
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A concise synthesis of (±)-7-O-galloyltricetiflavan†
Wenxuan Zhang ,
Wenjie Xue,
Yuqing Jia,
Gang Wen,
Xu Lian,
Jing Shen,
Ailin Liu and
Song Wu*
,
Wenjie Xue,
Yuqing Jia,
Gang Wen,
Xu Lian,
Jing Shen,
Ailin Liu and
Song Wu*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, People's Republic of China. E-mail: ws@imm.ac.cn
First published on 18th April 2018
Abstract
(±)-7-O-galloyltricetiflavan (1a) was synthesized successfully in five steps from the commercially available trihydroxyacetophenone (2) and trimethoxybenzoyl chloride (3). The flavone 4a was prepared in a one-pot reaction and it gave hex-O-methylflavan 6 followed by acylation and reduction. However, the demethylation of flavan 6, 5-O-acetylflavan 10 and 5-O-phenylacetylflavan 11 by BBr3 gave all the hydrolyzed fragments 7 and 8 as the major products. By contrast, in the same condition, hept-O-methylflavan 9 could provide the desired product (±)-7-O-galloyltricetiflavan (1a) in 91% yield. The additional 5-O-B-Br2 complex may stabilize the ester bond during the demethylation process.
(−)-7-O-Galloyltricetiflavan (1, Fig. 1) was isolated from a methanolic extract of the leaves of Pithecellobium clypearia by Ooi V. and coworkers in 2006.1 It is a catechin-like compound without an OH substituent at C-3, and it shows good antiviral activities against respiratory syncytial virus (RSV), influenza A (H1N1) virus, coxsackie B3 (Cox B3) virus and herpes simplex virus type 1 (HSV-1) as well as anti-inflammatory and anti-allergic activities.2–5 To date, the preparation of (−)-7-O-galloyltricetiflavan (1) still requires extraction and purification of plant material, and only a few synthetic examples of this type of flavan have been reported.6,7 Herein we report the first total synthesis of (±)-7-O-galloyltricetiflavan (1a) in five steps as well as an interesting discovery during the demethylation process.
