
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSm2O3/Co3O4 catalysts prepared by dealloying for low-temperature CO oxidation
Dong Duan ,
Chunxi Hao,
Wenyu Shi,
Haiyang Wang and
Zhanbo Sun*
,
Chunxi Hao,
Wenyu Shi,
Haiyang Wang and
Zhanbo Sun*
School of Science, MOE Key Laboratory for Non-Equilibrium Synthesis and Modulation of Condensed Matter, Key Laboratory of Shaanxi for Advanced Functional Materials and Mesoscopic Physics, Xi'an Jiaotong University, Xi'an 710049, PR China. E-mail: szb@mail.xjtu.edu.cn; Tel: +86 29 82665995
First published on 21st March 2018
Abstract
A series of Co3O4 catalysts modified by Sm were prepared by a combined dealloying and calcination approach, and the catalytic activities were evaluated using CO catalytic oxidation. The Sm2O3/Co3O4 catalysts were composed of a large number of nanorods and nanosheets, and exhibited a three-dimensional supporting structure with pores. The experimental results revealed that the addition of a small amount of Sm into the precursor AlCo alloy led to a dealloyed sample with improved catalytic activity, and the dealloyed Al90Co9.5Sm0.5 ribbons (0.5 Sm2O3/Co3O4) calcined at 300 °C showed the highest activity for CO oxidation with complete CO conversion at 135 °C, moreover, CO conversion almost no attenuation, even after 70 hours of catalytic oxidation, which is superior to that of Co3O4. The enhanced catalytic activity of the Sm2O3/Co3O4 catalyst can be attributed to the large specific surface area, more reactive oxygen species and Co3+ ion, as well as electronic interactions between Sm and Co.
1. Introduction
The catalytic oxidation of CO has drawn much attention due to its important applications in industrial catalysis,1 pollution control,2,3 gas masks,4,5 closed-cycle CO2 lasers6,7 and gas sensors.8 At present, the mainstream catalyst is a supported catalyst containing noble metal,9–11 but due to its scarce resources and expensive price, its large-scale industrial application is limited. Increasing the utilization of non-precious metals and the use of non-precious metal catalysts are the two basic ways to save costs. Oxides have become an important direction for the study of low-cost catalysts because of their cheap prices, abundant reserves and unique redox properties.12Transition metal oxides have been widely studied as catalysts for heterogeneous catalysis.13,14 Among the metal oxides, tricobalt tetraoxide with excellent redox property and rich reserves is the most active for CO oxidation and is a very promising substitute for precious metal catalysts.15 Xie et al.15 reported that Co3O4 nanorods not only catalyse CO completely at temperatures as low as −77 °C (the feed gas flow rate was 50 ml min−1) but also have high stability in the presence of steam in the feed gas. Song et al.16 reported that the synthesis of mesoporous tricobalt tetroxide by an inverse surfactant micelle method can achieve complete oxidization (100% conversion) of CO to CO2 at −60 °C under normal conditions (∼3–10 ppm of H2O and a flow rate of 20 ml min−1) and at 80 °C under moisture rich conditions (∼3% H2O and a flow rate of 20 ml min−1).
However, pure Co3O4 used as a catalyst still has some drawbacks, such as low thermal stability at high temperatures and water deactivation for CO oxidation at low temperatures.17 In view of this, researches on the modification of Co3O4-based catalytic materials have drawn much attention. It has been determined that the catalytic performance of Co3O4 can be significantly improved by doping appropriate rare earth metals as promoters in a Co3O4 catalyst.18,19 For example, Hou et al.18 reported that a CeO2/Co3O4 catalyst that was prepared by an impregnation method exhibits much better resistance to water vapour poisoning than that of a Co3O4 catalyst for CO oxidation. Additionally, the CeO2/Co3O4 catalyst possesses a higher dispersion degree, smaller particles, and a larger SBET, and the interaction between CeO2 and Co3O4 exists, which may contribute to the excellent water resistance for low-temperature CO oxidation. Liotta et al.19 found that doping a proper amount of CeO2 into the Co3O4 catalyst significantly improved the catalytic activity and thermal stability due to the presence of CeO2 promoting the efficiency of the Co3+–Co2+ redox couple.
Lanthanide oxides are well-known effective rare earth catalyst additives. Samarium oxide, one of the lanthanide oxides, has great promise as a good catalyst due to its special properties. Imamura et al.20 reported that the addition of a small amount of Sm (molar ratio Sm/Co = 0.6/10) to Co3O4 not only enhances its catalytic oxidation activity for toluene but also enhances the thermal stability of Co3O4. Xu et al.17 studied a series of Sm-modified Co3O4 catalysts by coprecipitation, and the results show that the addition of a small amount of Sm into Co3O4 led to an improvement in the catalytic activity for CH4 and CO oxidation. This improvement is due to the addition of a small amount of Sm resulting in the formation of spinel Co3O4 and amorphous SmCoO3, hence increasing the number of Co3+ and the active surface oxygen species, which are responsible for the improvement in the activity. However, only a few studies are reported on preparation of Sm2O3/Co3O4 composites, and the design of Sm2O3/Co3O4 catalyst with high activity is still highly essential and technologically important.
Dealloying is a promising method to prepare various nanoporous materials included noble metals.21 This method can also be extended to fabricate other oxide composite materials with high catalytic activities.22 In this work, the Sm2O3/Co3O4 catalyst for CO oxidation was prepared by using dealloying melt-spun Al–Co–Sm alloys in an alkaline solution and calcination in O2. The prepared catalysts exhibited a unique nanorod and nanosheet interconnected structure and showed enhanced catalytic activity for CO oxidation.
