
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed aerobic decarboxylative coupling between cyclic α-amino acids and diverse C–H nucleophiles with low catalyst loading†
Jing Guo,
Ying Xie,
Qiao-Lei Wu,
Wen-Tian Zeng,
Albert S. C. Chan,
Jiang Weng * and
Gui Lu*
* and
Gui Lu*
Institute of Medicinal Chemistry, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: lugui@mail.sysu.edu.cn; wengj2@mail.sysu.edu.cn; Fax: +86-20-3994-3048
First published on 1st May 2018
Abstract
An aerobic decarboxylative cross-coupling of α-amino acids with diverse C–H nucleophiles has been realized using Cu2(OH)2CO3 (1 mol%) as the catalyst under air. This protocol enables highly efficient formation of various C(sp3)–C(sp3), C(sp3)–C(sp2) and C(sp3)–C(sp) bonds under simple conditions without the use of any ligand or extra oxidant, providing a practical approach to numerous nitrogen-containing compounds in good to excellent yields. The efficiency and practicability were also demonstrated by the gram-scale experiment and three-step synthesis of a Rad51 inhibitor.
Introduction
Decarboxylative coupling has recently emerged as a powerful method for the construction of C–C and C–heteroatom bonds in organic synthesis.1,2 In contrast to conventional coupling reactions where expensive and sensitive organometallic reagents are usually required, decarboxylative coupling utilizes carboxylic acids that are readily available, bench-stable and simple to handle. Moreover, these reactions generate active carbon species in situ along with nontoxic carbon dioxide, which enables highly chemo- and regioselective cross-couplings with various coupling partners.Pyrrolidines are privileged structural motifs in a wide variety of natural and unnatural products.3 The highly pronounced biological activities of pyrrolidine-containing compounds have made them attractive synthetic targets in medicinal chemistry. In the past decade, redox-neutral C–H functionalization affords a useful and economically efficient synthetic approach to these active molecules.4 On the other hand, proline is among the most abundant biomass feedstock in nature. As such, site-specific decarboxylative functionalization of proline has received particular attention, since this approach provides a facile and versatile synthesis of pyrrolidine derivatives. Recently, several research groups have developed useful synthetic methods for these nitrogen heterocycles through decarboxylative coupling between proline and diverse C–H nucleophiles. Li and Liang5 cooperatively developed a copper-catalyzed oxidative decarboxylative C–C coupling of N-benzyl-proline with alkynes, indoles and nitromethane in the presence of peroxide and ligand (Fig. 1a). Later they expanded the substrate scope of C–H nucleophiles to naphthols via iron catalysis (Fig. 1b).6 Subsequently, Seidel's and other groups independently realized copper-catalyzed α-functionalization of proline under neutral conditions, which involved the aldehyde- and ketone-induced formation of the azomethine ylide as the key step (Fig. 1c–d).7 Chien and co-workers recently reported an intermolecular decarbonylative Mannich reaction of N-benzyl-proline with methyl ketones in the presence of oxalyl chloride (Fig. 1e).8 In addition, significant progress has been achieved in visible-light-induced radical decarboxylative functionalization of α-amino acids including proline derivatives (Fig. 1f).9 However, most of the above methods suffer from one or more drawbacks, such as the requirement of peroxide, organic ligand or transition metal catalyst with high loading, and harsh reaction conditions. From the viewpoint of sustainable chemistry, it is still highly desirable to develop simple and practical methods for decarboxylative functionalization of proline and its derivatives. Herein we wish to report a copper-catalyzed aerobic decarboxylative coupling between cyclic α-amino acids and diverse C–H nucleophiles (Fig. 1g). The major advantage of our findings including: (1) the use of earth-abundant copper catalyst with 1 mol% loading; (2) ligand peroxide-free conditions; (3) efficient construction of various C(sp3)–C(sp3), C(sp3)–C(sp2) and C(sp3)–C(sp) bonds (more than 40 examples, 54–92% yields); (4) gram-scale synthesis; (5) application in facile synthesis of a Rad51 inhibitor.
