DOI:
10.1039/C8RA01766B
(Paper)
RSC Adv., 2018,
8, 17044-17055
Tough interpenetrating polymer network of silicone containing polyurethane and polystyrene with self-healing, shape memory and self-cleaning attributes†
Received
28th February 2018
, Accepted 3rd May 2018
First published on 9th May 2018
Abstract
Smart biodegradable tough interpenetrating polymer networks (IPNs) of bio-based polyurethane containing a silicone moiety and polystyrene at three different compositions were synthesized for the first time by using simultaneous polymerization technique. The structures of the synthesized IPNs were interpreted by FTIR, NMR, and XRD analyses, while morphology was provided from a SEM study. The synthesized IPNs exhibited outstanding elongation at break (up to 1608%) along with good tensile strength (up to 12.6 MPa), toughness (up to 92.34 MJ m−3), impact resistance (up to 26.8 kJ m−1), scratch resistance (up to 6.5 kg) and durometer hardness (up to 86 Shore A). Furthermore, the synthesized IPNs exhibited good thermal stability up to 245 °C and chemical resistance. Interestingly, these IPNs showed multi-stimuli responsive self-healing (within 62 s at 450 W microwave and 6–8 min under sunlight) and shape memory (100% shape recovery within 48 s with a 450 W microwave and 7–13 min under direct sunlight) behavior. A self-cleaning attribute was also observed for the synthesized IPNs which showed a static contact angle up to 120.8° and angle of hysteresis <5°. Most interestingly, the synthesized IPNs also exhibited moderate bio-degradation under the exposure to a P. aeruginosa bacterial strain. Therefore, the synthesized smart bio-degradable tough IPNs with the above properties have great potential for different advanced multifaceted applications.
Introduction
Pure original polymers have very limited uses. They need to be adulterated to obtain a desired level of properties for their different high-end applications.1,2 Thus to improve some desired properties and to incorporate some new properties, modification of a polymer is a well-established approach. Both physical and chemical methods have received great interest in the scientific world, for this purpose. Among various types of modifications, interpenetrating polymer network (IPN) has achieved great interest to meet the specific needs for advanced applications of polymers.3 IPNs are produced as a special class of polymers in order to achieve compatibility between two immiscible polymers and thereby forming a more uniform system. They are interlaced at a molecular scale with each other without any chemical bond formation.4,5 These types of polymers exhibit excellent properties because of the synergistic effect of the individual polymers. For example, polystyrene (PS) is brittle, whereas polyurethane (PU) is flexible and may be elastomeric in nature. Thus the formation of a PU network and interpenetrating with PS may result in a unique material with properties superior to those of the individual components. All types of IPNs such as sequential, simultaneous and semi-IPN are reported in the literature. In this milieu, the literature reveals several efforts to obtain such IPNs. Synthesis of IPN was first reported by Miller using styrene–divinyl benzene.6 Kim et al. reported a detailed morphological study of the IPN of PU–poly(methyl methacrylate).7 Zhang et al. reported the synthesis of pH- and temperature-sensitive IPNs using poly(methacrylic acid) and poly(N-isopropyl acrylamide).8 Vlad et al. synthesized a series of IPNs from PU and polysiloxane and evaluated their structures.9 Begum et al. synthesized a IPN from PU and poly(butyl methacrylate) and evaluated its physico-mechanical, optical, thermal, microcrystalline and morphological behaviors.10 Tugui et al. synthesized IPNs from poly(urethane-urea) and polydimethylsiloxane as active components in electromechanical transducers.11 Again, there are several reports on green resources based IPNs. For example, Ku et al. synthesized simultaneous IPNs from castor oil based PU with PS and established the structure–property relationship with the help of solid-state high-resolution 13C NMR spectral analyses.12 Siddaramaiah and his co-workers synthesized an IPN from castor oil-based PU with PS which exhibited good tensile strength.4 Dave and his co-workers evaluated the thermal, damping and morphological properties of IPN films synthesized from transesterified castor oil based PU and PS.3 Literatures advocated that most of the IPNs exhibited good tensile strength but suffer from very poor flexibility.2–11 Thus modification of PS by an elastomeric silicone containing PU may be an apt choice. In this milieu bis(hydroxyalkyl)–poly(dimethylsiloxane) can be used as flexible and low surface energy material. In addition, IPNs with smart properties like self-healing, shape memory and self-cleaning are not reported yet. These smart or intelligent polymers are a unique class of polymers which can perform special function upon application of a required stimulus.13 The self-healing polymers can heal the crack or damage surface by themselves under the exposure of an appropriate stimulus.13 On other hand, shape memory polymers have the ability to recover their original shape from a deformed temporary shape upon exposure of a suitable stimulus.13 The stimuli generally used are heat, light, solvent, ultrasonication, MW, sunlight etc. Again self-cleaning polymers are those which have the ability to clean their surfaces from dirt by photocatalytic or super-hydrophobicity effect.13
Henceforth, authors wish to report the synthesis of smart IPNs from different wt% of bio-based PU (containing silicone moiety) with PS for the first time. Synthesized IPNs were characterized with the help of different spectroscopic and microscopic tools. Various properties such as physical, mechanical, thermal, chemical resistance and biodegradation were evaluated along with self-healing, shape memory and self-cleaning behaviors for the present IPNs.
Materials and methods
Materials
Poly(ε-caprolactone)diol (PCL, Mn = 2000 g mol−1, Sigma-Aldrich, USA), dimer acid (DA, Mn ∼ 570, Sigma-Aldrich, USA), glycerol (Merck, India) and sunflower oil (Sigma-Aldrich, USA) were dried for 12 h in an hot oven under vacuum before use. Bis(hydroxyalkyl)–poly(dimethylsiloxane) (PDMS, Mn ∼ 5600 g mol−1, Sigma-Aldrich, Japan), isophorone diisocyanate (IPDI, Sigma-Aldrich, USA), 2,4/2,6-toluene diisocyanate (TDI, Merck, Germany), oxalic acid (Rankem, India), potassium hydroxide (Rankem, India), para-toluene sulfonic acid (p-TSA, SRL, Mumbai, India), styrene (Sigma-Aldrich, USA) and benzoyl peroxide (BPO, Merck, India) were used as received. Xylene (Merck, India) was distilled and kept in 4A type of molecular sieves prior to use.
Preparation of dimer acid-glycerol modified polyol (DAGP)
DAGP was prepared by the esterification reaction of 3 mol of DA and 6 mol of glycerol in presence of p-TSA (0.5 wt% of DA) as reported earlier.14
Preparation of monoglyceride of sunflower oil (MG)
MG of sunflower oil was prepared as reported elsewhere.15 Briefly, one equivalent of sunflower oil was reacted with two equivalent of glycerol in presence of CaO (0.5 wt% of sunflower oil) at elevated temperature (220 ± 5) under N2 atmosphere.
Synthesis of interpenetrating polymer (IPN) network of PU and PS
IPN of PU and PS was synthesized using a two-step polymerization process. In the first step pre-polymer of PU was synthesized by using requisite amount PCL and PDMS as the macroglycol, DAGP as a branch generating unit and TDI as aromatic diisocyanate in a three neck round bottom flask equipped with a nitrogen gas inlet, a mechanical stirrer, a Teflon septum and a thermometer. The solid content of the reaction was maintained near about 75 wt% by the incorporation of distilled xylene (1 mL) in the reaction vessel using a syringe. However the amount of PDMS used for the synthesis of PU-prepolymer was based on our earlier work.14 On the basis of performance, 10 mol% of PDMS (with respect to PCL, another macroglycol) was taken for the synthesis of PU. The reaction was carried out for 4.5 h at 85–90 °C under N2 atmosphere to obtain a viscous mass, treated as pre-polymer. In the second step, calculated amount of chain extender, MG and aliphatic diisocyanate, IPDI were added to the reaction mixture maintaining 50% solid content. The reaction was carried out at 100–110 °C for 2 h with constant stirring. Then the pre-polymer of styrene was added to the above reaction mixture using syringe. This pre-polymer of styrene containing 80–85% monomer was obtained by free radical polymerization of the monomer using 0.5 wt% initiator, BPO. The mixture was heated at 110 °C for 2.5–3 h until optimum viscosity was obtained without gel formation. After cooling the reaction mixture at room temperature under constant stirring, PU network was cross-linked by adding measured amount of TDI (30% of the total diisocyanate). The IPNs with different compositions as mentioned in Table 1 were prepared by varying wt% of PU and PS. IPN3 without PDMS was also synthesized by following the same procedure to understand the role of PDMS.
Table 1 Compositions of the reactants for the synthesized IPNs
| Reagent/parameter |
IPN1 (PU![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PS = 65:35) :PS = 65:35) |
IPN2 (PU:PS = 50:50) |
IPN3 (PU:PS = 35:65) |
| PCL (mM) |
1 |
1 |
1 |
| PDMS (mM) |
0.1 |
0.1 |
0.1 |
| DAGP (mM) |
0.1 |
0.1 |
0.1 |
| MG (mM) |
2.3 |
2.3 |
2.3 |
| TDI (mM) |
1.6 |
1.6 |
1.6 |
| IPDI (mM) |
2 |
2 |
2 |
| Styrene (mM) |
21.5 |
40 |
74.3 |
| Hard segment (wt%) |
58.9 |
68.5 |
77.8 |
| Soft segment (wt%) |
41.1 |
31.5 |
22.2 |
| Bio-based content (wt%) |
13.87 |
10.66 |
5.33 |
Mild steel plate (150 mm × 50 mm × 0.61 mm), glass slide and a glass plate (10 mm × 60 mm × 5.9 mm) coated with viscous IPN solutions were left for 7 days at room temperature for proper drying. Then the dried strips of IPNs were kept in a vacuum oven at 60 °C for 48 h to remove the rest amount of volatiles. These IPN strips were used for further testing and analyses.