2. Experiment and method
2.1. Preparation
Al90Co10−xSmx (x = 0, 0.3, 0.5, 1, 1.5 and 3 at%) and Al90Sm10 alloys were prepared from pure Al (99.90 wt%), pure Sm (99.90 wt%) and pure Co (99.99 wt%) by arc-melting using high-purity Ar as the protective atmosphere. The ingot alloys were remelted in a quartz tube on the surface of a single roller with rapid solidification into ribbons at 33 m s−1. The as-quenched ribbons were immersed into a 10 wt% NaOH aqueous solution at room temperature for 3 h and then treated at 80 °C for another 10 h to remove residual Al. The acquired samples were repeatedly rinsed with reverse osmosis water and dehydrated alcohol, and dried at 50 °C for 3 h. For simplicity, the samples after dealloying can also be named according to the Sm content in the precursor alloy, that is, Sm2O3, Co3O4 and YSm2O3/Co3O4 (Y = 0.3, 0.5, 1, 1.5, and 3). All the dealloyed ribbons were pretreated at 100–500 °C for 2 h in O2 at a total flow rate of 18 ml min−1.2.2. Characterization
X-ray diffraction (XRD) patterns of the prepared samples were recorded by a Bruker D8 Advance diffractometer instrument using a Cu Kα target for X-ray irradiation at 20 kV and 40 mA. The microscopic morphology of the samples was observed by field emission scanning electron microscopy (SEM, JSM-7000F microscope) equipped with an INCA X-sight Oxford energy dispersive X-ray spectrometer (EDS) and high-resolution transmission electron microscopy (TEM, JEM-2100 microscope). The nitrogen sorption isotherms, specific surface areas and pore sizes of those prepared samples were measured with a Micromeritics ASAP 2020 apparatus at −77 K. Thermogravimetric analysis (TG) and differential scanning calorimetry (DSC) experiments were carried out on an STA 449C instrument with a heating rate of 10 °C min−1. X-ray photoelectron spectroscopy (XPS) was recorded on an Axis Ultra Kratos (UK) multifunctional spectrometer using monochromatic Al Kα radiation (1486.6 eV).2.3. Catalytic evaluation
The catalytic activities of the samples were evaluated using CO catalytic oxidation. One-hundred milligrams of the catalyst was placed into a quartz tube with an inner diameter of 6 mm for the measurements, and an asbestos column with a length of approximately 1 cm was used to fix the catalyst. Then, the quartz tube was placed into a variable temperature reaction furnace. A reaction gas mixture consisting of 1% CO, 10% O2 and 89% N2 (volume) was fed into the reactor using a mass flow metre (Brooks 5850E) at a total flow rate of 100 ml min−1 (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1). The concentration of produced CO2 and unreacted CO gas was analysed online with a GC-7900 gas chromatograph equipped with a flame ionization detector (FID). The CO conversion rate (%) was determined by the changes in the inlet and outlet CO concentration with the following formula:
000 h−1). The concentration of produced CO2 and unreacted CO gas was analysed online with a GC-7900 gas chromatograph equipped with a flame ionization detector (FID). The CO conversion rate (%) was determined by the changes in the inlet and outlet CO concentration with the following formula:where Cin represents the inlet CO concentration and Cout is the outlet CO concentration.
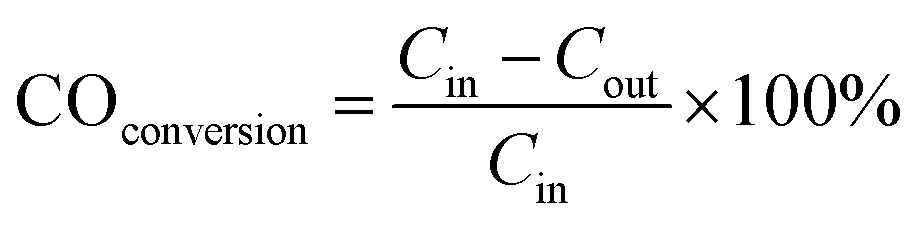
3. Results and discussion
3.1. Phase constituents and microstructure of the dealloyed samples
