereby binding isotherms were constructed as shown in Fig. 5a. The derived fitting parameters are presented in Table 3. Upon observation of the isotherms for the binding of GLIB on the corresponding imprinted and non-imprinted polymers, exceptionally strong binding is evident at the low concentration range, and virtually all of the template is removed from the supernatant by both polymers up to the concentration of 0.5 mmol L−1, following which point the isotherms level off rapidly, indicating that the saturation point has been reached. Furthermore, PGLIB greatly outperformed the previously reported PGLIB2 by nearly two orders of magnitude in affinity and over three-fold in binding capacity. Interestingly, the binding isotherms for GLIB on PGLIB and NP could not be fitted to the Langmuir model, in contrast to their previous counterparts PGLIB2 and NP2, and a bi-Langmuir model was used instead. Although both models offer a simplified approximation of the type of binding sites present in imprinted polymers, this result suggests the presence of very high affinity sites, in which methacrylate and quaternary ammonium groups bind GLIB synergistically, and lower affinity sites, where functionality orientation is sub-optimal. The chloride bridge mediated binding mechanism between GLIB and the quaternary ammonium moiety was verified by rebinding of GLIB on PGLIBTBA and PGLIBTBA(NTf2), both polymers containing only the positively charged quaternary ammonium monomer, with either chloride or bis-triflimide as counter anions. Thus, although significantly weaker binding and lower capacity for GLIB was observed on PGLIBTBA compared to PGLIB, when the chloride counterion of PGLIBTBA was exchanged with bis-triflimide (PGLIBTBA(NTf2)), GLIB binding capacity was further reduced by nearly a factor of two.
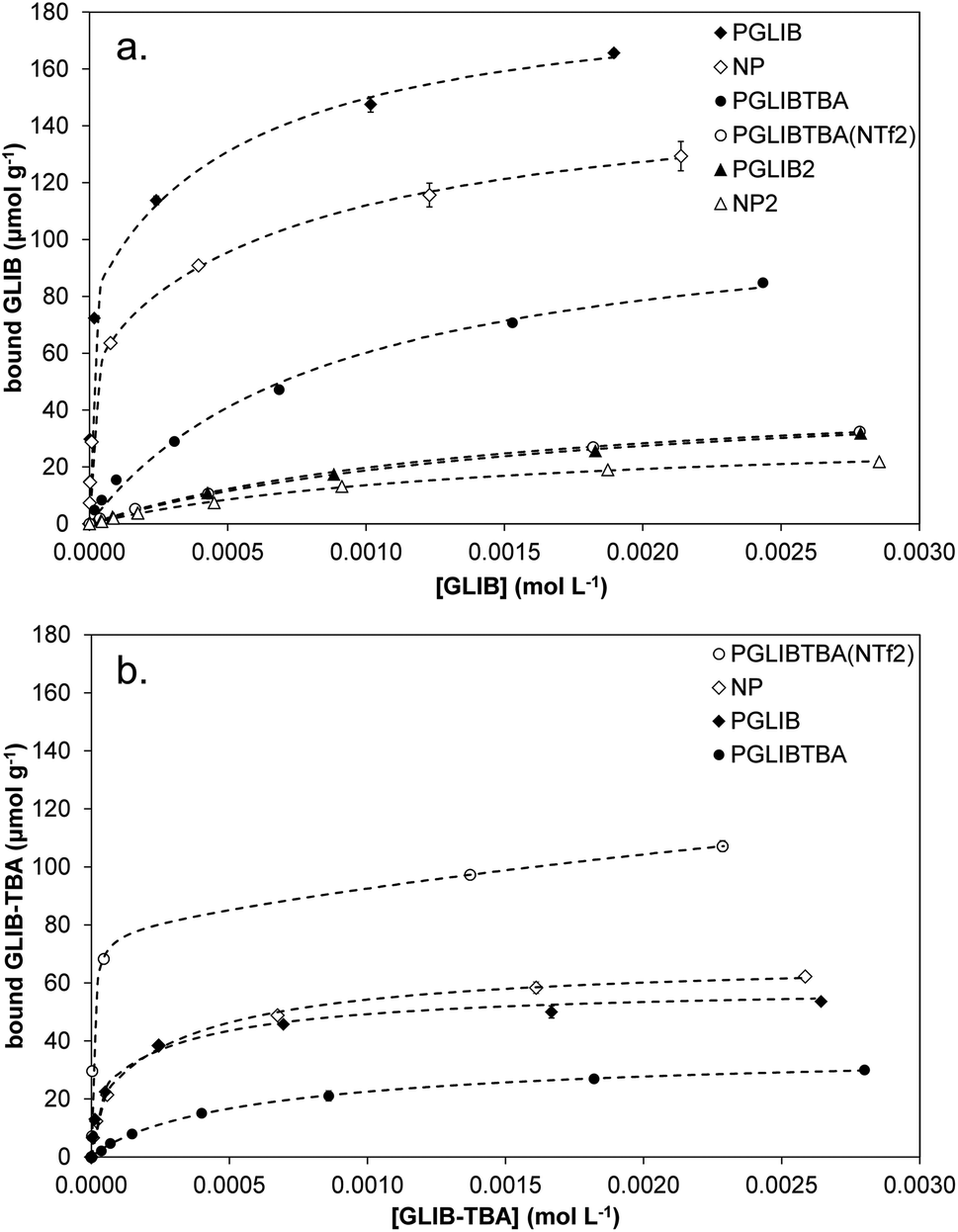 | ||
| Fig. 5 Equilibrium rebinding isotherms of (a) GLIB and (b) GLIB-TBA on the prepared polymers. | ||
| Polymer ID | GLIB | GLIB-TBA | ||
