
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring local solvation environments of a heme protein using the spectroscopic reporter 4-cyano-L-phenylalanine†
Caroline Kearney‡
,
Lukasz T. Olenginski‡,
Trexler D. Hirn,
Gwendolyn D. Fowler,
Daniyal Tariq,
Scott H. Brewer* and
Christine M. Phillips-Piro *
*
Department of Chemistry, Franklin & Marshall College, PO Box 3003, Lancaster, PA 17604-3003, USA. E-mail: sbrewer@fandm.edu; cpiro@fandm.edu
First published on 10th April 2018
Abstract
The vibrational reporter unnatural amino acid (UAA) 4-cyano-L-phenylalanine (pCNF) was genetically incorporated individually at three sites (5, 36, and 78) in the heme protein Caldanaerobacter subterraneus H-NOX to probe local hydration environments. The UAA pCNF was incorporated site-specifically using an engineered, orthogonal tRNA synthetase in E. coli. The ability of all of the pCNF-containing H-NOX proteins to form the ferrous CO, NO, or O2 ligated and unligated states was confirmed with UV-Vis spectroscopy. The solvation state at each site of the three sites of pCNF incorporation was assessed using temperature-dependent infrared spectroscopy. Specifically, the frequency–temperature line slope (FTLS) method was utilized to show that the nitrile group at site 36 was fully solvated and the nitrile group at site 78 was de-solvated (buried) in the heme pocket. The nitrile group at site 5 was found to be partially solvated suggesting that the nitrile group was involved in moderate strength hydrogen bonds. These results were confirmed by the determination of the X-ray crystal structure of the H-NOX protein construct containing pCNF at site 5.
Introduction
Site-specifically incorporated vibrational reporter unnatural amino acids (UAAs) provide an effective approach to examine local environments in peptides and proteins in solution.1–29 The vibrational reporter UAA 4-cyano-L-phenylalanine (pCNF, Fig. 1) has been genetically incorporated in numerous systems, such as myoglobin,2 N-terminal domain of the L9 protein (NTL9),6 cytochrome c,29 villin headpiece subdomain (HP35),30 superfolder green fluorescent protein (sfGFP),16,27 and N-terminal Src homology 3 domain of the murine adaptor protein Crk-II (nSH3)31 to probe local solvation environments of the nitrile group. This UAA is an effective probe of local solvation environments due, in part, to the sensitivity of the nitrile symmetric stretch frequency to local environment – especially hydrogen bonding interactions, the location of this vibration in a region devoid of vibrations from naturally occurring proteins, and the relatively strong excitation coefficient of this vibrational mode. This UAA can also be efficiently incorporated site-specifically into proteins utilizing the amber codon suppression methodology. The incorporation of the nitrile group into proteins results in minimal structural perturbations due to the small size and intermediate polarity of the nitrile group. For example, studies of pCNF genetically incorporated in either the sfGFP or the NTL9 protein illustrate that pCNF induces minimal structural perturbations of the local protein environment.6,27 Additionally, we recently used X-ray crystallography and solution infrared (IR) spectroscopy in concert to show that genetically incorporated pCNF reported on the local solvation environment at two sites in the model system sfGFP, supporting the use of pCNF as an effective vibrational reporter UAA.27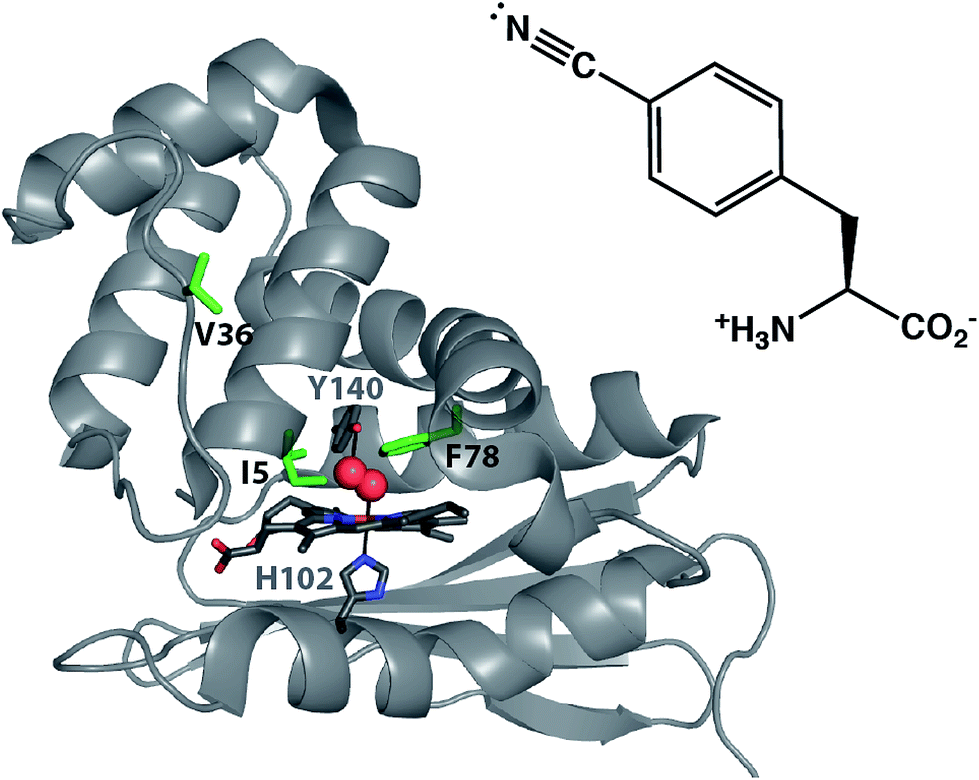 | ||
| Fig. 1 X-ray crystal structure of O2 bound Cs H-NOX (PDB ID 1U55). The distal pocket tyrosine stabilizing O2 binding (Y140) and the proximal histidine ligating the iron in the heme (H102) are shown in sticks. The three residues highlighted in green indicate the sites of pCNF incorporation in this study. The structure of pCNF is shown on the right. | ||
In this study, we examine the solvation environment in multiple sites, including the heme pocket of the biologically relevant heme nitric oxide and/or oxygen (H-NOX) protein from the thermophilic anaerobe Caldanaerobacter subterraneus (Cs H-NOX, formerly Thermoanaerobacter tengcongensis).32 The H-NOX proteins are an important class of gas binding heme proteins that include the mammalian nitric oxide-sensing protein soluble guanylate cyclase (sGC).33,34 Studies of H-NOX proteins illustrate that these proteins fall into two classes: the O2-binding H-NOXes and the non-O2-binding H-NOXes.35,36 Cs H-NOX is a member of the former class. Like other O2-binding H-NOX homologs, Cs H-NOX has a distal pocket tyrosine residue that acts as a hydrogen bond donor to stabilize O2 binding to the heme (Fig. 1, Y140).37 While O2 is thought to be the biologically relevant ligand for Cs H-NOX, some studies have illustrated that the ferrous form of this protein also binds NO and CO gasses, and the ferric heme binds water.34,37,38 The examination of local environments in Cs H-NOX for the ferrous CO, NO, O2 ligated and unligated states will be the focus of the current study.
Despite the studies that show that O2, CO, NO, and water can all access the heme pocket, X-ray crystal structures of wild-type (WT) Cs H-NOX indicate that the heme is considerably obscured at the center of the protein.37,38 There is no obvious tunnel from the solvent to the heme pocket present in any of the structures of Cs H-NOX and it is proposed that the gases are able to access the heme during conformational fluctuations of the protein.38,39 This study aims to understand the solvent accessibility of the H-NOX heme pocket compared to the surface of the protein in solution using the genetically incorporated pCNF as a reporter of local solvation environment. Here, we incorporated pCNF at three unique sites in a heme protein: one surface site (V36) and two sites near the heme (I5 and F78) (Fig. 1), to study the solvation environments at these locations under various gas ligation states. Temperature-dependent IR spectroscopy coupled with the frequency–temperature line slope (FTLS) method was effectively used to assess the relative solvation state of these three sites. The X-ray crystal structure of ferric hydroxy Cs H-NOX with pCNF incorporated at site 5 (I5pCNF) confirms that pCNF incorporation at this site does not perturb the protein structure. The structure also draws important connections with the FTIR results.
Experimental
Mutagenesis to create Cs H-NOX amber (TAG) mutants
C-terminal histidine (His6)-tagged WT Cs H-NOX in the pBAD expression vector was obtained from Michael Marletta, UC Berkeley. Site-directed mutagenesis was used to insert the amber TAG codon at the following sites – I5, V36, and F78 – in the Cs H-NOX construct using QuikChange®.Cs H-NOX expression
Cs H-NOX in pBAD vector and pDULE-pCNF vector containing the aminoacyl-tRNA synthetase for the incorporation of pCNF (obtained from Ryan Mehl, Oregon State University)40 were dual-transformed into chemically competent DH10B E. coli cells (Invitrogen). Cs H-NOX expressions were performed using the dual-transformed cells in 2 L baffled flasks containing 1 L of sterile media [45 g yeast extract, 10 mL glycerol, 900 mL 18 MΩ cm water and 100 mL of phosphate buffer (170 mM KH2PO4, 720 mM K2HPO4, pH 7.8)] with ampicillin and tetracycline (tetracycline excluded in WT Cs H-NOX expression) added to each expression culture. Expression cultures were inoculated with 10 mL of starter culture and grown up at 37 °C and 250 rpm. Once the OD600 reached 0.7–0.8 the incubation temperature was dropped to 18 °C and the cultures were induced with 2.5 mL of 20% arabinose. Then, pCNF (PepTech) and 5-aminolevulinic acid (ALA; RPI Corp) were added to a final concentration of 1 mM each one hour after induction (pCNF was not added to WT expression culture). Cultures grew at 18 °C and 250 rpm overnight and were spun down at 4000 rpm for 20 minutes. The cell pellets were collected, flash frozen in liquid nitrogen, and stored at −80 °C.Cs H-NOX purification
Cell pellets were thawed on ice, resuspended in 30 mL resuspension buffer (50 mM Tris, 300 mM NaCl, pH 7.8). Lysozyme (0.25 mg mL−1, Sigma-Aldrich), phenylmethylsulfonyl fluoride (0.5 mM, RPI), and DNAse (3.75 mM, Roche Diagnostics) were added to final concentrations indicated. The resuspended cells were sonicated for 2 minutes. Then the samples were heat-shocked at 70 °C for 30 minutes and spun down at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 45 minutes. The supernatant containing the protein was collected for further purification.
000 rpm for 45 minutes. The supernatant containing the protein was collected for further purification.
HisPur Ni-NTA resin (Thermo Scientific) (column volume of 2 mL for 1 L expression culture) was equilibrated with 20 column volumes of resuspension buffer. The supernatant was added to the equilibrated resin and batch-bound for approximately 30–40 minutes while nutating at 4 °C. After batch-binding, the flow-through was collected and 20 column volumes of resuspension buffer was added to the column. A second wash was completed with 10 column volumes of wash buffer (50 mM Tris, 300 mM NaCl, 30 mM imidazole, pH 7.8). Cs H-NOX was eluted with 5 column volumes of elution buffer (50 mM, Tris, 300 mM NaCl, 200 mM imidazole, pH 7.8). The protein was concentrated using a 5000 kDa membrane spin concentrator (Vivaspin, GE Healthcare) to between 4 and 17 mg mL−1 and buffer exchanged into the H-NOX storage buffer (50 mM TEA, 150 mM NaCl, 5% glycerol, pH 8.0) using a PD10 desalting column. Purified protein samples were flash frozen in liquid nitrogen and stored at −80 °C. ESI-Q-TOF and SDS-PAGE of WT and pCNF-incorporated Cs H-NOX confirmed the expression and purity of the protein (Fig. 2). The incorporation of pCNF individually at site 5, 36, and 78 yielded protein constructs I5pCNF (Cs H-NOX-5-pCNF), V36pCNF (Cs H-NOX-36-pCNF), and F78pCNF (Cs H-NOX-78-pCNF), respectively.
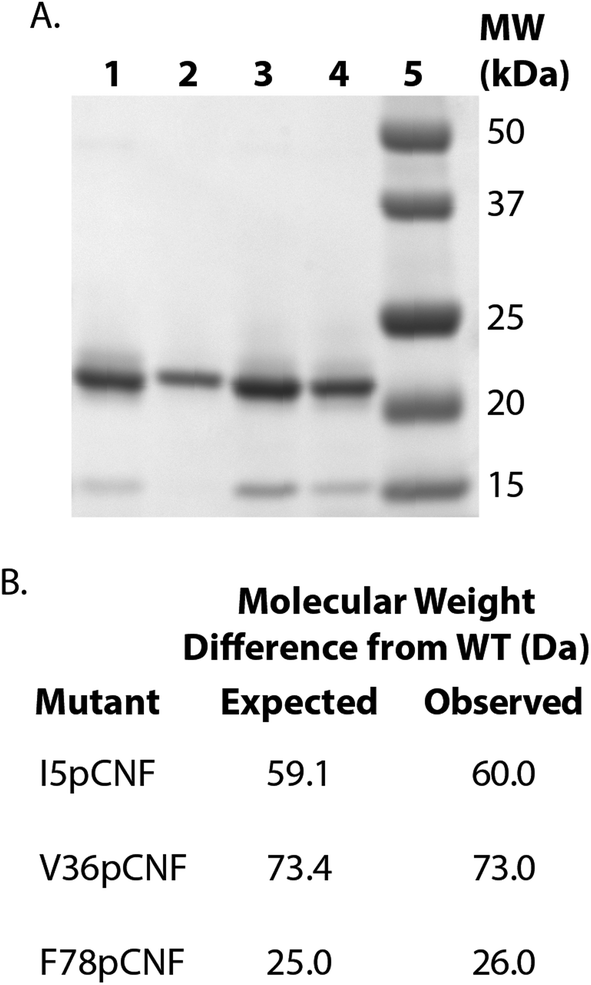 | ||
| Fig. 2 SDS-PAGE analysis of pCNF incorporated into H-NOX (A). The gel shows purified WT (lane 1), I5pCNF (lane 2), V36pCNF (lane 3), and F78pCNF (lane 4) while lane 5 contains molecular weight markers. ESI-Q-TOF mass spectrometry results for pCNF-incorporated H-NOX mutants (B). The table reports the calculated and observed molecular weight differences between each pCNF-incorporated mutant and the WT H-NOX protein. | ||
Preparation of mass spectrometry samples
Thawed protein samples were desalted into 20 mM ammonium acetate using PD10 gel filtration columns. The samples were then flash frozen and lyophilized. Lyophilized protein samples were sent to University of Illinois Urbana-Champaign Mass Spectrometry Facility for ESI-Q-TOF mass analysis. Samples were resuspended in 1:1H2O:CH3OH with 0.2% formic acid prior to analysis.