Instrumentation and testing methods
Structural analysis. The FT-IR spectra of the starting materials and synthesized IPNs were obtained by a FT-IR spectrometer (Perkin Elmer, Model: Frontier MIR-FIR, USA) over the scanning range of 400–4000 cm−1. The IPN samples were cut into very small pieces and mixed thoroughly with KBr and were used in the form of small pellets. The elemental compositions of the synthesized IPNs were supported by energy dispersive X-ray (EDX) analysis. The X-ray diffraction (XRD) study was carried out by using a D8 Focus XRD machine (Bruker AXS, Germany) at room temperature equipped with a Cu-Kα radiation source (λ = 1.54 Å) over the range of 2θ = 10–70° and a 2D detector were used to record the diffraction patterns. The procedure was automated by XRD commander 2 and Diffraction EVA software. Bragg's equation was used to evaluate inter-planner distance (d) of PCL moiety by the following equation.| | |
d = nλ/2sinθ
| (1) |
where n= integer (1), λ = wavelength of incident X-ray beam and θ = angle of incident X-ray beam. From the XRD spectral analyses the percent of crystallinity and amorphousness also evaluated. The ratio of the area under the crystalline peaks to the total (crystalline + amorphous) area represented the degree of crystallinity. The surface morphology of the IPNs before and after biodegradation test was studied by JEOL JSM-6390LV scanning electron microscope.
Determination of physical properties. The specific gravity of IPNs was evaluated using standard D7932 method. GPC chromatograms were obtained by a Waters Instrument (UK) equipped with a 2414 refractive index detector, linear polystyrene as the standard and a 515 HPLC pump. THF at a flow rate of 0.75 mL min−1 was used as the eluent. The concentration of PU solution was 2000 ppm and the injection volume was 50 μL.
Testing of mechanical properties. The mechanical properties of the IPN strips were evaluated with the help of Universal Testing Machine (UTM), model WDW-10 (JINAN, CHINA) with a 1.0 kN load cell and crosshead speed of 50 mm min−1 as per standard ASTM D 657 method. The scratch resistance of the IPN strips was measured using scratch hardness tester, model number 705 (Sheen Instrument Ltd, UK) with stylus accessory and a travel speed of 50 mm s−1. The impact resistance was measured by falling weight method using an impact tester (S.C. Dey & Co, Kolkata, 1 m is the maximum height) following the standard ASTM D1037 method. The energy per unit thickness of the IPN strip corresponding to the maximum height was taken as the impact strength.
Testing of thermal properties. The decomposition profiles of the synthesized IPN films were thermo gravimetrically analyzed with the help of a TGA-4000 (Perkin Elmer, USA) thermal instrument, under ultra pure nitrogen gas with flow rate of 30 mL min−1 at heating rate of 5 °C min−1 in a temperature range of 32–720 °C. Similarly, glass transition temperature (Tg) and melting temperature (Tm) of the IPNs were evaluated with the help of differential scanning calorimeter (DSC 6000, Perkin Elmer, USA) equipped with refrigerator cooling system at a heating rate of 10 °C min−1 from −70 to 120 °C by following a cycle of heating, cooling and heating under the flow of ultra pure nitrogen gas at a rate of 30 mL min−1.
Self-healing test. The self-healing ability of the IPN strips was evaluated under different stimuli like microwave, heat and sunlight. The IPN strips were cut in a transverse direction by a razor blade and exposed under the above mentioned stimulus. Tensile strength of the each IPN strips was evaluated with the help of UTM and healing efficiency of the strips was calculated as a ratio of tensile strength before and after healing. A domestic micro-oven (800 W) operating at a frequency of 2.45 GHz was used for healing. Sunlight healing was done at Tezpur University campus (altitude: 26.63 °N 92.8 °E) in the month of November with average temperature of (36 ± 1) °C, humidity (72 ± 2)% and light intensity: 1–1.1 × 105 lux. Healing efficiency of the strips was evaluated with the help of following equation.| | |
Healing efficiency = 100 × [tensile strength (healed)/tensile strength (initial)]
| (2) |
Self-cleaning test. Self-cleaning property of the synthesized IPN strips was investigated by measuring the contact angle (advancing angle, receding angle and angle of hysteresis) of the acidic, basic and neutral water droplets deposited on the IPN surfaces. Surface roughness of the synthesized IPNs was analyzed by the help of SEM images. Contact angles were measured by ImageJ2 software after capturing digital images of the droplets. 3D surface plot of the SEM images was also done by imageJ2 software. The contact angle values represented in manuscript were the average of ten measurements.
Shape memory test. To study the shape memory behavior of the IPN strips, spiral bending test was performed under the exposure of MW, heat and sunlight. At first the IPN strips were heated at 70 °C and folded in the form of spiral shape. Immediately the folded spiral strips were frozen in an ice-water-salt bath for 15 min to release the stress. After achieving the fixed shape, the IPN strips were dried under vacuum and kept in room temperature for 30 min. To achieve shape recovery from the temporary fixed shape, IPN strips were projected under MW, heat and sunlight. The time required to recover the original shape from fixed temporary shape was also noted. The shape fixity and shape recovery as two shape memory parameters were calculated from the following equations:| | |
Shape fixity (%) = {(90 − θf)/90} × 100
| (3) |
| | |
Shape recovery (%) = {(90 − θr)/90} × 100
| (4) |
where, θf represents the ‘fixed angle’ after cooling and θr represents the ‘recovered angle’.
Chemical resistance test. Chemical resistance of all IPN films was calculated by measuring the percentage of weight change upon exposure of the chemical media such as aqueous 20% EtOH, 10% HCl, 1% NaOH, 10% NaCl and tap water for 30 days as per the standard ASTM D 543-67 method.
Bio-degradation test. Bio-degradation study of the IPN strips was done by McFarland turbidity method against P. aeruginosa as the bacterial strain.16 Thus, a mineral salt solution containing 100 mg MnSO4·5H2O, 2.0 g (NH4)2SO4, 2.0 g Na2HPO4, 4.75 g KH2PO4, 0.5 mg CaCl2·2H2O, 1.2 g MgSO4·7H2O, 70 mg ZnSO4·7H2O, 10 mg H3BO3·5H2O, 100 mg CuSO4·7H2O, 10 mg MoO3, and 1 mg FeSO4·7H2O was prepared in 1 L demineralized water. The medium was sterilized in an autoclave at 120 °C under a pressure of 103 kPa for 15 min. Then P. aeruginosa bacterial strain was cultured for 48 h at 37 °C in the above sterilized medium. IPN strips (in triplicate) were submerged in test tubes with sterile salt medium (10 mL) containing 100 mL (108 microbes mL−1) of the cultured bacterial medium and incubated at 37 °C for a time period of 8 weeks. The increase in turbidity of the medium with exposure of time represents the bacterial growth. The optical density (OD) of the bacterial medium with respect to the control was observed by measuring the absorbance of the medium at 600 nm with the help of a UV-visible spectrophotometer. Surface images of bio-degraded IPN strips were taken by scanning electron microscopy. To study the functional changes of the bio-degraded films, FT-IR analyses were also conducted.
Results and discussion
Synthesis of IPNs
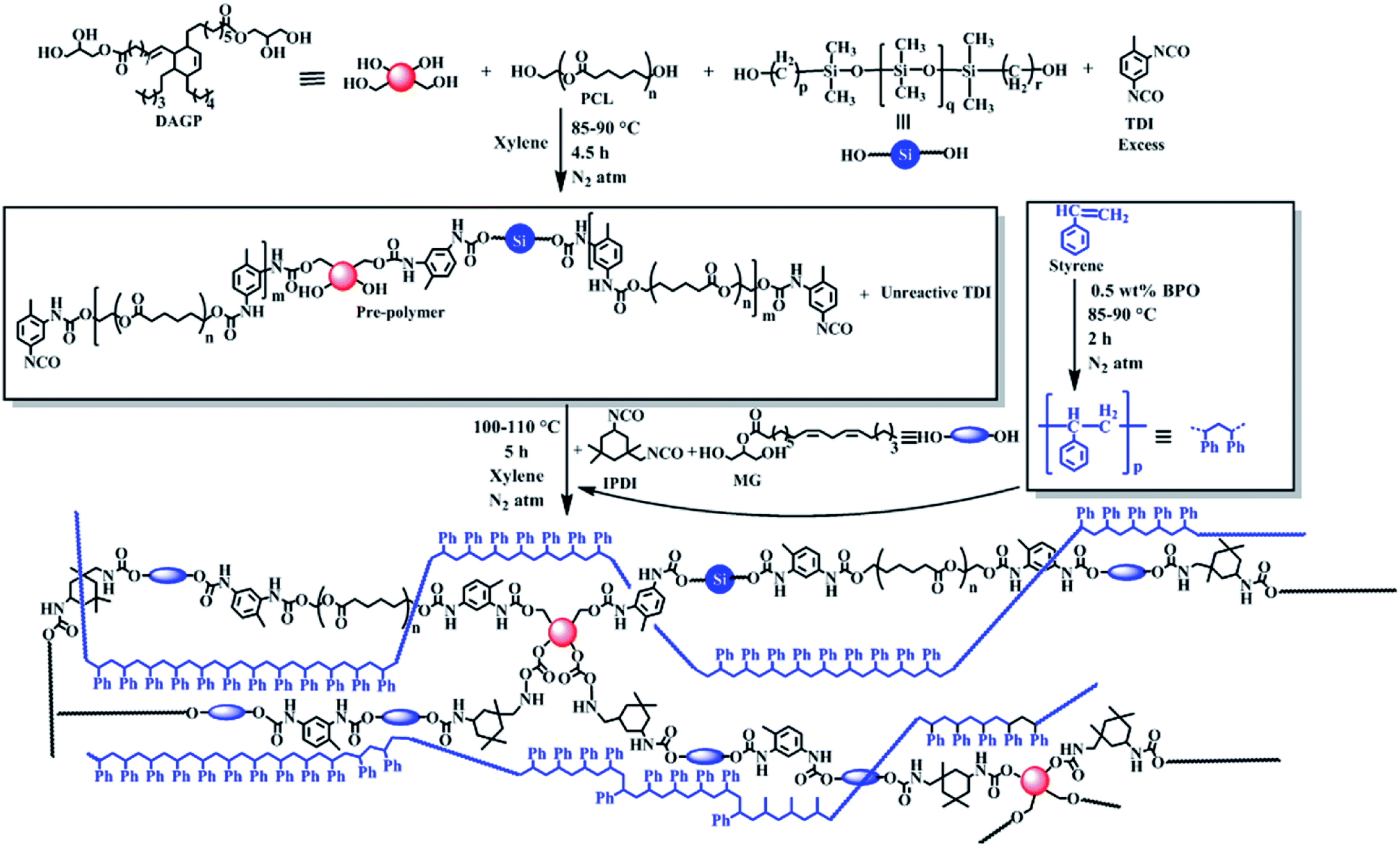
IPNs were synthesized using simultaneous polymerization technique of the pre-polymers as shown in Scheme 1.5 In the first step of synthesis, excess amount of TDI was added to prepare an isocyanate terminated pre-polymer of PU by the reaction with polyols like PCL, PDMS and DAGP. In second step monoglyceride of sunflower oil as a bio-based chain extender was reacted to the pre-polymer with requisite amount of IPDI to maintain the overall NCO and OH functionality equal and to control the reaction as the reactivity of two isocyanate groups of it, are different. In the second step a pre-polymer of PS was also further polymerized in the same reaction vessel. This pre-polymer of PS was prepared in another reactor just by heating with BPO initiator through free radical polymerization process, where along with 10–15% PS, 85–90% monomer of it was present. After completion of the polymerization reaction as indicated by increase of viscosity and absence of free isocyanate group and unreacted styrene monomer (as supported by FTIR study), the resultant PU was crosslinked by using TDI with an amount of 30 wt% of total isocyanate content of PU. The strong interlacing between PS and PU may be due to in situ formation of simultaneous IPN and π–π interactions among aromatic rings of both the polymers.
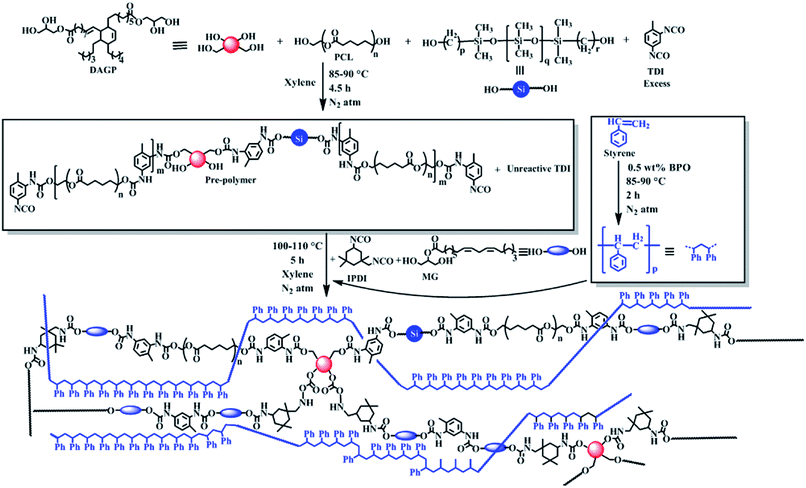 |
| | Scheme 1 Plausible synthetic route of IPN. | |
Structural analysis
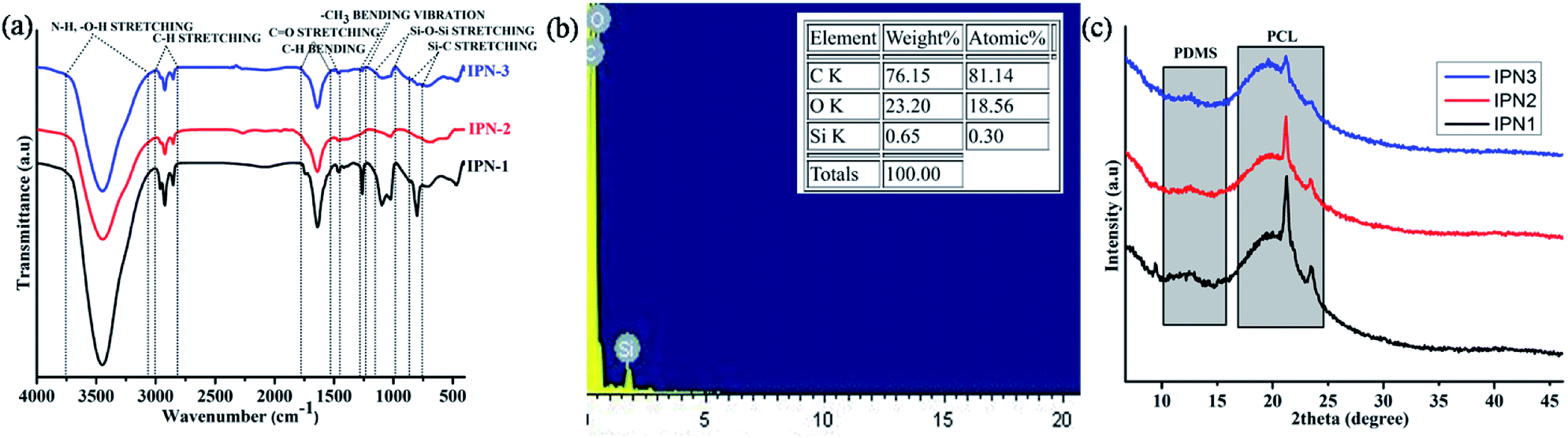
FT-IR spectra of the synthesized IPNs are displayed in Fig. 1a and analyses of which support the chemical linkages present in the IPNs. In addition FT-IR spectra of DAGP, PCL and PDMS along with styrene and pre-polymer of polystyrene are also shown in Fig. S1 of the ESI.† The detail analyses of these spectra are presented in ESI.† Absence of a characteristic band at 2270 cm−1 confirms the completion of the reaction. Absorption bands at 3450, 1731, 1638 and 1020 cm−1 correspond to the N–H, O–H, carbonyl of ester (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), carbonyl of amide (NH–CO) and O–C stretching frequencies. These are the characteristics bands of urethane linkage, –NH–(CO)–O–. Asymmetric and symmetric stretching vibrations of –C–H bond have appeared at 2925 cm−1 and 2846 cm−1. Backbone vibration of benzene ring of PS and TDI moiety was observed at 1497 and 1464 cm−1. The peak at 1267 cm−1 corresponds to the bending vibration of –CH3 in PDMS moiety. Asymmetric and symmetric stretching vibrations of Si–O–Si bond of PDMS moiety appear in the range of 1100–1020 cm−1. Intensity of this band decreases with the increase of wt% of PS in the polymer matrix. There is also overlap of bands in the region 760–800 cm−1 due to –CH3 rocking, Si–C stretching of PDMS and bending deformation of C–H bond.3,17 Similar decreasing pattern was also found in this region. Thus the FT-IR spectral analyses indicate the presence of both polystyrene and polyurethane in a same matrix of IPNs. Furthermore, the interaction of PS and PU was noticed in the IPNs as discussed in ESI (Fig. S1†).
O), carbonyl of amide (NH–CO) and O–C stretching frequencies. These are the characteristics bands of urethane linkage, –NH–(CO)–O–. Asymmetric and symmetric stretching vibrations of –C–H bond have appeared at 2925 cm−1 and 2846 cm−1. Backbone vibration of benzene ring of PS and TDI moiety was observed at 1497 and 1464 cm−1. The peak at 1267 cm−1 corresponds to the bending vibration of –CH3 in PDMS moiety. Asymmetric and symmetric stretching vibrations of Si–O–Si bond of PDMS moiety appear in the range of 1100–1020 cm−1. Intensity of this band decreases with the increase of wt% of PS in the polymer matrix. There is also overlap of bands in the region 760–800 cm−1 due to –CH3 rocking, Si–C stretching of PDMS and bending deformation of C–H bond.3,17 Similar decreasing pattern was also found in this region. Thus the FT-IR spectral analyses indicate the presence of both polystyrene and polyurethane in a same matrix of IPNs. Furthermore, the interaction of PS and PU was noticed in the IPNs as discussed in ESI (Fig. S1†).
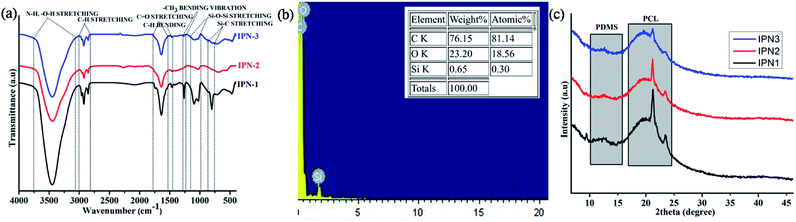 |
| | Fig. 1 (a) FT-IR spectra of IPNs, (b) EDX spectrum of IPN3 and (c) X-ray diffractograms of IPNs. | |
The energy dispersive X-ray spectroscopic analyses confirmed the presence of C, O and Si in the structure of IPN3 (Fig. 1b) with weight and atomic percentages of 76.15, 23.20 and 0.65 and 81.14, 18.56 and 0.30 respectively. The represented values of EDX are the average results of three different positions of IPN3.
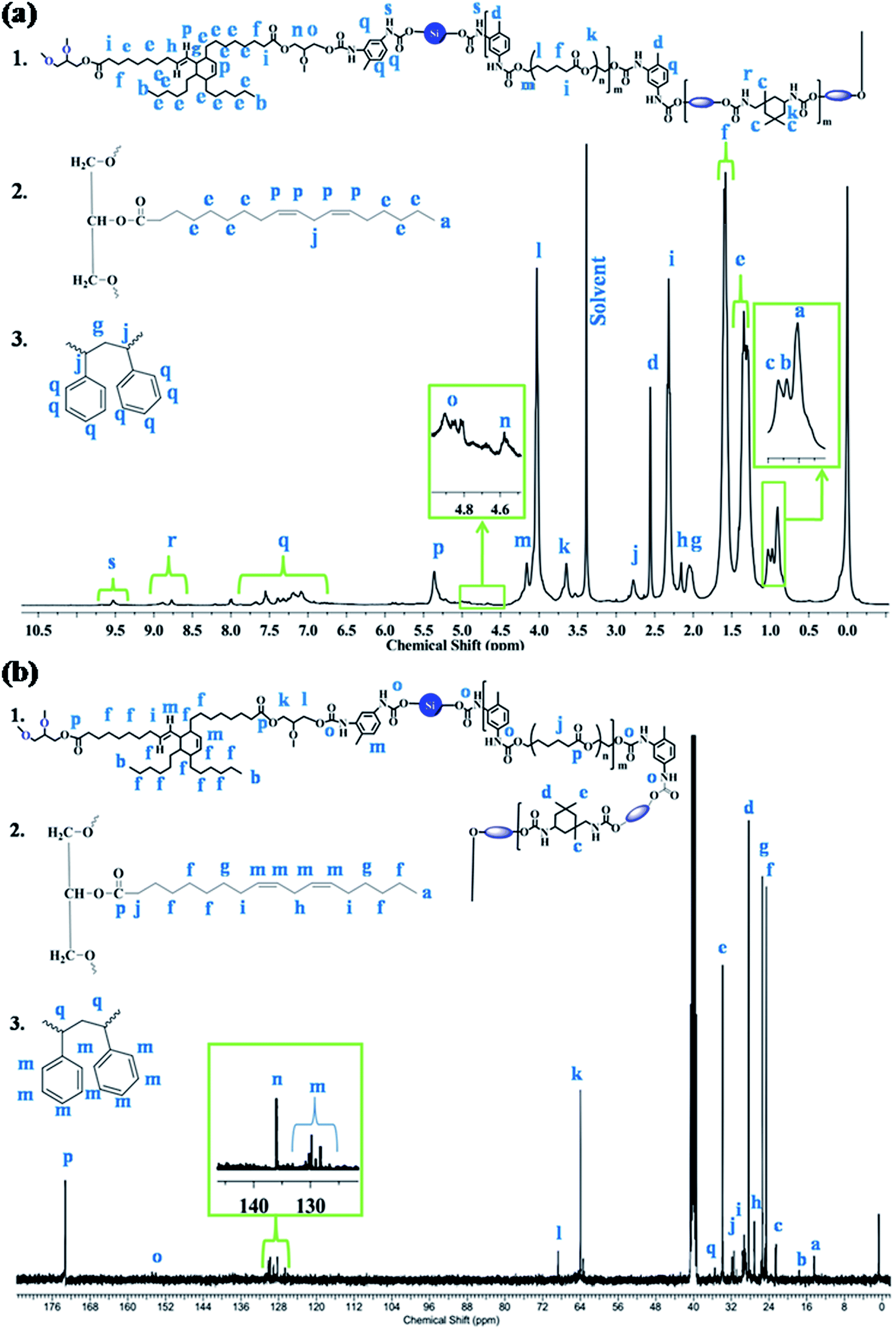
The macromolecular structure of the synthesized IPN1 was further analyzed with the help of 1H and 13C NMR spectra, as displayed in Fig. 2a and b. In 1H NMR spectrum, the peaks at 0.81 (a), 0.88 (b), 0.93 (c) and 2.46 (d) ppm are attributed to the terminal methyl group of MG, PDMS, DAGP and TDI moieties, respectively. Internal methylene protons of PCL, DAGP, MG and PDMS have appeared in the range of 1.19–1.24 (e) ppm. The β and α-methylene protons with respect to the ester group were found to appear at 1.56 (f) and 2.22 (i) ppm. The peak at 1.95 (g) ppm, is attributed to the -C-H proton of DAGP ring adjacent to the double bond and β methylene protons of PS with respect to the benzene ring. Methylene protons, α to the double bond were observed at 2.06 (h) ppm. The methylene protons between the two double bond of MG and α methine protons of PS ring with respect to the benzene ring were found at 2.68 (j) ppm. Protons attached to the urethane linkage in IPDI ring and the methylene protons between ester and urethane linkage in PCL moiety resulted signal at 3.55 (k) ppm. The signals at 3.93 (l) and 4.06 (m) are ascribed to the β and α-methylene protons which are directly connected with ester oxygen. The peaks at 4.59 (n) and 4.87 (o) ppm are credited to the methylene protons present in glycerol unit of DAGP moiety. The double bonded protons in MG and DAGP ring have appeared at 5.26 (p) ppm. Signals for the aromatic protons of TDI and styrene were observed in the range of 6.9–7.59 (q) ppm. The (N–H) urethane protons were found in the range of 8.6–9.5 ppm (Fig. 2a). In 13C NMR spectrum, the peaks at 14.43 (a) and 17.61 (b) ppm observed are due to the terminal methyl carbons of PDMS, MG and DAGP moieties. Signals for different type methyl carbons in IPDI ring were observed at 22.51 (c), 28.3 (d) and 33.87 (e) ppm. The signals for internal methylene carbons of PCL, MG, DAGP, PDMS, methylene carbons between two double bonds, and α to the double bond and ester group were found at 24.60 (f), 25.41 (g), 27.29 (h), 29.28 (i) and 33.87 (j) ppm respectively. Methylene carbons of glycerol unit of DAGP moiety were observed at 64 (k) and 68.74 (l) ppm. Peaks in the range of 128.64–130.25 (m) and 137.75 (n) are attributed to the double bonded and aromatic carbons. The signals at 154.06–154.83 (o) and 173.24 (p) correspond to the urethane and ester carbons of MG, and PCL moiety.14
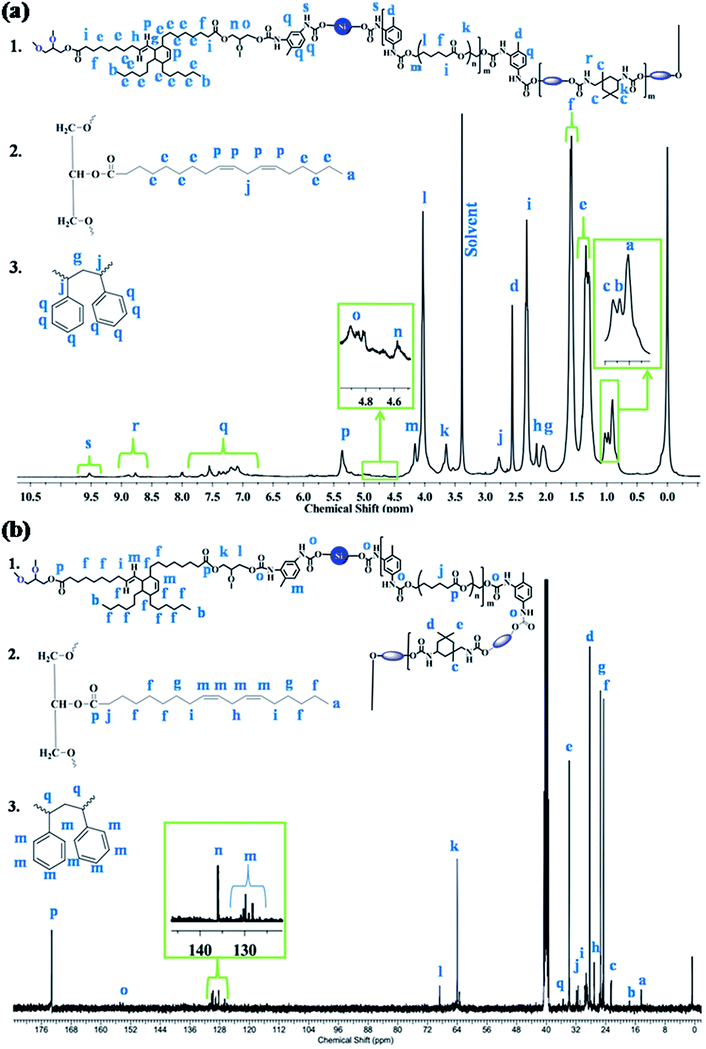 |
| | Fig. 2 (a) 1H and (b) 13C NMR spectra of IPN1. | |
In order to the study the arrangement of chain segments of the synthesized IPNs, XRD spectral analysis was performed. The degree of crystallinity associated with crystalline PCL and PDMS moieties for the IPNs differs in the range of 34.6–28.6%. From the X-ray diffractograms displayed in Fig. 1c, it is confirmed that with the increase of the amorphous PS content from IPN1 to IPN3 the overall crystalline area decreases, as reflected in the peak intensity. Spectral analysis also revealed that the IPNs exhibited three characteristic peaks at 12.3° (calculated d spacing of 7.18 Å) for the crystalline PDMS and 21.4° (calculated d spacing of 4.14 Å) and 23.5° (calculated d spacing of 3.78 Å) for crystalline PCL moiety corresponding to the lattice planes (110) and (200) (Fig. 1c) respectively. Similar, peaks were also observed in other reports.14,18
Physical and mechanical properties
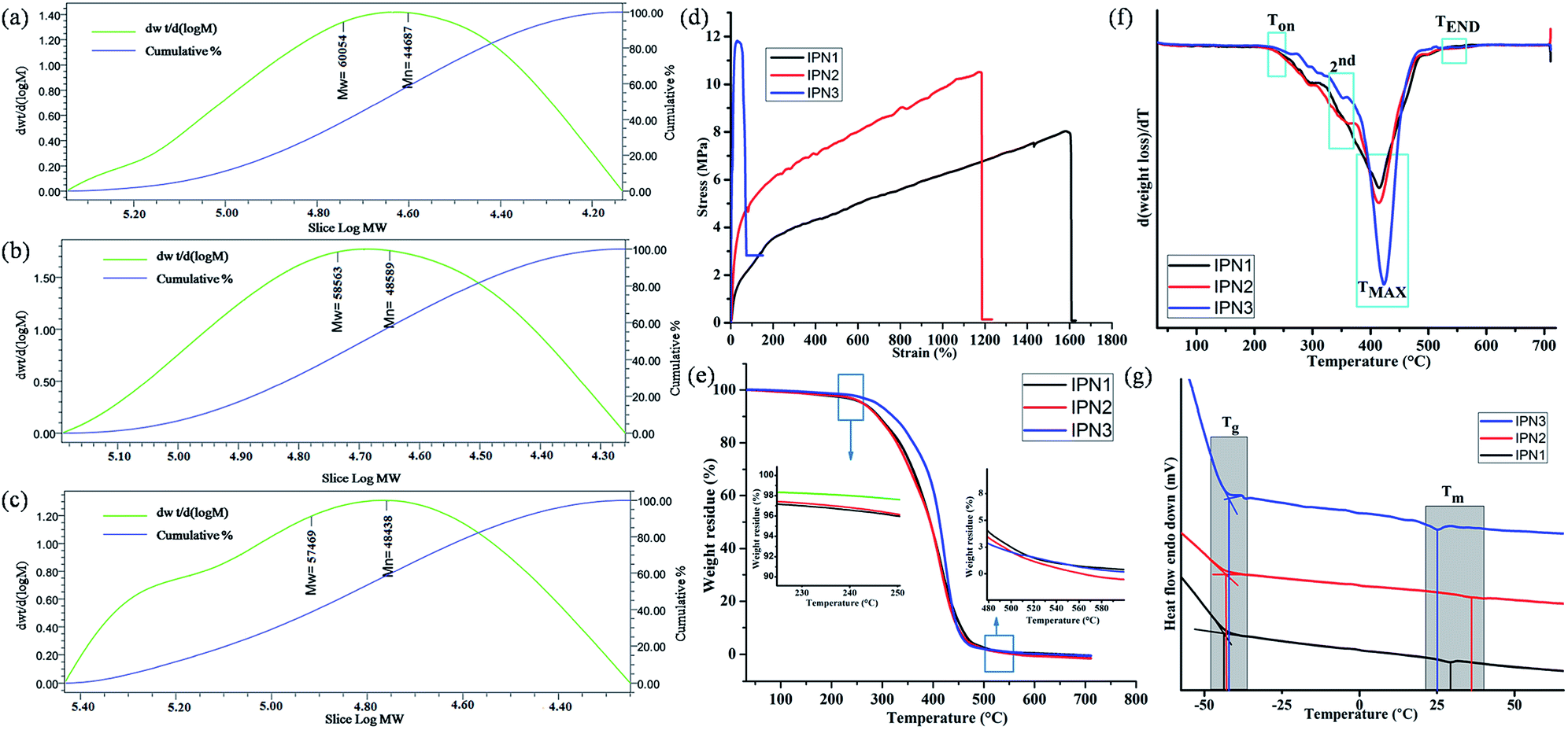
Different physical properties of the synthesized IPNs were tabulated in Table 2. The off white color of the IPNs might be due to the structural inhomogeneity of the components. Specific gravity values of the synthesized IPNs were found in the range of 1.36 to 1.08. Furthermore, similar types of results were also found when specific gravity of the IPNs was also calculated by weight fraction rule. With the increase of PS content from IPN1 to IPN3, overall amorphous character increases resulted low specific gravity.2 The number average (Mn) and weight average (Mw) molecular weight obtained from the GPC analyses for the synthesized IPNs before cross-linking were found to be in the order of 104, as summarized in Table 2 and representative chromatograms are shown in Fig. 3a–c. Furthermore, the polydispersity index (PDI) of the synthesized IPNs varies in the range of 1.2–1.42 which seems to be very low.
Table 2 Physical and mechanical properties of IPNs
| Properties |
IPN1 |
IPN2 |
IPN3 |
| Color |
Off white |
Off white |
Off white |
| Specific gravity |
1.36 |
1.28 |
1.08 |
| Mn (g mol−1) (uncrosslinked IPN) |
44687 |
48589 |
48438 |
| Mw (g mol−1) (uncrosslinked IPN) |
60054 |
58563 |
57469 |
| PDI |
1.34 |
1.2 |
1.42 |
| Tensile strength (MPa) |
8.03 ± 0.3 |
10.8 ± 0.5 |
12.6 ± 0.3 |
| Elongation (%) |
1608 ± 50 |
1184 ± 88 |
151 ± 25 |
| Toughness (MJ m−3) |
87.64 ± 1.3 |
92.34 ± 1.3 |
9.28 ± 1.0 |
| Impact resistance (kJ m−1) |
24.5 ± 0.5 |
26.8 ± 0.2 |
17.3 ± 0.8 |
| Scratch resistance (kg) |
5.6 ± 0.2 |
6 ± 0.2 |
6.5 ± 0.1 |
| Hardness (Shore A) |
71 ± 4 |
78 ± 2 |
86 ± 2 |
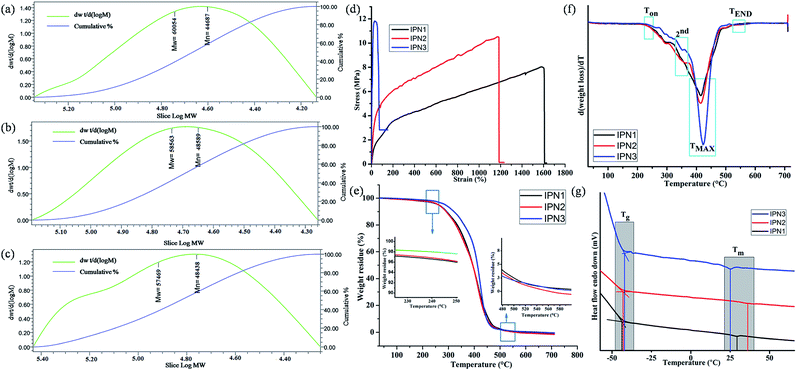 |
| | Fig. 3 GPC chromatograms of (a) IPN1, (b) IPN2 and (c) IPN3, (d) stress–strain profiles, (e) TGA thermograms, (f) derivative of TG (DTG) curves and (g) DSC curves with glass transition and melting temperature of IPNs. | |
Tensile strength, elongation at break and toughness of IPN strips were calculated from the stress–strain curves as displayed in Fig. 3d and the vales are summarized in Table 2. These properties were highly affected by the ratio of hard and soft segments, crystallinity, chain entanglement, physical cross linking, orientation of segments, hydrogen bonding, rigidity of aromatic units etc. From IPN1 to IPN3 the wt% of PS increases which enhances hard segment content in IPN matrix resulting good tensile strength. The enhancement of tensile strength for the IPN in phase separated systems is a combined result of reinforcement by the glassy dispersed phase and increased physical entanglement and adhesion at phase boundaries due to interpenetration. The increasing concentration of PS, restrict the rotation of flexible moieties of PU as well as decreases the secondary interaction present in the PU network which reduces the flexibility of the IPNs. Thus IPN3 showed the highest tensile strength but least flexibility. Interestingly, the flexibility of the prepared IPNs (IPN1 and IPN2) is greater than the other reported IPNs.3,10 The values of toughness of the IPNs were represented in Table 2 as the area under stress–strain curve, follows the same trend.19 The impact resistance of the synthesized IPNs was found to be very good. Literature advocated that the PS showed impact strength of ∼0.1 kJ m−1. But the interpenetration of PS into PU network resulted significant increase in impact strength. In addition, the scratch hardness i.e. resistance to deformation under dynamic condition increases with the increase of PS content. The static hardness of the IPN strips as measured by Durometer (Shore A) also follows an increase in trend as scratch hardness. Similar results were also found for PU-PS based reported IPN.7
Thermal properties
Thermal stability of the IPN films was analyzed with the help of TGA. TG thermograms and their derivatives (DTG) are displayed in Fig. 3e and f. From these figures, it is clear that the synthesized IPNs underwent a three step thermal degradation pattern. From the DTG curves it is also found that the thermo stability of all the IPNs were more than 235 °C but a rapid weight loss at the range of 414–422 °C was observed. An initial degradation (onset) temperature (Ton) of the synthesized IPNs varied in the range of 237–245 °C (Table 3). This mass loss is due to the thermal degradation of most thermolabile moieties present in the structure of the IPNs such as long aliphatic chains of MG and DAGP. On the other hand 2nd step of degradation in the range of 350–355 °C was due to the presence of relatively more thermostable moieties like urethane and ester linkages, cycloaliphatic ring of DAGP etc. While, the 3rd step of degradation at 414–422 °C corresponds to the thermo stable moieties present in the structure of the IPN such as aromatic unit of polystyrene and TDI. In this step maximum degradation was noticed which indicates that before attaining this temperature, the weight loss of the IPNs was not very high. From the DTG thermograms it is clearly evident that with the increase of the PS content from IPN1 to IPN3, the peak intensity for maximum degradation increased. End set degradation temperature found in the range of 538–543 °C is due to the degradation of most thermally stable Si–O–Si bond.4 However, there was no significant difference between the molecular weight of IPNs, thus the difference in the values of Ton, TMAX and TEND is marginal.
Table 3 Thermal properties, retention of tensile strength after UV aging, self-healing times, contact angles, shape memory of the synthesized IPNs
| Parameter |
IPN1 |
IPN2 |
IPN3 |
| Ton (°C) |
237 |
243 |
245 |
| TMAX (°C) |
414 |
414 |
422 |
| TEND (°C) |
538 |
539 |
543 |
| Tg (°C) |
−44.1 |
−43 |
−42.1 |
| Tm (°C) |
29.2 |
35.3 |
24.8 |
| Crystallinity (%) |
34.6 |
31.1 |
28.6 |
| Tensile strength retention (%) |
94.5 |
93.3 |
89.56 |
| Self-healing time under MW (s) |
62 ± 4 |
78 ± 6 |
123 ± 8 |
| Self-healing time under direct sunlight (s) |
450 ± 8 |
570 ± 4 |
780 ± 4 |
| Advancing angle (°) |
116.1 |
117.4 |
120.7 |
| Receding angle (°) |
113.8 |
114.3 |
116.8 |
| Angle of hysteresis (°) |
2.3 |
3.1 |
3.9 |
| Shape fixity (%) |
98 |
100 |
100 |
| Shape recovery (%) |
100 |
100 |
100 |
| Shape recovery time under MW (s) |
48 ± 2 |
56 ± 4 |
72 ± 3 |
| Shape recovery time under sunlight (s) |
369 ± 5 |
412 ± 10 |
442 ± 10 |
DSC is important analytical tool which also emphasis on the phase separated morphology of the synthesized IPNs. From the DSC curves, the glass transition temperature (Tg) and melting temperature (Tm) for the soft segment of the synthesized IPNs were evaluated (Fig. 3g) and the values are given in Table 3. The Tg values of the IPNs varies in the range of −44.1 to −42.1 °C. The increasing amount PS from IPN1 to IPN3, the amount of free volume decreases which leads to increase in the Tg values. In the same way the Tm values for the IPNs also increased. These are due to the fact that PS is a rigid polymer with high Tg (100 °C) and Tm (165 °C) values. However at very high content of PS the amount of polyurethane is seemed to be insufficient to obtain homogeneous mixture in IPN3, which resulted inferior properties including low melting point of the soft segment of PU.
Self healing study
Multi stimuli responsive self-healing ability signifies the durability, i.e. longer life span without high costs of active monitoring or external repair and thus to provide extended safety of the polymeric material for a broad range of applications. Optical images of the IPN strips and healing efficiency are manifested in Fig. 4a–j, which clearly evident that the IPN strips were effectively healed under different stimuli. Self-healing of IPN matrix strongly affected by ratio of PU and PS, molecular weight, flexibility, Tg of the soft segment, Tm, and percent crystalline of the matrix.20 IPN contains flexible PDMS and PCL moiety as the components of the synthesized PU. Presence of such moieties resulted high free volume in the polymeric matrix. This leads to decrease in Tg. DSC study also revealed that the synthesized IPNs exhibited Tg near −44 to 44.1 °C. Thus upon exposure of stimulus, some polar groups absorb the energy and started to oscillate their dipoles. Heat generated from the molecular vibration is greater than the Tg and Tm of the soft segments. Thus a rapid Brownian motion occurred in the soft segment of the IPN matrix resulting easy diffusion of the polymeric chain in the cracked surface. Inter diffusion of polymeric chain in the cracked surface re-entangled with each other resulting quick healing of the cracked surface.21
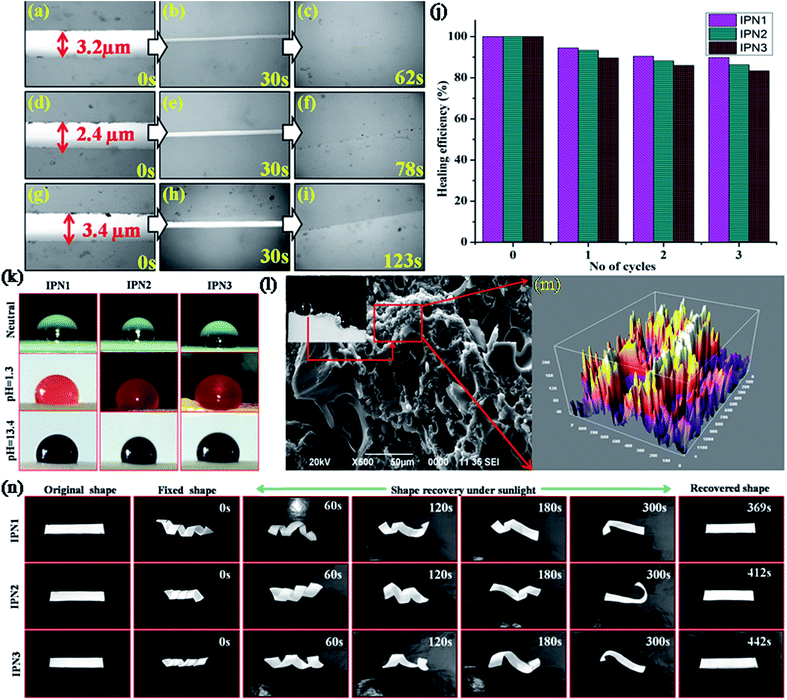 |
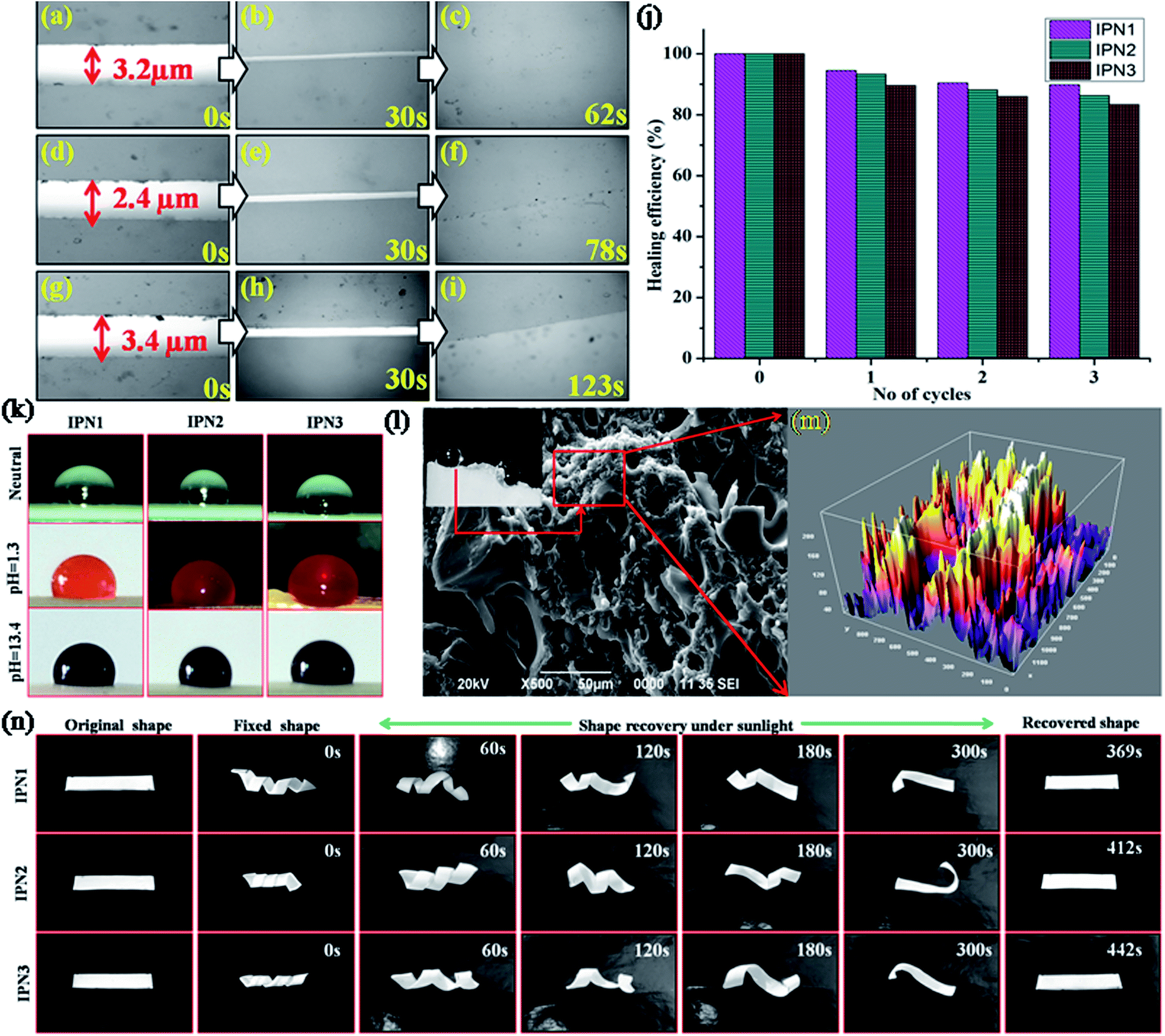
| | Fig. 4 (a) Optical images of mechanically damaged films; IPN1 (a, b and c after exposure of 0, 30 and 62 s of microwave radiation 450 W), IPN2 (d, e and f after exposure of 0, 30 and 78 s of microwave radiation 450 W), IPN3 (g, h and i after exposure of 0, 30 and 123 s of microwave radiation 450 W), (j) healing efficiency (%) of the IPNs for repeated cycles under microwave, (k) photos of water droplets at different pH, (l) water droplets at crack surface, SEM image of the crack surface of IPN3 (m) micro-roughness of crack surface captured by SEM and image is processed by ImageJ2 software and (n) shape memory behavior of IPNs under direct sunlight. | |
From self-healing study it was found that crystallinity plays a significant role in healing process. As IPN1 showed the highest degree of crystalline region compared to IPN2 and IPN3 thus IPN1 undergoes fastest healing compare to IPN2 and IPN3. In addition increasing percentage of PS inhibits the flow ability of the soft segments in the IPN matrix. Thus time requirement is higher as well as the healing efficiency is lower for IPN3 compared to IPN1 and IPN2.
Self-cleaning study
Self-cleaning property in the synthesized IPNs was achieved by creating hydrophobic surface. Surface chemical composition (intrinsic hydrophobicity) with low surface energy materials and the surface geometrical microstructures (roughness) are the governing factors for hydrophobic surface.22,23 Difference between the advancing angle and receding angle provides the angle of hysteresis of the water droplets and reported in Table 3.
The values of advancing angle, receding angle and the angle of hysteresis under different pH were found in the range of 116.14–120.78°, 113.8–116.8° and 2.3–3.9° respectively, which indicate a non-wetting surface of the synthesized IPNs and their applicability under all atmospheric conditions. Most of the compounds such as long aliphatic hydrocarbon chains of DAGP, MG and aromatic and aliphatic parts of diisocyanates in the structure of the IPNs are non-polar in nature which provided the intrinsic hydrophobic nature. In addition non polar nature of PS and low surface energy of the PDMS also responsible for enhancing the hydrophobicity reflected in the series of digital images of the contact angle provided in Fig. 4k. As IPN3 possesses the highest advancing angle among the studied IPNs, so the IPN3 without PDMS was synthesized to understand the role of the PDMS in hydrophobicity. From this study, a significant decrease in the contact angle (advancing angle: 102.6°) was observed as displayed in Fig. S3 of ESI.† Absence of PDMS moiety in IPN is the main reason for this decrease in contact angle. Again crystalline incompatibility of PCL and PDMS moiety in a same polymeric backbone along with interpenetration of hydrophobic PS into PU network created micro roughness which entrapped much more air on the polymer surface. This entrapped air under the water droplet act as cushion to maintain the round shape of the droplet.18 Most interestingly hydrophobic nature of the IPNs in the cracked surface increased drastically (contact angle 138–142°) due to increase in microroughness of the surface as supported by the SEM images of the cracked surface (Fig. 4l).24 Roughness of the surface was further verified by the processed SEM images of the crack surface displayed in Fig. 4m. From the SEM images displayed in Fig. S2 of ESI† of the IPNs it was confirmed that roughness of the surface increases from IPN1 to IPN3 due to increase in PS content as well as increase in incompatibility in IPN matrix due to interpenetration of the polymers.
Shape memory study
Traditional thermal induced shape memory behavior depends on the following factors such as hard to soft segment ratio and separation of hard and soft domains in a single polymeric network.21 The shape memory effect is assessed by two important parameters, shape fixity (%) and shape recovery (%). In the synthesized IPN; PCL and PDMS together act as the soft segment, whereas aromatic and aliphatic diisocyanates, MG and PS together act as the hard segment. The hard segment acts as net-points, which determine the capability of materials to remain in a temporary shape, thus provides shape fixity, while the soft segment acts as the switch to recover its original shape from the temporary fixed shape.25 Typical optical images of the shape memory study are displayed in Fig. 4n and the time required for shape recovery under different stimuli are given in Table 3.
In order to explain the shape fixity behavior of the IPNs, we have to understand the entropic change during this process. Brownian motion occurred in between the soft segments of IPN by which the entropy of the system increased. Thus applying an external force, these IPN strips can be easily deformed to the spiral shape. By subsequent freezing the deformed strips at −15 ± 5 °C, the chain mobility restricted which decreased the entropy of the system, resulting in temporary fixing of the spiral shape of the IPN and the internal stress is stored in between the net points. In the same time physical and chemical cross-linking in the hard segment provide dimensional stability of the fixed shape. The hard segment content increases from IPN1 to IPN3, which leads to high fixity. Upon reheating of the strips on switching temperature, segmental mobility of the soft segments in IPN matrix increased which released the internal stress and helped the IPN strips to regain its original shape very quickly. Shape recovery time under thermal heating and MW (450 W) showed effective results compared to recovery under direct sunlight. Though the shape recovery was gained by the elastic strain stored in the soft segments thus IPN1 showed quick recovery as it contains highest amount of soft segment compared to IPN2 and IPN3.
Chemical resistance
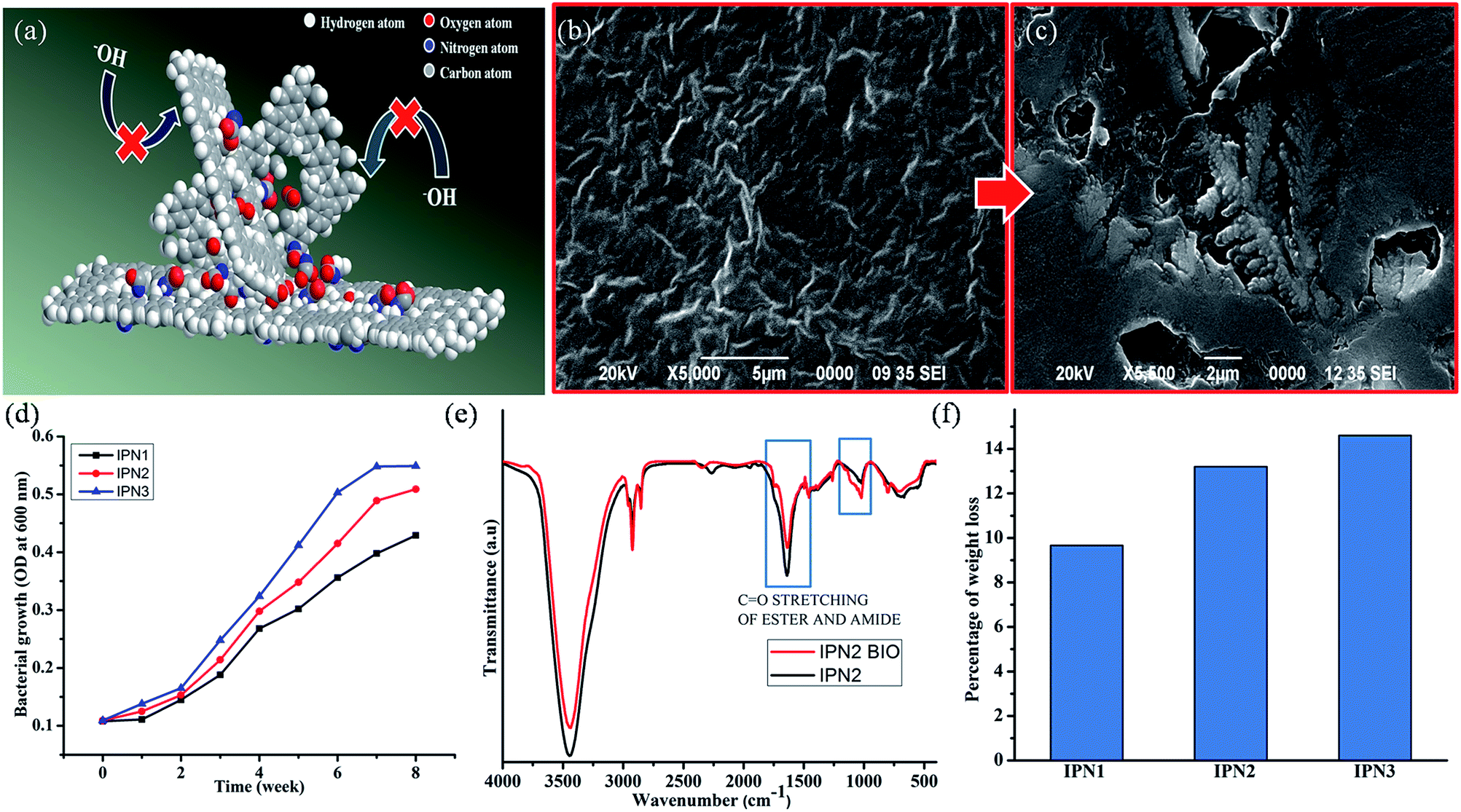
Chemical resistance of the IPN strips was performed to understand the durability and predictability of the films under different environmental conditions. As the natural weathering test was time consuming and produced bad reproducibility thus accelerated chemical resistance was performed. All the IPNs showed good chemical resistance under different chemical media such as aqueous 10% hydrochloric acid (v/v), 20% ethanol (v/v), 10% sodium chloride (w/v) and tap water; even in 1% aqueous sodium hydroxide (w/v) and the results are given in Table 4. Such good chemical resistance of IPN strips is attributed to the interpenetration of amorphous PS into PU, strong intra-/inter-molecular interactions such as hydrogen bonding, polar–polar interactions etc. In all the synthesized IPNs the hydrolysable ester bonds of PU were surrounded by amorphous PS which created a steric hindrance to the attacking group (e.g. H+, Cl−, −OH etc.), as displayed in cartoonish representation in Fig. 5a.
Table 4 Weight loss (%) of PU films in different chemical media
| IPN types |
1% NaOH |
10% HCl |
10% NaCl |
20% EtOH |
Tap water |
| IPN1 |
0.83 |
0.33 |
0.31 |
0.207 |
0 |
| IPN2 |
0.21 |
0.15 |
0.25 |
0.16 |
0 |
| IPN3 |
0.16 |
0.12 |
0.18 |
0.13 |
0 |
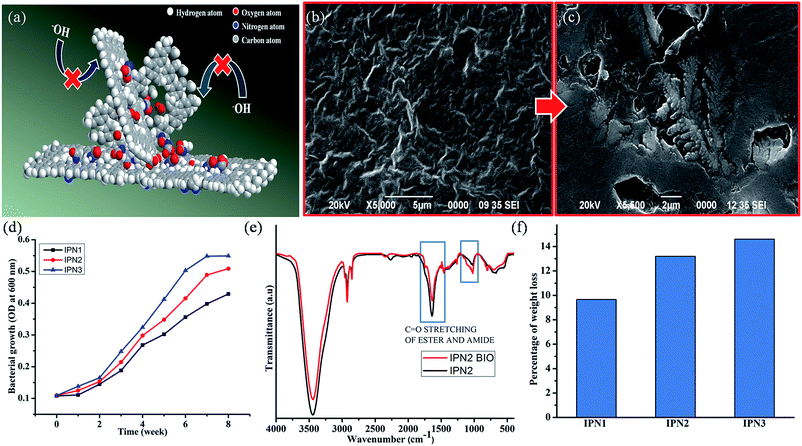 |
| | Fig. 5 (a) Cartoonish representation of alkali resistance property, (b and c) SEM images of the IPN3 surface before and after bio-degradation, (d) bacterial growth curve of P. aeruginosa on IPNs, (e) FTIR spectra of IPN2 before and after 8 weeks of bacterial exposure and (f) weight loss profiles of IPNs due to bacterial degradation. | |
Bio-degradation study
From the biodegradation study it was found that the IPN strips were degraded steadily with exposure of time by Gram negative P. aeruginosa bacterial strain as supported by the SEM images of the surfaces before and after biodegradation as well as plot of optical density (OD) against time of exposure (Fig. 4b–d). Rate of bio-degradation of the IPN strips also depends on several factors such as presence of bio-based units, hydrolyzable ester groups of MG and PCL, conformational flexibility of PDMS moiety and the ratio of hydrophilic/hydrophobic groups in the structure of the IPN.14,16 As the structure of the synthesized IPNs contain some amount of bio-degradable units like DAGP and MG they are prone towards bacterial degradation. From the literature survey it was also found that the degradation undergoes in a selective manner, amorphous region degraded prior to the crystalline region. As IPN3 contains highest amount of amorphous PS thus IPN3 showed the highest amount of bacterial growth shown in OD curve. In addition more crystalline region of IPN2 and IPN1 resulted less biodegradation. From the FT-IR spectra as displayed in Fig. 4e, it is also found that there is a decrease in intensity of the carbonyl group of ester moiety after 8 weeks of incubation under the exposure of the bacterial strain, which further supported the degradation of ester bonds of PU in IPN matrix. Thus about 9.66–14.6% of weight loss (Fig. 4f) of the IPN strips is confirmed by the bacterial degradation.
Conclusion
From this study, it can be concluded that a novel interpenetrating network of PU and PS with intrinsic multi stimuli responsive self-healing and shape memory attributes, was synthesized using simple pre-polymerization technique. FT-IR and NMR spectral analyses further support the structures of the IPNs. All the synthesized IPNs exhibited good mechanical properties, especially outstanding flexibility along with moderate bio-degradability, thermostability and chemical resistance. Most interestingly due to hydrophobic nature, IPNs showed self-cleaning behavior. Thus the above study solved the genuine problems like non bio-degradability, and brittleness character of the IPNs along with imparting some smart properties. Furthermore, to achieve high tensile strength along with outstanding flexibility in the polymeric material judicious choice of chemical components with their structures is very crucial. Thus the studied smart, bio-degradable IPNs with long service life span can hold significant promise as a sustainable material for its wide range of advance applications in automobile, footwear, textile, coating and bio-medical applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors express their gratitude to the research project assistance given by Department of Science and Technology (DST), India through the grant No. EMR/2016/001598, dated 04-Jan-2017.
References
- N. Karak, Fundamentals of Polymers-Raw Materials to Finish Products, PHI Private Ltd., New Delhi, India, 2009 Search PubMed.
- S. I. Hussain, M. M. Shahid, M. B. Sadique and A. Ali, International Journal of Scientific & Engineering Research, 2017, 8, 666–671 Search PubMed.
- V. J. Dave and H. S. Patel, J. Saudi Chem. Soc., 2017, 21, 18–24 CrossRef CAS.
- Siddaramaiah, P. Mallua and A. Varadarajulu, Polym. Degrad. Stab., 1999, 63, 305–309 CrossRef CAS.
- L. H. Sperling, Adv. Chem., 1994, 239, 3–38 CAS.
- J. R. Millar, J. Chem. Soc., 1960, 1311–1317 RSC.
- S. C. Kim, D. Klempner, K. E. Frisch, H. L. Frisch and H. Ghiradella, Polym. Eng. Sci., 1975, 15, 339–342 CAS.
- J. Zhang and N. A. Peppas, Macromolecules, 2000, 33, 102–107 CrossRef CAS.
- S. Vlad, A. Vlad and S. Oprea, Eur. Polym. J., 2002, 38, 829–835 CrossRef CAS.
- M. Begum and Siddaramaiah, J. Mater. Sci., 2004, 39, 4615–4623 CrossRef CAS.
- C. Tugui, S. Vlad, M. Iacob, C. D. Varganicia, L. Pricop and M. Cazacu, Polym. Chem., 2016, 7, 2709–2719 RSC.
- W. H. Ku, J. L. Liang, K. T. Wei and H. T. Liu, Macromolecules, 1991, 24, 4605–4610 CrossRef CAS.
- N. Karak, Biobased Smart Polyurethane Nanocomposites: From Synthesis to Applications, The Royal Society of Chemistry, Cambridge, 2017 Search PubMed.
- T. Ghosh and N. Karak, ACS Sustainable Chem. Eng., 2018, 6, 4370–4381 CrossRef CAS.
- H. Kalita and N. Karak, J. Appl. Polym. Sci., 2014, 131, 1–8 CrossRef.
- S. Gogoi and N. Karak, ACS Sustainable Chem. Eng., 2014, 2, 2730–2738 CrossRef CAS.
- W. Ningjing, H. Likan and Z. Anna, Front. Chem. China, 2006, 3, 350–356 Search PubMed.
- M. V. Pergal, V. V. Antic, M. N. Govedarica, D. Goaevac, S. Ostojic and J. Djonlagic, J. Appl. Polym. Sci., 2011, 122, 2715–2730 CrossRef CAS.
- M. Wang, K. P. Pramoda and S. H. Goh, Chem. Mater., 2004, 16, 3452–3456 CrossRef CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- H. U. Rehman, Y. Chen, M. S. Hedenqvist, H. Li, W. Xue, Y. Guo, Y. Guo, H. Duan and H. Liu, Adv. Funct. Mater., 2017, 1704109 Search PubMed.
- P. Ragesh, V. A. Ganesh, S. V. Nair and A. S. Nair, J. Mater. Chem. A, 2014, 2, 14773–14797 CAS.
- H. Zhou, H. Wang, H. Niu, Y. Zhao, Z. Xu and T. Lin, Adv. Funct. Mater., 2017, 1604261 CrossRef.
- S. Ramakrishna, K. S. S. Kumar, D. Mathew and C. P. R. Nair, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- Y. Wu, J. Hu, J. Han, Y. Zhu, H. Huang, J. Li and B. Tang, J. Mater. Chem. A, 2014, 44, 18816–18822 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR spectra of DAGP, PCL, PDMS, styrene and prepolymer of PS, SEM images and surface 3D plot of the IPNs, digital images of water droplets on PU without PDMS, PU with PDMS, IPN without PDMS and IPN with PDMS surface. See DOI: 10.1039/c8ra01766b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*