|---|---|---|---|---|
| Ka (L mol −1) | N (μmol g−1) | Ka (L mol −1) | N (μmol g−1) | |
| a Previously published data.8 | ||||
| PGLIB | 1.7 ± 0.4 × 103 | 108.8 ± 7.1 | 4.8 ± 1.2 × 103 | 37.0 ± 3.9 |
| 3.8 ± 0.6 × 105 | 80.8 ± 4.9 | 1.2 ± 0.5 × 105 | 18.2 ± 4.4 | |
| NP | 1.2 ± 0.3 × 103 | 93.7 ± 5.0 | 2.5 ± 0.6 × 103 | 47.9 ± 3.8 |
| 1.2 ± 0.2 × 105 | 62.0 ± 3.6 | 6.3 ± 2.5 × 104 | 20.4 ± 4.6 | |
| PGLIB2a | 6.0 ± 0.5 × 102 | 50.7 ± 2.3 | No binding | |
| NP2a | 7.0 ± 0.4 × 102 | 33.1 ± 1.0 | No binding | |
| PGLIBTBA | 1.1 ± 0.2 × 103 | 113.3 ± 6.9 | 1.9 ± 0.1 × 103 | 35.1 ± 0.7 |
| PGLIBTBA(NTf2) | 6.5 ± 0.6 × 102 | 50.2 ± 2.0 | 1.4 ± 0.2 × 102 | 115.3 ± 12.3 |
| 1.5 ± 0.8 × 105 | 78.2 ± 7.1 | |||
| PGLIBTBA(XL) | No binding | No binding | ||
Following the rebinding experiments of GLIB-TBA (Fig. 5b), the deprotonated analogue of GLIB, it was found that the overall binding affinity and capacity of both PGLIB and NP for GLIB-TBA was lower by a factor of three compared to GLIB, while imprinting selectivity was also lost. These results verify the proposition that the functional group responsible for selective binding of GLIB is methacrylate, as in its absence binding is diminished. Furthermore, the binding of GLIB-TBA on its corresponding imprinted polymer, PGLIBTBA, was significantly lower than on PGLIB, suggesting again that the presence of chloride results in repulsive forces between the interacting species. When exchanged with bis-triflimide, which does not hinder the interaction between the positively charged quaternary ammonium groups and the negatively charged deprotonated sulfonylurea, PGLIBTBA(NTf2) exhibits a five-fold higher binding capacity for GLIB-TBA, further supporting the proposed binding mechanism. As additional proof of the importance of the functional monomer in the binding process, a polymer prepared without any functional monomer, PGLIBTBA(XL), showed no binding for either the neutral or deprotonated form of the sulfonylurea drug.