Fig. 1 shows the XRD patterns and EDS results of the dealloyed samples calcined at different temperatures. Only the definite diffraction peaks of Co3O4 appear in the diffraction pattern after the Al90Co9.5Sm0.5 samples were dealloyed and calcined at 300 °C. After the samples were annealed at 700 °C or 800 °C, the diffraction peak of Sm2O3 still was not apparent (Fig. 1(a)). However, as shown in Fig. 1(b), the atomic ratio of Sm to Co is close to the mean composition in the precursor, indicating that the loss of Sm and Co was small during the dealloying, and the peaks cannot be detected by XRD due to the low Sm content. The residual Al contents were less than 3.93 at% (Fig. 1(b)) of the dealloyed samples obtained from the Al90Co9.5Sm0.5 alloys. At the same time, note that when the content of Sm is increased to 30%, a weak diffraction peak of Sm2O3 appears, and the diffraction peak of Co3O4 disappears, indicating that the addition of rare earth Sm may suppress the crystallization of Co3O4, which is similar to that of the oxide CeO2.12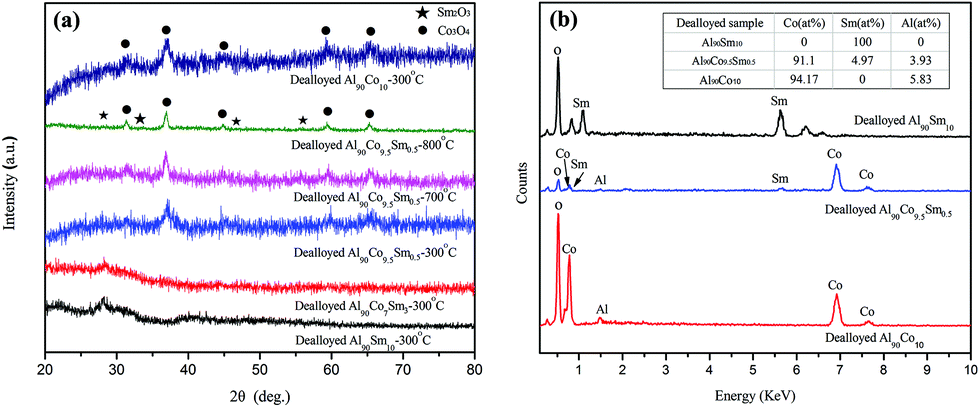 | ||
| Fig. 1 (a) XRD patterns of the dealloyed Al90Co10, Al90Sm10, Al90Co7Sm3 ribbons calcined at 300 °C and Al90Co9.5Sm0.5 ribbons calcined at 300, 700 and 800 °C. (b) EDS patterns of the dealloyed Al90Co10, Al90Sm10 and Al90Co9.5Sm0.5 ribbons calcined at 300 °C. | ||
The SEM images of dealloyed Al90Co10, Al90Sm10 and Al90Co9.5Sm0.5 ribbons calcined at 300 °C are shown in Fig. 2. For Sm2O3 and Co3O4, the samples exhibit a nanosheet and nanorod morphologies, respectively. The nanosheet thickness is approximately 30 nm, and the nanorod diameter is approximately 20–30 nm with excellent uniformity, which can be seen in Fig. 2(a) and (b). However, the plan-view image exhibited a unique nanorod and nanosheet interconnected structure for the Sm2O3/Co3O4 prepared from the dealloyed Al90Co9.5Sm0.5 ribbons calcined at 300 °C, see Fig. 2(c), and the cross-sectional image exhibited a three-dimensional porous ligament structure composed of many nanosheets and nanorods interpenetrated with each other, see Fig. 2(d).
 | ||
| Fig. 2 SEM images of dealloyed Al90Co10 (a), Al90Sm10 (b) and Al90Co9.5Sm0.5 (c); and (d) calcined at 300 °C. | ||
The TEM images and corresponding selected-area electron diffraction patterns of dealloyed Al90Co9.5Sm0.5 ribbons calcined at 300 °C are presented in Fig. 3. The sample is composed of nanorods and nanosheets, consistent with the SEM results, and the nanosheets contain many micropores, as shown in Fig. 3(a). The diffraction rings of the SAED pattern (Fig. 3(b)) were indexed to Co3O4 (440), (511), and (220) and Sm2O3 (400), (440), and (211), which verified the coexistence of Co3O4 and Sm2O3. Fig. 3(c) and (d) shows the HRTEM images of the nanorods and nanosheets in Fig. 3(a). The lattice spacing in Fig. 3(c) is approximately 0.286 nm, which corresponds to the (220) plane of Co3O4. The lattice spacings in Fig. 3(d) are approximately 0.269 nm and 0.193 nm, which correspond to the (400) and (440) planes of Sm2O3, respectively. According to these results, Sm2O3 is present in the form of nanorods, and Co3O4 is present in the form of nanosheets in the sample of dealloyed Al90Co9.5Sm0.5 ribbons. When calcined at 300 °C, the Sm2O3 nanorods and Co3O4 nanosheets stacked together constitute a rough interface, and the interface interaction is significantly enhanced between them.
 | ||
| Fig. 3 TEM image (a), corresponding selected-area electron diffraction pattern (b) and HRTEM images (c) and (d) of the dealloyed Al90Co9.5Sm0.5 ribbons calcined at 300 °C. | ||