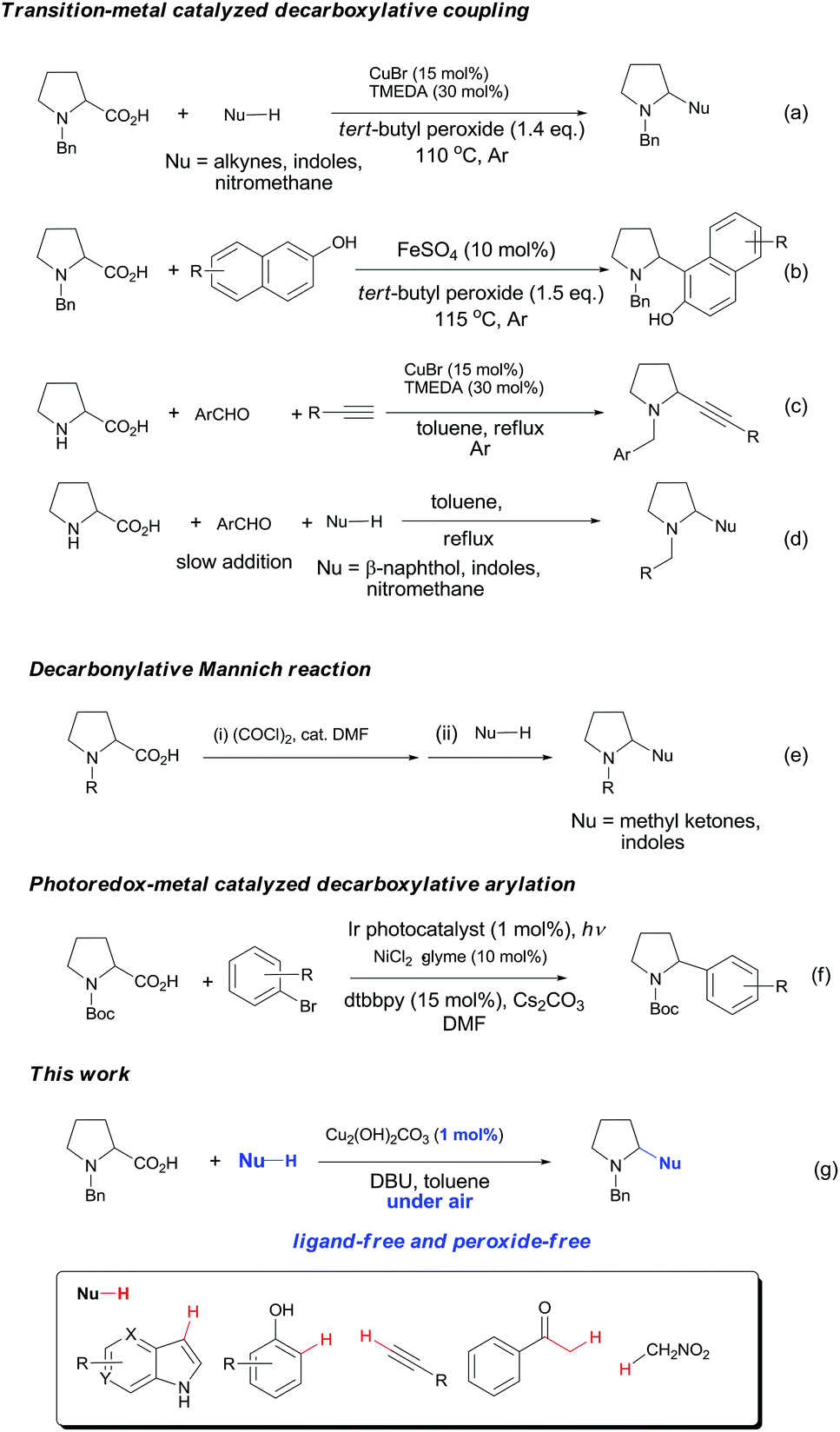 | ||
| Fig. 1 Decarboxylative functionalization of cyclic α-amino acids. | ||
Results and discussion
We chose N-benzyl proline (1a) and indole (2a) as model reaction substrates. In the presence of CuBr2 (10 mol%) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), the desired tertiary amino-indole product 3a was obtained in 48% isolated yield after 24 h (Table 1, entry 1). In order to improve the yield, a series of Cu(I), Cu(II), Fe(II) and Fe(III) salts were examined and Cu2(OH)2CO3 gave the best yield (entries 2–13). Comparing with Cu(I) catalyst, Cu(II) catalyst resulted in higher yield. Changing Cu catalyst to Fe catalyst could also generate the desired product, and FeCl3 afforded comparable yield with Cu2(OH)2CO3 (64% vs. 65%). In view of the lower price of Cu2(OH)2CO3, we chose Cu2(OH)2CO3 as optimal catalyst. Subsequently, different solvents, including N,N-dimethylformamide (DMF), chlorobenzene, dimethyl sulfoxide (DMSO) and mesitylene were screened with Cu2(OH)2CO3 as catalyst (entries 14–17). It was found that toluene was the optimal choice for this transformation (entry 7). The attempts using other bases such as N,N,N′,N′-tetramethylethylene diamine (TMEDA), triethylamine, Ag2CO3 and Cs2CO3 were also investigated, but all resulted in lower yields than DBU (entries 18–21). The amount of base showed obvious effect on the reaction, the use of 3 equiv. of DBU resulted in the highest yield (entries 23–24). To clarify whether DBU plays as a ligand or a base in this reaction, we have also carried out a control experiment with catalytic amount of DBU (20 mol%) and 3.0 equiv. of KOH, and obtained low yield (20%, entry 22 in Table 1). In view of the comparable basicity of DBU and KOH, we assumed that DBU might not be a ligand but mainly act as a base. To our delight, 1 mol% of Cu2(OH)2CO3 is sufficient to provide the desired product 3a in 65% yield within 24 h (entries 25–26). This cross-coupling did not occur in the absence of Cu2(OH)2CO3 catalyst (entry 27). We have also performed the reaction under N2 and O2 atmosphere respectively, and the yields were 5% and 35% (entries 28–29 in Table 1), which implied that O2 was essential for this transformation. Lower temperatures as 80 °C and 60 °C caused poor yields (<25%, entries 30–31), which indicated that high temperature was necessary for the conversion.