It is noteworthy that VBTAC has been previously used in conjunction with MAA for the preparation of ion exchange MIPs used in the extraction of anionic sweetener acesulfame K from wastewater samples, however, in that case VBTAC acted as a phase transfer agent and no ion pair was formed with MAA, resulting in overall poor selectivity in the presence of other anionic compounds.19
Solid phase extraction of blood serum samples
The exceptionally strong binding for GLIB exhibited by the novel materials presented here, especially at lower concentrations, suggests these imprints could act as molecular “traps”, able to selectively recognise the sulfonylurea drug in complex matrices, such as blood serum. We thus opted to use solid phase extraction (SPE) as a simple and rapid tool for the development of an optimised extraction protocol. Loading of blood serum solutions spiked with 5 mg L−1 of the drug on PGLIB and NP resulted in near quantitative capture of GLIB, suggesting the predominance of non-specific, hydrophobic interactions under these conditions. A systematic study of the so-called molecular recognition step was then conducted, whereby a wide range of solvent mixtures with acidic or basic modifiers were tested, aiming to find a solvent system that promotes specific interactions, without compromising the final recovery of the target. As seen in Fig. 6, mixtures of water with methanol, acetonitrile or triethylamine, resulted in final recoveries >80% from both polymers, however imprinting factors (IF), defined as the recovery of the drug on PGLIB over the recovery on NP, ranged from 0.99 to 1.07. When more polar solvents, such as methanol and acetonitrile, modified with triethylamine, were used in the washing step, a marginal improvement in selectivity was observed (1.18 and 1.45 respectively), although recoveries on PGLIB dropped below 80%, which was below the desirable level. Finally, chloroform was tested, as it is frequently hypothesised that using the polymerisation porogen as a washing step solvent will assist the polymer to recover the three-dimensional structure generated during the polymerisation. Thus, an IF of 1.13 was initially obtained, which increased to 1.47 when 0.01–0.1% acetic acid was added, but with a concomitant decrease in recovery to less than 30%. Addition of 0.01% TEA in chloroform resulted in IF of 1.59, which was further improved to a maximum value of 2.21 when the volume of the washing solution was increased to 2 mL. Under these optimised conditions, 98% of GLIB was recovered from blood serum using the imprinted polymer, and 46% using the non-imprinted polymer. The specificity of the prepared polymers was probed by analysis of blood serum samples spiked with equal concentrations of GLIB and two competing sulfonylureas, GLIC and GLIP, each at 5 mg L−1 (Fig. 7). Thus, it was shown that recoveries for GLIB on PGLIB remained unaffected in the presence of the competing analytes, whose recoveries were 13% and 43% respectively. Calculated corresponding imprinting factors were 0.86 and 1.07 respectively, proving that the imprinting process had generated predominantly GLIB-specific binding sites and not generic sulfonylurea binding sites.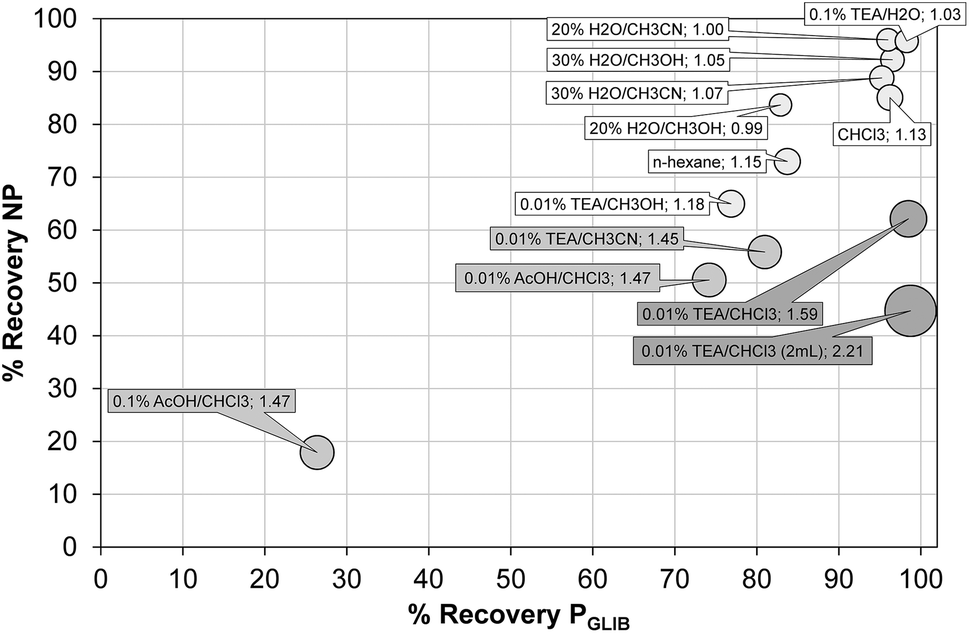 | ||
| Fig. 6 Optimisation of GLIB recovery using different SPE washing step conditions on PGLIB and NP polymers. Size of each circle is proportional to the corresponding imprinting factor. | ||
 | ||
| Fig. 7 Recovery (%) of GLIB from spiked blood serum samples, compared to recovery of GLIB, GLIC and GLIP from equimolar spiked blood serum samples, using the optimised SPE procedure (bars – left axis), and corresponding imprinting factors for each analyte (diamonds – right axis). | ||
It should be noted that the above optimisation process of over 100 blood serum extraction cycles was conducted using a set of three cartridges for each polymer and that, in contrast to our previously reported sulfonylurea binding materials, no regeneration steps were required after each extraction cycle.
Binding mechanism
The binding mechanism between the novel polymerisable ion pair and the sulfonylurea drug, as elucidated by a series of solution interaction and equilibrium rebinding experiments, can be summarised as follows: using VBTAC as the sole functional monomer to imprint the deprotonated form of GLIB, results in weak template binding due to electrostatic repulsion between chloride and the deprotonated sulfonylurea, but stronger binding for neutral GLIB, via formation of chloride bridges (Fig. 8). Subsequent exchange of chloride with bis-triflimide “switches on” the binding sites for recognition of GLIB-TBA, but “switches off” the binding of neutral GLIB. These observations support the hypothesis that following initial binding of methacrylate to the sulfonylurea moiety, which has been shown previously to result in deprotonation of the acidic group and formation of GLIB anions, the latter is captured by the adjacent positively charged quaternary ammonium units.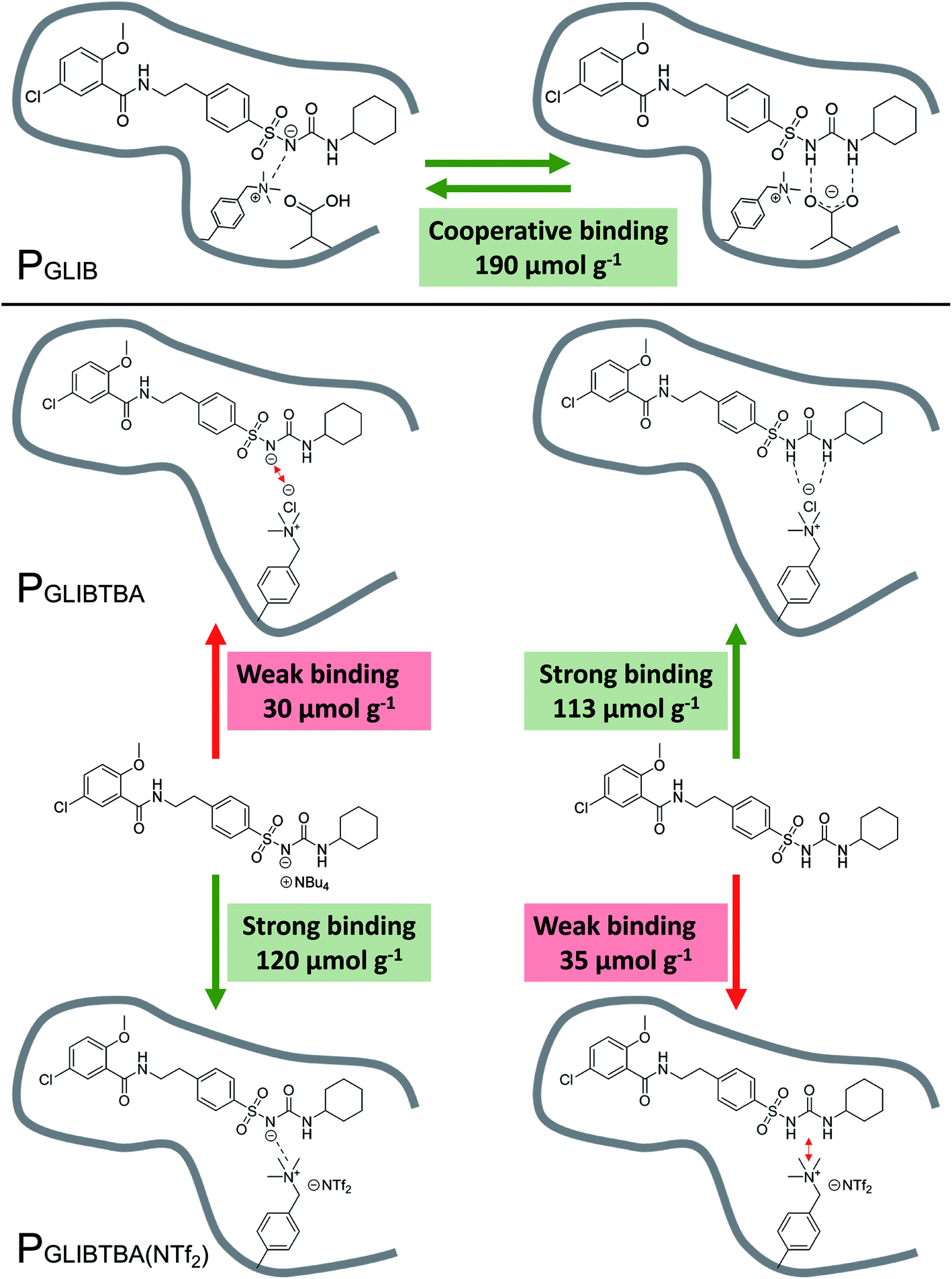 | ||
| Fig. 8 Overview of binding mechanism between the neutral or anionic forms of GLIB and PGLIB, PGLIBTBA and PGLIBTBA(NTf2), showing the corresponding binding capacities. | ||
This is the first report of a co-operative binding mechanism within an imprinted polymer, which is capable of binding both the neutral and dissociated form of a target substance, greatly enhancing the overall binding performance. It also offers direct evidence that the binding mechanism observed in solution by 1H NMR studies, still applies during the interaction of the drug with the imprinted polymer.
Conclusions
A new concept of molecular imprinting, and its application to the recognition of a sulfonylurea antidiabetic drug, were demonstrated in this report. We have introduced, for the first time, a polymerisable ion pair as a binding element comprising a negatively charged methacrylate group and a positively charged quaternary ammonium counterion. The ion pair monomer exhibited exceptionally strong affinity for GLIB in solution and formed 1:1 complexes with the drug with Ka > 105 M−1, while we have shown how the deprotonation of GLIB by methacrylate results in binding of GLIB anions by the positively charged partner of the ion pair. The new receptor motif was used in the preparation of stoichiometrically imprinted polymers for GLIB, which were capable of quantitative binding of the drug under static conditions up to concentrations of 0.5 mmol L−1 and total binding capacities improved by at least three-fold compared to previously reported materials.