Oxidation and reduction to form ferrous unligated Cs H-NOX constructs
Purified protein samples were thawed on ice and cycled into an anaerobic glovebag (Coy). The protein solutions were diluted with anaerobic, glycerol free H-NOX storage buffer (50 mM TEA, 150 mM NaCl, pH 8.0) to a final concentration of approximately 1–5 mg mL−1. A 500 mM potassium ferricyanide (Sigma-Aldrich) solution was added to a final concentration of 10 mM to oxidize the protein to the ferric hydroxy state. After 20 minutes, potassium ferricyanide was removed using a PD10 desalting column equilibrated with anaerobic, glycerol free H-NOX storage buffer. The column was re-equilibrated and the protein was desalted a second time. UV-Vis absorbance in the glovebag (Thermo Scientific NanoDrop 2000c) confirmed oxidation and complete removal of potassium ferricyanide. A 500 mM sodium dithionite (Sigma-Aldrich) solution was added to a final concentration of 5 mM to reduce the protein to the ferrous unligated state. After 15 minutes, sodium dithionite was removed in the same manner as the potassium ferricyanide. UV-Vis absorbance confirmed reduction.Preparation of CO, NO, and O2 bound Cs H-NOX constructs
Glycerol free H-NOX storage buffer was bubbled with CO or O2 gas (Praxair) for 20 minutes and cycled into the anaerobic glovebag. To generate the CO and O2 bound forms of the protein, ferrous unligated Cs H-NOX protein was diluted approximately 20-fold in either CO or O2 equilibrated buffer, respectively. The protein was concentrated using a 5000 kDa membrane spin concentrator and then re-diluted with the CO or O2 equilibrated buffer, respectively. The NO bound form was generated similarly except the protein was dissolved in a buffer containing DEA-NONOate (Cayman Chemical) to produce NO in situ. UV-Vis spectroscopy confirmed the CO, NO, or O2 bound forms of the protein.Infrared spectroscopy of Cs H-NOX pCNF constructs
Equilibrium FTIR absorbance spectra were recorded on a Bruker Vertex 70 FTIR spectrometer equipped with a globar source, KBr beamsplitter, and a liquid-nitrogen-cooled mercury cadmium telluride (MCT) detector. The spectra were the result of (multiple) 512 scans recorded at 1.0 cm−1 resolution using a temperature-controlled transmission cell consisting of calcium fluoride windows with a path length of approximately 50 μm. The temperature was measured using an embedded thermocouple in the transmission cell. Protein samples had a concentration of 0.5–1.5 mM in storage buffer. The intensity normalized and baseline corrected absorbance spectra were fit to a line shape function that consisted of a linear combination of a Gaussian and Lorentzian function41 to ascertain the central frequency of the corresponding IR absorbance band in Igor Pro (Wavemetrics).Ultraviolet-visible spectroscopy of Cs H-NOX constructs
UV-Vis spectra (Fig. 3 and S3†) were recorded with an Agilent Technologies Cary 100 UV-Vis spectrophotometer using sealed quartz cuvettes (Schott) scanning from 250 to 750 nm with a resolution of 0.5 nm and normalized based upon the absorbance of the Soret band in Igor Pro.Purification of untagged Cs H-NOX I5pCNF for crystallization
Following amber TAG codon insertion as described above, a TEV protease cleavage site, ENLYFQG, was inserted at the C-terminus before the His6-tag of the Cs H-NOX I5pCNF construct using Q5® site-directed mutagenesis. Cs H-NOX I5pCNF with a TEV protease cleavage site was expressed and purified as described above. Purified protein was buffer exchanged into a standard TEV reaction buffer (50 mM Tris–HCl, 0.5 mM EDTA, 1 mM DTT, pH 8.0)42 using a PD10 desalting column. A catalytic amount of the TEV protease (obtained from Yan Kung, Bryn Mawr College) was added to the purified protein and the reaction incubated overnight at 30 °C and 25 rpm. A 7 mL column volume of HisPur Ni-NTA resin (Thermo Scientific) was equilibrated with 20 column volumes of resuspension buffer (50 mM Tris, 300 mM NaCl, pH 7.8). The equilibrated resin was added to the protein, diluted with about 30 mL of resuspension buffer, and batch-bound for approximately 30 minutes while nutating at room temperature. After batch-binding, the non-His6-tagged Cs H-NOX I5pCNF was collected in the flow-through.Cs H-NOX I5pCNF protein crystallography
As-purified untagged Cs H-NOX I5pCNF protein was buffer exchanged into 20 mM TEA (pH 7.5) using a PD10 desalting column and concentrated to 25 mg mL−1 using a 5000 kDa membrane spin concentrator at 4000 rpm. The protein was crystallized via sitting drop vapor diffusion by mixing 1 μL of protein with 1 μL of precipitant solution (Hampton Index Condition 80:0.2 M ammonium acetate, 0.1 M HEPES, pH 7.5, 25% PEG 3350) and equilibrating the drop against 99 μL of precipitant solution. Red crystals formed within 4 days, were mounted on loops, and then flash frozen in liquid nitrogen for storage.
X-ray diffraction data were collected at the Advanced Photon Source (APS) at Argonne National Laboratory (ANL) on NE-CAT 24-ID-E beamline (λ = 0.979180 Å). Crystals diffracted to 1.85 Å and data were processed in HKL2000 (ref. 43) in space group P21. The protein crystallized with two protein monomers in the asymmetric unit. The protein component (heme and water molecules removed) of the WT structure (PDB ID 1U55) with I5 mutated to an alanine was used for molecular replacement in PHASER.44 The resulting structure was refined in Phenix45 against 1.85 Å data with alternate cycles of positional and atomic displacement parameter refinement with manual refitting in Coot.46 Following a round of refinement with alanine modeled at site 5, pCNF was modeled into the difference density at this site (Fig. S1†). The final model contains 2 chains with 185 residues in chain A and 187 residues in chain B. Two heme molecules and 197 water molecules were also modeled to produce the final refinement parameters of Rwork of 19.8% and Rfree of 25.8%.
