
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous Co-catalyzed direct 2-alkylation of azoles with ethers†
Ke Yang ,
Dashan Li,
Lei Zhang,
Qun Chen* and
Tiandi Tang*
,
Dashan Li,
Lei Zhang,
Qun Chen* and
Tiandi Tang*
Jiangsu Key Laboratory of Advanced Catalytic Materials & Technology, School of Petrochemical Engineering, Changzhou University, 1 Gehu Road, Changzhou, Jiangsu 213164, China. E-mail: tangtiandi@cczu.edu.cn; chenqunjpu@yahoo.com
First published on 12th April 2018
Abstract
The direct 2-alkylation of oxazoles and thiazoles with ethers through cross-dehydrogenative coupling reaction using Co-containing mesoporous zeolite ETS-10 as the heterogeneous catalyst is described. The basic Co-containing mesoporous zeolite ETS-10 catalyst facilitates this cross-dehydrogenative coupling reaction through metal-base synergy catalytic principle.
Transition metal-catalyzed C–H bond functionalization reaction has become one of the most efficient methods for selective C–C and C–X bond formation in synthetic chemistry.1 Current primarily transition metal-catalyzed C–H bond functionalization reactions rely on homogeneous metal catalysts, in most cases, which also need extra basic or acidic additives and catalyst ligands.2 Recently, considerable efforts have been devoted to heterogeneous catalysis,3 because heterogeneous transition metal catalysts not only provide easy product-catalyst separation and catalyst recycling features, but also afford additional important active sites through support materials in C–H bond functionalization. It is noteworthy that zeolites are crystalline porous support materials with good chemical and thermal stability. Especially, the unique frameworks of zeolites present basic or acidic properties in catalytic performance, which have been widely used in the organic transformations.4 Therefore, developing novel heterogeneous metal catalysts by using zeolites would be of prime synthetic value.
2-Substituted azoles are widely present in biological activities compounds, pharmaceuticals and organic functional materials.5 Most current methods for accessing 2-alkylazoles depend on the transition metal-catalyzed direct C-2-alkylation of azoles with alkylating reagents under basic conditions.6 However, the convenient and straightforward approach using non-functionalized ethers as alkylating reagents through cross-dehydrogenative coupling is still rare.7 Very recently, Lu and Li group also demonstrated the cobalt-catalyzed cross-dehydrogenative coupling reaction of oxazoles and ethers.7a It still suffered from certain limitations such as the restricted use of relatively harsh reaction conditions. One possible reason is that the cobalt catalyst is incompatible with base. For this reason, we believe that exploring the suitable bases could benefit this reaction.
Mesoporous zeolite ETS-10 has a three-dimensional 12-membered ring network and possesses relatively strong Lewis basicity. The basic characteristic is related to its framework that comprises corner sharing TiO62− octahedra and SiO4 tetrahedra as building units. In this manner, each TiO6 unit contributes two negative charges that are compensated by extra-framework cations (Na+ and K+).8 Considering the unique framework architectures and chemical compositions of zeolite ETS-10, we envisioned basic zeolite ETS-10 supported metal catalysts might facilitate this cross-dehydrogenative coupling reaction through metal-base synergy catalytic principle. Herein, we report the first example of Co-containing mesoporous zeolite ETS-10 catalyzed 2-alkylation of azoles with ethers through the cross-dehydrogenative coupling reaction.
We began our study with the preparation of 3 wt% Co-containing basic mesoporous zeolite ETS-10 (Co/METS-10). For comparison, 3 wt% Co-containing acidic mesoporous ZSM-5 (Co/MZSM-5) was also tested in this catalytic cycle. The cross-dehydrogenative coupling reaction of benzoxazole 1a and tetrahydrofuran 2a with Co/METS-10 was carried out in the presence of tert-butyl hydroperoxide (TBHP, 5.0–6.0 M in n-decane) at 100 °C under the N2 atmosphere, and the desired product 2-(tetrahydrofuran-2-yl)benzo[d]oxazole 3a could be obtained in 80% yield (Table 1, entry 1). Then acidic Co/MZSM-5 was examined in our reaction, however, this acidic heterogeneous catalyst Co/MZSM-5 was not compatible with our reaction system, indicating that the acidic support material MZSM-5 cannot promote this process (Table 1, entry 2). We also found that the yield of 3a was decreased using the reported CoCO3 catalyst under relatively mild conditions (Table 1, entry 3). Furthermore, basic mesoporous Cu/METS-10, Fe/METS-10 and Ni/METS-10 were also prepared to investigate in our catalytic cycle, and the results demonstrated that Co/METS-10 exhibited the higher catalytic efficiency, indicating that Co/METS-10 is the optimal catalyst (Table 1, entries 4–6). After that, replacement of TBHP with other additives, such as K2S2O8, 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ), di-tert-butyl peroxide (DTBP), dicumyl peroxide (DCP), benzoyl peroxide (BPO) and t-BuOOBz, caused either the low yield of desired product 3a or no reaction, suggesting that THBP is the optimal additives (Table 1, entries 7–12). In addition, no desired product 3a was detected in absence of additives (Table 1, entry 13). Finally, only 20% yield of 3a was examined when cobalt catalyst was absent in this process (Table 1, entry 14).
|
|||
|---|---|---|---|
| Entry | Catalyst | Additives | Yield (%)b |
| a Conditions: 1a (0.2 mmol), 2a (2.5 mL), 5 mol% metal in catalyst, additives (0.4 mmol), 100 °C, 24 h, N2, yield.b Yields are based on 1a, determined by crude 1H NMR using dibromomethane as the internal standard.c Isolated yield. | |||
| 1 | Co/METS-10 (5 mol%) | TBHP | 82 (80)c |
| 2 | Co/MZSM-5 (5 mol%) | TBHP | 35 |
| 3 | CoCO3 (5 mol%) | TBHP | 56 |
| 4 | Cu/METS-10 (5 mol%) | TBHP | 50 |
| 5 | Fe/METS-10 (5 mol%) | TBHP | 65 |
| 6 | Ni/METS-10 (5 mol%) | TBHP | 38 |
| 7 | Co/METS-10 (5 mol%) | K2S2O8 | 0 |
| 8 | Co/METS-10 (5 mol%) | DDQ | 0 |
| 9 | Co/METS-10 (5 mol%) | DTBP | Trace |
| 10 | Co/METS-10 (5 mol%) | DCP | 0 |
| 11 | Co/METS-10 (5 mol%) | BPO | 42 |
| 12 | Co/METS-10 (5 mol%) | t-BuOOBz | 60 |
| 13 | Co/METS-10 (5 mol%) | — | 0 |
| 14 | — | TBHP | 20 |
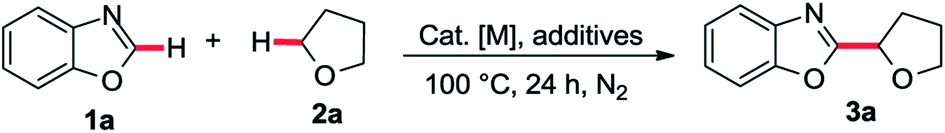
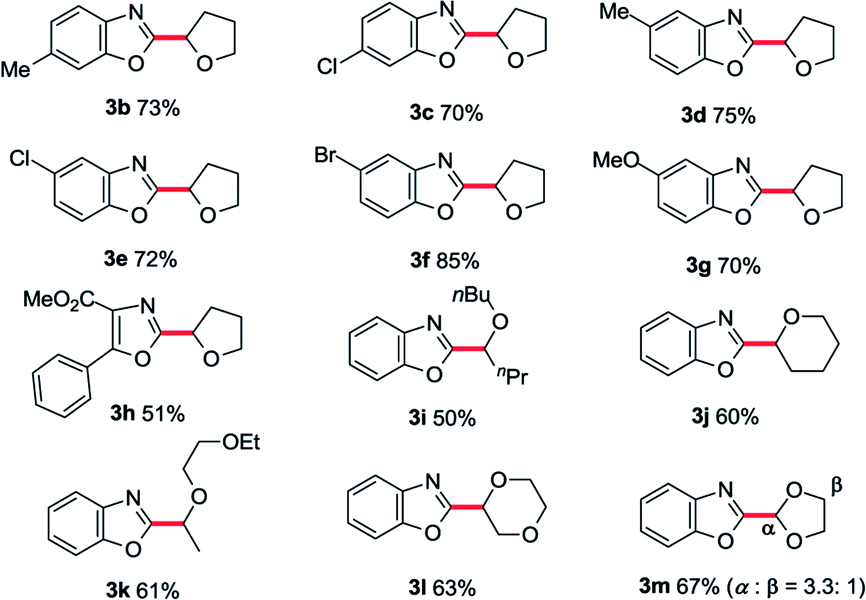
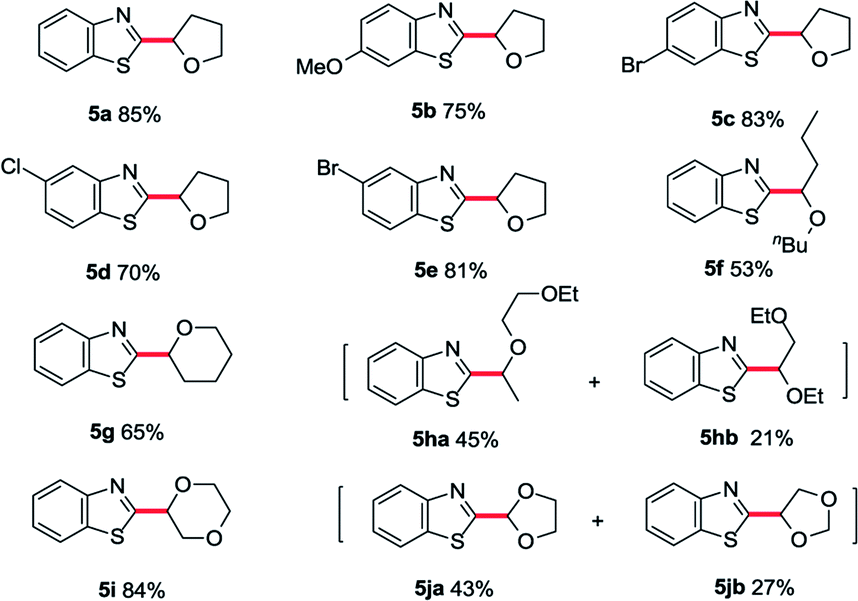
With the optimized conditions in hand, a substrate scope study of azole moiety was carried out firstly (Table 2). As we expected, the current catalytic system proceeded in good yields for the substituted benzoxazoles (3b–g). Both electron-donating and electron-withdrawing groups on the phenyl ring were tolerated in this catalytic cycle. Moreover, employing di-substituted oxazole 1h as the coupling partner, the desired product 3h was isolated in moderate yield. Besides, the scope study of aliphatic ethers was also tested in our cobalt catalysis system. Both linear aliphatic ethers and cyclic aliphatic ethers could be used to prepare the corresponding products in moderate to good yields (3i–m). Next, we sought the substrate scope study of thiazoles and aliphatic ethers in the presence of basic Co/METS-10 catalyst. As shown in Table 3, different substituted benzo[d]thiazoles generated the desired products in good yield (5a–e). Furthermore, different kinds of aliphatic ethers were also well tolerated in this catalytic cycle (5f–j).
