
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMn–Fe nanoparticles on a reduced graphene oxide catalyst for enhanced olefin production from syngas in a slurry reactor†
AL-Hassan Nasser abc,
Lisheng Guoc,
Hamada ELnaggara,
Yang Wangc,
Xiaoyu Guoc,
Ahmed AbdelMoneim*a and
Noritatsu Tsubaki*c
abc,
Lisheng Guoc,
Hamada ELnaggara,
Yang Wangc,
Xiaoyu Guoc,
Ahmed AbdelMoneim*a and
Noritatsu Tsubaki*c
aMaterials Science and Engineering Department, Egypt-Japan University of Science and Technology, New Borg El-Arab, Alexandria 21934, Egypt. E-mail: ahmed.abdelmoneim@ejust.edu.eg
bChemical Engineering Department, Faculty of Engineering, Alexandria University, Alexandria 11432, Egypt
cDepartment of Applied Chemistry, School of Engineering, University of Toyama, Gofuku 3190, Toyama city, 930-8555, Japan. E-mail: tsubaki@eng.u-toyama.ac.jp
First published on 19th April 2018
Abstract
Fe nanoparticles (NPs) supported on reduced graphene oxide (rGO) nano-sheets were promoted with Mn and used for the production of light olefins in Fischer–Tropsch reactions carried out in a slurry bed reactor (SBR). The prepared catalysts were characterized by X-ray fluorescence (XRF), X-ray diffraction (XRD), transmission electron microscope (TEM), Raman spectroscopy, N2 physisorption, temperature programmed reduction (TPR) and X-ray photoelectron spectroscopic (XPS) methods. Mn was shown to preferentially migrate to the Fe NP surface, forming a Mn-rich shell encapsulating a core rich in Fe. The Mn shell regulated the diffusion of molecules to and from the catalyst core, and preserved the metallic Fe phase by lowering magnetite formation and carburization, so decreasing water gas shift reaction (WGSR) activity and CO conversion, respectively. Furthermore, the Mn shell reduced H2 adsorption and increased CO dissociative adsorption which enhanced olefin selectivity by limiting hydrogenation reactions. Modification of the Mn shell thickness regulated the catalytic activity and olefin selectivity. Simultaneously the weak metal–support interaction further increased the migration ability owing to the utilization of a graphene-based support. Space velocities, pressures and operating temperatures were also tested in the reactor to further enhance light olefin production. A balanced Mn shell thickness produced with a Mn concentration of 16 mol Mn/100 mol Fe was found to give a good olefin yield of 19% with an olefin/paraffin (O/P) ratio of 0.77. Higher Mn concentrations shielded the active sites and reduced the conversion dramatically, causing a fall in olefin production. The optimum operating conditions were found to be 300 °C, 2 MPa and 4.2 L g−1 h−1 of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2:CO syngas flow; these gave the olefin yield of 19%.
:1 H2:CO syngas flow; these gave the olefin yield of 19%.
1 Introduction
The use of Fischer–Tropsch synthesis (FTS) technology for the conversion of syngas into useful end products is an important process. Light olefins are essential components in the petrochemical industry, acting as a starting point for many production lines.1–4 Fe catalysts commonly used in FTS industrial plants are characterized by their high selectivities for the light olefin fractions.5–7 They also give high water gas shift reaction (WGSR) activities, rendering them more flexible in processing syngas streams with various H2:CO ratios.8–11 However, further enhancement of their orientation towards light olefins is required if more competitive systems are to be designed. The common strategy for achieving this goal is via promotion of the Fe catalyst systems.
The most common promoters for the enhancement of light olefin production are the alkali metals, especially Na and K, which increase the selectivity of Fe systems towards light olefins dramatically.12–14 Mn is comparably effective in achieving this target. Extensive research has been done over the years for various mixtures and combinations of Mn with alkali metals on various supporting materials for this purpose.5,8,15,16 Other promoters used for olefin enhancement include Zn and Mg.6,7,12,17
Mn is known to increase the olefin selectivity by increasing dissociative CO adsorption on the catalyst surface whilst reducing H2 adsorption.16,18 This chokes off the hydrogenation reactions which are responsible for converting the olefins to paraffins. However, Mn at very high levels was reported to have a detrimental effect on the CO conversion in some cases, especially when carbon supports were used.5,16,19 In reality, the rate of carburization of metallic Fe was retarded by an excess of Mn oxide which led to a significant decrease in FTS activity.5 Thus it was found that an optimum level exists at which there is a balance between the reduced CO conversion and the enhanced olefin selectivity. Support materials including ceramic-silica,20 alumina,21 zeolites,22 and titania23 were intensively used to support Fe NPs. Considering the strong metal–support interactions (SMSI),8 other supports were developed using carbon-based materials like activated carbon, carbon nanotubes (CNTs)5,16 and graphene.24,25 Carbon supports are linked to the Fe particles by weaker bonds which allow for higher catalyst activity and stability at the right set of operating conditions.
Graphene is a relatively new material which has emerged as a promising candidate for many applications in the past decade. It is characterized by a high specific surface area, 2D structural features, and high thermal and electrical conductivities caused by the delocalized π electrons migrating freely on the sheet surface; this all renders this material an interesting choice for electronic, electrochemical and catalytic applications.6,26–29 Common pathways for the production of graphene are chemical exfoliation methods like the Hummers' method,30 modified Hummers' method24,26,31,32 and the improved Hummers' method.33 In these methods, strong oxidizing mediums are used to introduce oxygenated groups between the graphite layers to form graphite oxide (GtO). The GtO can then be easily exfoliated mechanically or ultrasonically to produce the low layer number graphene oxide sheets, which can then be reduced into graphene, usually referred to as reduced graphene oxide (rGO) or chemically reduced graphene oxide.
In catalytic applications, it is important to provide stable positions for the NPs planted on the graphene sheets. A very smooth graphene sheet with no defects will allow the NPs to migrate freely on heating during reduction or reaction stages which can cause sintering and segregation of the catalyst particles. So rough sheets with high defect densities are preferred for such applications where the defects act as anchor points for the catalyst particles, hindering their motion and increasing their dispersion.7,24,26
In this study a series of Fe-based catalysts loaded on rGO sheets were prepared with various Mn loading levels. These catalysts were used in an FTS slurry bed reactor (SBR) to test their performance against that of a Mn-free Fe/rGO catalyst. Then the promotional effect of Mn was investigated in detail based on the catalytic performance as well as characterization results. At the same time, the optimum conditions were investigated to gain a high yield of light olefins.
2 Materials and methods
2.1 Graphite oxide preparation
Graphite oxide (GtO) was prepared by the modified Hummers' method, the details of which are given elsewhere24,32 and described in the ESI.†2.2 Fe/rGO preparation by solvothermal coprecipitation
The Fe/rGO catalyst was prepared by the coprecipitation method,6,7 and the Mn promoter was introduced into the Fe/rGO via ultrasonic assisted impregnation. The details are described in the ESI.†2.3 Characterization
The XRD patterns of the as prepared, calcined (300 °C, 3 h, He 100%, 30 ml min−1), reduced, and spent catalysts were obtained using a Rigaku Ultima IV X-ray diffractometer. The samples were exposed to Cu-Kα radiation (λ = 0.154 nm) at 40 kV and 20 mA within the 5–80° 2θ range at a rate of 0.02° min−1. The strongest intensity peak of the phase being studied was chosen for crystallite size calculation by the Scherrer equation. TEM images of the samples were obtained by a Hitachi H-7650 TEM instrument operating at 100 kV. The images were used to calculate the average particle size of each catalyst, by analyzing at least 100 particles. The Raman spectra of the as prepared catalysts were collected using a Renishaw Invia Raman microscope equipped with a 532 nm green laser source. N2 physisorption tests were carried out in a NOVA-2200e Quantachrome Instruments analyzer in which the samples (50–60 mg) were first degassed at 200 °C for 2 h. Afterwards the N2 adsorption isotherms were recorded at 77 K to get the BET (Brunauer–Emmett–Teller) area, total pore volume and the average pore size by the BJH (Barrett–Joyner–Halenda) method. TPR tests were conducted on the as prepared catalyst samples (30 mg) using a BELCAT-II, MicrotracBEL instrument. The samples were loaded into the quartz cell, heated up to 300 °C (10° min−1), kept at this temperature for 2 h in an inert He atmosphere (30 ml min−1), and then the temperature was cooled down to 50 °C. The temperature was then ramped up to 800 °C (5° min−1) and finally kept at 800 °C for 30 min while the sample was exposed to a flow of 5% vol H2/Ar at 30 ml min−1. H2 in the effluent gas stream was analyzed by an online TCD detector to observe the reduction behavior of the samples. XRF measurements were carried out on a Philips PW 2404 wave dispersive X-ray Fluorescence instrument to analyze the composition of as prepared catalysts. The XPS spectra were observed using a Thermo Fischer Scientific ESCALAB 250Xi instrument equipped with an Al-Kα irradiation source. All binding energies were corrected by using the C1s line at 284.5 eV as an internal standard.2.4 FTS SBR performance evaluation
The catalyst FTS performance tests were carried out in a SBR reaction system; full details are given in the ESI as shown in Fig. S1.†3 Results and discussion
3.1 XRD and TEM
The XRD patterns for GtO and rGO are shown in Fig. S2.† The results for GtO show a sharp peak at about 10° indicating an interlayer spacing of 8.5 Å. The rGO shows a broad low intensity peak at 24° which is common when graphene nano-sheets are amorphously stacked with a small number of graphene layers per nano-sheet, as reported by others.24,34 The XRD spectra for the as prepared catalysts after hydrazine hydrate (HH) reduction, and where necessary, incipient wetness impregnation (IWI), are shown in Fig. 1(A). A very good match is observed when comparing them with the hematite (α-Fe2O3) JCPDS card, which shows that hematite is the major Fe phase in the as prepared catalysts.6,7 Some distortions are observed in the spectra of FeMn16 (concentration of 16 mol Mn/100 mol Fe) and FeMn29 (concentration of 29 mol Mn/100 mol Fe) but these are not clear enough to indicate the explicit presence of Mn-rich separate phases. The Mn is either in an amorphous form or is highly dispersed with very small crystallite sizes, thus Mn cannot be clearly detected by XRD. All this is in agreement with our previous investigation of a similar system,24 as well as with other work on FeMn FTS catalysts.8–10,13,15,17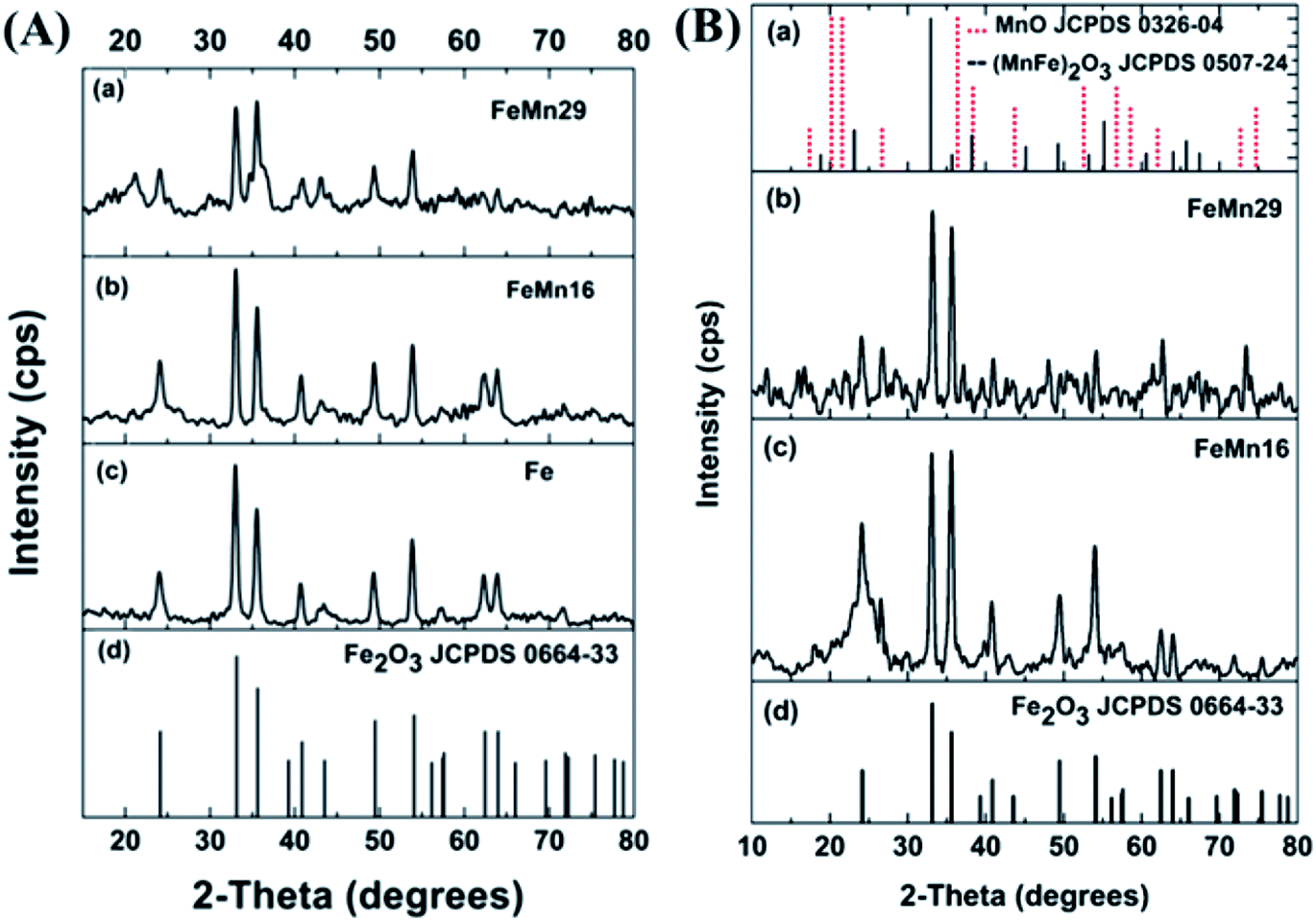 | ||
| Fig. 1 XRD spectra of (A) as prepared catalysts, and (B) catalysts after calcination for 3 h. | ||
The hematite crystallite sizes for the as prepared catalysts calculated from the Scherrer equation are of similar values (about 18.5–19 nm), as shown in Table 1. The same particle size is expected for the different catalysts since, without calcination, the IWI loaded promoters will not have any significant effect on the catalyst particle size. Generally, the agglomeration and sintering of Fe-based catalysts lead to low catalytic activity. It has been reported that CO conversion in the FTS is dependent on particle size, and hydrocarbon selectivity is strongly affected by the catalyst particle size in the range of 2.0–12.0 nm for Fe/Al2O3 catalysts.35 Therefore, an appropriate nanoparticle size for the FeMn catalyst is beneficial for improving catalytic performance.
| Catalyst | Phase | Method | 2θ/° | Size (nm) | ||
|---|---|---|---|---|---|---|
| Fe | FeMn16 | FeMn29 | ||||
| As prepared | Fe2O3 | XRD | 33 | 18.9 | 19.0 | 18.4 |
| Calcined | Fe2O3 | XRD | 33 | 18.9 | 18.1 | 20.2 |
| TEM | — | 19.9 | 18.2 | 14.7 | ||
| Carburized | Fe3O4 | XRD | 35 | 14.2 | 14.1 | 8.0 |
| TEM | — | 14.7 | 9.0 | 8.6 | ||
| Carbides | XRD | 57 | 12.8 | 10.6 | 8.8 | |
While hematite is still the dominant component in the calcined catalysts, the spectra in Fig. 1(B) have some minor peaks which match well to peaks in the bixbyite Fe(2−x)MnxO3 standard card as reported by Maiti et al. for FeMn catalysts.36 A similar FeMn solid solution was also found according to Mössbauer results; the solid solution contained a mixture of Fe2O3 and Mn2O3 as in other reports.9,13,15,37,38 It is worth noticing that the same behavior is also expected with systems that form spinel magnetite Fe3O4 in which mixed spinels of F(3−x)MnxO4 single phase oxides formed.5 According to the XRD data, the grain sizes did not show any significant changes after calcination, with basically the same average value of 19 nm for the hematite crystallites (Table 1).
After TPR of the catalysts, the reduced samples were transferred to the XRD equipment to record their diffraction spectra. The samples gave clear peaks resembling those of α-Fe0 as shown in Fig. 2(A).20,36 There were no other peaks observed besides those of metallic iron in the Mn-free sample (Fe). However, after Mn addition, some additional peaks were clearly visible as small bumps at 35°, 40° and 43° which resembled those of MnO (manganosite) and FeO (wustite) when compared with their respective JCPDS cards. This proves that Mn has a restricting effect on the Fe reduction by preventing some FeO from being reduced to Fe0. This is in agreement with a lot of previous investigations which state clearly that separate MnO and FeO phases as well as mixed solid solutions between the two were detected by various characterization techniques.15,36,38,39
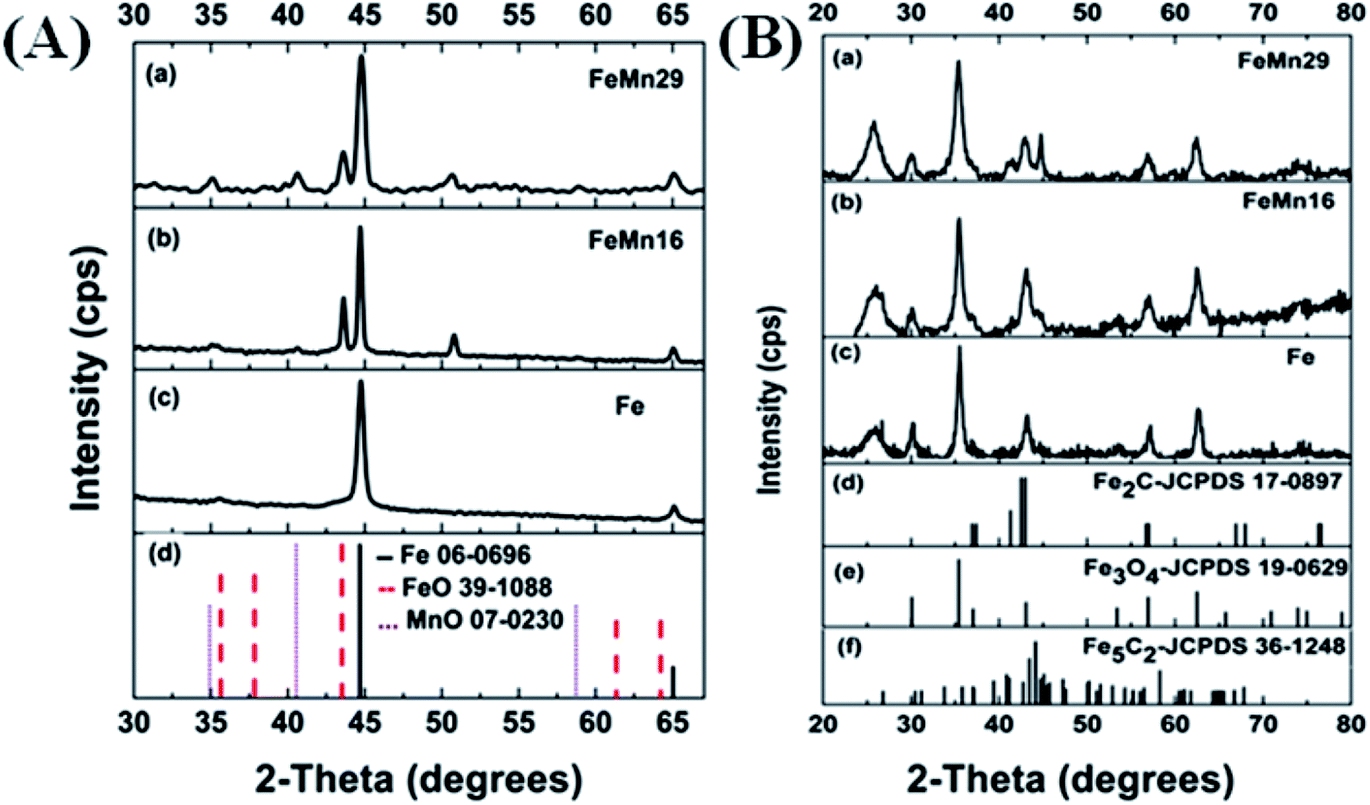 | ||
| Fig. 2 XRD spectra of (A) reduced, and (B) spent catalysts. | ||
The spectra for the carburized catalysts after syngas treatment are shown in Fig. 2(B). The largest peak (35°) is from the oxygen-poor Fe3O4 magnetite6 produced by the oxidation of metallic Fe on exposure to the H2O produced from the FTS activity.15 The formed magnetite is shielded from further reduction into carbides by the layers of carbides being formed at the catalyst's surface. These layers impede the seeping out of oxygen from the magnetite core.9,15
The presence of α-Fe0 (the peak at 45°) after carburization increased as the Mn loading increased. In the Fe catalyst (0% Mn) the metallic peak nearly disappeared completely, showing that the Fe had no resistance to carburization. However, a significant shoulder peak could be detected in the FeMn16 catalyst and a clear distinct peak of metallic iron was even more obvious with FeMn29. All these findings imply that Mn impeded the carburization of Fe.5,15,38 It can be deduced that high Mn levels in FeMn16 and especially FeMn29 hindered the carburization reactions and preserved some Fe0 in the NP core.
The peaks of Fe-carbides, probably Fe5C2 and Fe2C, appeared after the reaction. Fe5C2 (also known as the Hagg carbide) is the main active phase responsible for the FTS activity of the catalyst.5,8,10,15 The carbide peaks (at 43° and 56°) increased in intensity as the molar ratio Mn/100Fe increased from 0 to 16, but the increase in Mn molar content to 29/100Fe caused these peaks to fade again significantly showing the reduced crystallinity and grain sizes in this sample. In addition, different iron carbides are reported to exhibit distinct influences on the product selectivity. Fe2C produced lower CH4 and higher C5+ selectivity than χ-Fe5C2,40 thus the appearance of Fe2C owing to the addition of Mn is beneficial for excellent performance. This observation will be important in explaining the FTS activity trends later on. The same observation can be drawn from the crystallites' sizes calculated for the carburized catalysts for both the magnetite and the carbide components (Table 1). The size of the magnetite crystallites decreased steadily (14.2 to 8 nm) with the rise in Mn content, and the grain size of the carbide phase decreased from 12.8 for the Mn-free sample down to 8 nm when the Mn content reached 29 mol/100 mol Fe. This again implies that a very high Mn content exhibited negative effects on the carburization of Fe and the subsequent carbide crystallization,5 and it also decreased magnetite formation.
The TEM pictures along with the particle size distribution (PSD) histograms of the catalysts before and after the FTS reaction are compared in Fig. S3 and S4.† The PSD results are in good agreement with the XRD results except in two cases: the calcined FeMn29 and the carburized FeMn16 (Table 1). In both cases the XRD overestimates the grain size slightly by 5 nm.
Accordingly, the TEM PSD values differed from the XRD in the cases of the hematite phase after calcination as well as the magnetite phase after carburization, both of which decreased in average particle size as the Mn level increased. It was shown earlier from the XRD results that Mn forms solid solutions in both the hematite and spinel phases. It was also demonstrated that a high Mn content rendered higher stability of the metallic Fe phase, shielding the Fe-rich core from the carburizing species which hindered carburization and retarded the carbide crystallization.5,15,16,41 The magnetite content decreased with Mn doping since the outer Mn-rich layer restricted H2O formation during early FTS reaction stages which preserved the inner Fe0 metallic core formed after reduction.15 Therefore, the falling particle sizes with FeMn29 are not at all unexpected after taking these observations into consideration.
The fall in the carbide phase particle size as stated by the TEM and XRD results at the same Mn level can also explained by the high carbide mass density compared to that of the other precursory phases,9,42,43 which causes the decrease in particle size regardless of other effects.
3.2 Raman
The Raman spectra for the samples are given in Fig. S5.† The characteristic D and G bands appear at about 1350 and 1600 cm−1 respectively. The D band corresponds to the vibrations associated with defects or sheet edges. It is thus expected to increase in intensity as the degree of disorder and number of defects in the sheets increase, while the G band corresponds to the degree of graphitization and increases with the number of graphene layers per nano-sheet.44,45The band intensity ratio ID/IG can be taken as a measure of the degree of defects and exfoliation of the graphene sheets.7,44 It can be seen from Fig. 3 that the ratio increases from 0.73 in GtO to reach 1.01 after ultrasonic exfoliation and reduction with rGO, which shows that the ultrasonic exfoliation is successful in reducing the number of layers per sheet significantly. Fe and Mn decoration had a negligible effect on the ratio with it showing only a slight fall to 0.93 with FeMn16. The results of the 2D band in the vicinity of 2600–2800 cm−1 are compared in Fig. S6.† It has been mentioned that the 2D band will shift to values higher than 2700 cm−1 if the number of layers per sheet is high;26,44,45 the results here show that the peaks are below 2700 cm−1 demonstrating that the prepared graphene has low graphitization.
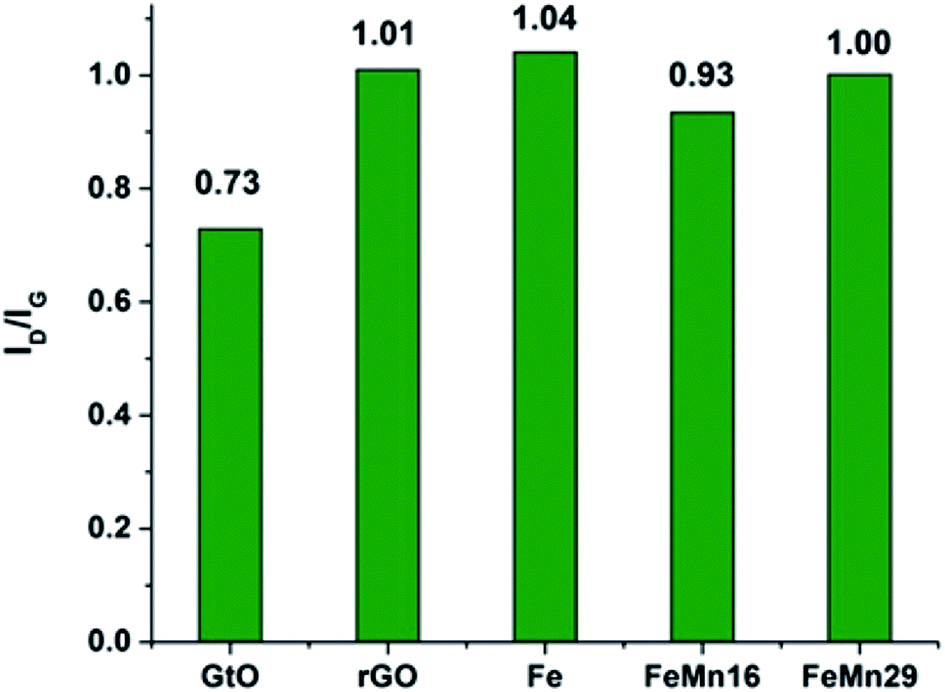 | ||
| Fig. 3 The ID/IG band intensity ratios for GtO, rGO and the catalysts before the FTS reaction. | ||
3.3 Nitrogen physisorption
The BET areas along with the average pore sizes and volumes are listed in Table 2. The coprecipitation of Fe on the rGO sheets caused a severe decrease in area from 395 to 218 m2 g−1 and a very slight increase in pore volume, while the average pore size decreased by 2 Å. The area and pore volume decreased steadily with the rise in Mn content to reach minimums of 169 m2 g−1 and 0.26 cm3 g−1, respectively, with 29 mol Mn/100 mol Fe. The pore size decreased from 16.5 Å for rGO to an average uniform value of about 14.9 Å for the three catalysts. In reality, the introduction of suitable Mn additives does not change surface physical properties significantly according to Table 2. This is in agreement with previous work done on similar systems.24,25| Catalyst | BET area (m2 g−1) | Pore volume (cm3 g−1) | Average pore size (Å) |
|---|---|---|---|
| rGO | 395.3 | 0.34 | 16.5 |
| Fe | 218.1 | 0.35 | 14.81 |
| FeMn16 | 194.6 | 0.33 | 15.01 |
| FeM29 | 169.0 | 0.26 | 14.86 |
3.4 H2 TPR
H2 TPR profiles are compared in Fig. 4, and resemble the typical scheme15,36,38,41,46 for Fe reduction with a three step transformation process in which hematite α-Fe2O3 is reduced to magnetite Fe3O4, then to FeO, and finally to the metallic Fe.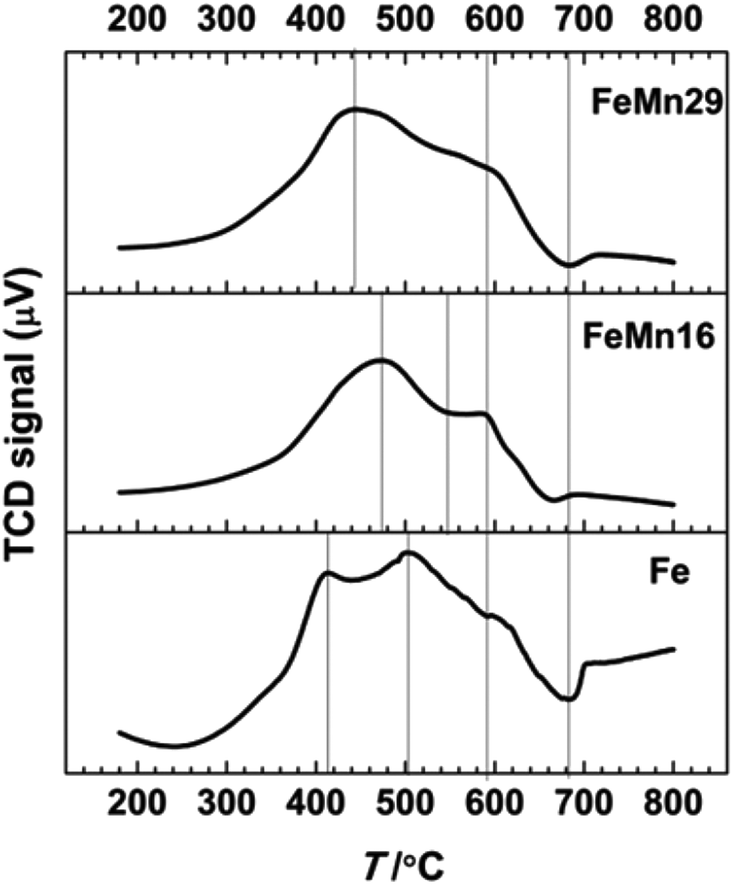 | ||
| Fig. 4 H2 TPR results for the catalysts Fe, FeMn16 and FeMn29. | ||
Although Mn can stabilize the phases preceding the metallic Fe phase by forming various solid solutions with them,47 this stabilization hinders the reduction of α-Fe2O3 to Fe0 as indicated by the TPR results. The introduction of Mn results in a delay in the reduction of hematite to magnetite, and so the first peak merges in with the magnetite reduction peak in a much smaller peak that is on average 40–60 °C higher than that of Fe. The shoulder FeO reduction peak which starts at about 550 °C is smoother and more diminished after Mn addition. The retarding effect results from the formation of the bixbyite solid solution along with hematite after calcination, which was observed in the XRD patterns and is also confirmed from the Fe2O3–Mn2O3 phase diagrams.36,48,49 It is followed by the formation of spinel solid solution comprising two phases, one Mn-rich and the other Fe-rich.36 On further reduction these are transformed to the MnO and FeO phases to form a solid solution manganowustite with 20% FeO.15,36 These solid solutions are formed when Mn migrates preferentially at high temperatures to the particle surface forming a Mn-rich crust15,16,38,47 that isolates the Fe species from the reducing agents and eventually causes the segregation of Fe-rich phases in the core of the particles.15,16,38,47
3.5 XPS
The surface compositions of the as prepared, calcined and carburized catalysts as atomic ratios obtained from the XPS data are compared in Table 3. The Mn/Fe molar ratio is higher on the surface than the bulk values measured by XRF methods (Table S1†), and there is a decrease in the Fe/C and a rise in the Mn/C molar ratios as the overall Mn content is increased. This gives further evidence that Mn preferentially migrates to the catalyst particle surface to form the Mn-rich mixed oxide phase which was described in the XRD discussion. The migration of Mn leads to Mn-rich shell formation which will play a critical role during the FTS activation and reaction stages, as will be clarified later on. A similar promoter promotional effect derived from preferential migration was demonstrated by different researchers.50,51 More recently, Guo et al. reported the enhanced surface migration ability of the K promoter being applied to improve CO2 hydrogenation performance via influencing surface hydrogenation ability and carbide content.52 The sharp increase of carbide content with the K promoter addition led to weaker hydrogenation ability and stronger C–C coupling ability. Besides, it was reported that the selectivity of olefins is generally positively correlated with the strength of the surface basicity of the catalyst.53 Therefore, the increase of surface basicity owing to Mn migration is beneficial to olefin formation. Furthermore, the addition of Mn was able to increase the dissociative adsorption of CO and decrease the H2 adsorption.16,18 Consequently the appropriate surface content of Mn resulting from the migration phenomenon indeed changed the surface physicochemical properties of the catalyst, such that it realized a favorable performance for olefin formation.| As prepared | Calcined | |||
|---|---|---|---|---|
| Atomic ratio (%) | Fe | Fe | FeMn16 | FeMn29 |
| Mn/Fe | 0.00 | 0.00 | 69.28 | 72.99 |
| Fe/C | 1.54 | 1.50 | 1.17 | 1.24 |
| Mn/C | 0.00 | 0.00 | 0.81 | 0.90 |
3.6 FTS SBR performance evaluation
| Fea | FeMn16a | FeMn29a | |
|---|---|---|---|
| a H2/CO = 1, 2 MPa, 340 °C, 4.2 L g−1 h−1, Time On Stream (TOS) = 8 h. | |||
| CO conversion % | 83 | 84 | 74 |
| Fraction | Selectivity C mol% | ||
| CO2 | 38 | 34 | 35 |
| CH4 | 22 | 24 | 23 |
| C2–4 olefin | 8 | 12 | 10 |
| Total olefin | 14 | 21 | 22 |
| C9+ HC | 22 | 5 | 2 |
| Total paraffin | 70 | 72 | 73 |
| Total iso | 16 | 7 | 5 |
| Total par + iso | 86 | 79 | 78 |
| O/P | 0.169 | 0.271 | 0.278 |
| Olefin yield | 7 | 12 | 10 |
| α | 0.57 | 0.55 | 0.51 |
A thicker Mn-rich/Fe-poor shell obstructs diffusion, regulating the chemical interaction of the catalyst core with the surrounding chemicals. During the reduction reaction, it hinders the full reduction of FeO by forming a manganowustite mixed oxide phase that is not easily reduced to metallic Fe0. This was confirmed by the TPR results which showed a diminished shoulder peak corresponding to the FeO reduction. Another feature of this shell was observed during the FTS reaction itself, in which the Mn-rich shell restricted the diffusion of water from the reaction medium to the Fe-rich core and caused the slow formation of the magnetite phase,15 as observed from the TEM and XRD data which showed a fall in magnetite particle size and peak intensity. The XRD data also showed an increase in the metallic Fe0 shoulder peak at 45° giving strong evidence that the increasing thickness of the Mn-rich outer crust preserved more Fe0 and decreased Fe3O4 formation.5 This is important since magnetite is the main phase responsible for CO2 production via the WGSR reaction and consequently Mn should indirectly decrease the WGSR activity; this was observed to a mild extent in our FTS reaction data with rising Mn levels.
On the other hand, the Mn shell has the very beneficial effect of increasing the selectivity towards olefin production; this effect is caused by the enhancement of dissociative CO adsorption and decreased H2 adsorption.16,18 This renders the reaction medium more favorable for olefin production by decreasing the hydrogenation activity. Therefore, the modification of the Mn shell thickness plays a crucial role in catalytic activity and olefin selectivity. Simultaneously the weak interaction between the metal and the graphene-based support further increases the migration ability. This regulatory Mn–Fe core–shell structure has not previously been reported in graphene-based supported catalysts.
To date, reports detailing the use of graphene and graphene derivatives in the FTS industry are limited in number6,7,11,19,21,24,25,56–60 and more systematic and detailed studies are required to give a clearer role of these materials in the FTS reaction. Some of the published works used graphene as a support for Fe NPs,6,7,11,19,24,56–58 while others utilized Co catalyst systems.21,25,59,60 Cheng et al.6,7 aimed at improving light olefin selectivities by using Mg and K promoters. They also provided a comparison between the alkaline earth metal promoters. Moussa et al.19 provided some insight into the role of K and Mn promoters in the general performance of Fe/rGO catalysts but did not discuss in detail the effects on olefin selectivities. The rest of the publications concentrated on comparing graphene prepared with different methods or other support materials. Lastly, all of the literature dealing with graphene supports carried out the FTS reaction in fixed bed reactors. Therefore, this is the first work to our knowledge that tries to provide a description of the Fe–Mn NPs on the rGO support, and reports the use of rGO supported catalysts in SBRs.
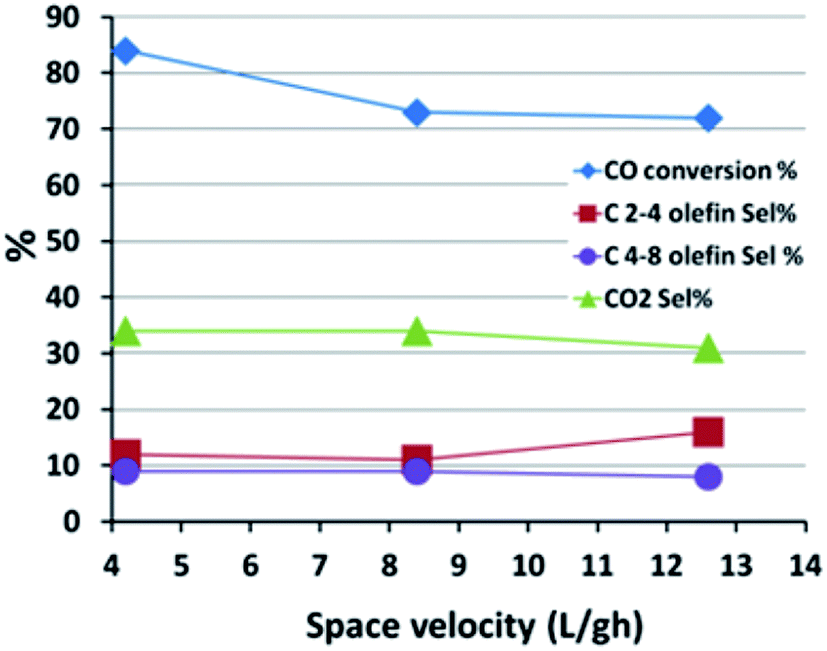 | ||
| Fig. 5 The effect of space velocity on the FTS reaction performance. | ||
Generally, olefin double bond hydrogenation is enhanced by an increase in pressure, thus it is expected that the olefin selectivity will be adversely affected by rising pressures.61,62 The effect of pressure on the FTS catalytic performance of the FeMn16 catalyst was investigated and is shown in Fig. 6 and Table S3.†
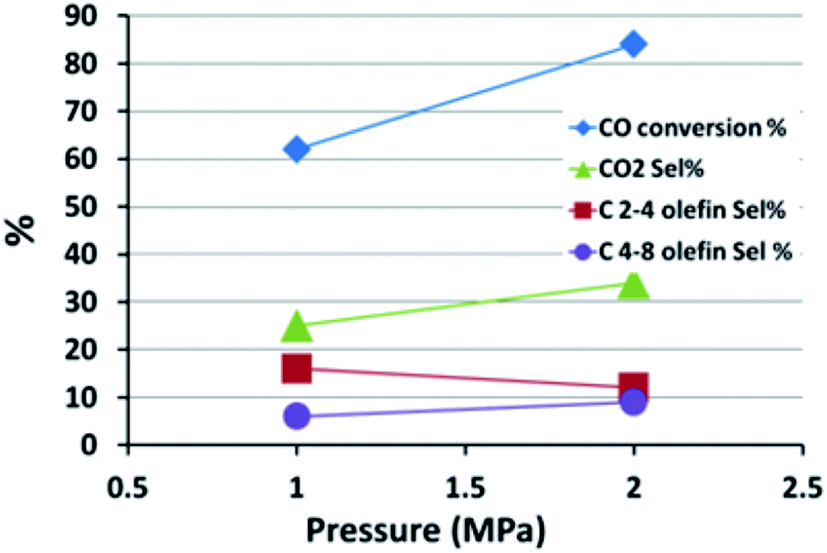 | ||
| Fig. 6 The effect of pressure on the FTS reaction performance. | ||
When the pressure was lowered from 2 MPa to 1 MPa, a lower FTS activity was observed and the CO conversion fell from 84 to 62%, accompanied by a surge in CH4 selectivity from 24% up to 38%. There was a slight decline in WGSR activity with a fall in CO2 selectivity down from 34 to 25%. The WGSR activity is intrinsically related to the FTS activity. The WGSR reaction depends on the presence of water which is a side product of FTS, therefore a low FTS activity means a low water content and hence reduced WGSR activity and low CO2 selectivity.
The heavy fractions (C9+) remained almost constant at 6%, although the reduced pressure levels did shift the ASF parameter to lower values, falling from 0.55 to 0.49, which is a typical response to the fall in pressure. This decrease is attributed to the increased methane formation rather than decreased C9+ selectivity obviously.
The FTS performance of FeMn16 was evaluated at various temperatures to find the best temperature for light olefin production; the results are shown in Fig. 7 and Table S4.† The conversion decreased sharply from 84 to 67% as the temperature fell from 340 to 320 °C, but only decreased to 64% at 300 °C after which it collapsed down to 35% at 280 °C. An increase in conversion with increased temperature will provide more water as a side product from the FTS reaction which will in turn increase the WGSR activity and the CO2 selectivity as shown in Fig. 7. Methane selectivity rises with temperature due to the reduced stability of long chain hydrocarbons which can undergo chain fission (cracking) reactions.18
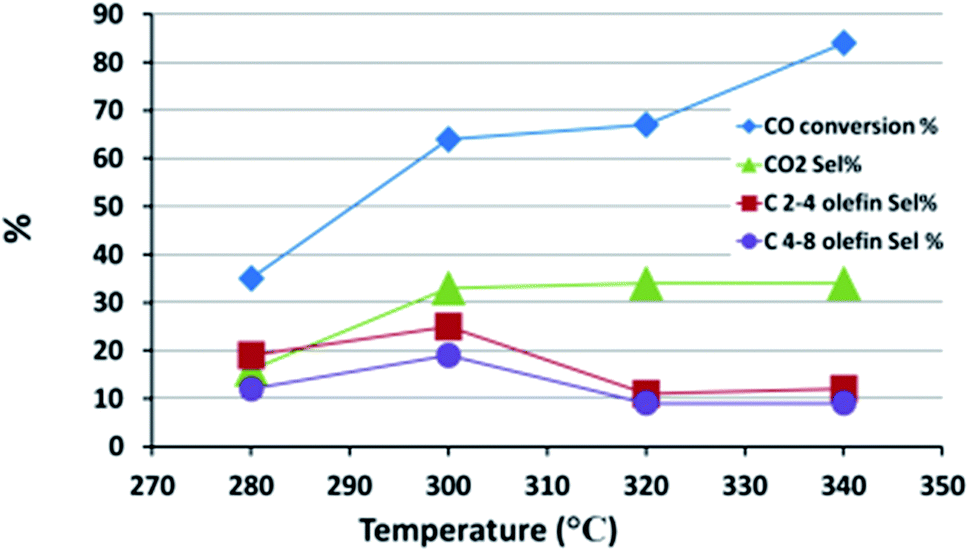 | ||
| Fig. 7 The effect of temperature on the FTS reaction performance. | ||
The main reactions influencing the selectivity of the paraffins and olefins are chain growth and olefin hydrogenation reactions. While chain growth, or the main FTS reaction, is the first step in the process, the hydrogenation reactions occur afterwards to increase the paraffin selectivities. Higher temperatures provide enough activation energy for the hydrogenation of double bonds and thus a fall in olefin selectivity is found as the temperature rises from 280 to 340 °C. In our work, the olefin yield depends on both CO conversion and olefin selectivity and so the slight stabilization in conversion level between 320 and 300 °C and the continuous rise in olefin selectivity in this temperature range led to an optimum olefin yield of 19% at 300 °C. A very similar result was found for the FTS performance of the Fe–Mn–K FTS SBR system in the 260–300 °C range; this had the highest O/P ratios at 300 °C.61 A coprecipitated Fe–Mn system tested in a fixed bed reactor also gave an optimum O/P ratio at 280 °C.13
In general paraffin selectivity experienced an increase from 59% to 72% with rising temperatures except at 300 °C where it fell to 49%; this fall gives further evidence that 300 °C is the best condition to suppress all saturated products in favor of olefins. The fall may be attributed to hydrogenation activity at 300 °C which is insufficient to convert the hydrocarbon chains into paraffins before desorption from the active site, especially given the increased desorption due to heating.61 Once this bottle neck temperature level is surpassed, the hydrogenation rate is accelerated and higher paraffin selectivities are reached.
The heavy hydrocarbon (C9+) fraction selectivity was very high at 23% at 280 °C, as would be expected typically in an FTS system, and then it decreased sharply as the temperature increased to stabilize at about 6%. Isoparaffin selectivity is more intimately related to the C9+ selectivity since the susceptibility to branching and isomerization increases with chain length due to the larger number of possible isomers. That is the reason why the isoparaffin selectivity mirrored that of C9+ with a max of 10% at 280 °C and a slight fall to 7% at higher temperatures.
The chain growth probability parameter (α) behaved as expected, showing a slight decrease with the rise in temperature, and indicating the obvious effect of heat in reducing the average product chain length due to faster chain desorption from the active sites of the catalyst.
4 Conclusion
Manganese acted as a promoter of an Fe NP catalyst by forming a Mn-rich layer around a core rich in Fe. The outer layer of Mn is beneficial for the formation of olefins via tuning the gas adsorption ability and surface properties, as verified by XPS and TPR, etc. However, excessively high Mn levels shielded the Fe active sites and hindered diffusion of the reactants to the catalyst particle core which caused a decrease in catalyst activity. Modification of the surface Mn shell thickness could regulate catalytic activity as well as product selectivity. The tunable properties are of great significance to the fabrication of high efficiency carbon material supported Fe NP catalysts, and promote the catalytic performance. Besides that, suitable operational conditions further enhanced the production of olefins. The best process conditions for high olefin production using the FeMn16 catalyst were at 2 MPa, 300 °C and 4.2 L g−1 h−1 using syngas with a 1:1 H2:CO ratio. At these conditions, the O/P ratio was 0.7 and the olefin yield was 19%.
Conflicts of interest
There are no conflicts to declare.References
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS.
- D. Gao, X. Qiu, Y. Zhang and P. Liu, Comput. Chem. Eng., 2018, 109, 112–118 CrossRef CAS.
- A. Boulamanti and J. A. Moya, Renewable Sustainable Energy Rev., 2017, 68, 1205–1212 CrossRef CAS.
- Q. Zhang, S. Hu and D. Chen, J. Cleaner Prod., 2017, 165, 1351–1360 CrossRef CAS.
- J.-D. Xu, K.-T. Zhu, X.-F. Weng, W.-Z. Weng, C.-J. Huang and H.-L. Wan, Catal. Today, 2013, 215, 86–94 CrossRef CAS.
- Y. Cheng, J. Lin, T. Wu, H. Wang, S. Xie, Y. Pei, S. Yan, M. Qiao and B. Zong, Appl. Catal., B, 2017, 204, 475–485 CrossRef CAS.
- Y. Cheng, J. Lin, K. Xu, H. Wang, X. Yao, Y. Pei, S. Yan, M. Qiao and B. Zong, ACS Catal., 2016, 6, 389–399 CrossRef CAS.
- Q. Chen, G. Liu, S. Ding, M. Chanmiya Sheikh, D. Long, Y. Yoneyama and N. Tsubaki, Chem. Eng. J., 2018, 334, 714–724 CrossRef CAS.
- T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, Appl. Catal., A, 2006, 311, 66–75 CrossRef CAS.
- N. Lohitharn, J. G. Goodwin Jr and E. Lotero, J. Catal., 2008, 255, 104–113 CrossRef CAS.
- F. Jiang, B. Liu, W. Li, M. Zhang, Z. Li and X. Liu, Catal. Sci. Technol., 2017, 7, 4609–4621 CAS.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y.-W. Li, Q. Zhang, X.-D. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed.
- M. Feyzi, M. Irandoust and A. A. Mirzaei, Fuel Process. Technol., 2011, 92, 1136–1143 CrossRef CAS.
- Ş. Özkara-Aydinoĝlu, Ö. Ataç, Ö. F. Gül, Ş. Kinayyiĝit, S. Şal, M. Baranak and I. Boz, Chem. Eng. J., 2012, 181–182, 581–589 CrossRef.
- M.-D. Lee, J.-F. Lee, C.-S. Chang and T.-Y. Dong, Appl. Catal., 1991, 72, 267–281 CrossRef CAS.
- Z. Yang, X. Pan, J. Wang and X. Bao, Catal. Today, 2012, 186, 121–127 CrossRef CAS.
- S. Li, A. Li, S. Krishnamoorthy and E. Iglesia, Catal. Lett., 2001, 77, 197–205 CrossRef CAS.
- C. H. Bartholomew, Catal. Lett., 1990, 7, 303–315 CrossRef CAS.
- S. O. Moussa, L. S. Panchakarla, M. Q. Ho and M. S. El-Shall, ACS Catal., 2014, 4, 535–545 CrossRef CAS.
- C. H. Zhang, H. J. Wan, Y. Yang, H. W. Xiang and Y. W. Li, Catal. Commun., 2006, 7, 733–738 CrossRef CAS.
- Z. Hajjar, M. Doroudian Rad and S. Soltanali, Res. Chem. Intermed., 2017, 43, 1341–1353 CrossRef CAS.
- J. Bao, G. Yang, C. Okada, Y. Yoneyama and N. Tsubaki, Appl. Catal., A, 2011, 394, 195–200 CrossRef CAS.
- M. A. Haider, M. R. Gogate and R. J. Davis, J. Catal., 2009, 261, 9–16 CrossRef CAS.
- A. H. M. Nasser, H. M. Elbery, H. N. Anwar, I. K. Basha, H. A. Elnaggar, K. Nakamura and A. A. El-Moneim, Key Eng. Mater., 2017, 735, 143–147 CrossRef.
- S. Karimi, A. Tavasoli, Y. Mortazavi and A. Karimi, Appl. Catal., A, 2015, 499, 188–196 CrossRef CAS.
- H. M. a. Hassan, V. Abdelsayed, A. E. R. S. Khder, K. M. AbouZeid, J. Terner, M. S. El-Shall, S. I. Al-Resayes and A. a. El-Azhary, J. Mater. Chem., 2009, 19, 3832 RSC.
- S. Sayed, M. Gamil, A. Fath El-Bab, K. Nakamura, T. Tsuchiya, O. Tabata and A. Abd El-Moneim, Sens. Rev., 2016, 36, 140–147 CrossRef.
- E. Ghoniem, S. Mori and A. Abdel-Moniem, J. Power Sources, 2016, 324, 272–281 CrossRef CAS.
- Y. Gao, X. Chen, J. Zhang, H. Asakura, T. Tanaka, K. Teramura, D. Ma and N. Yan, Adv. Mater., 2015, 27, 4688–4694 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- J. Xu, S. Gai, F. He, N. Niu, P. Gao, Y. Chen and P. Yang, Dalton Trans., 2014, 43, 11667–11675 RSC.
- J.-Y. Park, Y.-J. Lee, P. K. Khanna, K.-W. Jun, J. W. Bae and Y. H. Kim, J. Mol. Cata., 2010, A 323, 84–90 CrossRef.
- G. C. Maiti, R. Malessa and M. Baerns, Appl. Catal., 1983, 5, 151–170 CrossRef CAS.
- M. C. Ribeiro, G. Jacobs, R. Pendyala, B. H. Davis, D. C. Cronauer, A. J. Kropf and C. L. Marshall, J. Phys. Chem. C, 2011, 115, 4783–4792 CAS.
- M.-D. Lee, J.-F. Lee and C.-S. Chang, Bull. Chem. Soc. Jpn., 1989, 62, 2756–2758 CrossRef CAS.
- I. R. Leith and M. G. Howden, Appl. Catal., 1988, 37, 75–92 CrossRef CAS.
- Q. Chang, C. Zhang, C. Liu, u. Wei, A. V. Cheruvathur, A. I. Dugulan, J. W. Niemantsverdriet, X. Liu, Y. He, M. Qing, L. Zheng, Y. Yun, Y. Yang and Y. Li, ACS Catal., 2018, 8, 3304–3316 CrossRef CAS.
- T. Grzybek, H. Papp and N. Baerns, Appl. Catal., 1987, 29, 335–350 CrossRef CAS.
- S. Li, W. Ding, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2002, 106, 85–91 CrossRef CAS.
- S. Li, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2001, 105, 5743–5750 CrossRef CAS.
- Z. Ni, Y. Wang, T. Yu and Z. Shen, Nano Res., 2008, 1, 273–291 CrossRef CAS.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS.
- C. Wang, Q. Wang, X. Sun and L. Xu, Catal. Lett., 2005, 105, 93–101 CrossRef CAS.
- K. B. Jensen and F. E. Massoth, J. Catal., 1985, 92, 98–108 CrossRef CAS.
- H. H. Kedesdy, A. Tauber and J. Am, Ceram. Soc., 1956, 39, 425–431 CrossRef CAS.
- A. Muan and S. Somiya, Am. J. Sci., 1962, 260, 230–240 CrossRef CAS.
- P. S. Sai Prasad, J. W. Bae, K.-W. Jun and K.-W. Lee, Catal. Surv. Asia, 2008, 12, 170–183 CrossRef.
- J. Li, X. Cheng, C. Zhang, J. Wang, W. Dong, Y. Yang and Y. Li, J. Chem. Technol. Biotechnol., 2017, 92, 1472–1480 CrossRef CAS.
- L. Guo, J. Sun, X. Ji, J. Wei, Z. Wen, R. Yao, H. Xu and Q. Ge, ChemComm, 2018, 1, 1–8 CrossRef.
- H. Ando, Q. Xu, M. Fujiwara, Y. Matsumura, M. Tanaka and Y. Souma, Catal. Today, 1998, 45, 229–234 CrossRef CAS.
- M. Dad, R. J. Lancee, M. Janse van Vuuren, J. van de Loosdrecht, J. W. H. Niemantsverdriet and H. O. A. Fredriksson, Appl. Catal., A, 2017, 537, 83–92 CrossRef CAS.
- E. Ø. Pedersen, I.-H. Svenum and E. A. Blekkan, J. Catal., 2018, 361, 23–32 CrossRef CAS.
- H. Zhao, Q. Zhu, Y. Gao, P. Zhai and D. Ma, Appl. Catal., A, 2013, 456, 233–239 CrossRef CAS.
- B. Sun, Z. Jiang, D. Fang, K. Xu, Y. Pei, S. Yan, M. Qiao, K. Fan and B. Zong, ChemCatChem, 2013, 5, 714–719 CrossRef CAS.
- X. Chen, D. Deng, X. Pan, Y. Hu and X. Bao, Chem. Commun., 2015, 51, 217–220 RSC.
- S. Taghavi, A. Asghari and A. Tavasoli, Chem. Eng. Res. Des., 2017, 119, 198–208 CrossRef CAS.
- J. Huang, W. Qian, H. Ma, H. Zhang and W. Ying, RSC Adv., 2017, 7, 33441–33449 RSC.
- L. Bai, H. Xiang, Y. Li, Y. Han and B. Zhong, Fuel, 2002, 81, 1577–1581 CrossRef CAS.
- F. A. N. Fernandes, Ind. Eng. Chem. Res., 2006, 45, 1047–1057 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02193g |
| This journal is © The Royal Society of Chemistry 2018 |