Results and discussion
Successful incorporation of pCNF in H-NOX
H-NOX proteins with pCNF site-specifically incorporated were expressed using the amber stop codon expression technology with an engineered, orthogonal aminoacyl-tRNA synthetase. The His6-tagged proteins were purified using affinity chromatography, assessed for purity by SDS-PAGE, and verified by ESI-Q-TOF mass spectrometry. Fig. 2 shows the SDS-PAGE illustrating the successful incorporation of pCNF individually in three sites of the protein (site 5, 36, or 78) when compared to the band position of the wild type (WT) protein and the molecular weight markers. Fig. 2 also shows the expected molecular weight differences resulting from pCNF incorporation compared to WT and measured mass differences from ESI-Q-TOF mass spectrometry. For example, the mass of isoleucine is 131 Da and the mass of pCNF is 190 Da, therefore the expected mass difference between WT and I5pCNF H-NOX proteins is 59 Da. Following mass spectral analysis of WT H-NOX and I5pCNF H-NOX individually, we calculated the an observed mass difference of 60 Da (Fig. 2B). The molecular weight differences for all of the pCNF-containing samples compared to WT were within experimental error (±2 Daltons) of their expected mass difference, confirming the site-specific incorporation of pCNF in each of our samples.Verification of heme incorporation in pCNF H-NOX constructs
The pCNF incorporated H-NOX proteins mark the first literature report of this heme protein with UAAs; thus, proteins were characterized by UV-Vis spectroscopy to assess heme incorporation and ligation state. Fig. 3 shows the electronic absorption spectra from 250–650 nm for ferrous carbon monoxide (CO) bound protein constructs of WT, I5pCNF, V36pCNF, and F78pCNF. The oxidation and subsequent reduction of the iron in the heme cofactor followed by exposure to CO gas to form this ligation state is detailed in the Experimental section. Each of the spectra contained a Soret band (∼424 nm) indicative of successful heme incorporation. Specifically, the position of the Soret band for WT, I5pCNF, V36pCNF, and F78pCNF was 424.0, 424.5, 424.0 and 424.5 nm, respectively. These positions are indicative of the ferrous CO bound form of the heme in each of these constructs based upon previous studies of Cs H-NOX with naturally occurring amino acids at various sites 5 and 78 that showed successful heme incorporation.47–49 The unligated, NO bound, and O2 bound forms of the protein were also successfully obtained for each pCNF construct (see Table 1, Fig. S2,† and discussion below).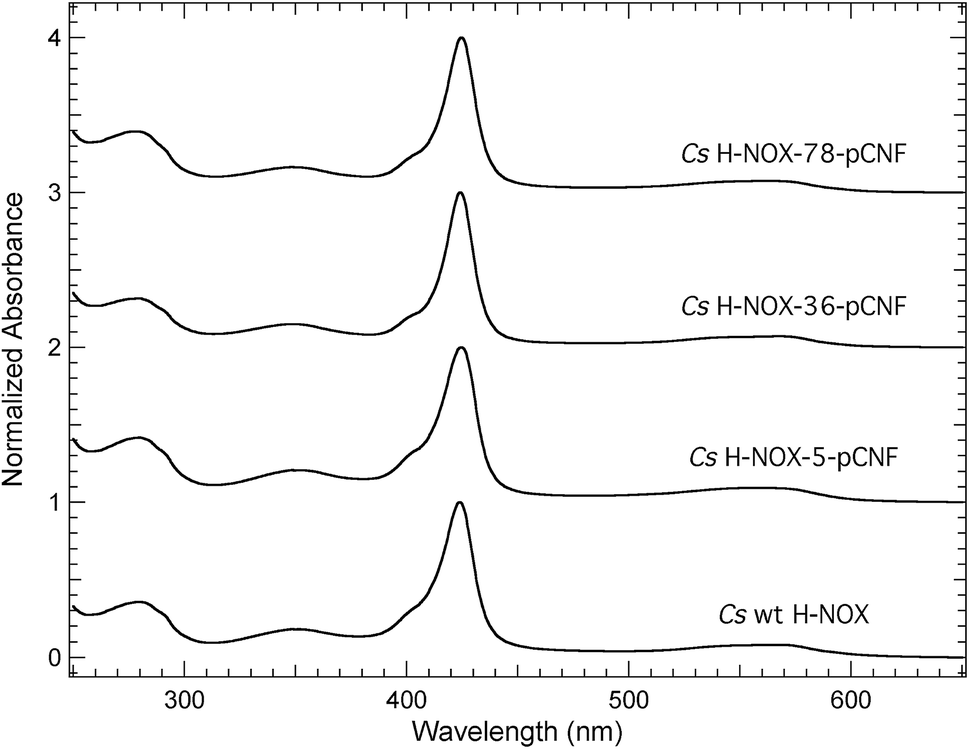 | ||
| Fig. 3 UV-Vis spectra (250–650 nm) of CO bound WT, I5pCNF, V36pCNF, and F78pCNF H-NOX constructs recorded in an aqueous pH 8.0 buffer solution consisting of 50 mM TEA and 150 mM NaCl at 25 °C. Spectra were intensity normalized to the Soret band maximum. | ||
| Soret Band Position (nm) | ||||
|---|---|---|---|---|
| Construct | CO | NO | O2 | Unligated |
| WT | 424.0 | 419.5 | 416.5 | 431.5 |
| I5pCNF | 424.5 | 420.0 | 414.5 | 431.5 |
| V36pCNF | 424.0 | 420.0 | 417.0 | 431.0 |
| F78pCNF | 424.5 | 420.0 | 416.0 | 433.5 |
The Soret band absorbance of the pCNF incorporated CO bound H-NOX proteins in Fig. 3 was greater than the protein absorbance at 280 nm in each case, consistent with near stoichiometric heme incorporation as described previously for H-NOX proteins.34 Earlier H-NOX studies illustrated that mutagenesis with naturally occurring amino acids at sites 5 and 78 did not lead to any significant structural perturbations as assessed by X-ray crystallography, suggesting that the mutations used herein are not significantly disruptive to the protein structure, which is an important result given the location of these residues in the protein interior.47–49 The X-ray crystal structure of the I5pCNF H-NOX construct obtained here is consistent with these earlier results and is discussed in detail below.
Probing local solvation states in H-NOX with pCNF
The frequency of the nitrile symmetric stretch of pCNF has been shown previously to be dependent upon local environment.1,16,27 Specifically, solution studies of the free UAA in water or DMSO have mimicked a solvated or buried site in a protein, respectively.27 These studies illustrate a red shift of the CN stretching frequency in pCNF from 2236.7 cm−1 in water to 2225.8 cm−1 in DMSO.27 These probes have also been used in a few proteins to report on the local solvation environment; for instance, at two sites in sfGFP,16,27 two sites in cytochrome C,29 HP35,13,30 nSH3,31 and two sites in NTL9 protein.6 Here, the solvation environments of three sites (sites 5, 36, 78) in H-NOX were studied using pCNF. X-ray crystal structure analysis of WT Cs H-NOX suggests that the nitrile group of pCNF would be in the protein interior near the heme pocket at sites 5 and 78, while site 36 would place pCNF on the surface.37,38 Fig. 4 shows the room-temperature FTIR spectra of each of these constructs. Each spectra shows a single, symmetric absorbance band in the region 2180–2280 cm−1 resulting from the nitrile symmetric stretch of pCNF incorporated into the protein. The position of the nitrile IR absorbance bands for I5pCNF, V36pCNF, and F78pCNF are at 2230.0, 2233.6, and 2230.7 cm−1 for the ferrous CO bound form of the proteins, respectively. The nitrile symmetric stretching frequency of V36pCNF is indicative of a solvated position in the protein given the similarity of this frequency to that of the free UAA in water and to observed frequencies at solvent accessible positions in other proteins.1,16,27 The assignment of the nitrile stretching frequency was verified based upon the similarity of the expected and experimentally observed isotopic shift between pCNF and 13C labeled pCNF (p13CNF) separately incorporated into the protein (see Fig. S3†). The nitrile stretching frequencies of I5pCNF and F78pCNF suggest that the nitrile group is in a relatively buried position in the protein, given the red shifted frequency from the V36pCNF frequency and a frequency closer to the frequency of pCNF in DMSO. Overall, these results suggest that the heme pocket around sites 5 and 78 represent a relatively buried region of the protein, consistent with the X-ray crystal structure of I5pCNF determined here (see below, Fig. 6).37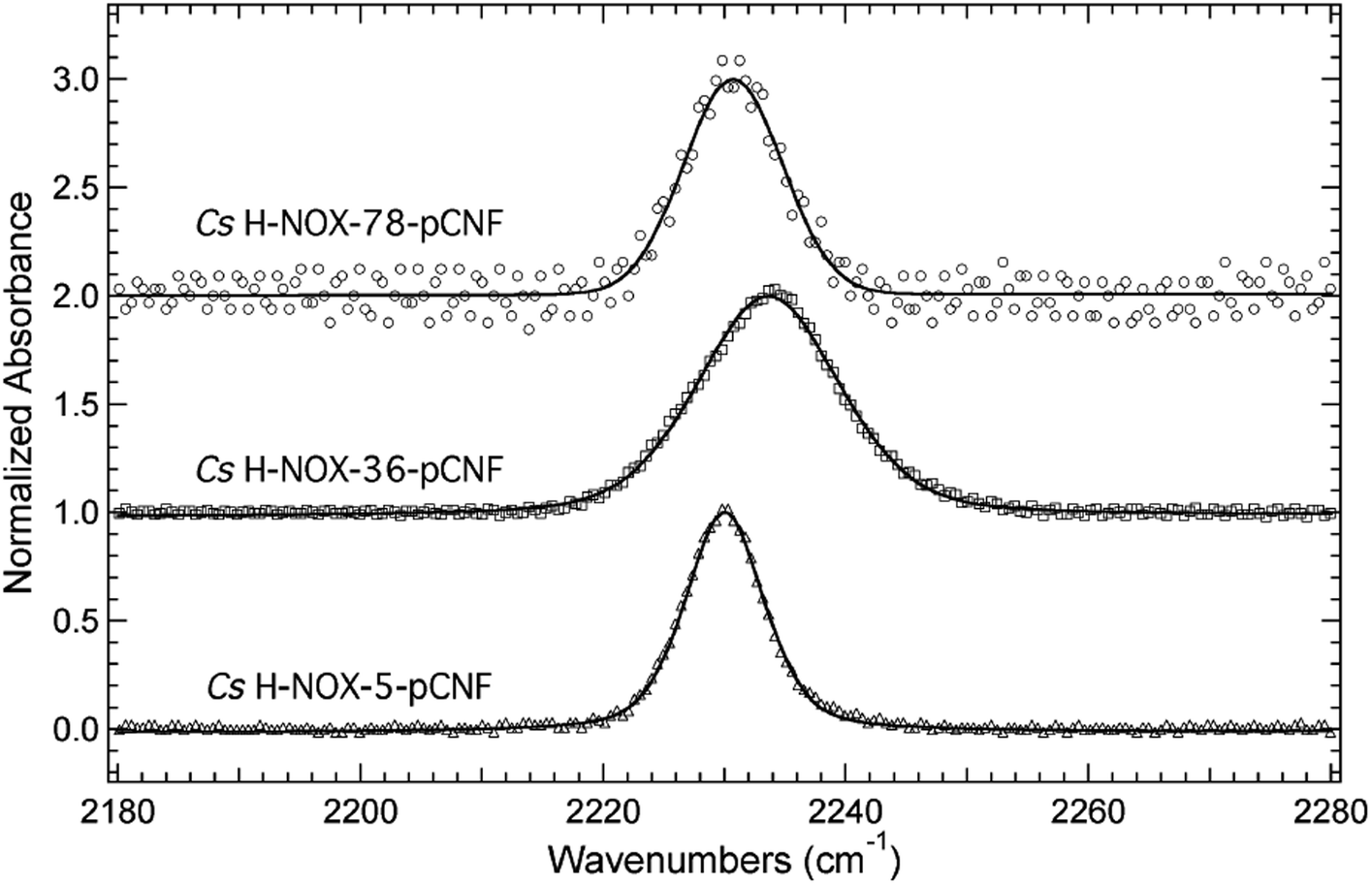 | ||
| Fig. 4 FTIR spectra of CO bound H-NOX proteins I5pCNF (open triangles), V36pCNF (open squares), and F78pCNF (open circles) in the region 2180–2280 cm−1. The H-NOX proteins were dissolved in a pH 8.0 aqueous buffer solution containing 50 mM TEA and 150 mM NaCl. All spectra were recorded at 25 °C, baseline corrected, intensity normalized, and fitted (solid curves) to a linear combination of Gaussian and Lorentzian functions. | ||
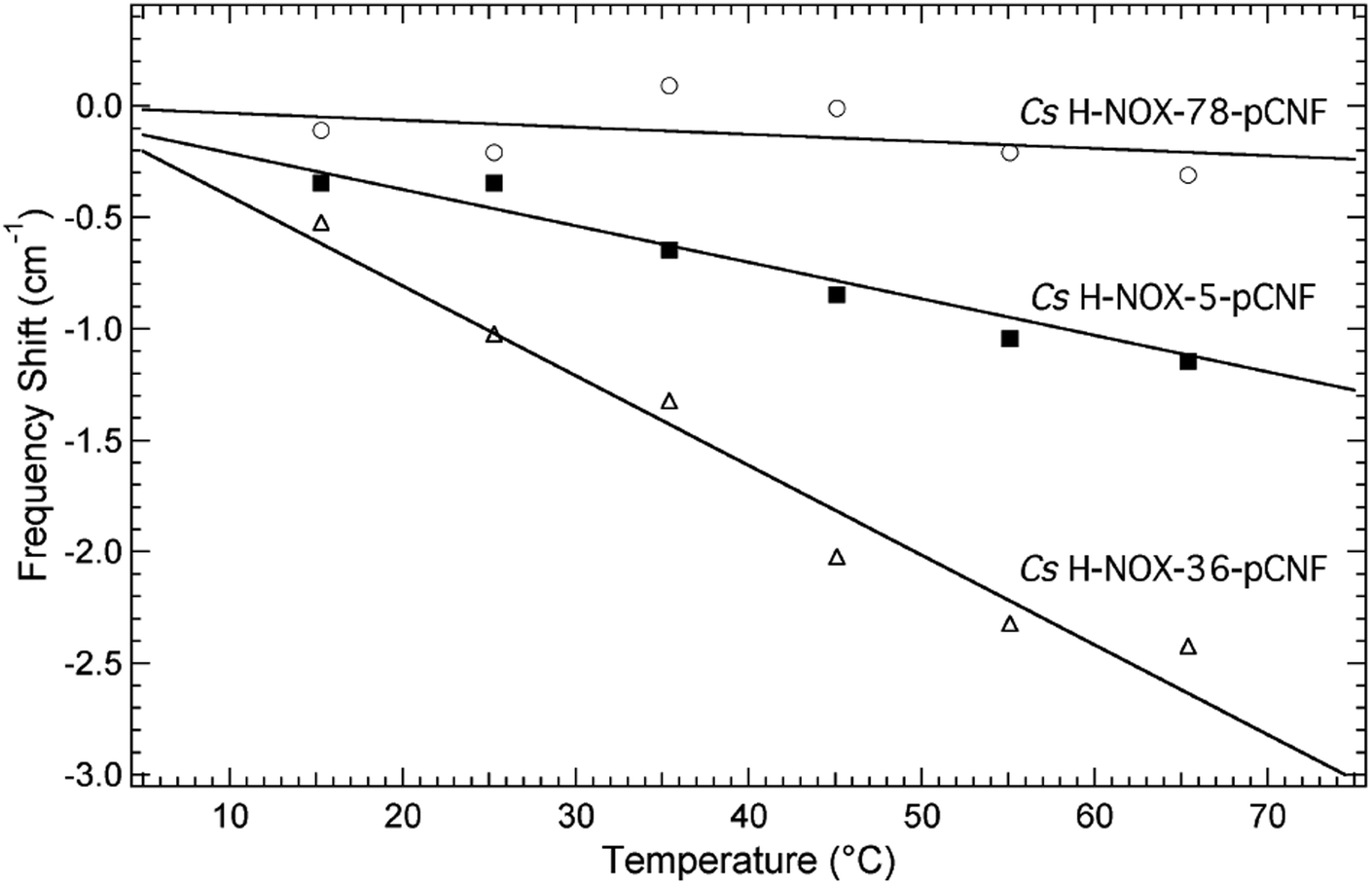 | ||
| Fig. 5 Temperature-dependence shifts in the nitrile stretching frequency in the range 15–65 °C for the CO bound H-NOX protein constructs V36pCNF (open triangles), I5pCNF (solid squares), and F78pCNF (open circles) relative to the frequency at 15 °C. The temperature-dependent frequency shifts were fit to a straight line. The H-NOX proteins were dissolved in a pH 8.0 aqueous buffer solution containing 50 mM TEA and 150 mM NaCl. | ||
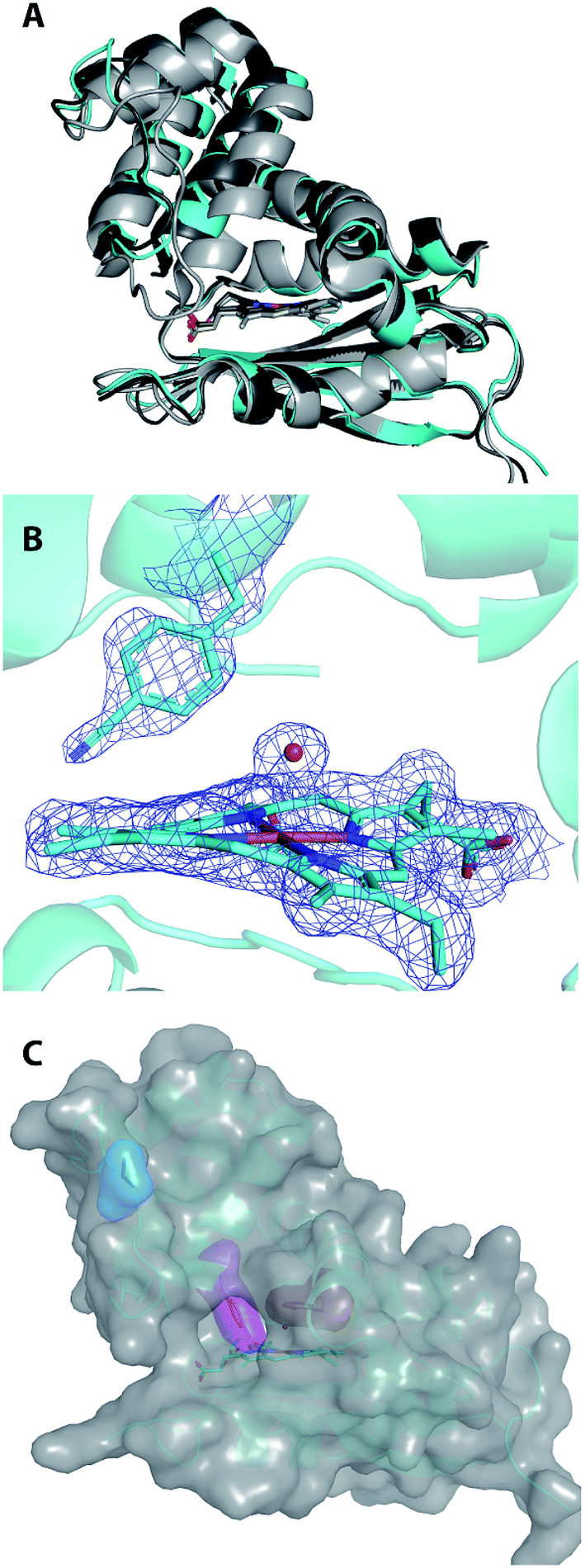 | ||
| Fig. 6 Structural alignment of ferric, water-ligated WT Cs H-NOX (shown in light grey; PDB ID 1U56), Cs H-NOX I5F (shown in dark grey; PDB ID 3SJ5), and chain A of Cs H-NOX I5pCNF (shown in cyan) (A). The protein folds are represented by ribbon diagrams and the hemes are shown in sticks. Final 2FO-FC density map at 1.0 σ shown in blue mesh for I5pCNF, heme, and water molecule in Cs H-NOX (B). The pCNF residue, heme, and coordinated water molecule are explicitly shown in sticks or balls with carbon represented by cyan, nitrogen by dark blue, iron by orange, and oxygen by red. Structure of Cs H-NOX I5pCNF with van der Waals surface shown in transparent grey (C). The protein is represented by a ribbon diagram (cyan) with the heme (sticks), I5pCNF (magenta), V36 (blue), and F78 (red) residues represented by sticks with their van der Waals surfaces in the same colors as stick representations. | ||
The local solvation state of pCNF incorporated in H-NOX at the three sites was further assessed using the frequency–temperature line slope (FTLS) method (Fig. 5 and S4†).23,50,51 This method compares the temperature dependent shift of the nitrile stretching frequency of pCNF incorporated in the protein with either free pCNF or p-tolunitrile dissolved in a range of solvents selected to mimic potential local solvation states present in proteins. The selective use of p-tolunitrile is due to solubility limits of pCNF. This method allows the relative strength of hydrogen-bonding between the solvent (or neighboring amino acid side-chain) and the nitrile group of the incorporated pCNF to be assessed without solely relying on the frequency of the nitrile stretch of pCNF measured at 25 °C. This analysis is needed given the sensitivity of the nitrile stretch of pCNF to the geometric orientation of the hydrogen bonds between the UAA and water molecules.52,53
Fig. 5 shows the temperature dependent shift of the nitrile stretching frequency of pCNF incorporated at site 5 (solid squares), 36 (open triangles), or 78 (open circles) from 15–65 °C. The frequency shifts are relative to the frequency measured at 15 °C. The data are fit to a straight line to compare the FTLS to previous data on model compounds in various solvents. The slope of V36pCNF is the largest and is in agreement to the FTLS of free pCNF dissolved in aqueous buffer.23,50 This agreement reinforces that pCNF at site 36 is solvent exposed. The nitrile stretch frequency of F78pCNF is essentially not temperature-dependent, similar to the FTLS of p-tolunitrile dissolved in THF.23,50 This result also suggest that pCNF at site 78 is buried. Lastly the magnitude of the FTLS for I5pCNF is intermediate between the FTLS of F78pCNF and V36pCNF. This slope is similar to the FTLS of pCNF dissolved in formamide,23,50 indicative of a moderate hydrogen-bonding environment. This results suggest that pCNF at site 5 is involved in hydrogen-bond interactions with either water molecules, amino acid side chains, or the peptide backbone. The I5pCNF crystal structure determined here (see below) does not show amino acid side chains or backbone amine groups in hydrogen-bonding distance to the CN group of pCNF at this site (see Fig. S5†). Therefore pCNF at site 5 is likely involved in moderate hydrogen-bonding with water molecules although the crystal structure does not show structural water molecules in close proximity of the CN group. Thus the hydrogen-bonding of the CN group is likely with bulk water molecules. The nitrile stretching frequency of pCNF at site 5 recorded at 25 °C (Fig. 4) suggested that the CN group was buried however the FTLS analysis importantly shows that the CN group is in a moderate hydrogen bonding environment. This partially solvated nature of pCNF at site 5 is in agreement with the solvent accessible surface areas (SASA) analysis of the I5pCNF crystal structure determined here (see below). Thus the local solvation environment of sites 5, 36, and 78 was able to be effectively probed with the vibrational reporter UAA pCNF.
X-ray crystal structure of I5pCNF Cs H-NOX
The 1.85 Å resolution X-ray crystal structure of I5pCNF H-NOX is shown in Fig. 6 and the data collection and refinement statistics are shown in Table S1.† Alignment of I5pCNF H-NOX with WT (RMSD of 1.82 Å for Cα and 2.25 Å for all atom), or the previously published I5F structure (RMSD of 0.79 Å for Cα and 1.37 Å for all atom), indicates little change in structure upon pCNF incorporation (Fig. 6A). In contrast to the I5F H-NOX structure that showed two conformers of the phenylalanine residue at site 5,48 the electron density surrounding the pCNF residue in the I5pCNF H-NOX structure supports a single conformation of the pCNF residue at the side of the heme pocket, corresponding to the major conformer in the I5F structure (Fig. 6B and S1†). The van der Waals surface of the I5pCNF protein is shown in grey in Fig. 6C with the van der Waals surface for V36, I5pCNF, and F78 highlighted in blue, magenta, and red, respectively, to emphasize the relative solvent accessibility of these sites.The small RMSD of I5pCNF H-NOX with the two previous crystal structures (Fig. 6A) illustrates that pCNF is a relatively non-perturbative probe, supporting the use of the UAA to serve as an effective reporter of local solvation environment. The I5pCNF structure is in agreement with the FTLS analysis (see above) for site 5, in addition to sites 36 and 78. Specifically, the I5pCNF structure shows the solvated nature of the 36 site, as supported by the fully visible side chain of V36 (in blue), and the buried nature of the 78 site, as supported by the side chain for F78 (in red) being fully covered by the van der Waals surface of the protein (Fig. 6C). However, the partially visible I5pCNF surface (in magenta) illustrates the partially buried nature of the 5 site (Fig. 6C). In addition, the H-NOX structure with tyrosine residues substituted as close pCNF analogs at the 5, 36, and 78 sites indicated solvent accessible surface areas for each tyrosine as 49 Å2, 174 Å2, and 35 Å2, respectively.54 These calculated solvent accessible surface areas also parallel the findings that the solvent accessibility of the sites probed decreased from 36 to 5 to 78. The parallel between structure and spectroscopic data emphasizes the complementarity of solution IR spectroscopy and X-ray crystallography. Notably, the FTLS analysis (Fig. 5) was able to resolve the partially buried and fully buried nature of sites 5 and 78, respectively, while the room temperature IR data alone suggested that I5pCNF and F78pCNF were in nearly identical environments. This final analysis of the local environments of site 5 is supported by both the FTLS method and the I5pCNF crystal structure determined here, illustrating the power of IR spectroscopy and protein crystallography coupled with pCNF to effectively assess local solvation environments.
Exploring nitrile stretch dependence on ligation state
The Soret band positions for WT, I5pCNF, V36pCNF, and F78pCNF H-NOX protein constructs for various ligation states, including unligated, and CO, NO, or O2 bound proteins are shown in Table 1. For all CO bound constructs, the Soret band position (∼424 nm) was within the resolution of the instrument. Similarly, all of the NO bound constructs had a Soret band position within the resolution of the instrument (∼420 nm). The O2 bound proteins had Soret band positions ranging from 414.5–417.0 nm. Specifically, the Soret band for the O2 bound I5pCNF construct was 414.5 nm, while the Soret band position for the other O2 bound constructs were 416–417 nm. The Soret band position of the unligated proteins ranged from 431.0–433.5 nm. The Soret band position for each unligated protein construct was approximately 431 nm with the exception of the Soret band position of the unligated F78pCNF construct that appeared at 433.5 nm. The electronic absorbance spectra for the NO, O2, or unligated forms of the proteins are shown in Fig. S2.† These results illustrate that pCNF incorporation at each of the three sites did not prevent the different ligation states of the protein from being accessed.Table 2 contains the nitrile stretching frequencies of I5pCNF, V36pCNF, and F78pCNF for the CO, NO, and O2 ligation states, in addition to the unligated state. The stretching frequency for V36pCNF for each of the ligation states is within the range 2233.6–2234.3 cm−1. These results suggest that the nitrile group of the incorporated pCNF is fully solvated due to the similarity of these stretching frequencies with the nitrile stretch of free pCNF in aqueous buffer. This is not a surprising result, given the earlier data for the CO bound form of V36pCNF (Fig. 4) and the I5pCNF crystal structure (Fig. 6C). This data also suggests that the nitrile stretch vibrational energy of pCNF at site 36 is independent of the ligation state of the heme, which was the expected result, since site 36 is on the surface of the protein, well removed from the heme pocket (see Fig. 1 and 6C).