3.2. Physical and surface properties
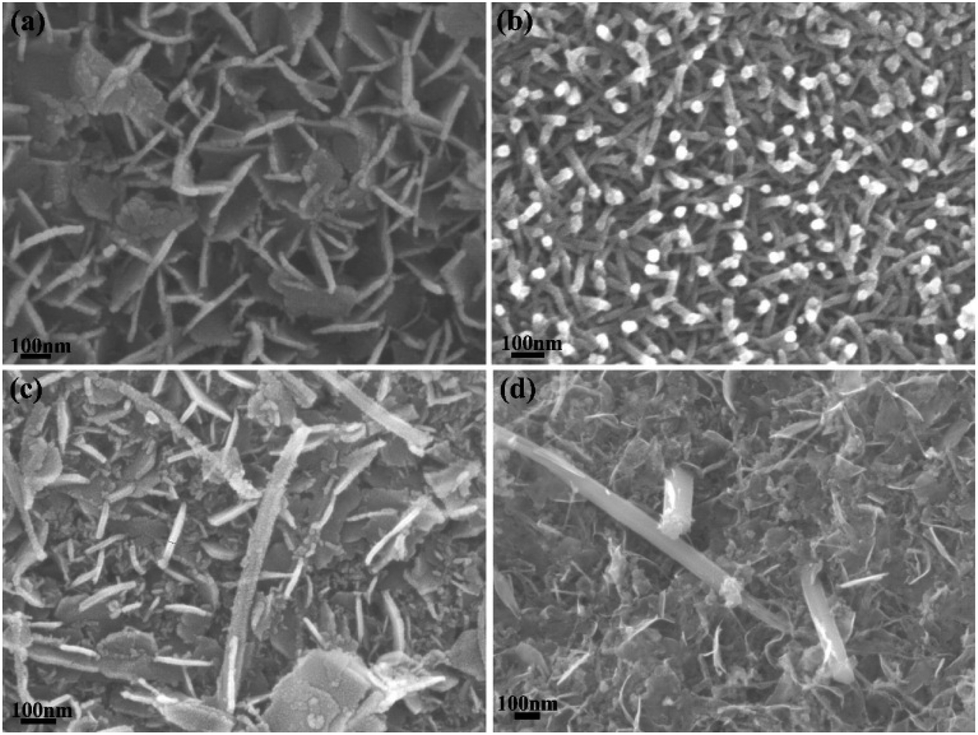
Fig. 4 shows the N2 adsorption–desorption isotherms and the corresponding pore size distribution curves for the dealloyed Al90Co10, Al90Sm10 and Al90Co9.5Sm0.5 ribbons calcined at 300 °C. The N2 sorption isotherms of all the samples were type IV with hysteresis loops, indicative of their mesoporous structures. The BET surface area (SBET), pore size (DP) and pore volume (VP) of these samples are listed in Table 1. As seen from Table 1, the BET surface area and pore volume of all the Sm2O3/Co3O4 composites are larger than those of the Co3O4 and Sm2O3, and the 0.5 Sm2O3/Co3O4 composite has the largest specific surface area, 95.72 m2 g−1, which is approximately three times of that of Sm2O3 and Co3O4. The results show that the addition of the Sm2O3 nanorods can effectively refine the porous structure. Fig. 4(b) is the corresponding pore size distribution curve for the three catalysts. Compared to Sm2O3 and Co3O4, the 0.5 Sm2O3/Co3O4 composite has a narrower pore size distribution and a smaller pore size with an average pore size of approximately 8.355 nm. This structure is a typical porous structure, which is suitable for CO catalytic oxidation. | ||
| Fig. 4 N2 adsorption–desorption isotherms and corresponding pore size distribution curves at 77 K of the dealloyed Al90Co10, Al90Sm10 and Al90Co9.5Sm0.5 ribbons calcined at 300 °C. | ||
| Sample | SBET/m2 g−1 | Dp/nm | Vp/cm3 g−1 |
|---|---|---|---|
| Co3O4 | 30.38 | 20.628 | 0.157 |
| 0.3 Sm2O3/Co3O4 | 58.34 | 11.442 | 0.168 |
| 0.5 Sm2O3/Co3O4 | 95.72 | 8.355 | 0.200 |
| 1 Sm2O3/Co3O4 | 86.82 | 11.140 | 0.242 |
| 1.5 Sm2O3/Co3O4 | 55.62 | 16.350 | 0.227 |
| 3 Sm2O3/Co3O4 | 41.55 | 22.148 | 0.230 |
| Sm2O3 | 33.68 | 30.672 | 0.258 |
Fig. 5 shows the TGA–DSC curves for the dealloyed Al90Co9.5Sm0.5 ribbons in O2/Ar atmosphere. The sample has mainly three weight loss stages. The first stage exhibits a 10.01% weight loss below 250 °C, which is attributed to the removal of physically adsorbed H2O. Note that there is a pronounced exothermic peak at 213 °C due to the oxidation of Co and the crystallization of Co3O4.17 In the temperature range of 250–950 °C, corresponding to the slow crystallization process of Co3O4 and Sm2O3 formed, the TG curve weight decreased by approximately 4.27%. At 962 °C, there is a distinct endothermic peak, which corresponds to a sharp decrease of approximately 4.22% in the TG curve due to the decomposition of Co3O4 into CoO and O2, and the corresponding equation is 2Co3O4 = 6CoO + O2↑.17,38
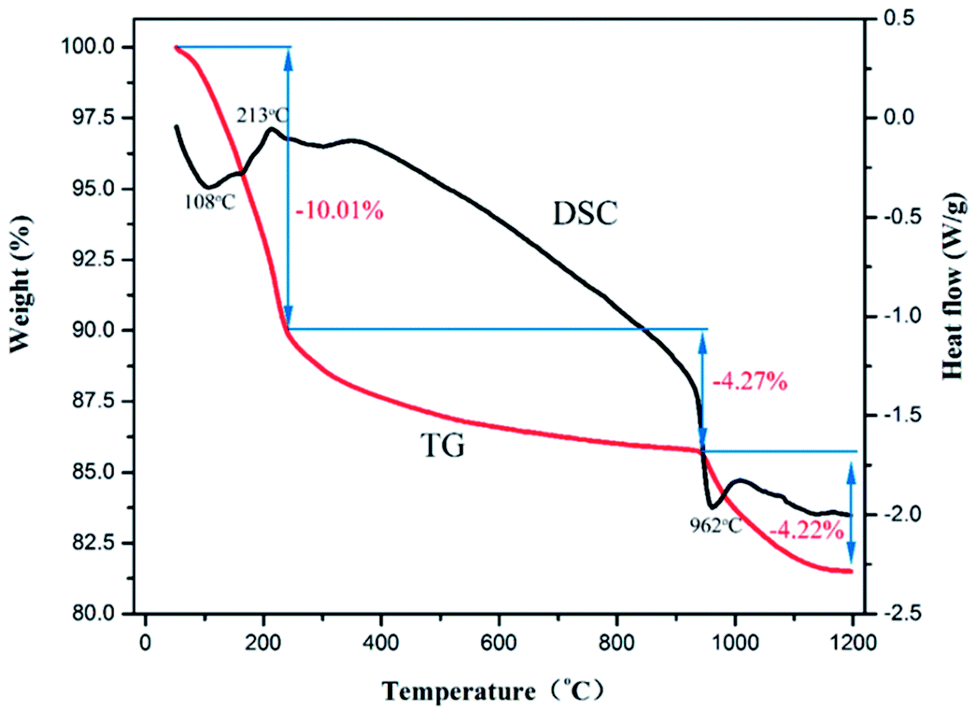 | ||
| Fig. 5 TGA–DSC curves for the dealloyed Al90Co9.5Sm0.5 ribbons in O2/Ar atmosphere. | ||
Fig. 6 shows the O 1s, Sm 3d and Co 2p XPS spectra of the Sm2O3 nanorod, 0.5 Sm2O3/Co3O4 composite and Co3O4 nanosheet. The O 1s spectrum of the Sm2O3/Co3O4 composite consisted of three peaks. The peaks centred at ∼529.95 eV (Olat), ∼531.3 eV (Osur), and ∼532.9 eV (OH2O) were assigned to lattice oxygen species, surface adsorbed and weakly bonded oxygen species, and adsorbed water, respectively, according to ref. 23 and 24. It is generally accepted that surface oxygen species are usually active oxygen species for catalytic reactions, which could be explained using the relative quantity of Osur species, i.e., 31.27% and 47.79% for Co3O4 and Sm2O3/Co3O4, respectively. The results show that there were more active oxygen species existed on the surface of the Sm2O3/Co3O4 composite compared with Co3O4.
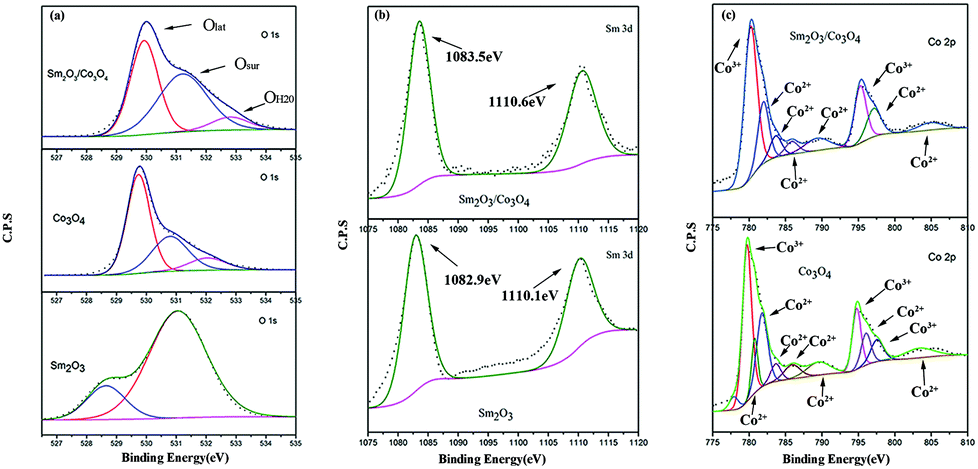 | ||
| Fig. 6 XPS spectra of Sm2O3, Co3O4 and Sm2O3/Co3O4 for the O 1s peaks (a), Sm 3d peaks (b) and Co 2p peaks (c). | ||
Fig. 6(b) presents Sm 3d XPS spectra that is deconvolved into two peaks. According to the literature,25 the low-energy peak (Sm 3d5/2) at 1083.5 eV and the high-energy peak (Sm 3d3/2) at 1110.6 eV were attributed to Sm3+ (Sm2O3), according to the literature.25 Note that the Sm 3d spectra of the Sm2O3/Co3O4 composite shifts nearly 0.6 eV compared to Sm2O3, indicating that there is an electronic interaction between Sm and Co. The XPS pattern of Co 2p is shown in Fig. 6(c). For the Sm2O3/Co3O4 composite, the peaks at 780.2 eV and 795.25 eV are assigned to Co3+ and the other six peaks (781.95 eV; 797.1 eV; 783.65 eV; 785.85 eV; 789.35 eV; and 804.80 eV) represent the presence of Co2+.26–31 According to the literature,15,17 Co3+ was responsible for the improvement in the activity, and the concentration of Co3+ ions can be expressed using the ratio of their corresponding spectral areas to the total area (Co2+ + Co3+). The ratios were 56.5% for Sm2O3/Co3O4 and only 46.6% for Co3O4, indicating that there were more Co3+ ions on the surface in the Sm2O3/Co3O4 catalyst. Therefore, the introduction of Sm2O3 to the Co3O4 resulted in an increase in Co3+ species and active oxygen species that facilitates an electronic interaction between Sm and Co, which improves the redox potential of the Co3O4-based catalysts.
3.3. CO catalytic oxidation
Fig. 7(a) shows the catalytic performance of the Sm2O3, Co3O4 and Sm2O3/Co3O4 catalysts with different Sm2O3 contents for the CO catalytic oxidation. The results show that the catalytic activity of all the Sm2O3/Co3O4 catalysts were greater than that of Sm2O3 and Co3O4. The CO conversion increased progressively as the content of Sm increased up to 0.5 at% in the precursory alloy, and then decreased with a Sm content above 0.5 at%. The CO conversion over Sm2O3/Co3O4 from the Al90Co9.5Sm0.5 alloy was more than 99% at 135 °C, while the conversions of Co3O4 and Sm2O3 were only approximately 35% and 3%, respectively. The temperature at which the conversion was 50% (T50) for Sm2O3/Co3O4 from the Al90Co9.5Sm0.5 alloys was 85 °C, which was much lower than that of Co3O4 (147 °C) and Sm2O3 (285 °C). These results indicated that the Sm2O3/Co3O4 catalyst from the Al90Co9.5Sm0.5 alloys exhibited an excellent ability to oxidize CO compared with the pure metal oxide counterparts.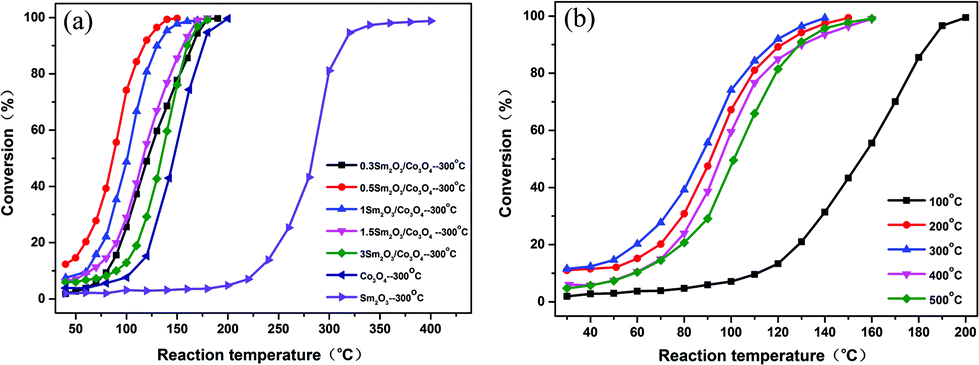 | ||
| Fig. 7 CO conversion as a function of reaction temperature over the Sm2O3, Co3O4 and Sm2O3/Co3O4 catalysts with different Sm2O3 contents (a) and the 0.5 Sm2O3/Co3O4 catalyst calcined at different temperatures (b). | ||
The CO conversion plots as a function of reaction temperature over the 0.5 Sm2O3/Co3O4 catalyst calcined at different temperatures are shown in Fig. 7(b). The catalytic activity was significantly enhanced when the sample was calcined above 200 °C. For the 0.5 Sm2O3/Co3O4 samples calcinated at different temperatures, the order of catalytic activity is obtained as follows: calcined at 100 °C < calcined at 500 °C < calcined at 400 °C < calcined at 200 °C < calcined at 300 °C. The experimental results indicated that the calcination temperature has an important influence on the catalytic activity of the sample.