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Base | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), 2a (0.2 mmol), catalyst (10 mol%), base (4 equiv.), solvent (2 mL), 110 °C, under air for 24 h.b Isolated yield.c DBU (3 equiv.).d DBU (2 equiv.).e Cu2(OH)2CO3 (5 mol%).f Cu2(OH)2CO3 (1 mol%).g Under N2.h Under O2.i Using 20 mol% DBU as ligand and KOH (3 equiv.) as base.j Under 80 °C.k Under 60 °C. | ||||
| 1 | CuBr2 | Toluene | DBU | 48 |
| 2 | Cu(OAc)2 | Toluene | DBU | 48 |
| 3 | Cu(OTf)2 | Toluene | DBU | 36 |
| 4 | CuCl2 | Toluene | DBU | 32 |
| 5 | CuO | Toluene | DBU | 15 |
| 6 | Cu(OH)2 | Toluene | DBU | 10 |
| 7 | Cu2(OH)2CO3 | Toluene | DBU | 65 |
| 8 | CuI | Toluene | DBU | 35 |
| 9 | CuCl | Toluene | DBU | 27 |
| 10 | CuBr | Toluene | DBU | 34 |
| 11 | FeCl3 | Toluene | DBU | 64 |
| 12 | FeSO4 | Toluene | DBU | 44 |
| 13 | Fe(OAc)3 | Toluene | DBU | 42 |
| 14 | Cu2(OH)2CO3 | DMF | DBU | 43 |
| 15 | Cu2(OH)2CO3 | PhCl | DBU | 42 |
| 16 | Cu2(OH)2CO3 | Mesitylene | DBU | 37 |
| 17 | Cu2(OH)2CO3 | DMSO | DBU | 32 |
| 18 | Cu2(OH)2CO3 | Toluene | TMEDA | 35 |
| 19 | Cu2(OH)2CO3 | Toluene | Et3N | 35 |
| 20 | Cu2(OH)2CO3 | Toluene | Ag2CO3 | 25 |
| 21 | Cu2(OH)2CO3 | Toluene | Cs2CO3 | 38 |
| 22i | Cu2(OH)2CO3 | Toluene | KOH | 20 |
| 23c | Cu2(OH)2CO3 | Toluene | DBU | 68 |
| 24d | Cu2(OH)2CO3 | Toluene | DBU | 45 |
| 25c,e | Cu2(OH)2CO3 | Toluene | DBU | 64 |
| 26c,f | Cu2(OH)2CO3 | Toluene | DBU | 65 |
| 27c | — | Toluene | DBU | 0 |
| 28c,f,g | Cu2(OH)2CO3 | Toluene | DBU | 5 |
| 29c,f,h | Cu2(OH)2CO3 | Toluene | DBU | 35 |
| 30c,f,j | Cu2(OH)2CO3 | Toluene | DBU | 25 |
| 31c,f,k | Cu2(OH)2CO3 | Toluene | DBU | 5 |
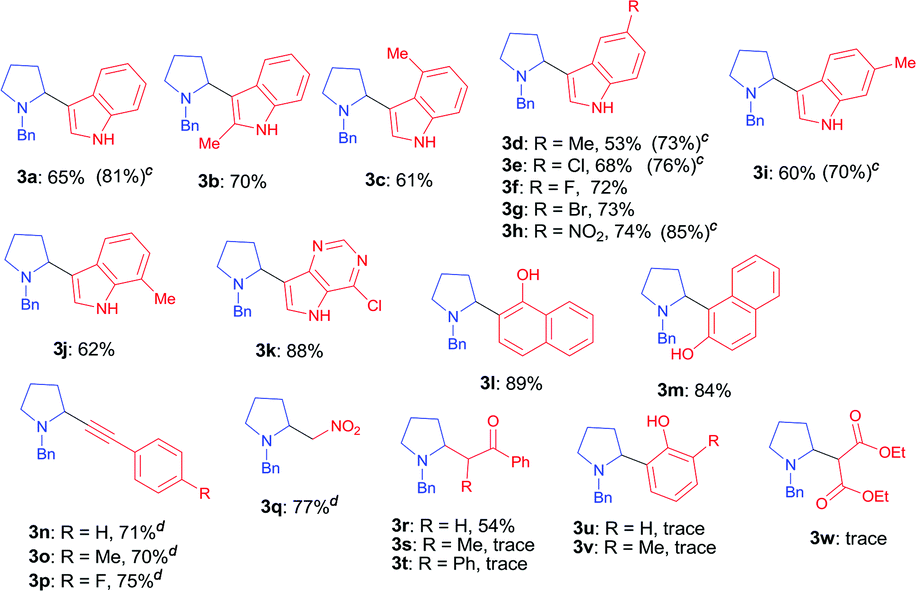
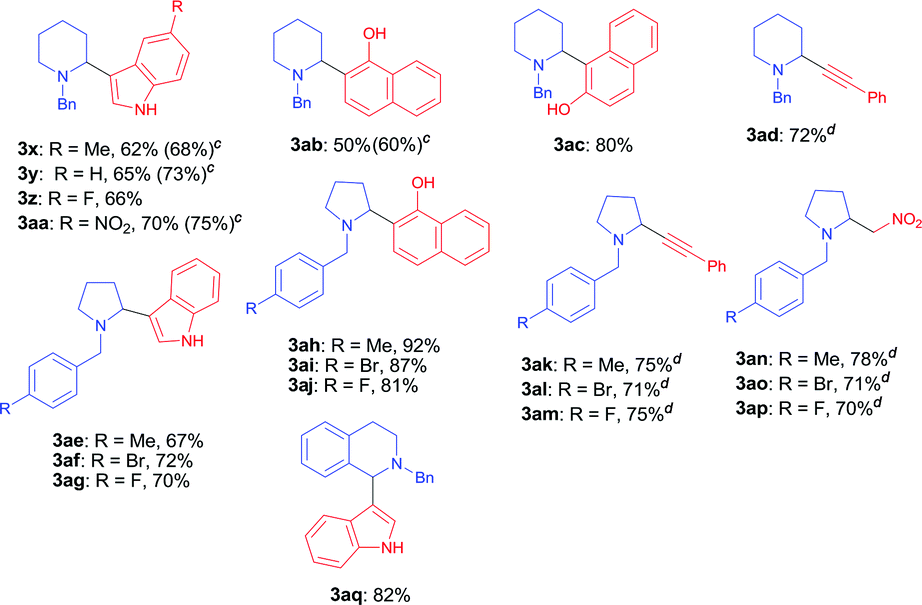
With the optimal reaction condition in hand (entry 26 in Table 1), we next set out to examine the scope of the new protocol with different C–H nucleophiles and the results were shown in Table 2. To our delight, this reaction exhibited broad substrate scope with respect to a wide range of sp2, sp3 and sp C–H nucleophiles. Indoles with either electron-withdrawing groups (5-F, 5-Cl, 5-Br, 5-NO2) or electron-donating groups (4-Me, 5-Me, 6-Me and 7-Me) reacted smoothly with N-benzyl proline 1a and afforded the corresponding C(sp2)–C(sp3) coupling products 3a–3j in moderate to good yields (52–74%), albeit indoles bearing electron-withdrawing substituents (3f, 3g, 3h) provided slightly higher yields than that with electron-donating groups. It should be noted that increasing the catalyst loading to 10 mol% could further improve the yields to 73–81% (see the yields in parentheses). The coupling of 1a with 4-chloro-5H-pyrrolo[3,2-d]pyrimidine also proceeded smoothly, affording 3k in 88% yield. Moreover, α- and β-naphthols were suitable coupling partners, the corresponding C(sp2)–C(sp3) coupling products 3l and 3m can be obtained in 89% and 84% yields, respectively. In addition, C(sp)–C(sp3) bond could also be constructed via the reaction of various aromatic alkynes with N-benzyl proline, affording the related coupling products 3n–3p in good yields (70–75%). Furthermore, this method was also suitable for the decarboxylative coupling of C(sp3)–C(sp3) bonds, and the reactions proceeded smoothly for nitromethane and acetophenone nucleophiles. Among them, products 3q and 3r are synthetic precursors for some bioactive natural products.10 The hindered C–H nucleophiles including propiophenone, 1,2-diphenylethan-1-one and diethyl malonate were also tested in this reaction with N-benzyl proline 1a under standard conditions. Unfortunately, these nucleophiles were found to be incompatible with the developed protocol, and gave complex mixture with only trace products (detected by mass spectroscopy). Furthermore, o-cresol and phenol were also tested, but only trace products were obtained (detected by mass spectroscopy).