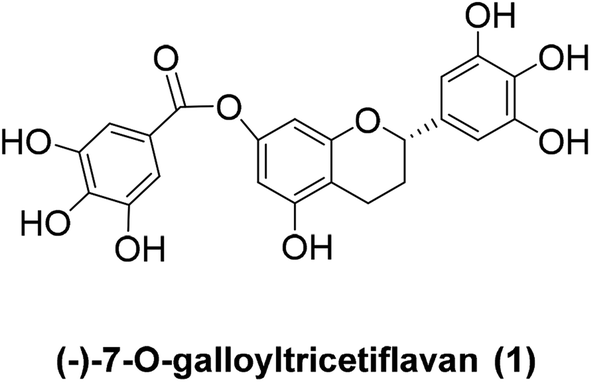 | ||
| Fig. 1 The structure of 7-O-galloyltricetiflavan (1). | ||
The synthesis of (±)-7-O-galloyltricetiflavan (1a) was started from the preparation of the flavone derivative 4a as shown in Scheme 1. Buckle's group reported an efficient one-pot synthesis of flavones by the treatment of 2-hydroxyacetophenones with the corresponding aroyl chloride in wet K2CO3/acetone (1% w/w water),8 but the reaction proceeded very slowly because the trihydroxyacetophenone (2) was insoluble in acetone. With water-toluene as the solvent, in the presence of K2CO3 and tetrabutylammonium hydrogen sulfate,9 the reaction could provide flavone 4a in 30% yield and 3-acylated product 4b in 50% yield in one-pot in about two hours. Many efforts to improve the yield of 4a failed, but 4b could be converted into 4a by hydrolysis in 50% yield.10 Afterwards, acylation of 4a with trimethoxybenzoyl chloride (3) and K2CO3 gave 7-O-galloylflavone 5 in 91% yield, which was then reduced to the flavan 6 by hydrogenation with palladium on carbon as the catalyst for 3 days in 62% yield.11
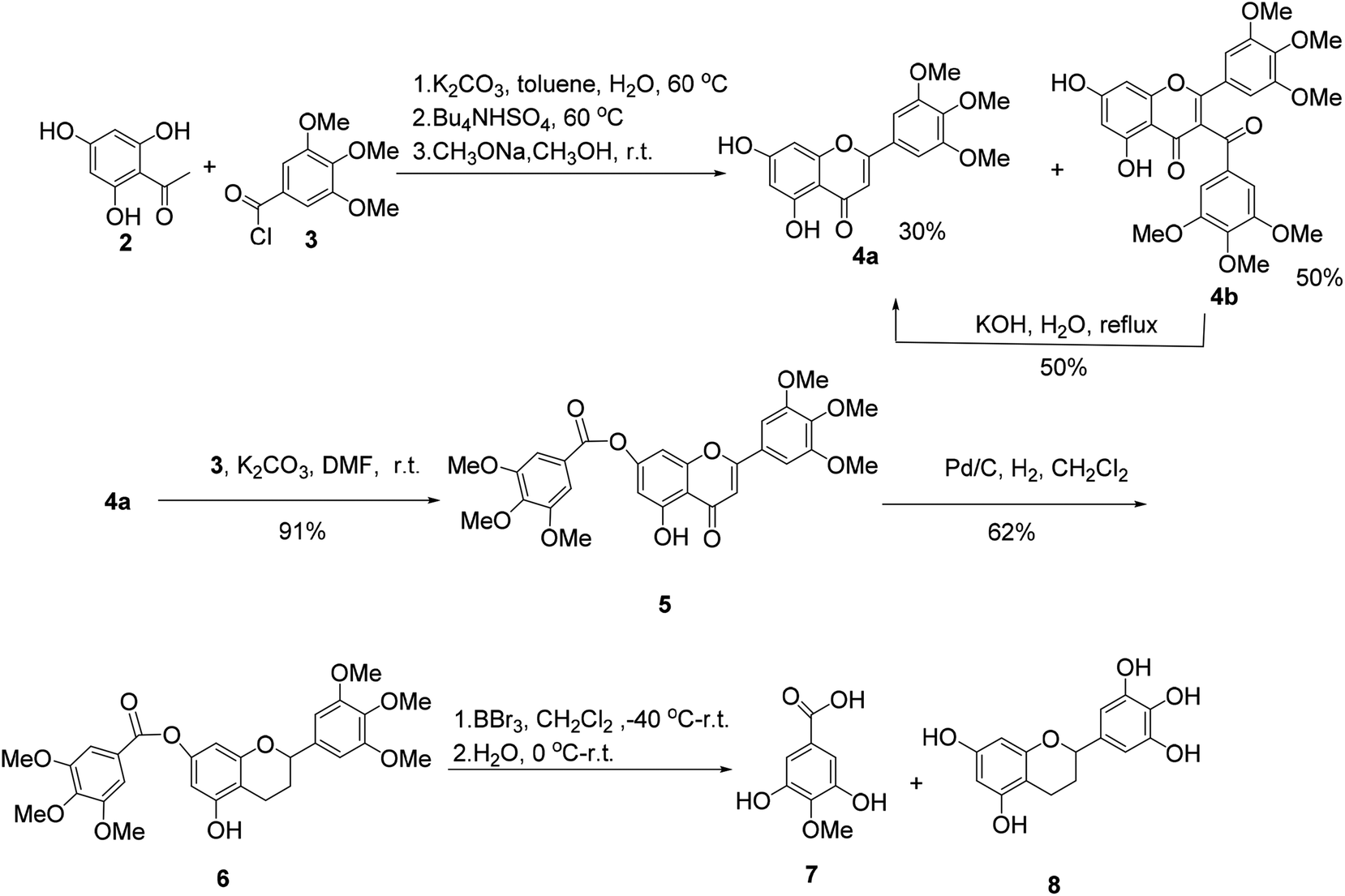 | ||
| Scheme 1 The synthesis of intermediates of (±)-7-O-galloyltricetiflavan (1a). | ||
When flavan 6 was treated with BBr3 in dichloromethane at −40 °C or −78 °C,12 the desired product 1a was generated in only 3% yield (based on HPLC-MS analysis), accompanied with 4-O-methyl gallic acid (7) and flavan 8 as the major products, indicating the ester bond of hex-O-methylflavan 6 is highly unstable under acidic conditions (Scheme 1).