With both counterparts of the ion pair monomer being permanently immobilised in the polymer matrix, we have overcome prior limitations imposed by the mobility of the counter-cation, which resulted in unstable materials that required regeneration after each application cycle. Indeed, we have shown that the new materials were capable of recoveries of GLIB from spiked blood serum up to 98% with imprinting factors of 2.21, while we were able to complete a study of over 100 blood serum extraction cycles using just three polymer cartridges, and without the need for intermediate regeneration steps, demonstrating the remarkable robustness of the new ion-pair based imprints. The approach presented here represents a paradigm shift in polymer based molecular recognition, and we are currently exploring the use of ion pair monomers in the recovery of sulfonylureas and related substances from a variety of complex matrices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Joseph Magill Research Scholarship (FP) and the Directorate of Research and Community Services Universitas Padjadjaran (ANH) is gratefully acknowledged.Notes and references
- S. AbuRuz, J. Millership and J. McElnay, J. Chromatogr. B, 2005, 817, 277–286 CrossRef CAS PubMed.
- E. P. C. Lai and S. Y. Feng, J. Chromatogr. B, 2006, 843, 94–99 CrossRef CAS PubMed.
- H. S. Yang, A. H. B. Wu, K. L. Johnson-Davis and K. L. Lynch, Clin. Chim. Acta, 2016, 454, 130–134 CrossRef CAS PubMed.
- Q.-Z. Zhu, P. Degelmann, R. Niessner and D. Knopp, Environ. Sci. Technol., 2002, 36, 5411–5420 CrossRef CAS PubMed.
- X. Wu, J. Liu, J. Wu, Y. Wang, C. Xue, R. Wang, J. Hong and X. Zhou, J. Sep. Sci., 2012, 35, 3593–3599 CrossRef CAS PubMed.
- R. Wang, Y. Wang, C. Xue, T. Wen, J. Wu, J. Hong and X. Zhou, J. Sep. Sci., 2013, 36, 1015–1021 CrossRef CAS PubMed.
- A. Ostovan, M. Ghaedi, M. Arabi and A. Asfaram, J. Chromatogr. A, 2017, 1520, 65–74 CrossRef CAS PubMed.
- A. N. Hasanah, F. Pessagno, R. E. Kartasasmita, S. Ibrahim and P. Manesiotis, J. Mater. Chem. B, 2015, 3, 8577–8583 RSC.
- A. J. Hall, P. Manesiotis, M. Emgenbroich, M. Quaglia, E. De Lorenzi and B. Sellergren, J. Org. Chem., 2005, 70, 1732–1736 CrossRef CAS PubMed.
- A. J. Hall, M. Quaglia, P. Manesiotis, E. De Lorenzi and B. Sellergren, Anal. Chem., 2006, 78, 8362–8367 CrossRef CAS PubMed.
- P. A. Gale, E. N. W. Howe and X. Wu, Chem, 2016, 1, 351–422 CAS.
- S. Camiolo, P. A. Gale, M. B. Hursthouse, M. E. Light and A. J. Shi, Chem. Commun., 2002, 758–759 RSC.
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC.
- M. Pei, X. Zhu and X. Huang, J. Chromatogr. A, 2018, 1531, 13–21 CrossRef CAS PubMed.
- H. Yan, M. Gao and J. Qiao, J. Agric. Food Chem., 2012, 60, 6907–6912 CrossRef CAS PubMed.
- J. Yuan and M. Antonietti, in Applications of Ionic Liquids in Polymer Science and Technology, 2015, pp. 47–67, DOI:10.1007/978-3-662-44903-5_3.
- W. Bi, M. Tian and K. H. Row, J. Chromatogr. B, 2013, 913–914, 61–68 CrossRef CAS PubMed.
- J. P. Fan, Z. Y. Tian, S. Tong, X. H. Zhang, Y. L. Xie, R. Xu, Y. Qin, L. Li, J. H. Zhu and X. K. Ouyang, Food Chem., 2013, 141, 3578–3585 CrossRef CAS PubMed.
- M. Tokuda and H. Minami, J. Colloid Interface Sci., 2013, 398, 120–125 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation data, 1H NMR titrations, FT-IR spectra. See DOI: 10.1039/c8ra01135d |
| This journal is © The Royal Society of Chemistry 2018 |