We next explored the scope of cyclic α-amino acids and the results were summarized in Table 3. Besides pyrrolidine, piperidine is also common heterocyclic skeleton existing in numerous natural products11 albeit the decarboxylative coupling of piperidine carboxylic acid has less been explored.5,6,7a We examined the decarboxylative coupling of N-benzyl piperidine carboxylic acid with diverse C–H nucleophiles including indoles, naphthols and alkynes. A range of indoles bearing electron-donating groups (–Me) or electron-withdrawing groups (–F and –NO2) at the C5 position reacted smoothly with N-benzyl piperidine-2-carboxylic acid, affording the coupling products 3x–3aa with moderate to good yields (62–70%). Increasing the amount of copper catalyst to 10 mol% resulted in higher yields (68–75%). This coupling reaction also proceeded successfully with α-naphthol and β-naphthol, leading to 3ab and 3ac in 50% and 80% yields, respectively. Moreover, phenylacetylene readily underwent the coupling to afford the corresponding piperidine-alkyne product 3ad with good yield. A range of N-benzyl prolines bearing different substituents (F-, Br- and Me-) at the para-position of the phenyl ring were also investigated, giving various products including indolyl pyrrolidines 3aq–3as, tertiary aminonaphthols 3ah–3aj, propargylamines 3ak–3am and β-nitroamines 3an–3ap in moderate to high yields (67–92%). In particular, tetrahydroisoquinoline acid, an important moiety in many pharmaceuticals and natural products, was also found to be competent substrate under the standard decarboxylative coupling conditions (3aq, 82%). Moreover, when N-benzyl L-proline was used in this reaction, only racemic product 3a was obtained, which was in agreement with the oxidative decarboxylation of L-proline and subsequent racemization of product.
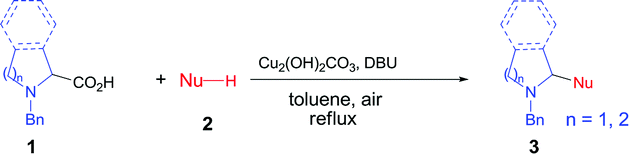
To explore the practical utility of this method, a gram-scale experiment was performed with N-benzyl proline 1a and α-naphthol 2l (Scheme 1). The coupling product 3l could be obtained in 88% yield with only 1 mol% of copper catalyst, which demonstrated that this protocol was scalable and practical.