Then, 5-O-methylflavan 9, 5-O-acetylflavan 10 and 5-O-phenylacetylflavan 11 were prepared as substrates to explore if they provided different results (Scheme 2). Similarly, 5-O-acetylflavan 10 and 5-O-phenylacetylflavan 11 were not tolerated under these reaction conditions, which gave the hydrolyzed products 7 and 8 as major products. In contrast, when flavan 9 was treated with BBr3 in dichloromethane at −40 °C to room temperature, the desired product (±)-7-O-galloyltricetiflavan (1a) was generated in 91% yield and no hydrolyzed product was detected after 24 h. The structure of (±)-7-O-galloyltricetiflavan (1a) were confirmed by 1H NMR, 13C NMR, and HR-MS spectrum, and they are consistent with the literature's report.1
We presumed that when BBr3 was added to the additional 5-O-methyl group to form the 5-O-B-Br2 complex, it may stabilizes the ester bond of 7-phenolic hydroxyl group. By contrast, the 5-O-acetyl or 5-O-phenylacetyl groups was more easily hydrolyzed and could not help stabilize the ester bond.
In conclusion, (±)-7-O-galloyltricetiflavan (1a) was synthesized successfully in five steps from commercial available trihydroxyacetophenone (2) and trimethoxybenzoyl chloride (3). Flavone 4a was prepared in a one-pot reaction and it gave hex-O-methylflavan 6 followed by acylation and reduction. However, the demethylation of flavan 6 by BBr3 gave the hydrolyzed fragments 7 and 8 as major products. Similarly, neither 5-O-acetylflavan 10 nor 5-O-phenylacetylflavan 11 could provide the desired product. In contrast, hept-O-methylflavan 9 could give the desired product (±)-7-O-galloyltricetiflavan (1a) in 91% yield. The additional 5-O-B-Br2 complex may stabilize the ester bond during the demethylation process. Our method could also provide an efficiently pathway to prepare other 7-O-acylated flavans.
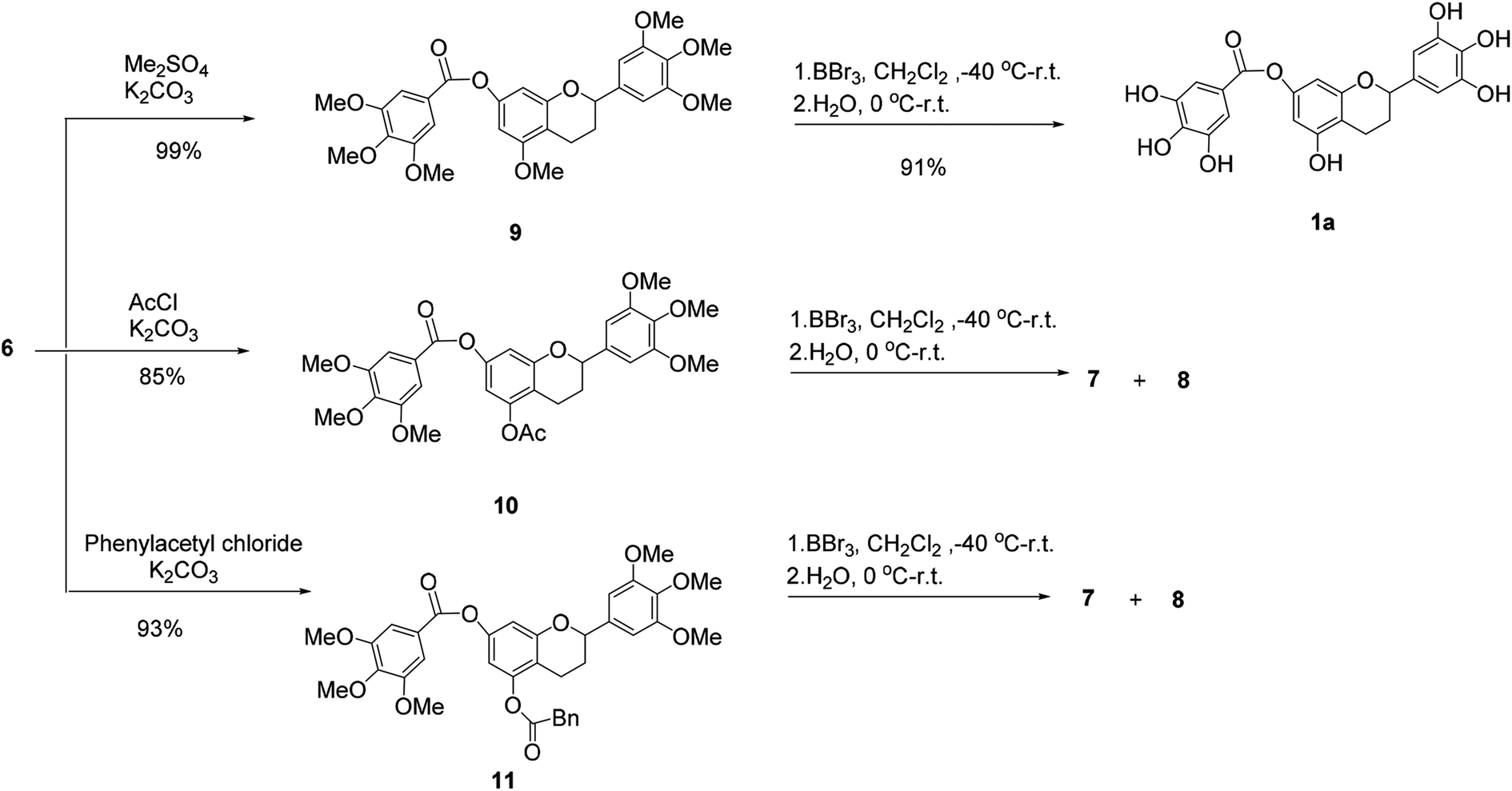 | ||
| Scheme 2 The demethylation of flavan derivatives. | ||
Experimental section
General experimental procedures
All reactions were performed in glassware containing a Teflon-coated stir bar. Solvents and chemical reagents were obtained from commercial sources and used without further purification. 1H and 13C NMR spectra were recorded on Varian Mercury 500 MHz or 400 MHz, and the data were recorded using DMSO-d6, CDCl3 and CD3OD as the solvents. Chemical shifts (δ) are reported in ppm downfield from an internal TMS standard. The reactions and products were analyzed by HPLC-MS. High-resolution mass spectra were obtained in ESI mode on a hybrid IT-TOF mass spectrometer. Flash column chromatography on silica gel (200–300 mesh) was used for the routine purification of reaction products. The column output was monitored by TLC on silica gel (100–200 mesh) precoated on glass plates (15 × 50 mm), and spots were visualized by a 5% vanillin sulfuric acid/ethanol solution.1H NMR (500 MHz, CDCl3) δ 12.78 (s, 1H), 7.44 (s, 2H), 7.10 (s, 2H), 7.01 (s, 1H), 6.70 (d, J = 3.7 Hz, 2H), 3.95 (d, J = 6.6 Hz, 18H). 13C NMR (101 MHz, CDCl3) δ 182.8, 164.6, 164.0, 162.0, 156.7, 156.3, 153.7, 153.2, 141.7, 126.1, 123.5, 108.9, 107.6, 105.9, 105.7, 103.8, 101.3, 61.1, 61.1, 56.4. HRMS-ESI (m/z): [M + Na+] calcd for C28H26NaO11 561.1367; found 561.1373.
1H NMR (500 MHz, CDCl3) δ 7.47 (s, 2H), 6.69 (s, 2H), 6.45 (s, 1H), 6.31 (s, 1H), 4.98 (d, J = 10.5 Hz, 1H), 3.97 (s, 6H), 3.92 (s, 6H), 3.90 (s, 3H), 2.87 (d, J = 16.0 Hz, 1H), 2.82–2.70 (m, 2H), 2.29 (d, J = 13.1 Hz, 1H), 2.15–2.03 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 165.2, 156.6, 154.7, 153.4, 153.1, 149.8, 142.9, 137.6, 137.0, 124.3, 107.5, 103.1, 102.8, 101.3, 78.0, 61.0, 60.9, 56.4, 56.2, 29.5, 19.6. HRMS-ESI (m/z): [M + Na+] calcd for C28H30NaO10 549.1731; found 549.1758.
1H NMR (400 MHz, CDCl3) δ 7.48 (s, 2H), 6.70 (s, 2H), 6.49 (d, J = 2.2 Hz, 1H), 6.36 (d, J = 2.2 Hz, 1H), 4.98 (dd, J = 10.8, 2.1 Hz, 1H), 3.98 (d, J = 1.0 Hz, 9H), 3.92 (s, 6H), 3.89 (s, 3H), 3.87 (s, 3H), 2.97–2.82 (m, 1H), 2.82–2.67 (m, 2H), 2.34–2.21 (m, 1H), 2.08–1.99 (m, 2H).13C NMR (100 MHz, CDCl3) δ 165.0, 158.4, 156.1, 153.4, 153.1, 150.2, 142.8, 137.6, 137.2, 137.2, 124.5, 108.7, 107.4, 103.2, 103.1, 96.9, 78.0, 61.0, 60.9, 60.4, 56.4, 56.2, 55.7, 29.7, 29.6, 19.8, 14.2. HRMS-ESI (m/z): [M + Na+] calcd for C29H32NaO11 563.1888; found 563.1892.
1H NMR (500 MHz, CD3OD) δ 7.21 (s, 2H), 6.47 (s, 2H), 6.22 (d, J = 11.9 Hz, 2H), 4.84 (s, 1H), 2.79 (d, J = 17.2 Hz, 1H), 2.74–2.65 (m, 1H), 2.27–2.15 (m, 1H), 2.09–1.95 (m, 1H). 13C NMR (100 MHz, CD3OD) δ 167.0, 157.7, 157.2, 151.4, 146.8, 146.5, 140.3, 133.9, 133.6, 120.7, 110.4, 108.5, 106.1, 102.3, 101.4, 79.0, 30.4, 20.3. HRMS-ESI (m/z): [M − H−] calcd for C22H19O10 441.0827; found 441.0825.
1H NMR (500 MHz, CDCl3) δ 7.42 (s, 2H), 6.74 (d, J = 2.2 Hz, 1H), 6.64 (s, 2H), 6.61 (d, J = 2.3 Hz, 1H), 4.98 (d, J = 10.6 Hz, 1H), 3.99–3.80 (m, 18H), 2.78–2.67 (m, 2H), 2.22 (d, J = 14.0 Hz, 1H), 2.06 (dt, J = 23.4, 9.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 168.8, 164.5, 156.4, 153.4, 153.1, 149.7, 149.5, 142.9, 137.7, 136.7, 124.2, 113.0, 108.5, 108.1, 107.4, 103.1, 78.2, 77.3, 61.0, 60.9, 56.4, 56.2, 56.2, 29.2, 20.8, 20.1. HRMS-ESI (m/z): [M + Na+] calcd for C30H32NaO11 591.1837; found 591.1849.
1H NMR (500 MHz, CDCl3) δ 7.54–7.33 (m, 7H), 6.76 (d, J = 2.3 Hz, 1H), 6.66 (s, 2H), 6.62 (d, J = 2.1 Hz, 1H), 4.97 (d, J = 10.3 Hz, 1H), 4.04–3.86 (m, 18H), 2.59 (dd, J = 10.7, 5.8 Hz, 2H), 2.23–2.11 (m, 1H), 2.09–1.94 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 169.3, 164.6, 156.4, 153.4, 153.1, 149.6, 149.4, 142.9, 137.7, 136.6, 133.3, 129.4, 128.8, 128.8, 127.5, 124.2, 113.0, 108.5, 108.1, 107.4, 103.1, 78.2, 77.3, 61.0, 60.9, 56.4, 56.2, 41.4, 29.2, 19.9. HRMS-ESI (m/z): [M + Na+] calcd for C36H36NaO11 667.2150; found 667.2165.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank The National Natural Science Fund (81602958) and CAMS Innovation Fund for Medical Sciences (CIFMS 2017-I2M-1-011) for financial support.References
- Y. Li, K.-T. Leung, F. Yao, L. S. M. Ooi and V. E. C. Ooi, J. Nat. Prod., 2006, 69(5), 833–835 CrossRef CAS PubMed.
- L. Bao, X.-S. Yao, J.-K. Xu, X.-Y. Guo, H.-W. Liu and H. Kurihara, Fitoterapia, 2009, 80(6), 349–353 CrossRef CAS PubMed.
- A. G. Mercader and A. B. Pomilio, Eur. J. Med. Chem., 2010, 45(5), 1724–1730 CrossRef CAS PubMed.
- J. Kang, C. Liu, H.-Q. Wang, B.-M. Li, C. Li, R.-Y. Chen and A. L. Liu, Molecules, 2014, 19(4), 4479–4490 CrossRef PubMed.
- P. T. Nguyen, B. T. T. Luyen, J. H. Kim, A. R. Jo, T. D. Nguyen, P. V. Kiem, C. V. Minh and Y. H. Kim, Bioorg. Med. Chem., 2016, 24(14), 3125–3132 CrossRef CAS PubMed.
- J. G. Sweeny and G. A. Iacobucci, Tetrahedron Lett., 1977, 33(22), 2927–2932 CrossRef CAS.
- G. Lewin, M. Bert, J. C. Dauguet, C. Schaeffer, J. L. Guinamant and J. P. Volland, Tetrahedron Lett., 1989, 30(50), 7049–7052 CrossRef CAS.
- C. F. Chee, M. J. C. Buckle and N. A. Rahman, Tetrahedron Lett., 2011, 52(24), 3120–3123 CrossRef CAS.
- S. Saxena, J. K. Makrandi and S. K. Grover, Synthesis, 1985,(6/7), 697 CrossRef CAS.
- A. K. Ganguly, S. Kaur, P. K. Mahata, D. Biswas, B. N. Pramanik and T. M. Chan, Tetrahedron Lett., 2005, 46(23), 4119–4121 CrossRef CAS.
- Z.-P. Xiao, Z.-Y. Peng, J.-J. Dong, J. He, H. Ouyang, Y.-T. Feng, C.-L. Lu, W. Q. Lin, J.-X. Wang, Y.-P. Xiang and H.-L. Zhu, Eur. J. Med. Chem., 2013, 63, 685–695 CrossRef CAS PubMed.
- H. Yuan, K. J. Bi, B. Li, R. C. Yue, J. Ye, Y.-H. Shen, L. Shan, H.-Z. Jin, Q.-Y. Sun and W.-D. Zhang, Org. Lett., 2013, 15(18), 4742–4745 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01606b |
| This journal is © The Royal Society of Chemistry 2018 |