| Nitrile Stretching Frequency (cm−1) | ||||
|---|---|---|---|---|
| Construct | CO | NO | O2 | Unligated |
| I5pCNF | 2230.0 | 2229.4 | 2229.3 (85%), 2238.3 (15%) | 2229.8 |
| V36pCNF | 2233.6 | 2233.5 | 2234.2 | 2234.3 |
| F78pCNF | 2230.7 | 2230.8 | 2231.4 | 2229.2 |
The nitrile stretching frequency of I5pCNF for the CO, NO and unligated forms of the protein is in the range 2229.4–2230.0 cm−1. The nitrile IR absorbance band of O2 bound I5pCNF contained two components (see Tables 2 and 2S, Fig. S6†). The major component (85% of total band area) is located at 2229.3 cm−1 while the minor component (15% of total band area) is located at 2238.3 cm−1 (Table 2). The primary component has a frequency similar to other I5pCNF ligation states. The presence of the minor component could be the result of two conformations of pCNF at site 5 in solution. The minor component could correspond to a conformation resembling the minor conformer of phenylalanine incorporated at site 5 in the I5F H-NOX crystal structure.48 In this conformation, the nitrile group would be pointed towards the heme group, providing a different environment of the nitrile group of pCNF at site 5 thus explaining the altered frequency. Alternatively, a different conformation of pCNF could be present, increasing the strength of hydrogen bonding between the solvent water molecules and the nitrile group of pCNF at site 5. However, the I5pCNF crystal structure only observes a single conformation of pCNF at site 5 (Fig. 6B), which is consistent with the 2229.3 cm−1 frequency (see discussion above). Overall, these results suggest that the nitrile stretching frequency of pCNF incorporated at site 5 is not dependent upon ligation state. This result is not surprising given the position of pCNF in the I5pCNF crystal structure indicating that the nitrile group is not pointed towards the heme and is not in direct proximity to the distal gas binding site in the protein (Fig. 6B).
Lastly, the nitrile stretching frequency of F78pCNF was observed at 2230.7 and 2230.8 cm−1 for the CO and NO bound protein constructs, respectively, suggesting that the local environment of the nitrile group is similar for both constructs. The nitrile stretching frequency is slightly blue shifted for the O2 bound state (2231.4 cm−1), potentially suggesting an altered polarity of the heme pocket sensed by the nitrile vibrational reporter. However, this shift is within the resolution of the instrument. Similarly, the nitrile stretch vibration of the unligated form (2229.2 cm−1) is slightly red-shifted from the CO and NO bound forms, possibly indicative of an altered polarity, but this shift is also within the instrument resolution. Notably, the shift between the O2 and unligated forms is outside the resolution of the instrument, suggesting a different local environment of the nitrile group in these two constructs. Thus, pCNF is potentially serving as a reporter of the heme pocket environment at site F78, which is consistent with its location in the distal pocket of the heme (Fig. 1 and 6C). However, the geometric orientation of the nitrile group of pCNF at site 78 in H-NOX to the heme compared to the geometric orientation of pCNF incorporated at site 64 in myoglobin2 affects the magnitude of the observed shifts due to different ligation states in H-NOX.
Conclusions
Here we have successfully incorporated the vibrational reporter unnatural amino acid pCNF individually at three sites in the heme protein H-NOX using an engineered, orthogonal tRNA synthetase in E. coli. The nitrile symmetric stretch frequency of pCNF was utilized to report on local hydration environments at each of the three sites in the protein under four ligation states of the heme cofactor. UV-Vis spectroscopy confirmed that each UAA containing protein construct could form the ferrous CO, NO, or O2 bound, or unligated state. The room temperature nitrile symmetric stretching frequencies of pCNF incorporated protein constructs of these various ligation states showed that sites 5 and 78 were relatively de-solvated compared to the hydrated site 36. The nitrile symmetric stretch vibration assignment was verified based upon the isotopic shift of this vibration in pCNF compared to p13CNF incorporated in the protein.To further assess the hydration state at each of the three sites, the FTLS method was utilized on each of the CO bound protein constructs. The FTLS analysis confirmed that V36pCNF is in a highly solvated environment and F78pCNF is in a buried, or solvent inaccessible, environment. F78pCNF is located in the distal pocket above the heme and this result indicates that the heme pocket is de-solvated. The FTLS analysis of I5pCNF suggests that it is in a partially buried environment where the nitrile group is involved in hydrogen bonds of moderate strength. Further, the crystal structure of pCNF incorporated at site 5 in H-NOX was determined to further correlate the observed temperature dependence of the nitrile stretching frequency to local environment. The environment of the pCNF at site 5 in the crystal structure of I5pCNF H-NOX confirmed that this site is partially buried at the side of the heme pocket, suggesting that the nitrile group could be involved in hydrogen bonding interactions with the solvent. Additionally, the nitrile stretching frequency of F78pCNF was slightly sensitive to the ligation state of the heme.
Thus temperature dependent IR spectroscopy and X-ray crystallography were effectively used in concert to assess the solvation environments in H-NOX utilizing the UAA pCNF. This combined spectroscopic and crystallographic approach is powerful methodology to probe local solvation environments in proteins, especially when used in conjunction with the vibrational reporter UAA pCNF.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Lisa Mertzman for obtaining materials and supplies and Emily E. Weinert and the Marletta lab (UC Berkeley) for cloning of WT Cs H-NOX into the pBAD vector. This work was supported by F&M Hackman funds, F&M Eyler Funds, Fred A. Snavely Award to CK, NSF (CHE-1053946) to SHB, Henry Dreyfus Teacher-Scholar Award (TH-15-009) to SHB, and NIH (R15GM121984) to CMPP. Data for the I5pCNF Cs H-NOX structure was collected at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Eiger 16M detector on 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract no DE-AC02-06CH11357.References
- Z. Getahun, C.-Y. Huang, T. Wang, B. De León, W. F. DeGrado and F. Gai, J. Am. Chem. Soc., 2003, 125, 405–411 CrossRef CAS PubMed.
- K. C. Schultz, L. Supekova, Y. Ryu, J. Xie, R. Perera and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 13984–13985 CrossRef CAS PubMed.
- J. M. Glasscock, Y. Zhu, P. Chowdhury, J. Tang and F. Gai, Biochemistry, 2008, 47, 11070–11076 CrossRef CAS PubMed.
- K.-I. Oh, J.-H. Lee, C. Joo, H. Han and M. Cho, J. Phys. Chem. B, 2008, 112, 10352–10357 CrossRef CAS PubMed.
- C. L. Weeks, A. Polishchuk, Z. Getahun, W. F. DeGrado and T. G. Spiro, J. Raman Spectrosc., 2008, 39, 1606–1613 CrossRef CAS PubMed.
- H. Taskent-Sezgin, J. Chung, V. Patsalo, S. J. Miyake-Stoner, A. M. Miller, S. H. Brewer, R. A. Mehl, D. F. Green, D. P. Raleigh and I. Carrico, Biochemistry, 2009, 48, 9040–9046 CrossRef CAS PubMed.
- H. Taskent-Sezgin, J. Chung, P. S. Banerjee, S. Nagarajan, R. B. Dyer, I. Carrico and D. P. Raleigh, Angew. Chem., Int. Ed. Engl., 2010, 49, 7473–7475 CrossRef CAS PubMed.
- M. M. Waegele, M. J. Tucker and F. Gai, Chem. Phys. Lett., 2009, 478, 249–253 CrossRef CAS PubMed.
- M. M. Waegele, R. M. Culik and F. Gai, J. Phys. Chem. Lett., 2011, 2, 2598–2609 CrossRef CAS PubMed.