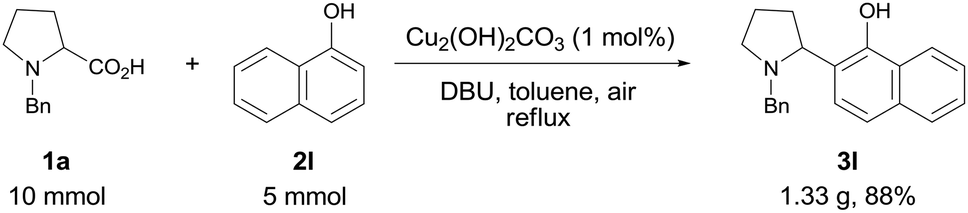 | ||
| Scheme 1 Gram-scale preparation of 3l. | ||
The efficiency and convenience of this copper-catalyzed decarboxylative coupling reaction was further demonstrated in the facile synthesis of IBR2 analogue 4a, which is a Rad51 inhibitor for the treatment of triple-negative human breast cancer (Scheme 2).12,13 The decarboxylative coupling of tetrahydroisoquinoline acid with indole was employed as the key step to afford intermediate 3aq, subsequent N-deprotection and N-sulfonylation led to the IBR2 analogue 4a (70% yield, 3 steps). This new synthetic route is a significant simplification when comparing with the previous eight to thirteen steps using conventional synthetic approaches.12,13a
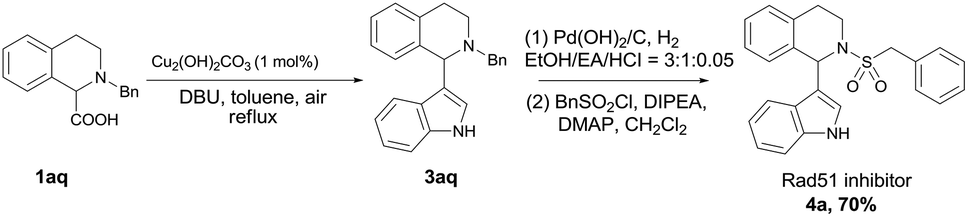 | ||
| Scheme 2 New synthetic route of Rad51 inhibitor. | ||
In order to gain mechanistic insight into this transformation, some control experiments were performed. The reaction proceeded well in the presence of radical scavenger TEMPO (2,2,6,6-tetramethylpiperidinooxy), affording the desired product 3a in 64% yield (Scheme 3a), which ruled out the involvement of a radical coupling pathway. The function of N-protective group was also explored. When N-Boc-proline or N–H free proline was employed under the standard conditions with indole, no desired product was observed (Scheme 3b), which implied the necessity of benzyl group for the formation of azomethine ylide intermediate.5,6,7a To test whether the azomethine ylide intermediate 5a was involved in this reaction, we have prepared the deuterium-labeled N-benzylproline (1a-d7) and subjected it to the reaction with indole under our optimized conditions. The 1H NMR spectrum of 3a-d7 indicated that all deuterium atoms were retained, no deuterium–hydrogen exchange occurred at the benzylic position, which excluded the possibility of azomethine ylide intermediate 5a (Scheme 3c). To our delight, N-benzyl iminium intermediate 5b (m/z = 160) could be detected by ESI-MS from the reaction mixture, which demonstrated that N-alkyl iminium might be the reactive intermediate (Scheme 3d). We also tried to prepare the possible N-benzyliminium intermediate 5d in situ from 5c, then carried out the subsequently Mannich reaction with indole in the presence of DBU. The desired product 3y can be isolated in 21% yield (Scheme 3e).
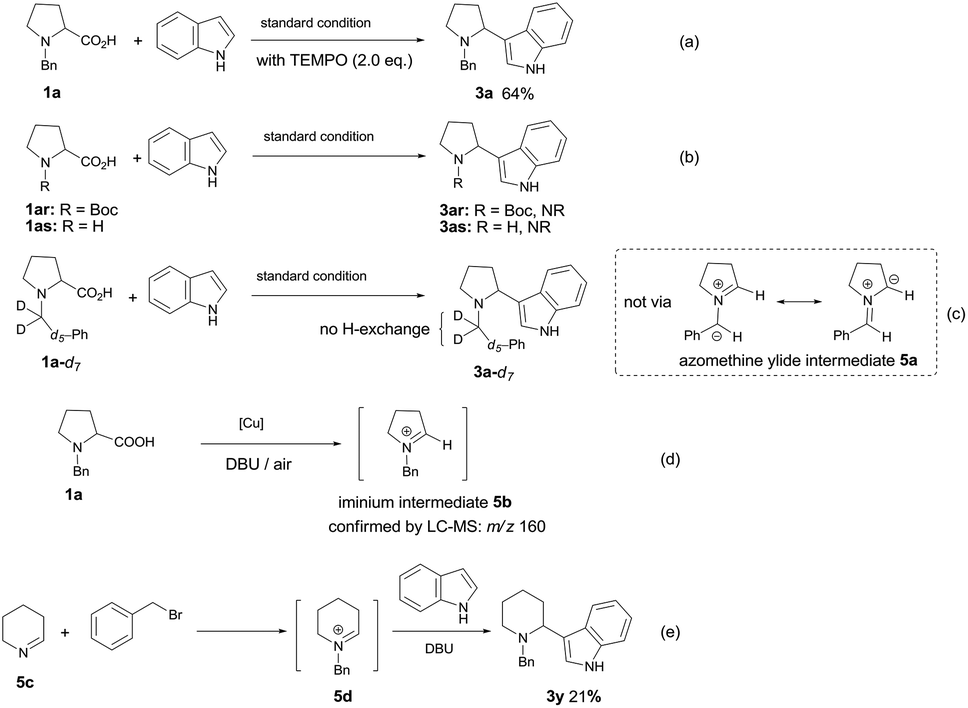 | ||
| Scheme 3 Control experiments. | ||
On the basis of literatures5,6 and our experimental observations, a tentative mechanism has been depicted in Scheme 4. Initially, the deprotonation of N-Bn-proline 1a by DBU generated intermediate A, then the oxidative decarboxylation of A catalyzed by Cu2(OH)2CO3 afforded N-benzyl iminium intermediate B, which underwent a Mannich reaction with carbon nucleophile to deliver the desired product. The regeneration of the Cu catalyst was realized via the oxidation of O2 in air. It should be mentioned that this mechanism is different from Li's work, where an azomethine ylide was proposed as reactive intermediate.5
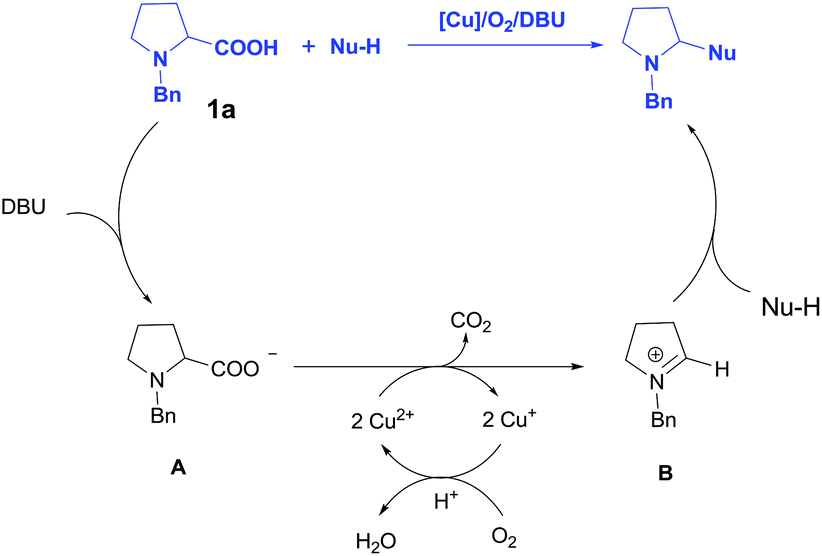 | ||
| Scheme 4 Proposed mechanism for the copper-catalyzed decarboxylative C–H cross-coupling. | ||
Conclusions
In summary, we have developed a ligand-free, copper-catalyzed aerobic decarboxylative coupling of cyclic α-amino acids and diverse C–H nucleophiles (indoles, naphthols, alkynes, ketones and nitroalkanes) with extremely low catalyst loading (1 mol%). This simple catalytic system enabled the direct construction of various C(sp3)–C(sp3), C(sp3)–C(sp2) and C(sp3)–C(sp) bonds in numerous nitrogen-containing heterocycles. The efficiency and practicability of this protocol was also demonstrated by gram-scale preparation and three-step synthesis of a Rad51 inhibitor.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21502240), the Guangdong Natural Science Foundation (No. 2015A030313184), Science and Technology Program of Guangzhou (No. 201607010118).Notes and references
- For selected reviews, see: (a) Y. Wei, P. Hu, M. Zhang and W.-P. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed; (b) N. Rodrguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (c) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed.
- For selected recent examples, see: (a) J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS PubMed; (b) T. Qin, J. Cornella, C. Li, L. P. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed.
- (a) F.-X. Felpin and J. Lebreton, Eur. J. Org. Chem., 2003, 19, 3693–3712 CrossRef; (b) J. R. Liddell, Nat. Prod. Rep., 2002, 19, 773–781 RSC; (c) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435–446 RSC; (d) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (e) P. A. Procopiou, C. Browning, P. M. Gore, S. M. Lynn, S. A. Richards, R. J. Slack and S. L. Sollis, Bioorg. Med. Chem., 2012, 20, 6097–7108 CrossRef CAS PubMed; (f) P. Ruggeri, L. Fumagalli, M. Binda, V. Mucchietto, M. Sciaccaluga, R. Budriesi, S. Fucile and M. Pallavicini, J. Med. Chem., 2015, 58, 6665–6677 CrossRef PubMed.
- (a) D. Das and D. Seidel, Org. Lett., 2013, 15, 4358–4361 CrossRef CAS PubMed; (b) W. Chen, R. G. Wilde and D. Seidel, Org. Lett., 2014, 16, 730–732 CrossRef CAS PubMed; (c) W. Chen and D. Seidel, Org. Lett., 2014, 16, 3158–3161 CrossRef CAS PubMed; (d) D. Seidel, Acc. Chem. Res., 2015, 48, 317–328 CrossRef CAS PubMed.
- H.-P. Bi, L. Zhao, Y.-M. Liang and C.-J. Li, Angew. Chem., Int. Ed., 2009, 48, 792–795 (Angew. Chem., 2009, 121, 806–809) CrossRef CAS PubMed.
- H.-P. Bi, W.-W. Chen, Y.-M. Liang and C.-J. Li, Org. Lett., 2009, 11, 3246–3249 CrossRef CAS PubMed.
- (a) C. Zhang and D. Seidel, J. Am. Chem. Soc., 2010, 132, 1798–1799 CrossRef CAS PubMed; (b) H.-P. Bi, Q. Teng, M. Guan, W.-W. Chen, Y.-M. Liang, X. Yao and C.-J. Li, J. Org. Chem., 2010, 75, 783–788 CrossRef CAS PubMed; (c) D.-X. Yang, D.-P. Zhao, L.-J. Mao, L.-Q. Wang and R. Wang, J. Org. Chem., 2011, 76, 6426–6431 CrossRef CAS PubMed; (d) S. U. Dighe, A. K. KS, S. Srivastava, P. Shukla, S. Singh, M. Dikshit and S. Batra, J. Org. Chem., 2015, 80, 99–108 CrossRef CAS PubMed.
- Y.-C. Shih, P.-H. Tsai, C.-C. Hsu, C.-W. Chang, Y. Jhong, Y.-C. Chen and T.-C. Chien, J. Org. Chem., 2015, 80, 6669–6678 CrossRef CAS PubMed.
- (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed; (b) Z.-W. Zuo, H. Cong, W. Li, J.-W. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed; (c) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 (Angew. Chem., 2015, 127, 15854–15863) CrossRef CAS PubMed.
- (a) D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 (Angew. Chem., 1998, 110, 2724–2772) CrossRef CAS; (b) F. Roessler, D. Ganzinger, S. Johne, E. Schoepp and M. Hesse, Helv. Chim. Acta, 1978, 61, 1200–1206 CrossRef CAS.
- (a) S. Nara, R. Tanaka, J. Eishima, M. Hara, Y. Takahashi, S. Otaki, R. J. Foglesong, P. F. Hughes, S. Turkington and Y. Kanda, J. Med. Chem., 2003, 46, 2467–2473 CrossRef CAS PubMed; (b) R. He, T. Kurome, K. M. Giberson, K. M. Johnson and A. P. Kozikowski, J. Med. Chem., 2005, 48, 7970–7979 CrossRef CAS PubMed.
- J. Zhu, H. Chen, X. E. Guo, X.-L. Qiu, C.-M. Hu, A. R. Chamberlin and W.-H. Lee, Eur. J. Med. Chem., 2015, 96, 196–208 CrossRef CAS PubMed.
- (a) X.-L. Qiu, J. Zhu, G. Wu, W.-H. Lee and A. R. Chamberlin, J. Org. Chem., 2009, 74, 2018–2027 CrossRef CAS PubMed; (b) T.-W. Chung, Y.-T. Hung, T. Thikekar, V. V. Paike, F. Y. Lo, P.-H. Tsai, M.-C. Liang and C.-M. Sun, ACS Comb. Sci., 2015, 17, 442–451 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C NMR spectra. See DOI: 10.1039/c8ra02340a |
| This journal is © The Royal Society of Chemistry 2018 |