|
|---|
| a Conditions: 1 (0.2 mmol), 2 (2.5 mL), 3 wt% Co/METS-10 (5 mol%, 20 mg), TBHP (0.4 mmol), 100 °C, 24 h, N2, Isolated yield. |
 |
|
|---|
| a Conditions: 4 (0.2 mmol), 2 (2.5 mL), 3 wt% Co/METS-10 (5 mol%, 20 mg), TBHP (0.4 mmol), 100 °C, 24 h, N2, Isolated yield. |
 |
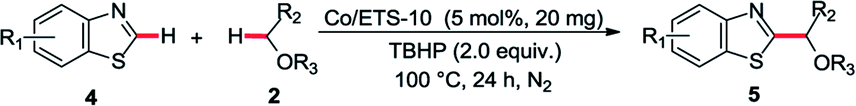
In order to provide some insights into the possible mechanism, a series of control experiments were carried out. The radical trapping experiment was firstly performed, which was found that the addition of TEMPO inhibited this reaction (Scheme 1a). It suggests that this reaction may be involving a radical pathway. Next, the investigations of TBHP and tetrahydrofuran with or without Co/METS-10 catalyst using TEMPO as a radical trapping reagent were also performed (Scheme 1b). In the presence of Co/ETS-10, the alkyl radical trapping product 6 could be isolated in 89% yield when 2.0 equivalent of TBHP was added. It suggests that Co/METS-10 catalyst may play an important role in the process of alkyl radical generation.9 Moreover, some intermolecular H/D exchange of azole (9% D) was observed when additional D2O was added in reaction system, suggesting a metal-mediated C–H bond cleavage process is involved (see Scheme S1 in ESI†). Finally, the intermolecular competition experiment was also carried out, indicating that electron density in phenyl rings of azoles does not affect the reaction rate (Scheme 2).
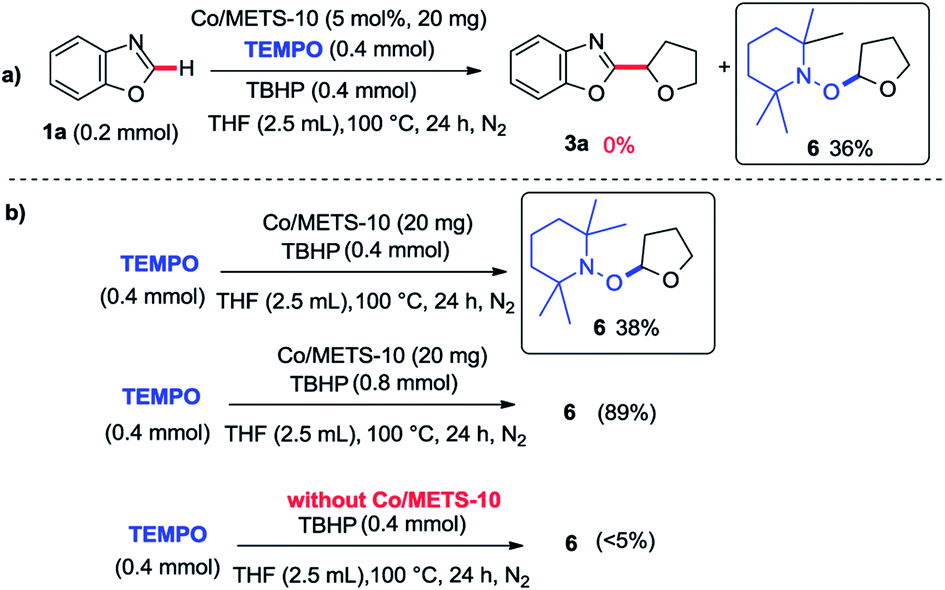 | ||
| Scheme 1 The radical trapping experiments. | ||
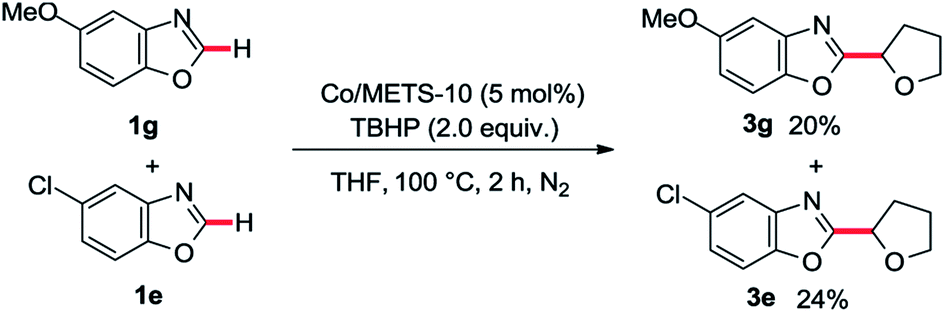 | ||
| Scheme 2 The intermolecular competition experiment. | ||
To gain further insight into this reaction mechanism, the electron state and the location as well as the dispersion of the Co species in the Co/METS-10 catalyst were analyzed by X-ray photoelectron spectroscopy (XPS) and the transmission electron microscopy (TEM) techniques, respectively (see Fig. S1–S3 in ESI†). The results indicate that the CoII species are highly dispersed in the micropore and mesoporous channels of the Co/METS-10 catalyst (the details are described in the ESI†).
On the basis of above results and the previous literatures,7a,9–11 the following plausible mechanistic pathway has been proposed in Scheme 3. It is believed that the Co(II)/METS-10 catalyst is transformed to the Co(III)/METS-10 in the presence of TBHP via a SET process with producing the corresponding t-BuO radical.10 Then alkyl radical B can be generated through t-BuO radical abstracts hydrogen atoms from 2a. The metallization of 1a in the presence of Co(III)/METS-10 provides the CoIII intermediate A, and subsequent oxidative addition of the radical B gives the CoIV intermediate C,11 leading to the desired product 3a and regeneration of Co(II)/METS-10 catalyst through reductive elimination.
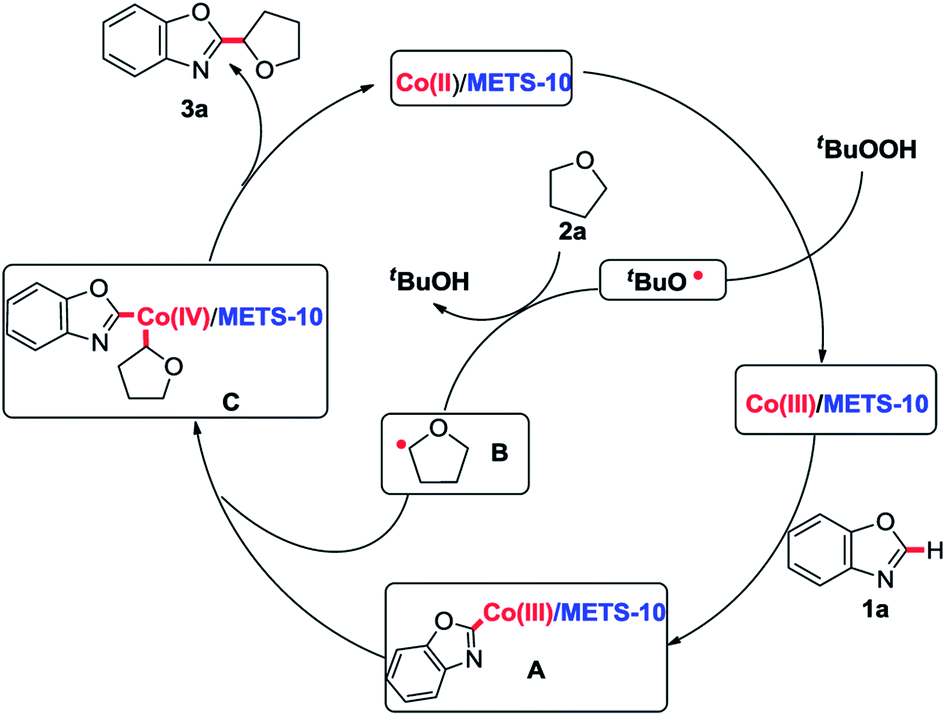 | ||
| Scheme 3 A plausible catalytic cycle. | ||
To probe the recyclability of the Co/METS-10 catalyst in this process, five round consecutive reactions of 1a with 2a were carried out under the recovered catalyst. We discovered that the catalytic activity of Co/METS-10 slightly decreased after five consecutive rounds with 75% NMR yield (Scheme 4).
 | ||
| Scheme 4 The Co/METS-10 catalyst recyclability experiments. | ||
Conclusions
In summary, we have developed an efficient basic Co-containing mesoporous zeolite ETS-10 catalyzed direct 2-alkylation of azoles with aliphatic ethers. This transformation is the first example of cross-dehydrogenative coupling reactions through a basic heterogeneous Co/METS-10 catalyst. This catalytic system tolerates a variety of functional groups with good yields. Furthermore, this novel method provides an important complementary approach to access the important bioactive 2-alkylated azoles derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (21676030, 21776022 and 21702019). We are also grateful to Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University for financial support.Notes and references
- For selected reviews, see: (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (b) Q. Lu and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 49 CrossRef CAS PubMed; (c) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed; (d) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed; (e) Q.-Z. Zheng and N. Jiao, Chem. Soc. Rev., 2016, 45, 4590 RSC; (f) H. Kim and S. Chang, ACS Catal., 2016, 6, 2341 CrossRef CAS; (g) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC; (h) J. Miao and H. Ge, Eur. J. Org. Chem., 2015, 2015, 7859 CrossRef CAS; (i) B. Liu, F. Hu and B.-F. Shi, ACS Catal., 2015, 5, 1863 CrossRef CAS; (j) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC; (k) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (l) S. D. Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef; (m) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (n) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (o) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (p) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed.
- For selected recent examples, see: (a) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. Yeung and J.-Q. Yu, Nature, 2017, 551, 489 CrossRef CAS PubMed; (b) Y. Liu and H. Ge, Nature Chem., 2017, 9, 26 CAS; (c) J. M. Ahn, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 18101 CrossRef CAS PubMed; (d) S. Asghar, S. B. Tailor, D. Elorriaga and R. B. Bedford, Angew. Chem., Int. Ed., 2017, 56, 16367 CrossRef CAS PubMed; (e) K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775 CrossRef CAS PubMed; (f) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed; (g) Y. Xu, M. Young, C. Wang, D. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084 CrossRef CAS PubMed.
- For selected recent reviews see: (a) S. Santoro, S. I. Kozhushkov, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 3471 RSC; (b) A. J. Reay and I. J. S. Fairlamb, Chem. Commun., 2015, 51, 16289 RSC ; For selected recent examples, see; (c) B. Paplal, S. Nagaraju, K. Sathish and D. Kashinath, Catal. Commun., 2018, 103, 110 CrossRef CAS; (d) S. Warratz, D. J. Burns, C. Zhu, K. Korvorapun, T. Rogge, J. Scholz, C. Jooss, D. Gelman and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1557 CrossRef CAS PubMed; (e) Q. Hu, X. Liu, G. Wang, F. Wang, Q. Li and W. Zhang, Chem.–Eur. J., 2017, 23, 17659 CrossRef CAS PubMed; (f) S. Vásquez-Céspedes, A. Ferry, L. Candish and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 5772 CrossRef PubMed; (g) C. Bai, X. Yao and Y. Li, ACS Catal., 2015, 5, 884 CrossRef CAS; (h) M. Cao, D. Wu, W. Su and R. Cao, J. Catal., 2015, 321, 62 CrossRef CAS.
- For selected recent examples, see: (a) L. Wang, G. Wang, J. Zhang, C. Bian, X. Meng and F.-S. Xiao, Nature Commun., 2017, 8, 15240 CrossRef PubMed; (b) J. Zhang, L. Wang, Y. Shao, Y. Wang, B. Gates and F.-S. Xiao, Angew. Chem., Int. Ed., 2017, 56, 9747 CrossRef CAS PubMed; (c) C. Wang, L. Wang, J. Zhang, H. Wang, J. P. Lewis and F.-S. Xiao, J. Am. Chem. Soc., 2016, 138, 7880 CrossRef CAS PubMed; (d) S. Chen, Z. Shao, Z. Fang, Q. Chen, T. Tang, W. Fu, L. Zhang and T. Tang, J. Catal., 2016, 338, 38 CrossRef CAS; (e) W. Fu, Y. Feng, Z. Fang, Q. Chen, T. Tang, Q. Yu and T. Tang, Chem. Commun., 2016, 52, 3115 RSC; (f) W. Fu, T. Liu, Z. Fang, Y. Ma, X. Zheng, W. Wang, X. Ni, M. Hu and T. Tang, Chem. Commun., 2015, 51, 5890 RSC.
- For selected examples, see: (a) J. V. Faria, P. F. Vegi, A. G. C. Miguita, M. S. dos Santos, N. Boechat and A. M. R. Bernardino, Bioorg. Med. Chem., 2017, 25, 5891 CrossRef CAS PubMed; (b) H. S. Kumbhar, S. S. Deshpande and G. S. Shankarling, Dyes Pigm., 2016, 127, 161 CrossRef CAS; (c) M. Kim, J. Jeon, J. Song, K. H. Suh, Y. H. Kim, K. H. Min and K. O. Lee, Bioorg. Med. Chem. Lett., 2013, 23, 3140 CrossRef CAS PubMed; (d) S. R. Mandadapu, P. M. Weerawarna, M. R. Gunnam, K. R. Alliston, G. H. Lushington, Y. Kim, K.-O. Chang and W. C. Groutas, Bioorg. Med. Chem. Lett., 2012, 22, 4820 CrossRef CAS PubMed; (e) I. K. Mangion, B. D. Sherry, J. Yin and F. J. Fleitz, Org. Lett., 2012, 14, 3458 CrossRef CAS PubMed; (f) R. E. Martin, L. G. Green, W. Guba, N. Kratochwil and A. Christ, J. Med. Chem., 2007, 50, 6291 CrossRef CAS PubMed.
- For selected examples, see: (a) G. Tan, L. Zhang, X. Liao, Y. Shi, Y. Wu, Y. Yang and J. You, Org. Lett., 2017, 19, 4830 CrossRef CAS PubMed; (b) C. M. Filloux and T. Rovis, J. Am. Chem. Soc., 2015, 137, 508 CrossRef CAS PubMed; (c) X. Wu, C. Lei, G. Yue and J. S. Zhou, Angew. Chem., Int. Ed., 2015, 54, 9601 CrossRef CAS PubMed; (d) X. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J. Hierso and J. S. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13573 CrossRef CAS PubMed; (e) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 775 CrossRef CAS PubMed.
- (a) Y. Li, M. Wang, W. Fan, F. Qian, G. Li and H. Lu, J. Org. Chem., 2016, 81, 11743 CrossRef CAS PubMed; (b) A. Correa, B. Fiserb and E. Gómez-Bengoa, Chem. Commun., 2015, 51, 13365 RSC; (c) Z. Xie, Y. Cai, H. Hu, C. Lin, J. Jiang, Z. Chen, L. Wang and Y. Pan, Org. Lett., 2013, 15, 4600 CrossRef CAS PubMed; (d) T. He, L. Yu, L. Zhang, L. Wang and M. Wang, Org. Lett., 2011, 13, 5016 CrossRef CAS PubMed.
- (a) M. W. Anderson, O. Terasaki, T. Ohsuna, A. Philippou, S. P. MacKay, A. Ferreira, J. Rocha and S. Lidin, Nature, 1994, 367, 347 CrossRef CAS; (b) M. Xiang, X. Ni, X. Yi, A. Zheng, W. Wang, M. He, J. Xiong, T. Liu, Y. Ma, P. Zhu, X. Zheng and T. Tang, ChemCatChem, 2015, 7, 521 CrossRef CAS.
- (a) Y. Li, F. Qian, M. Wang, H. Lu and G. Li, Org. Lett., 2017, 19, 5589 CrossRef CAS PubMed; (b) K. Yang, X. Chen, Y. Wang, W. Li, A. A. Kadi, H.-K. Fun, H. Sun, Y. Zhang, G. Li and H. Lu, J. Org. Chem., 2015, 80, 11065 CrossRef CAS PubMed.
- (a) J. Kim, S. Cho, J. Joseph and S. Chang, Angew. Chem., Int. Ed., 2010, 49, 9899 CrossRef CAS PubMed; (b) N. Turrà, U. Neuenschwander, A. Baiker, J. Peeters and I. Hermans, Chem.–Eur. J., 2010, 16, 13226 CrossRef PubMed.
- (a) X. Wu, K. Yang, Y. Zhao, H. Sun, G. Li and H. Ge, Nat. Commun., 2015, 6, 6462 CrossRef CAS PubMed; (b) J. Zhang, H. Chen, C. Lin, Z. X. Liu, C. Wang and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 12990 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01796d |
| This journal is © The Royal Society of Chemistry 2018 |