- S. Ye, T. Huber, R. Vogel and T. P. Sakmar, Nat. Chem. Biol., 2009, 5, 397–399 CrossRef CAS PubMed.
- S. Ye, E. Zaitseva, G. Caltabiano, G. F. X. Schertler, T. P. Sakmar, X. Deupi and R. Vogel, Nature, 2010, 464, 1386–1389 CrossRef CAS PubMed.
- A. T. Fafarman, P. A. Sigala, D. Herschlag and S. G. Boxer, J. Am. Chem. Soc., 2010, 132, 12811–12813 CrossRef CAS PubMed.
- D. C. Urbanek, D. Y. Vorobyev, A. L. Serrano, F. Gai and R. M. Hochstrasser, J. Phys. Chem. Lett., 2010, 1, 3311–3315 CrossRef CAS PubMed.
- S. Nagarajan, H. Taskent-Sezgin, D. Parul, I. Carrico, D. P. Raleigh and R. B. Dyer, J. Am. Chem. Soc., 2011, 133, 20335–20340 CrossRef CAS PubMed.
- M. C. Thielges, J. Y. Axup, D. Wong, H. S. Lee, J. K. Chung, P. G. Schultz and M. D. Fayer, J. Phys. Chem. B, 2011, 115, 11294–11304 CrossRef CAS PubMed.
- C. G. Bazewicz, J. S. Lipkin, E. E. Smith, M. T. Liskov and S. H. Brewer, J. Phys. Chem. B, 2012, 116, 10824–10831 CrossRef CAS PubMed.
- C. G. Bazewicz, M. T. Liskov, K. J. Hines and S. H. Brewer, J. Phys. Chem. B, 2013, 117, 8987–8993 CrossRef CAS PubMed.
- S. Bagchi, S. D. Fried and S. G. Boxer, J. Am. Chem. Soc., 2012, 134, 10373–10376 CrossRef CAS PubMed.
- S. Bagchi, S. G. Boxer and M. D. Fayer, J. Phys. Chem. B, 2012, 116, 4034–4042 CrossRef CAS PubMed.
- I. Peran, T. Oudenhoven, A. M. Woys, M. D. Watson, T. O. Zhang, I. Carrico, M. T. Zanni and D. P. Raleigh, J. Phys. Chem. B, 2014, 118, 7946–7953 CrossRef CAS PubMed.
- A. W. Ritchie and L. J. Webb, J. Phys. Chem. B, 2015, 119, 13945–13957 CrossRef CAS PubMed.
- W. Hu and L. J. Webb, J. Phys. Chem. Lett., 2011, 2, 1925–1930 CrossRef CAS.
- J. D. Slocum and L. J. Webb, J. Am. Chem. Soc., 2016, 138, 6561–6570 CrossRef CAS PubMed.
- E. M. Tookmanian, C. M. Phillips-Piro, E. E. Fenlon and S. H. Brewer, Chem.–Eur. J., 2015, 21, 19096–19103 CrossRef CAS PubMed.
- E. M. Tookmanian, E. E. Fenlon and S. H. Brewer, RSC Adv., 2015, 5, 1274–1281 RSC.
- E. E. Smith, B. Y. Linderman, A. C. Luskin and S. H. Brewer, J. Phys. Chem. B, 2011, 115, 2380–2385 CrossRef CAS PubMed.
- A. B. Dippel, G. M. Olenginski, N. Maurici, M. T. Liskov, S. H. Brewer and C. M. Phillips-Piro, Acta Crystallogr., Sect. D: Struct. Biol., 2016, 72, 121–130 CAS.
- S. D. Fried, S. Bagchi and S. G. Boxer, J. Am. Chem. Soc., 2013, 135, 11181–11192 CrossRef CAS PubMed.
- J. Zimmermann, M. C. Thielges, Y. J. Seo, P. E. Dawson and F. E. Romesberg, Angew. Chem., Int. Ed. Engl., 2011, 50, 8333–8337 CrossRef CAS PubMed.
- J. K. Chung, M. C. Thielges and M. D. Fayer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3578–3583 CrossRef CAS PubMed.
- R. Adhikary, J. Zimmermann, P. E. Dawson and F. E. Romesberg, ChemPhysChem, 2014, 15, 849–853 CrossRef CAS PubMed.
- M.-L. Fardeau, M. B. Salinas, S. L'Haridon, C. Jeanthon, F. Verhé, J.-L. Cayol, B. K. C. Patel, J.-L. Garcia and B. Ollivier, Int. J. Syst. Evol. Microbiol., 2004, 54, 467–474 CrossRef CAS PubMed.
- L. M. Iyer, V. Anantharaman and L. Aravind, BMC Genomics, 2003, 4, 1–8 CrossRef.
- D. S. Karow, D. Pan, R. Tran, P. Pellicena, A. Presley, R. A. Mathies and M. A. Marletta, Biochemistry, 2004, 43, 10203–10211 CrossRef CAS PubMed.
- E. M. Boon and M. A. Marletta, Curr. Opin. Chem. Biol., 2005, 9, 441–446 CrossRef CAS PubMed.
- E. M. Boon and M. A. Marletta, J. Inorg. Biochem., 2005, 99, 892–902 CrossRef CAS PubMed.
- P. Pellicena, D. S. Karow, E. M. Boon, M. A. Marletta and J. Kuriyan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12854–12859 CrossRef CAS PubMed.
- C. W. Hespen, J. J. Bruegger, C. M. Phillips-Piro and M. A. Marletta, ACS Chem. Biol., 2016, 11, 2337–2346 CrossRef CAS PubMed.
- M. B. Winter, M. A. Herzik, J. Kuriyan and M. A. Marletta, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E881–E889 CrossRef CAS PubMed.
- S. J. Miyake-Stoner, A. M. Miller, J. T. Hammill, J. C. Peeler, K. R. Hess, R. A. Mehl and S. H. Brewer, Biochemistry, 2009, 48, 5953–5962 CrossRef CAS PubMed.
- X. S. Gai, E. E. Fenlon and S. H. Brewer, J. Phys. Chem. B, 2010, 114, 7958–7966 CrossRef CAS PubMed.
- S. Nallamsetty, R. B. Kapust, J. Tözsér, S. Cherry, J. E. Tropea, T. D. Copeland and D. S. Waugh, Protein Expression Purif., 2004, 38, 108–115 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- L. C. Storoni, A. J. McCoy and R. J. Read, Acta Crystallogr., Sect. D: Struct. Biol., 2004, 60, 432–438 CrossRef PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Struct. Biol., 2010, D66, 213–221 CrossRef PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Struct. Biol., 2004, 60, 2126–2132 CrossRef PubMed.
- E. E. Weinert, L. Plate, C. A. Whited, C. Olea Jr and M. A. Marletta, Angew. Chem., Int. Ed., 2009, 49, 720–723 CrossRef PubMed.
- E. E. Weinert, C. M. Phillips-Piro, R. Tran, R. A. Mathies and M. A. Marletta, Biochemistry, 2011, 50, 6832–6840 CrossRef CAS PubMed.
- E. E. Weinert, C. M. Phillips-Piro and M. A. Marletta, J. Inorg. Biochem., 2013, 127, 7–12 CrossRef CAS PubMed.
- R. Adhikary, J. Zimmermann, P. E. Dawson and F. E. Romesberg, Anal. Chem., 2015, 87, 11561–11567 CrossRef CAS PubMed.
- R. Adhikary, J. Zimmermann and F. E. Romesberg, Chem. Rev., 2017, 117, 1927–1969 CrossRef CAS PubMed.
- J.-H. Choi, K.-I. Oh, H. Lee, C. Lee and M. Cho, J. Chem. Phys., 2008, 128, 134506 CrossRef PubMed.
- P. Deb, T. Haldar, S. M. Kashid, S. Banerjee, S. Chakrabarty and S. Bagchi, J. Phys. Chem. B, 2016, 120, 4034–4046 CrossRef CAS PubMed.
- R. Fraczkiewicz and W. Braun, J. Comput. Chem., 1998, 19, 319–333 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02000k |
| ‡ Indicates authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |