
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid formation of polyimide nanofiber membranes via hot-press treatment and their performance as Li-ion battery separators†
Jian Houa,
Wongi Jangab,
Sungyul Kimc,
Jun-Hyun Kimb and
Hongsik Byun *a
*a
aDepartment of Chemical Engineering, Keimyung University, Daegu, 42601, South Korea. E-mail: hsbyun@kmu.ac.kr
bDepartment of Chemistry, Illinois State University, Normal, Illinois 61790-4160, USA
cDepartment of Electronic and Electrical Engineering, Keimyung University, Deagu, 42601, South Korea
First published on 19th April 2018
Abstract
We describe a new strategy to prepare thermally- and electrochemically-stable polyimide (PI) nanofiber membranes by the hot-press treatment of polyamic acid (PAA) nanofiber sheets in situ and examine their performance as Li-ion battery separators. Typical thermal imidization of PAA to PI membranes using sequential high temperature treatments in an oven takes a long time, but our method readily completes this conversion process at a mild temperature in 30 min while generating a high probability of inter-nanofiber imidization. Along with the improved electrolyte uptake capability and uniform distribution of the pore size and porosity caused by the dense and compact arrangements, the hot-press-induced PI membrane exhibits relatively thin sheets and a much greater mechanical strength than the membrane prepared by the thermal treatment. Subsequently, these PI-based membranes are installed in Li-ion full coin cells as battery separators whose C-rate (charging and discharging) performances are comparable to a commercial polyethylene (PE) separator. In addition, the highly improved thermal stabilities of these PI separators over PE separators are observed during thermal shrinkage and hot-box tests. Overall, our strategy can allow for the manufacture of diverse PI-based membranes with minimal preparation time and cost that can be utilized in high power portable devices requiring thermal and electrochemical stability.
Introduction
With the growing use of portable electronic devices, improving the performance of currently available rechargeable battery systems based on lithium (Li)- and nickel (Ni)-derived materials is of great interest.1–5 Portable electronic systems designed with Li-ion batteries are very attractive due to their high energy density, stable cyclability, low maintenance, ability to be miniaturized, and environmental safety characteristics.2–4 However, the functions of Li-based batteries are strongly dependent on the proper combination of components, including electrodes, electrolytes, and separators, which still require further improvement.5 The battery separator plays an essential role in achieving a high power density and maintaining the safe operation of the battery, keeping it free from short circuits and overheating problems; separators serve as a barrier to prevent the physical contact between two electrodes and freely transport electrolytes (i.e., Li ions) to complete the battery circuits.1,4,6–9Most commonly available separators for Li-ion batteries are made of porous polymer membranes, such as polyethylene (PE), polypropylene (PP), and PE-PP blends, due to their relatively low processing cost and good mechanical properties.10 However, these polyolefin-based separators have limited applications because of their inherent low porosity and relatively poor electrolyte uptake capabilities. In addition, these separators have safety concerns because their thermal stabilities are not high enough to manage the high Joule heating caused by an extensive current flow, which could lead to a fire and/or the explosion of the batteries. Thus, electrochemically- and thermally-stable separators with improved electrolyte uptake capabilities and better safety at a low cost are necessary to replace these polyolefin-based separators.4,7,10 Furthermore, these types of membrane separators can also allow for their utilization in high power portable battery systems with a greater reliability.
Polymer-nanofiber-based membranes are one of the most promising separator systems because their preparation process requires a minimum use of solvent (i.e., they are eco-friendly and cost effective), and their nanoscale polymer network renders highly increased surface areas.11–13 The reliable preparation of nanoscale fibers with a tunable pore size and porosity can be achieved by properly selecting thermally- and electrochemically-stable polymer precursors and precisely controlling the electrospinning conditions. Given the increased surface area of the fiber network, the resulting materials can ideally exhibit a superior electrolyte uptake capability. In this research, we prepared polyimide (PI) nanofiber membranes possessing a high thermal stability by electrospinning polyamic acid (PAA) precursors followed by a hot-press treatment of the resulting PAA nanofibers. Although electrospun PAA nanofibers typically undergo a thermal imidization process to form PI membranes via sequential high temperature treatments in an oven over a long period of time,7,9,10,12–14 our rapid imidization approach works by introducing pressure under mild temperature conditions (i.e., the hot-press treatment). This approach reduced the overall imidization process to 30 min, and the resulting PI membranes exhibited a uniform distribution of pore sizes and increased porosity, as well as an improved electrolyte uptake capability over the conventional PI membranes prepared by a thermal treatment and commercially available PE membranes. In addition, the PI-based membranes exhibited much higher thermal stabilities than the commercial PE membranes observed by a temperature-induced shrinkage test and thermal analyses. Subsequently, Li-ion full coin cells were manufactured with these membranes as battery separators, which exhibited a comparable C-rate (i.e., charging and discharging) performance and a much higher thermal stability in the hot-box test than the PE membrane separators. Our strategy is a simple and cost-efficient approach for modifying nanoscale-polymer-fiber-based membranes as novel separators possessing improved thermal and mechanical properties that can be used in battery systems that require excellent safety, reliability, and high power/rechargeable capabilities.
Experimental section
Materials
Pyromellitic dianhydride (PMDA, 97%), 4,4′-oxydianiline (ODA, 97%), and N,N-dimethylformamide (DMF, 99.5%) were purchased from Sigma-Aldrich. The commercially available polyethylene (PE) membranes were obtained from Toray Tonen Chemical Corp. (Japan) and served as the reference separators for the Li-ion batteries. Conductive materials [NCM523, (L&F Co., Ltd., South Korea), SGO-5 (SEC Carbon, Japan), and Super P (IMERYS, France)] and a binder (PVDF, Kureha, Japan) were used to prepare a cathode of coin cells, while a mixture of A-graphite (Showa Denko, Japan), Super P, and PVDF were used to prepare a carbon-based anode electrode. The PuriEL electrolyte containing 1.1 M of LiPF6 in a mixture of ethylene carbonate (EC), dimethyl carbonate (DMC), and diethyl carbonate (DEC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 v%) solution was used as purchased (Soulbrain Co. Ltd., South Korea). Stainless steel cases (SUS Cr2032) were obtained from Wellcos Corporation (South Korea) and used to assemble the coin cells.
:1:1 v%) solution was used as purchased (Soulbrain Co. Ltd., South Korea). Stainless steel cases (SUS Cr2032) were obtained from Wellcos Corporation (South Korea) and used to assemble the coin cells.
Preparation of PAA nanofibers and their conversion to PI membranes under various conditions
The formation of PAA nanofibers involved in two steps (Scheme S1 in the ESI section†); the first step was the preparation of the viscous PAA precursor by mixing the ODA and PMDA monomers.7,12,13,15,16 ODA (5.006 g, 0.025 mol) was completely dissolved in DMF (41.836 g) in a three-neck round-bottom flask under an N2 environment for 5 min, followed by adding a small amount of PMDA five times (for a total of 5.453 g, 0.025 mol) at 0 °C under continuous stirring. The viscosity of the mixture was measured as a function of time at 25 °C using a viscometer (DV-II + Pro, Brookfield Ametek Inc.) equipped with a CPE-52 spindle. The second step involved the optimization of the electrospinning precursor solution under various conditions. The viscous precursor solution was transferred to a 5 mL plastic syringe with a 23-gauge needle placed in an automated syringe pump (KDS100, KD Scientific Inc.) which was subjected to electrospinning at room temperature under the following conditions for 6 h: the ejection speeds were 0.3, 0.4, and 0.6 mL h−1, the voltage was 18 kV, the tip-to-collector distance (TCD) was 20 cm, the relative humidity was 50–60%, and a rotating metal drum covered with aluminum foil served as the counter electrode. The resulting electrospun nanofibers were then dried at 60 °C in a vacuum oven (JSVO-60T, JSR Corp.) for 24 h to completely remove the remaining solvent, which led to the formation of the PAA nanofiber mats. After folding these mats, the pressure (3000 psi) was gently applied at room temperature to form the membrane type of PAA sheets.Subsequently, the formation of the PI nanofiber membranes was carried out by the imidization of PAA nanofiber sheets under various conditions (representative images are shown in Fig. S1 in ESI†). Our strategy involved the introduction of both pressure and temperature (hot-press machine shown in Fig. S1g,† Heating Press DHP-2, Dae Heung Sci., South Korea) to minimize the tedious imidization process. The electrospun PAA nanofibers were placed either on a glass (or metal) or paper (or polymer) surface during the hot-press treatment. As the pressure was applied to the PAA nanofibers, a high temperature (over 100 °C) readily resulted in the partial melting of the PAA during the formation of the PI membranes (Fig. S1c†). The slightly mild temperatures resulted in uniform PI membranes whose water contact angles were comparable to that of a thermally-treated PI membrane, as monitored by the contact angle analyzer (PHOENIX 300, SEO Inc.). In addition, the selection of the substrates was found to be important to the preparation of thin PI membranes without wrinkles; the hard substrates (e.g., glass or metal) often generated wrinkles throughout the sheets (Fig. S1f†), but soft substrates (e.g., paper) allowed for the formation of wrinkle-free PI membranes. The treatment at 80 °C under 3000 psi for 30 min resulted in the reliable formation of PI membranes with distinctive color changes (from white to yellow) and an increased water contact angle of 84°. A further increase in the temperature (e.g., 90 °C) under the same pressure led to a successful imidization of the membranes (Fig. S1d†), but it became more difficult to separate the PI membranes from the paper substrates. The typical formation of PI membranes was achieved by the stepwise heat-treated imidization process in an oven (at 100 °C for 2 h, 200 °C for 2 h, and 300 °C for 2 h) and these standard PI membranes served as the reference separators.
Characterizations of PI nanofiber membranes as battery separators
The surface morphology and nanofiber diameter distribution were examined by scanning electron microscope (SEM, JSM5410, JEOL Inc.) after the samples were coated with a gold sputter to improve their electron conductivity.The Fourier transform infrared spectroscopy (FT-IR) spectra of the PAA and PI membranes were obtained in the scan range of 4000 to 400 cm−1 using a FT/IR-620V spectroscopy (JASCO). All samples were completely dried in a vacuum oven overnight prior to analysis.
The tensile strength of the membranes was examined by the UTM-2020 (Myungji Tech., South Korea) based on the ASTM D882 test method using 100 mm × 20 mm samples and a cross-head speed of 500 mm min−1.
The pore properties of the membranes (e.g., the bubble point and mean flow pore (MFP)) were examined by a capillary flow porometer (Porolux 1000, IB-FT Inc.) using a Porewick standard solution with a 16.0 dynes cm−1 surface tension. The wet and dry method was utilized, with an effective diameter of the membranes of ∼1.85 cm.17,18
The porosity of the samples (5.0 cm × 5.0 cm) was evaluated by comparing the dry and wet weights of the membranes after fully soaking them in n-butanol for 1 h. To measure the weight of the wet sample, the membrane was briefly wiped with filter paper to remove excess n-butanol from the surface. This process was repeated three times after completely drying the membranes for 24 h at room temperature. The porosity of the membranes was then calculated using the following equation:
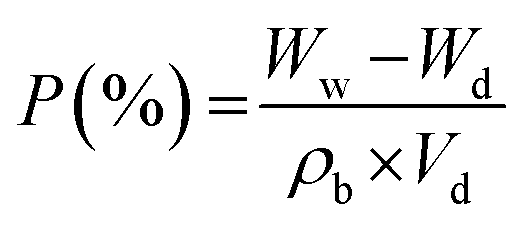 | (1) |
To examine the electrolyte uptake capability, the membrane samples (3.0 cm × 3.0 cm) were soaked in an electrolyte solution consisting of 1 M of LiPF6 dissolved in a mixture of EC and DMC (1:1 v%) for 1 h. The dry weight of the membranes was initially measured prior to soaking them in the electrolyte solution. The electrolyte uptake of the membranes was then calculated using the following equation.
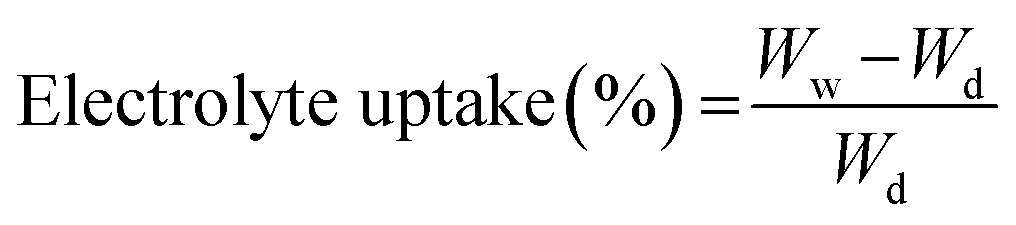 | (2) |
The differential scanning calorimeter (DSC, Q20, TA instruments) and thermogravimetric analysis (TGA, TAQ-500) were used to examine the thermal stability of the membranes. For the DSC measurements, the membrane samples (3–5 mg) were placed in an aluminum pan and preheated at 50 °C for 10 min to remove the remaining organic solvents, followed by heating at a rate of 10 °C min−1 from 50 to 350 °C under N2 gas. For the TGA analyses, the samples were placed in an alumina pan and heated at a rate of 10 °C min−1 from 30 to 900 °C under N2 gas.
The thermal shrinkage was monitored by the size changes (MD: machine direction and TD: transverse direction) of the membrane samples (3.0 cm × 3.0 cm) before and after the treatment in an oven at two different temperatures for 1 h.
Preparation of Li-ion coin cells
To test the membrane performance in battery systems, Li-ion coin cells were manufactured using the PI membranes and a commercial PE membrane. Two electrodes were prepared prior to designing the full coin cell batteries. The active electrode (cathode) was prepared by mixing the NCM523 cathode material (95 wt%), SGO-5, Super P conducting materials (1.5 wt% each), and PVDF binder (2 wt%) in NMP (42 wt% of NMP, N-methyl-2-pyrrolidone, relative to the total amount of cathode materials) using a disper mixer at 1600 rpm and a planetary mixer at 30 rpm for 90 min. The resulting mixture was coated with ∼100 μm thickness onto Al foil (10 μm thick) using a Comma Coater. The electrode was then dried at three different temperatures (i.e., three-zone drying at 100 °C, 105 °C, and 120 °C) and pressed with a compression rate of 30% (i.e., the thickness reduction of the electrode reached ∼70% of its original sample after the pressing step). A-graphite was the active material in the anode, and it was mixed with the Super P conducting material and PVDF binder with the ratio of 90:3:7 wt%. They were then thoroughly mixed with 5 wt% excess of NMP solvent (i.e., 105 wt%). The mixture was coated with an approximatively 110 μm thickness on Cu foil (10 μm thick) which was then completely dried. The resulting electrode was then pressed with a compression rate of 25%.
Subsequently, the assembly of the full coin cells was completed in an Ar glove box with less than 0.3% humidity. The SUS case was used to manufacture the coin cells along with 4-point welding to reduce the contact resistance of the electrodes. The PI membranes were inserted into the system containing 2.0 g of electrolytes, which was a commercially available source consisting of 1.1 M of LiPF6 in EC/DMC/DEC (1:1:1 v%). The coin cells with a PE membrane were also prepared and used for comparison.
Electrochemical performance of the full coin cells
The capacity of the coin cells was tested using the conditions shown in Table S1† in ESI.† The C-rate test of the prepared coin cells was performed using a battery testing system (WBCS3000S, WonATech Co. Ltd., South Korea) to compare the charge and discharge rates under the conditions shown in Table S2 in ESI.†The thermal stability of the coin cells was evaluated by a hot-box test. The fully charged cells were placed in a chamber of charging and discharging equipment (BIGWAVE Inc., South Korea) capable of maintaining a constant temperature and humidity. The voltages of the cells were measured under the charging and discharging conditions. In addition, the voltages of the cells were also monitored under conditions where the chamber temperature was gradually raised from 25 to 150 °C at a rate of 5 °C min−1 and maintained for 1 h.
Results and discussion
Prior to the formation of the PI membranes, a mixture of ODA and PMDA in DMF was carefully electrospun to form PAA nanofiber mats. Electrospinning a precursor solution with low viscosities (<10 kcP) generally did not lead to the formation of nanofiber features (e.g., it was difficult to accumulate fibrous layers),19 but the collectable layers of the PAA nanofibers were obtained with the polymer precursor mixture with a minimum viscosity of 10 kcP. The viscosity of the precursor solution was simply developed and measured as a function of the stirring time (Fig. 1a). After finding an appropriate viscosity for the precursor solution, the ejection speed was controlled to examine the quality of the nanofibers during electrospinning. The random formation of numerous polymer beads throughout the nanofiber mats was observed when the ejection speed was relatively fast. In addition, this fast ejection of the PAA precursors with a viscosity of 12 kcP or higher often caused the clogging of the needle tip under our electrospinning conditions. The formation of the beads was notably reduced with the decrease in the ejection speed, which readily allowed for the formation of uniform strands of PAA nanofibers (SEM images in Fig. 1b–d).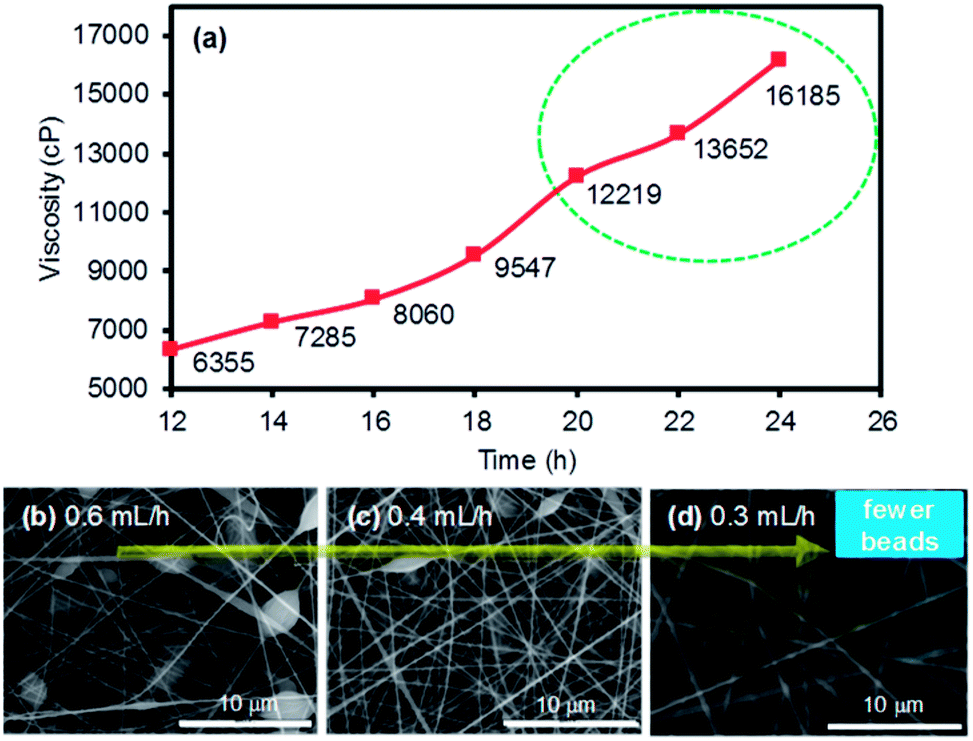 | ||
| Fig. 1 Viscosity development of the polymer precursor solution composed of ODA and PMDA in DMF as (a) a function of the stirring time, and (b–d) the SEM images of the electrospun nanofibers at different ejection speeds. | ||
Upon the reliable formation of the PAA nanofiber mats, imidization was optimized to form PI membranes at a mild temperature and high pressure (i.e., the hot-press treatment). While the conventional imidization process requires tedious and stepwise thermal treatments from 100 °C to over 300 °C, followed by the subsequent processing of the nanofiber mats into membranes, our hot-press strategy can simultaneously complete imidization and the formation of the PI membranes in 30 min. Fig. S1 in ESI† shows the various imidization conditions for preparing the PI membranes via the hot-press treatment, which were compared to those prepared with the conventional thermal approach. The progress of imidization was easily monitored by the contact angle changes of the final PI membranes (i.e., the wettability of the membrane surface using a water droplet). The PAA nanofiber mat initially exhibited a very low water contact angle (i.e., hydrophilic characteristics) due to the presence of numerous COOH functional groups, but the angle dramatically increased as the imidization proceeded (i.e., the cyclization of amic acid into anhydrous). The PI membranes prepared by the conventional heat treatment using an oven exhibited a contact angle of 83.8°. The hot-pressed membranes prepared at 80 °C under 3000 psi for 30 min exhibited a contact angle of 83.5°. The membranes prepared with an extended hot-press time (e.g., 60 min) remained at the contact angle of 83.5°, implying that 30 min of the hot-press treatment was sufficient to complete the imidization process. In addition, the structural features of the membranes were thoroughly examined by SEM (Fig. 2). While the commercial PE membrane somewhat shows detectable pores at high magnification, the prepared PAA and the PI membranes clearly exhibited multiple layers of nanofibers with porous network. Unlike the typical thermal conversion of PAA to PI that involves either a slight or notable reduction of the nanofiber diameter caused by the elimination of water,10,20–22 the diameter of our PI membranes slightly increased after imidization, particularly the PI membranes prepared by the hot-press treatment. Prior to imidization, the initial PAA nanofiber mats (∼0.54 μm) were pressed (e.g., 3000 psi for 30 min) at room temperature to form PAA membranes (∼0.57 μm, data not shown). This process could result in the compressed contact of the nanofibers, possibly causing greater inter-nanofiber imidization (i.e., nanofiber–nanofiber interactions) rather than intra-nanofiber interactions (i.e., within nanofibers) during the thermal and hot-press treatment conditions. The conversion of PAA to PI during the hot-press induced imidization process could result in more inter-nanofiber interactions than those that occur during the heat-induced process (∼0.55 μm), thereby notably increasing the nanofiber diameters (∼0.61 μm). This speculation is partially supported by the improved tensile strength of the hot-press induced PI membranes (vide infra, Fig. 5). More thorough studies are underway to precisely understand this unusual trend in the diameter changes from PAA to PI at our imidization conditions.
 | ||
| Fig. 2 SEM images of PE, as prepared PAA, and PI membranes prepared by the thermal and hot-press treatments (the average diameters of the nanofibers are shown in the images). | ||
In a separate study, the change of the water contact angles was monitored under three different storage conditions as the imidization process of PAA to PI could slowly occur at ambient conditions (Fig. 3 and S2†). The PAA nanofiber mat was initially dried overnight in a vacuum oven to remove any residual solvent, and this was followed by the formation of the membranes via a pressure treatment. The PAA membranes were then stored at ambient conditions and clearly showed the gradual increase in the water contact angle as a function of time. However, the angle reached a plateau after 13 days, implying the maximum imidization had occurred under the ambient storage conditions. In addition, the degree of imidization for the PAA membranes was monitored using FT-IR spectra (Fig. S2†), which still showed negligible changes. The discrepancy between the slightly increased contact angles and the insignificant changes of FT-IR spectra as a function time could possibly be explained by the partial imidization of the PAA nanofibers only around the surface areas. When the storage temperature decreased to 4 °C (the samples were placed in a refrigerator), imidization still slowly proceeded. The contact angles of these two samples were far different from those of the fully imidized PI membranes (dashed line in Fig. 3).23–25 When the PAA sample was stored at room temperature under vacuum, the water contact angles remained the same, which implied the importance of the storage conditions for maintaining the features of the PAA nanofiber membranes.
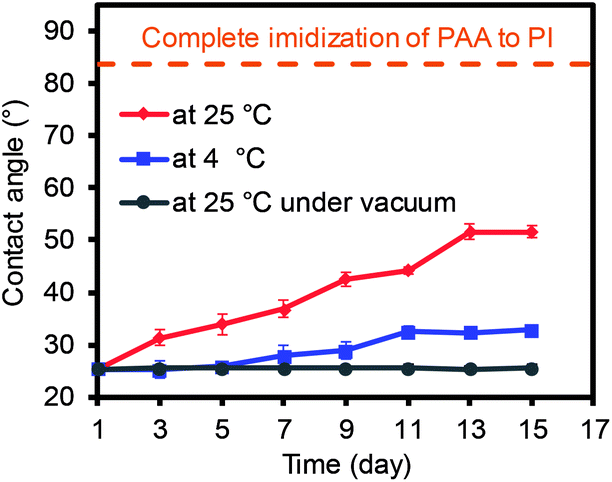 | ||
| Fig. 3 Water contact angle changes of the PAA nanofiber membranes under various storage conditions. | ||
The FT-IR spectra were also compared before and after the imidization process of PAA to confirm the formation of the PI membranes (Fig. 4). The representative PAA membrane exhibited peaks at 1652 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from CONH), 1710 cm−1 (CO from COOH), and 1550 cm−1 (C–N from C–N–H), along with a broad peak over 3000 cm−1 (OH from COOH, not shown). Both PI membranes prepared by the thermal and hot-press treatments exhibited almost identical spectra, showing slightly shifted peaks at 1773 cm−1 and 1710 cm−1 for the asymmetric and symmetric stretching modes of CO, respectively. In addition, two new peaks at 1374 cm−1 and 723 cm−1 (C–N and CO from the imide ring) and the absence of the broad band over 3000 cm−1 confirmed the successful conversion of PAA to PI.26–29
O from CONH), 1710 cm−1 (CO from COOH), and 1550 cm−1 (C–N from C–N–H), along with a broad peak over 3000 cm−1 (OH from COOH, not shown). Both PI membranes prepared by the thermal and hot-press treatments exhibited almost identical spectra, showing slightly shifted peaks at 1773 cm−1 and 1710 cm−1 for the asymmetric and symmetric stretching modes of CO, respectively. In addition, two new peaks at 1374 cm−1 and 723 cm−1 (C–N and CO from the imide ring) and the absence of the broad band over 3000 cm−1 confirmed the successful conversion of PAA to PI.26–29
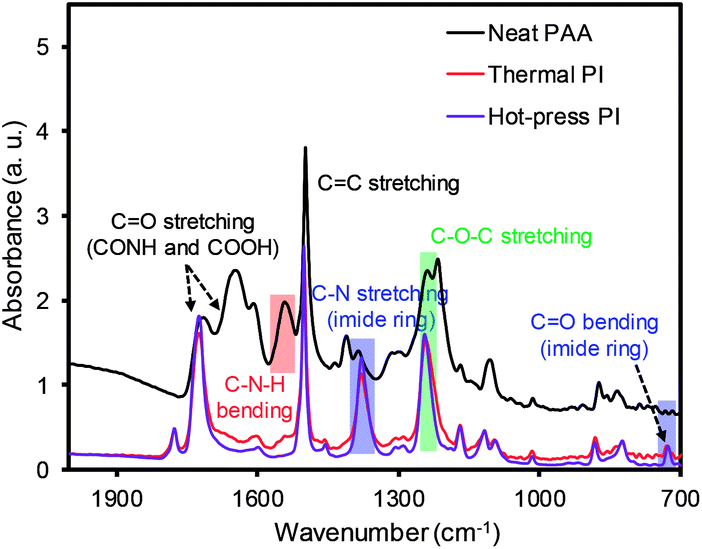 | ||
| Fig. 4 FT-IR of the PAA nanofiber and PI membranes after the thermal and hot-press treatments. | ||
The mechanical property of the PI membranes (via the thermal and hot-press treatments) was carefully compared to the commercial PE membrane (Fig. 5). Both PI membranes exhibited a much higher stress than the neat PAA membrane, ∼29 kgf cm−2, due to imidization.15,30–32 In particular, the hot-press-induced PI membrane (∼220 kgf cm−2) displayed a notably increased stress compared to the thermally-treated PI membranes (∼107 kgf cm−2). This higher value for the mechanical property could be explained by the dense packing of the nanofiber layers and the inter-nanofiber interactions, as briefly mentioned above. This conjecture is due to the fact that our hot-press-induced PI membrane was a thinner sheet with a slightly larger nanofiber diameter than that of the thermally-treated PI membrane (Fig. 2 and Table 1), where the mechanical strength generally improved with a thicker layer and smaller fiber diameter.12,33 Although the commercial PE membranes showed a much higher tensile strength (∼1138 kgf cm−2) than that of the PI membranes prepared by both methods, the tensile strength values of these PI membranes were still found to be applicable to coin cells and pouch-type battery systems.34
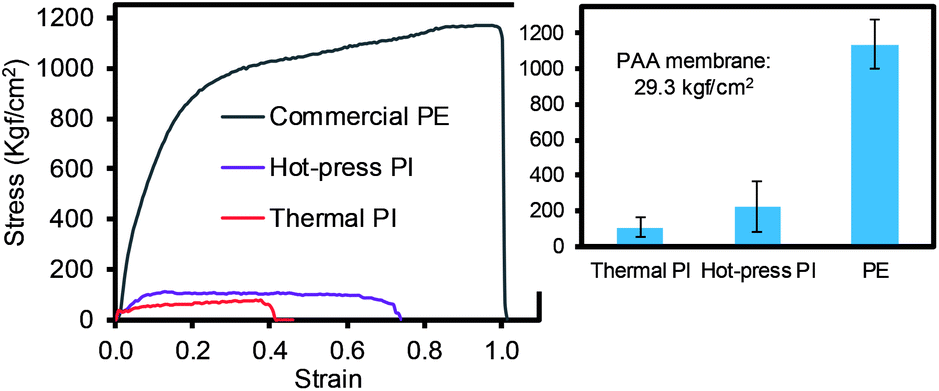 | ||
| Fig. 5 Tensile strength of different types of membranes (inset shows the average of the minimum three measurements). | ||
| Sample | Thickness (μm) | Bubble point (nm) | MFP (nm) | Smallest pore size (nm) | Porosity (%) | Electrolyte uptake (%) |
|---|---|---|---|---|---|---|
| a PAA: 29.7 + 2.5 μm thickness. | ||||||
| Thermal PI | 24.2 ± 2.2 | 958.1 | 465.3 | 303.1 | 38.60 | 56.2 ± 1.5 |
| Hot-press PI | 23.3 ± 1.5 | 440.4 | 294.0 | 194.8 | 41.50 | 61.2 ± 1.9 |
| Commercial PE | 15.6 ± 0.5 | 46.71 | 38.40 | 37.98 | 35.72 | 48.8 ± 2.4 |
The integrity of the membranes was then evaluated to validate the possibility of utilizing PI-based membranes as separators in battery systems. The pore size and distribution of the membranes play important roles in the mobility of Li ions and strongly impact the quality of battery systems. The summary of the overall properties is shown in Table 1. The PI membranes generally exhibited large pore sizes and distributions, as well as porosities, because of the nature of the structures designed from multiple layers of nanoscale fibers. The hot-press-induced PI membrane exhibited a relatively smaller MFP size with a higher porosity than the thermally-treated PI membrane. The hot-press treatment presumably resulted in the compact packing of each of the PI nanofiber strands, which allowed for the formation of many small mean free pores with uniform pore variations, possibly explaining the improved mechanical strength. An additional interesting finding was that the hot-press-induced PI membrane (23.3 ± 1.5 μm) exhibited a significantly reduced thickness and its variation was lower than that found in the thermally-induced PI membrane (24.2 ± 2.2 μm) and original PAA membrane (29.7 μm ± 2.5 μm). These properties of the PI membranes were measured and found to be suitable to serve as membrane separators in Li-ion battery systems because the commercially-available membrane-based battery separators typically require pore sizes in the range of 0.03–0.1 μm with a 30–50% porosity and 10–30 μm thickness.35 The electrolyte uptake capability was also examined using a solution with Li ions. The PI membranes showed a higher electrolyte uptake capability than that of the commercial PE membrane (with a water contact angle over 95°), which may be due to their higher porosity and hydrophilicity.36,37 The hot-press-induced PI membrane showed a higher electrolyte uptake efficiency than that of the thermally-treated PI membrane because of the higher porosity, along with the dense, uniform arrangement of the nanofibers.
The thermal behaviors of the commercial PE, neat PAA, and PI membranes were monitored by TGA under the N2 environment (Fig. 6). The commercial PE membrane showed one sharp weight loss at ∼450 °C, corresponding to the decomposition temperature. However, the PAA membrane clearly displayed three weight loss patterns: the first drop at ∼70 °C for the water and/or solvent from the polymer network, the second drop at ∼200 °C for imidization, and the third drop at ∼575 °C for the decomposition of the PI main chains.11,21,38 The PI membrane prepared by the hot-press treatment only showed one sharp weight loss at ∼575 °C, completely overlapping with the third drop of the PAA membrane.32,39,40 Additionally, the thermal shrinkage of the PI and PE membranes was compared after the heat treatment in an oven (at three different temperatures) for 1 h (see the digital photos in Fig. 6 and S3†). As this experiment was performed in air, the PE membrane underwent ∼24% (at 130 °C), ∼60% (at 160 °C), and over 90% (at 300 °C) shrinkage of its dimensions, respectively.41,42 The PI membrane, however, did not show a detectable reduction in size and shape under these conditions, implying a good thermal resistance and stability. These DSC curves also supported the thermal behavior of all samples obtained from the TGA patterns. The PI membrane did not show endo- or exo-thermic peak patterns under our measurement conditions up to 350 °C,43 while the PAA membrane showed two distinctive endothermic patterns for the removal of the solvent at 70 °C and imidization at 200 °C, respectively.16 The commercial PE membrane displayed only one endothermic peak at 140 °C, corresponding to the melting temperature of typical PE materials. Due to the DSC temperature limitation, the decomposition temperature was not observed at ∼450 °C. Unlike other polymers exhibiting a simultaneous melting section (i.e., from a solid to liquid phase) accompanied by a decomposition segment (i.e., chemical decomposition by heat), a crystalline PE polymer matrix often displays two distinctive peaks for the melting and decomposition temperatures. Thus, the commercial PE membrane did not show any weight loss at this melting temperature (140 °C) from the TGA curve, but underwent a significant reduction in dimensions during the shrinkage test at temperatures over 130 °C.
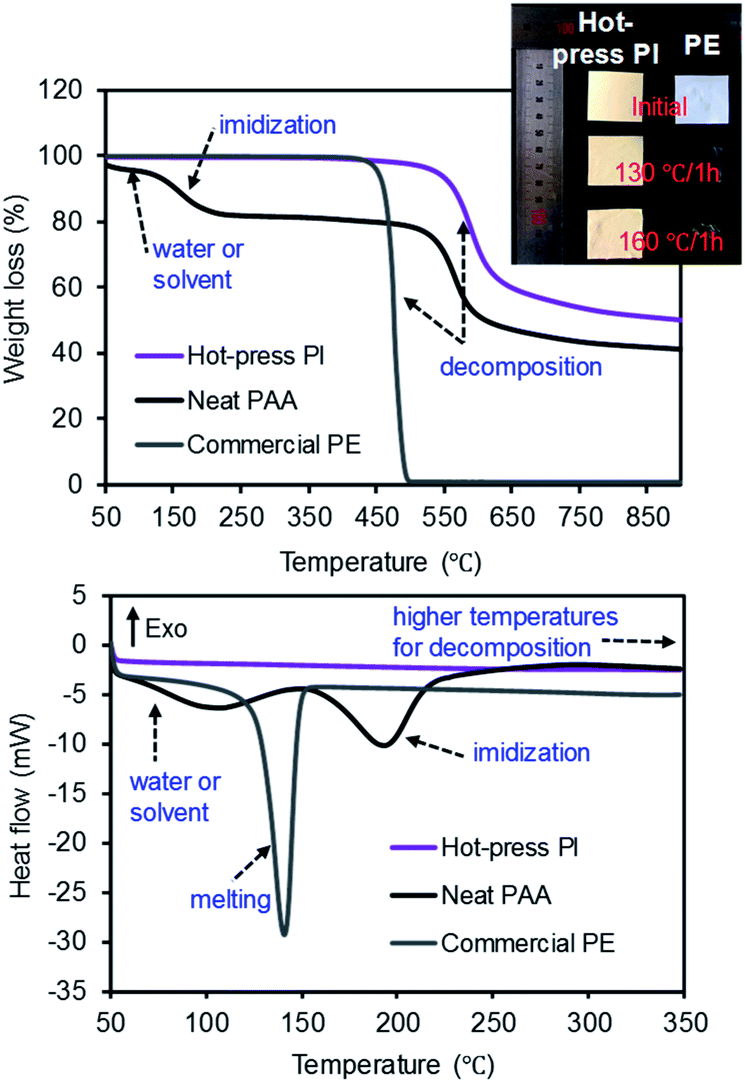 | ||
| Fig. 6 TGA (top) and DSC (bottom) curves of the PE, PAA, and PI membranes and a digital photo of the PI and PE membranes before and after treatment in a hot-box. | ||
To examine electrical performance of the full coin cell batteries, the C-rate test was carried out using the membrane separators containing the same active material ratios under various charging and discharging conditions (Fig. 7 and S4 in ESI†). All cells exhibited similar patterns of ohmic polarization under various discharge current densities from 0.2C to 5C, where the PI membrane prepared by the hot-press treatment exhibited comparable performance to the commercial PE membrane (Fig. S4a and S4b†), which was slightly better than the thermally-treated PI membrane (Fig. S4c†). Fig. 7 displays the summary of the cycle stabilities of three different coin cells as a function of the discharge rates, which is a common validation process for rechargeable battery systems. The cells were initially stabilized by discharging three cycles at 0.1C, and the efficiency was calculated using the initial capacity at 0.2C. All of the coin cells generally decreased with the increase of the current rate from 0.2C to 5C, but regained a higher initial discharge capacity upon returning to the current density of 0.2C. Interestingly, the initial discharging current of the PI membrane at 0.2C was slightly lower than that of the commercial PE membrane. This observation could be caused by the relatively easy deintercalation and intercalation of the Li ions within the nanofiber-based separators with larger pore sizes, which might result in a self-discharge after a long storage time. As the PI membranes with a large pore size and porosity could exhibit the increased mobility of the Li ions, this high mobility would also lead to the loss of Li ions from the active materials near the electrodes.7,10 After the slightly low initial C-rate performance, the PI membrane separator prepared by the hot-press treatment showed comparable capacity retention ratios to those of the commercial PE membrane separator, regardless of the number of cycles (numerical values shown in the table). The PI membrane prepared by the thermal treatment with a relatively low discharge capacity at all current densities (0.2–5C) could have originated from the structural features with an inferior uniform pore size distribution and a slightly thicker layer of the separator, hindering the mobility of the Li ions during the charging and discharging process (i.e., differentiating Li-ion channeling process). As these two PI membranes exhibited slightly different patterns of rate capability, possibly associated with the thickness and porosity variations, more thorough studies are underway to fully understand these phenomena. The hot-press-induced imidization strategy can allow for the reliable design of PI-based-nanofiber membranes as battery separators that can significantly minimize the processing time and reduce the preparation cost of materials without sacrificing their integrity.
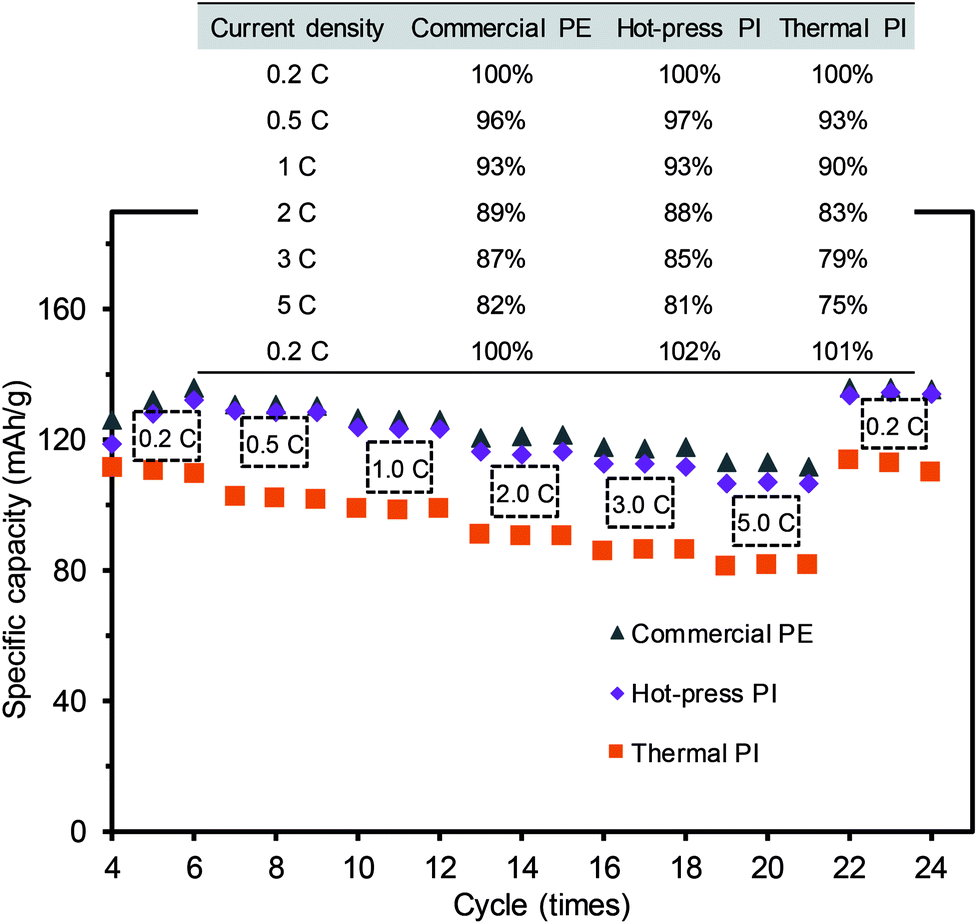 | ||
| Fig. 7 Discharge voltage profiles of the full coin cells with three different separators at a constant current charge and discharge rate (three cycles at each C-rate with a design capacity of 130 mAh). | ||
Three coin cells were gradually heated to 150 °C in a hot-box test machine and maintained at this temperature for 1 h to examine the thermal damage of the full coin cell batteries (Fig. 8). Over the course of the temperature increase, the voltage of the cell with the commercial PE membrane dropped to 0 V at 70 °C. This incidence of a short circuit was from the contact between the positive electrode and the negative electrode, presumably caused by the shrinkage of the membrane in the coin cell.7,44 Although the coin cells with the PI membranes prepared using the thermal and hot-press procedures gradually decreased their voltages, these cells were maintained at above the battery drive voltage during the entire test, implying a high thermal stability without the shrinkage of the membranes. The inset digital photo taken after the hot-box test shows that the coin cells equipped with the PI membranes remained unchanged, clearly supporting the importance of the thermal stability of the separators for preventing the voltage drop and damage of the coin cell batteries. This hot-box test could also explain the important relationship between the stability of the membrane separator and the safety of the overall battery system. The C-rate and hot-box tests indicated that the PI-based membranes exhibited greatly improved electrochemical and thermal stabilities while maintaining a comparable Li-ion channeling performance compared to that of commercial PE membranes.
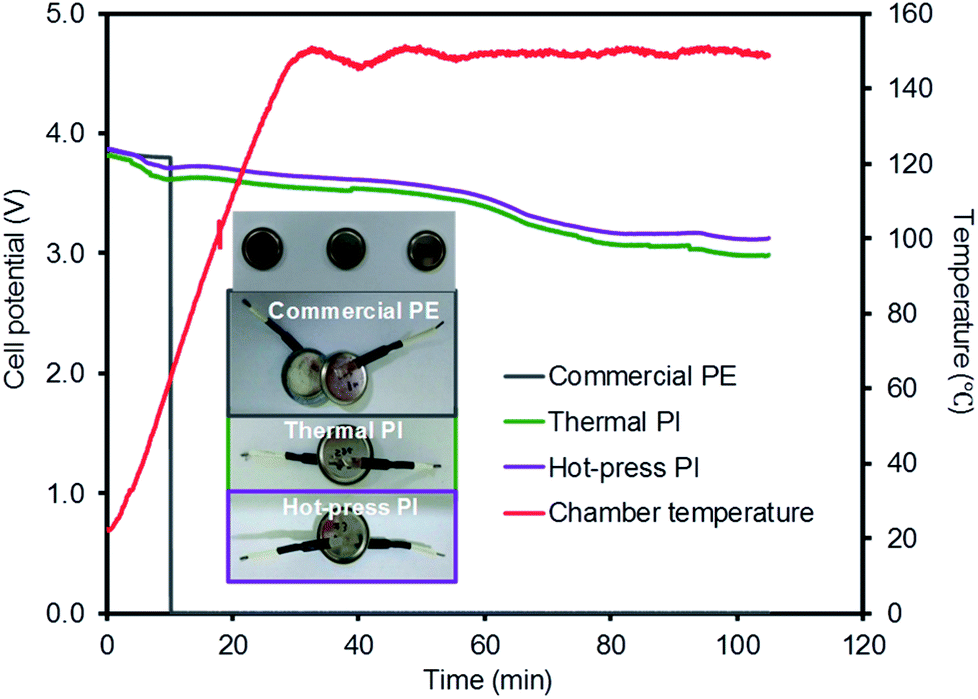 | ||
| Fig. 8 Hot-box test of coin cells with three different membrane separators (inset: digital photos of the coin cells after the hot-box test). | ||
Conclusions
We demonstrated a simple, rapid strategy for preparing PI membranes from PAA nanofiber mats via a hot-press treatment. Unlike the thermally-treated PI membrane, the hot-pressed PI membrane exhibited a generally uniform pore size distribution and improved porosity and electrolyte uptake capability. In addition, the hot-press-induced imidization process allowed for the high possibility of inter-nanofiber interactions to enhance the mechanical property of the PI membrane compared to that of the PI membranes prepared by the thermal process. The subsequent utilization of these PI membranes as separators in Li-ion coin cell batteries verified the significant improvement of their thermal stabilities over the typical PE membrane, as examined by the shrinkage and hot-box tests. In addition, the hot-press-induced PI membrane separator exhibited a higher C-rate (charging and discharging) performance than the thermally-treated PI membrane and was comparable to the commercial PE membrane. The establishment of an easy and reliable strategy to utilize nanofiber membranes as separators possessing highly improved thermal and electrical properties can broaden their potential applications in many portable battery systems requiring a high power capability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2018R1A2B6008854).References
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 Search PubMed.
- V. Deimede and C. Elmasides, Energy Technol., 2015, 3, 453–468 Search PubMed.
- X. Huang, J. Solid State Electrochem., 2011, 15, 649–662 Search PubMed.
- S. S. Zhang, J. Power Sources, 2007, 164, 351–364 Search PubMed.
- Y. Xiang, J. Li, J. Lei, D. Liu, Z. Xie, D. Qu, K. Li, T. Deng and H. Tang, ChemSusChem, 2016, 9, 3023–3039 CrossRef CAS PubMed.
- Y. Saito, W. Morimura, R. Kuratani and S. Nishikawa, J. Phys. Chem. C, 2015, 119, 4702–4708 CAS.
- L. Cao, P. An, Z. Xu and J. Huang, J. Electroanal. Chem., 2016, 767, 34–39 CrossRef CAS.
- M.-N. Huang, Z.-Q. Jang, F.-B. Li, H. Yang and Z.-L. Xu, New J. Chem., 2017, 41, 7544–7552 RSC.
- W. Jiang, Z. Liu, Q. Kong, J. Yao, C. Zhang, P. Han and G. Cui, Solid State Ionics, 2013, 232, 44–48 CrossRef CAS.
- J. Shayapat, O. H. Chung and J. S. Park, Electrochim. Acta, 2015, 170, 110–121 CrossRef CAS.
- S. Jiang, H. Hou, S. Agarwal and A. Greiner, ACS Sustainable Chem. Eng., 2016, 4, 4797–4804 CrossRef CAS.
- Y. Ding, H. Hou, Y. Zhao, Z. Zhu and H. Fong, Prog. Polym. Sci., 2016, 61, 67–103 CrossRef CAS.
- Q. Wang, W.-L. Song, L. Wang, Y. Song, Q. Shi and L.-Z. Fan, Electrochim. Acta, 2014, 132, 538–544 CrossRef CAS.
- W. Chen, Y. Liu, Y. Ma, J. Liu and X. Liu, Mater. Lett., 2014, 133, 67–70 CrossRef CAS.
- C. Huang, S. Wang, H. Zhang, T. Li, S. Chen, C. Lai and H. Hou, Eur. Polym. J., 2006, 42, 1099–1104 CrossRef CAS.
- M. Kotera, T. Nishino and K. Nakamae, Polymer, 2000, 41, 3615–3619 CrossRef CAS.
- J. Lee, H.-R. Chae, Y. J. Won, K. Lee, C.-H. Lee, H. H. Lee, I.-C. Kim and J.-M. Lee, J. Membr. Sci., 2013, 448, 223–230 CrossRef CAS.
- M. J. J. Erniza, A. L. Ahmad and S. C. Low, J. Membr. Sci. Technol., 2016, 2, 102–108 Search PubMed.
- M. K. Leach, Z.-Q. Feng, S. J. Tuck and J. M. Corey, J. Visualized Exp., 2011, 47, e2494 Search PubMed.
- X. Li, X. Wang, Q. Wang and Q. Wei, J. Eng. Fibers Fabr., 2014, 9, 33–38 CAS.
- S. Ramakrishnan, M. Dhakshnamoorthy, E. J. Jelmy, R. Vasanthakumari and N. K. Kothurkar, RSC Adv., 2014, 4, 9743–9749 RSC.
- K. S. Yang, D. D. Edie, D. Y. Lim, Y. M. Kim and Y. O. Choi, Carbon, 2003, 41, 2039–2046 CrossRef CAS.
- I. Sava and S. Chisca, Mater. Chem. Phys., 2012, 134, 116–121 CrossRef CAS.
- C. J. Wohl, M. A. Belcher, S. Ghose and J. W. Connell, Appl. Surf. Sci., 2009, 255, 8135–8144 CrossRef CAS.
- M. Zuo, T. Takeichi, A. Matsumoto and K. Tsutsumi, Colloid Polym. Sci., 1998, 276, 555–564 CAS.
- L. Ding, Y. Zhang, L. Liu, J. Hu and F. Lv, RSC Adv., 2016, 6, 56812–56818 RSC.
- T. J. Shin and M. Ree, J. Phys. Chem. B, 2007, 111, 13894–13900 CrossRef CAS PubMed.
- J. Zhang, Q. Lu, J. Fang, J. Wang, J. Yang and Y. NuLi, ACS Appl. Mater. Interfaces, 2014, 6, 17965–17973 CAS.
- H. Kizil, M. O. Pehlivaner and L. Trabzon, Adv. Mater. Res., 2014, 970, 132–135 CrossRef.
- Y. He, D. Han, J. Chen, Y. Ding, S. Jiang, C. Hu, S. Chen and H. Hou, RSC Adv., 2014, 4, 59936–59942 RSC.
- Y. Cheng, S. Zhang, J. Li, J. Sun, J. Wang, C. Qin and L. Dai, RSC Adv., 2017, 7, 21953–21961 RSC.
- H. W. Ha, A. Choudhury, T. Kamal, D.-H. Kim and S.-Y. Park, ACS Appl. Mater. Interfaces, 2012, 4, 4623–4630 CAS.
- J. Yao, M. F. Pantano, N. M. Pugno and C. W. M. Bastiaansen, Polymer, 2015, 76, 105–112 CrossRef CAS.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS PubMed.
- M. Yang and J. Hou, Membranes, 2012, 2, 367–383 CrossRef CAS PubMed.
- L. Eykens, K. De Sitter, C. Dotremont, W. De Schepper, L. Pinoy and B. Van Der Bruggen, Appl. Sci., 2017, 7, 118 CrossRef.
- M. Tsuchida and Z. Osawa, Colloid Polym. Sci., 1994, 272, 770–776 CAS.
- D. Chao, J. Zhang, X. Liu, X. Lu, C. Wang, W. Zhang and Y. Wei, Polymer, 2010, 51, 4518–4524 CrossRef CAS.
- I. A. Al-Ajaj and A. A. Kareem, Mater. Sci.-Pol., 2016, 34, 132–136 CAS.
- O. Jirsak, P. Sysel, F. Sanetrnik, J. Hruza and J. Chaloupek, J. Nanomater., 2010, 2010, 842831 Search PubMed.
- P. K. Roy, P. Surekha, C. Rajagopal and V. Choudhary, eXPRESS Polym. Lett., 2007, 1, 208–216 CrossRef CAS.
- J. D. Peterson, S. Vyazovkin and C. A. Wight, Macromol. Chem. Phys., 2001, 202, 775–784 CrossRef CAS.
- Y. Mansoori and K. Darvishi, J. Mex. Chem. Soc., 2014, 58, 126–136 CAS.
- D. Lin, D. Zhuo, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2016, 138, 11044–11050 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction scheme for PI formation and experimental details of coin cell tests. See DOI: 10.1039/c8ra01556b |
| This journal is © The Royal Society of Chemistry 2018 |