The long-term stability of the Co3O4 and 0.5 Sm2O3/Co3O4 catalysts were evaluated and the results is are in Fig. 8(a). The Sm2O3/Co3O4 catalyst exhibited near 100% CO conversion without a noticeable deterioration in activity, even at high reaction rate, CO conversion only decays from 37% to 33%. However, CO conversion of the Co3O4 catalyst decreased from 37% to 23% when the holding time was 70 h at 140 °C. This result implied that the addition of Sm2O3 is conducive to maintaining a high stability.
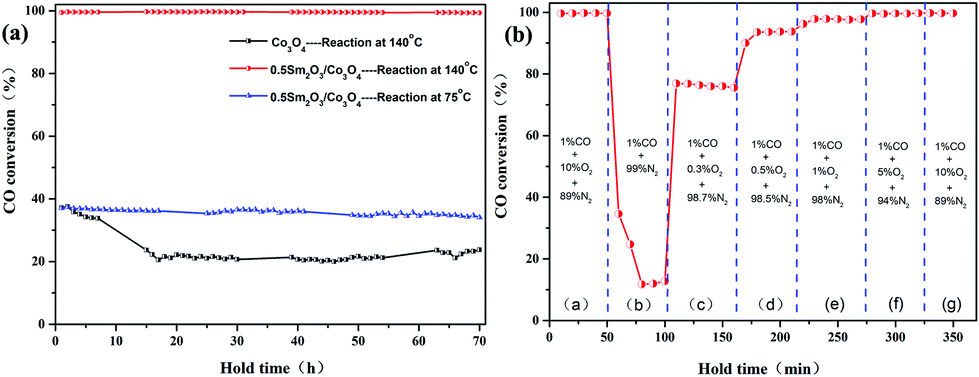 | ||
| Fig. 8 Long-term stability of the Co3O4 and 0.5 Sm2O3/Co3O4 catalysts for 70 h (reaction conditions: 1% CO, 10% O2 and 89% N2) (a) and the catalytic performance of 0.5 Sm2O3/Co3O4 catalysts under different oxygen concentrations at 140 °C (flow rate: 100 ml min−1) (b). | ||
Fig. 8(b) shows the effect of the oxygen concentration on the CO conversion over the 0.5 Sm2O3/Co3O4 catalyst calcined at a 300–140 °C constant temperature. As for the catalyst, the CO/O2 ratio has an important effect on the catalytic activity for CO oxidation. The effects of changing from O2-rich (10% O2), CO-rich (0–0.5% O2) to a close-to-stoichiometric (1% O2) and O2-rich gas mixtures were investigated. The catalyst was tested directly with the reactant gas (1% CO/10% O2/89% N2) at 140 °C for 50 min. As the O2 concentration decreased from 10% to 0%, as the residual O2 continued to be consumed, the CO conversion decreased sharply from 99% to 12%, then tended to be steady. The reason for the phenomenon maybe is caused by the surface lattice oxygen of catalyst reacted with CO and conforms to the Mars–van Krevelen type mechanism. When O2 concentration increased to 0.3%, the CO conversion increased abruptly from 12% to 76%, according to equation 2CO + O2 = 2CO2. The complete reaction cannot be achieved because 0.3% of O2 can only react with 0.6% of CO. As the oxygen content continued to increase to an O2-rich condition, the final CO conversion reached the initial 99%. During the increase in the O2 concentration from 0.5% to 10%, a new steady-state can be rapidly rebuilt. These results show that during CO oxidation, the activity is strongly affected by the reactant gas mixture composition. Above results clearly indicated that the prepared Sm2O3/Co3O4 catalyst can exhibit enhanced catalytic activity and higher catalytic stability without a deactivation in activity after an extended time test.
3.4. Analysis and discussion
In the present work, the Sm2O3/Co3O4 catalyst with a high catalytic activity was obtained using a facile chemical dealloying and calcination method. During the dealloying process, melt-spun Al–Co–Sm alloys can preserve the structure and monolithic characteristics of the parent alloy at a macroscale because diffusion only occurs in the domain size.21 When the melt-spun Al–Co–Sm alloys were immersed in a NaOH solution, the Al atoms were progressively removed, and the Co atoms and Sm atoms on the surface were exposed to the alkaline solution. These lower coordinated metal atoms are highly active,32 which were likely to combine with OH− ions from H2O to form hydroxide at 80 °C. During this process, Sm(OH)3 can form nanorods due to the anisotropic growth of different nuclei under the basic hydrothermal condition, and Co(OH)3 aggregates and grows into nanosheet structures. Co(OH)3 and Sm(OH)3 could easily transform into Co3O4 and Sm2O3 by dehydration during calcination in O2, and due to the calcination, the nanorods and nanosheets are closely interconnected, forming a rough interface and enhancing the synergistic effect between the Sm2O3 and Co3O4. In addition to its inherent simplicity, the dealloying method can effectively avoid surface contamination of the preparative nanomaterials by organic chemicals or other surface-directing molecules, which is often seen in wet chemical synthesis at elevated temperatures.33Thus, this preparation method is a promising strategy for large-scale production.The BET and XPS analyses suggested that the introduction of trace Sm2O3 greatly enhanced the specific surface area of the catalyst, and created more active oxygen species and Co3+ ions, according to the literature.15 Additionally, the more Co3+ ions are created, the more active catalytic sites exist. On the other hand, the Sm2O3 nanorods and Co3O4 nanosheets are tightly connected to create many nanoscale interfaces, where the generated free electrons can easily migrate at the interfaces37 (Fig. 6(b)). These electrons not only transform molecular oxygen into an active oxygen species to interact with CO molecules adsorbed at the interfaces but also weaken the CO2 bond and accelerate CO2 dissociation, leading to an increase in the reaction rate.12 In addition, the Sm2O3/Co3O4 catalyst possessed a higher specific surface area (95.72 m2 g−1), narrower pore size distribution and more uniform pores than those of Co3O4 and Sm2O3 (Fig. 4), which also enlarged the contact interface accessibility or adsorbing molecules. All of these factors play a very positive role in the catalytic oxidation of CO.
Experimental and theoretical studies have widely discussed the CO oxidation reaction mechanism. Behm et al.34 showed that surface lattice oxygen is the active oxygen species for the CO oxidation, which follows an Au-assisted Mars–van Krevelen mechanism. Zhang et al.35 showed that the molecularly oxygen absorbed on both surface vacancies and lattice oxygen should participate in the CO oxidation. Zhang et al.36 reported that both oxygen on PTA (phosphotungstic acid) and oxygen molecules in the reaction gas are involved in the CO catalytic oxidation reaction. A possible CO oxidation reaction pathway on Sm2O3/Co3O4 catalyst is proposed in Fig. 9, by piecing together information from different techniques. Firstly, the surface lattice oxygen reacts with adsorbed CO and generates oxygen vacancies, and then reconnects with the molecular oxygen in the reaction gas, part of the molecular oxygen replenishes the missing surface lattice oxygen, and the other part adsorbed on the surface of the catalyst becomes the surface active oxygen, which directly reacts with CO, so the catalytic oxidation of CO can be long-term and stable.
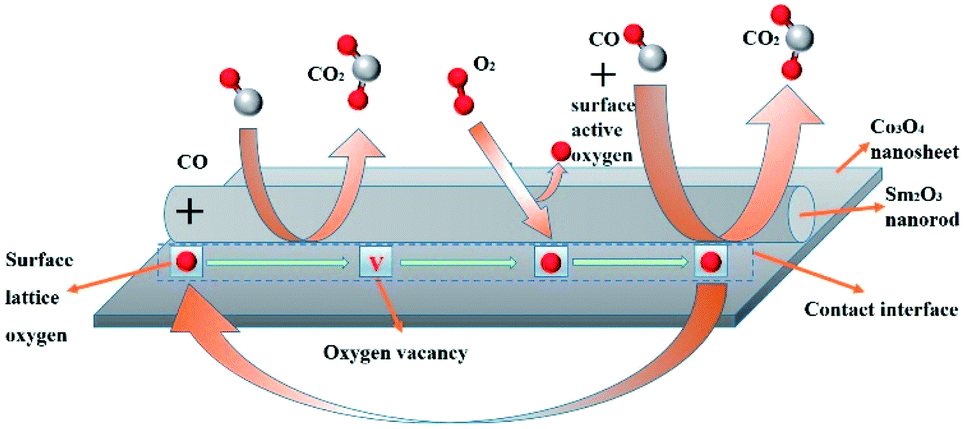 | ||
| Fig. 9 Possible CO oxidation reaction pathway on Sm2O3/Co3O4 catalyst. | ||
4. Conclusions
We have developed a simple and effective method for fabricating a Sm2O3/Co3O4 catalyst by means of dealloying in a 10 wt% NaOH aqueous solution and calcination in O2. Our results indicated that the Sm2O3/Co3O4 catalyst exhibits excellent catalytic activity for CO oxidation at ambient temperature. The enhanced catalytic activity of the Sm2O3/Co3O4 catalyst can be attributed to the large specific surface area, more reactive oxygen species and Co3+ ions, and electronic interactions between Sm and Co. In addition, the Sm2O3/Co3O4 catalyst showed good long-term catalytic stability and even after 70 hours, CO conversion still almost no attenuation. It is expected that many other useful metal oxide composites can be fabricated in a similar manner.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51771141 and 51371135 and 51671155).Notes and references
- A. Mart Nez-Arias, M. Fernández-Garc A, O. Gálvez, J. M. Coronado, J. A. Anderson, J. C. Conesa, J. Soria and G. Munuera, J. Catal., 2000, 195, 207–216 CrossRef.
- S. Jirawongnuson, W. Wachirapan, T. Suthiprasert and E. Wirojsakunchai, Key Eng. Mater., 2015, 656–657, 538–543 CrossRef.
- T. Suthiprasert, T. Limpurimongkol, S. Jirawongnuson, T. Aroonsrisopon and E. Wirojsakunchai, Eng. J., 2017, 21, 93–103 CrossRef.
- D. K. Kim, B. I. Kim, C. H. Shin and C. S. Shin, Journal of the Korean Society of Safety, 2006, 35–45 Search PubMed.
- H. S. Kong and S. H. Kim, Int. J. Pharm. Technol., 2016, 8, 26870–26875 CAS.
- D. R. Schryer, B. T. Upchurch, R. V. Hess, G. M. Wood, B. D. Sidney, I. M. Miller, K. G. Brown, J. D. Vannorman, J. Schryer and D. R. Brown, Low-Temperature CO-Oxidation Catalysts for Long-Life CO2 Lasers, NASA Conf. Publ., 1990, 1, 41–53 Search PubMed.
- A. V. Bondarenko, V. D. Gavrilyuk, V. S. Golubev, F. V. Lebedev and M. M. Smakotin, Quantum Electron., 1980, 10, 775–780 Search PubMed.
- N. Barsan and U. Weimar, J. Phys.: Condens. Matter, 2003, 15, R813 CrossRef CAS.
- S. A. Nikolaev, E. V. Golubina, I. N. Krotova, M. I. Shilina, A. V. Chistyakov and V. V. Kriventsov, Appl. Catal., B, 2015, 168–169, 303–312 CrossRef CAS.
- A. Luengnaruemitchai, K. Srihamat, C. Pojanavaraphan and R. Wanchanthuek, Int. J. Hydrogen Energy, 2015, 40, 13443–13455 CrossRef CAS.
- H. Zhu, Z. Qin, W. Shan, W. Shen and J. Wang, J. Catal., 2004, 225, 267–277 CrossRef CAS.
- X. Zhang, K. Li, W. Shi, C. Wei, X. Song, S. Yang and Z. Sun, Nanotechnology, 2017, 28, 045602 CrossRef PubMed.
- C. Stampfl and M. Scheffler, Surf. Sci., 1999, 433–435, 119–126 CrossRef CAS.
- J. Jansson and C. T. H. Gskola, Chalmers University of Technology, 2002.
- X. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746 CrossRef CAS PubMed.
- W. Song, A. S. Poyraz, Y. Meng, Z. Ren, S. Y. Chen and S. L. Suib, Chem. Mater., 2012 Search PubMed.
- X. Xu, H. Han, J. Liu, W. Liu, W. Li and X. Wang, J. Rare Earths, 2014, 32, 159–169 CrossRef CAS.
- X. D. Hou, Y. Z. Wang and Y. X. Zhao, Catal. Lett., 2008, 123, 321–326 CrossRef CAS.
- L. F. Liotta, G. D. Carlo, G. Pantaleo, A. M. Venezia and G. Deganello, Appl. Catal., B, 2006, 66, 217–227 CrossRef CAS.
- S. Imamura, K. Fukuda, T. Nishida and T. Inui, J. Catal., 1985, 93, 186–191 CrossRef CAS.
- I. Mccue, E. Benn, B. Gaskey and J. Erlebacher, Annu. Rev. Mater. Res., 2016, 46, 263–286 CrossRef CAS.
- X. Zhang, G. Li, S. Yang, X. Song and Z. Sun, Microporous Mesoporous Mater., 2016, 226, 61–70 CrossRef CAS.
- N. S. Mcintyre, D. D. Johnston, L. L. Coatsworth, R. D. Davidson and J. R. Brown, Surf. Interface Anal., 1990, 15, 265–272 CrossRef CAS.
- L. Armelao, D. Barreca, S. Gross and E. Tondello, Surf. Sci. Spectra, 2001, 8, 14–23 CrossRef CAS.
- N. Sasmal, M. Garai and B. Karmakar, Journal of Asian Ceramic Societies, 2016, 4, 29–38 CrossRef.
- C. A. Strydom and H. J. Strydom, ChemInform, 1989, 20, 191–195 Search PubMed.
- J. P. Bonnelle, J. Grimblot and A. D'Huysser, J. Electron Spectrosc. Relat. Phenom., 1975, 7, 151–162 CrossRef CAS.
- C. V. Schenck, J. G. Dillard and J. W. Murray, J. Colloid Interface Sci., 1983, 95, 398–409 CrossRef CAS.
- J. C. Carver, G. K. Schweitzer and T. A. Carlson, J. Chem. Phys., 1972, 57, 973–982 CrossRef CAS.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855–861 CrossRef CAS.
- G. Mattogno, C. Ferragina, M. A. Massucci, P. Patrono and A. L. Ginestra, J. Electron Spectrosc. Relat. Phenom., 1988, 46, 285–295 CrossRef CAS.
- C. Xu, Y. Liu, C. Zhou, L. Wang, P. H. Geng and P. Y. Ding, Chemcatchem, 2011, 3, 399–407 CrossRef CAS.
- B. Varghese, C. H. Teo, Y. Zhu, M. V. Reddy, B. V. R. Chowdari, A. T. S. Wee, V. B. C. Tan, C. T. Lim and C. H. Sow, Adv. Funct. Mater., 2007, 17, 1932–1939 CrossRef CAS.
- D. Widmann and R. J. Behm, Acc. Chem. Res., 2014, 47, 740–749 CrossRef CAS PubMed.
- X. Zhang, D. Duan, G. Li, W. Feng, S. Yang and Z. Sun, Nanotechnology, 2018, 29(9), 095606 CrossRef PubMed.
- B. Zhang, H. Asakura and N. Yan, Ind. Eng. Chem. Res., 2017, 56, 3578–3587 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang and T. Zhang, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- T. Zhang, P. Hing, H. Huang and J. Kilner, J. Eur. Ceram. Soc., 2002, 22, 27–34 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |