
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCerium oxide based active catalyst for hydroxylammonium nitrate (HAN) fueled monopropellant thrusters†
Ruchika Agnihotri * and
Charlie Oommen
* and
Charlie Oommen
Department of Aerospace Engineering, Indian Institute of Science, Bangalore 560012, India. E-mail: ruchikaagni@gmail.com; Tel: +91 80 2293 3141
First published on 19th June 2018
Abstract
Hydroxylammonium nitrate (HAN) is an energetic ionic liquid which is fast emerging as a promising environmentally friendly, high performing monopropellant for space propulsion application. The high performance due to the higher adiabatic temperature for HAN based compositions also poses challenges as high temperature tolerant catalysts have to be developed for its decomposition. A novel cobalt doped cerium oxide based catalyst has been prepared by the co-precipitation route and characterized by SEM/EDS, XRD, and XPS. The effectiveness of the catalyst in decomposing HAN has been tested using thermo-analytical techniques. An evolved gas analysis (EGA) to examine decomposition products and the possible reaction mechanism was also performed using the hyphenated DTA-TG-FTIR technique. Formation of an in situ Ce3+/Ce4+ ion couple in ceria during co-precipitation was found to be critical in deciding the reactivity of HAN decomposition over the catalyst. The activity of the catalyst was also examined in a batch reactor for its longevity. The prepared catalyst was found to be more versatile and durable than a hitherto reported alumina supported iridium catalyst in the present studies.
Introduction
Hydroxylamine nitrate (HAN) has assumed significant importance in recent times as a promising greener substitute to hydrazine which currently is the most often used monopropellant in space propulsion. Hydrazine, while offering excellent performance and reliability, has serious issues due to its established carcinogenic nature, which escalates the overall cost of the mission due to exorbitant handling charges. HAN on the other hand is safe to handle besides offering plenty of other advantages. This compound has multiple advantages over hydrazine in terms of sensitivity, toxicity and volatility.1–4 It is generally employed as a ternary system comprising HAN, water and fuel. While an aqueous solution facilitates versatile handling, the positive oxygen balance of the compound allows inclusion of compatible fuel components, which raises the performance level of the propellant formulations. HAN formulations are reported to have lower crystallization points, higher density and volumetric impulse. Apart from its projected use as a monopropellant, it is also used as an insensitive liquid oxidizer in gun propellants. Besides, HAN along with nitric acid is used as a reducing agent in nuclear processing industry for plutonium extraction.5,6In a typical monopropellant thruster, usually a single chemical compound is injected over a heated catalytic bed which assists decomposition of the compound with short ignition delays. The decomposed products are subsequently expanded through a nozzle to produce desired thrust required for orbital and attitude correction. Schematic of a monopropellant thruster is shown in Fig. 1.
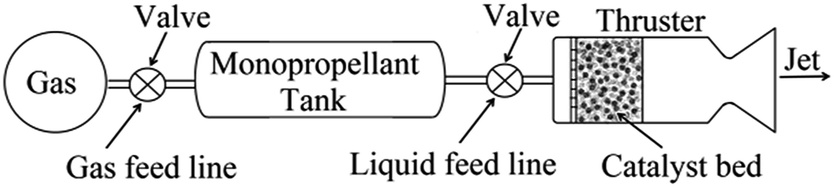 | ||
| Fig. 1 Schematic of a monopropellant thruster. | ||
Transition from hydrazine to HAN is not straightforward as challenges to a successful and reliable thruster technology are plenty. Among these challenges, development of a high temperature tolerant catalyst is foremost as the temperature levels in HAN thrusters are much higher unlike hydrazine thruster which generates a much lower temperature. An optimally designed catalyst bed manages and optimizes the hydrazine thruster performance. Though the same hydrazine catalyst, Ir/γ-Al2O3, facilitates a low temperature decomposition when used in HAN thrusters, the physical integrity and catalyst efficiency over a longer period of operation decline which renders it unfit for long duration mission.7 Loss of catalytic activity through poisoning or attrition leads to propellant pooling and hard start or explosion which is not conducive for a reliable space mission. Oxidation of iridium, loss of active metal surface and sintering caused due to high temperatures are considered to be the major indicators for catalytic deactivation. Besides, the γ-Al2O3 support traditionally used for hydrazine thrusters can undergo phase transition to other phases and are reported to be unfit for high temperature applications.7,8 There have been attempts to make more temperature tolerant active catalysts based on platinum metal supported over alumina doped with various elements like Si, Ba etc.8,9 Generally it is the platinum group metals like iridium, platinum, rhodium, rhenium etc. which are reported to be effective catalyst for HAN decomposition. Apart from alumina and silica, either in doped or pure phase, many other refractory materials also have been tried and tested in the past as catalyst support.3,10–13 It was observed that the autocatalytic nature of HAN decomposition triggered by nitric acid formed in situ and thermal shock during decomposition renders many of these catalyst ineffective over longer duration.13,14 Rocket Research Corporation (USA) has developed many catalysts in the past which are presumed to be effective for HAN decomposition.12,15
The catalyst in the thruster can be in the form of supported metal pellets, wire mesh or as monolith.14,16 Besides the bed geometry that decides the optimum pressure drop, the cost of active metal is critical in the development of monopropellant thruster. In recent times there have been many attempts to replace precious metal catalysts with low cost metal oxides in many combustion applications. Of these, two oxides namely, cobalt oxides and cerium oxides have demonstrated their catalytic effectiveness in many high temperature reaction systems in the recent times.17–19 Cobalt–cerium mixed oxides are reported to be good catalysts for soot and CO combustion.17 Co3O4 over ceria support is considered to be an effective catalyst in methane reduction.18 It is also known for its catalytic activity in NOx decomposition.19 The multivalent oxidation states of cobalt (II & III) and cerium (III & IV) oxide along with active oxygen species in these oxides play significant roles in the redox reactions. It was perceived during our endeavour to develop high temperature tolerant catalyst for HAN decomposition that cerium oxide, one of the widely known heterogeneous catalysts used in three way catalytic convertor, could be a good self-adjusting catalyst in combination with previously known active precious metals. Incidentally the products of a ternary propellant system consisting of HAN-TEAN-water, a reported propellant formulation, may produce products mainly consisting of NOx and COx along with other products. NOx being a primary product of decomposition of HAN, ceria could presumably convert NOx to nitrogen, and thereby alter the molecular weight of products which has direct bearing on the characteristic velocity.20 Interestingly, ceria catalysts are also known for water gas shift reaction which involves carbon monoxide.21 Synergistic promotional effects induced by doping are well known in the domain of catalysis. Since ceria can become non stoichiometric in the presence of dopants in its matrix and alter reaction routes,21–25 it was perceived that the compound could alter the product profile of HAN monopropellant systems in ways similar to automobile catalytic convertor. The catalytic traits of cerium oxide are mainly attributed to two properties of ceria: first, the high mobility and storage capacity of oxygen within the lattice and second, the ease with which cerium changes between Ce4+ and Ce3+. These properties combined with abundance of cerium on earth makes them attractive low cost alternative to precious metals. The fluorite structure of the oxide allows easy diffusion of oxygen on the surface and facilitates reduction of the surface with relative ease.21–24
Incidentally, cobalt oxides are known as sintering aids for ceria.26,27 Ceria which normally has a high temperature requirement for sintering can be sintered at lower temperatures in the presence of cobalt oxides. Apparently, these two oxides in a combined form present an alternate novel catalytic system with added advantages and appears to have been unexplored for HAN decomposition. In the present study we have explored the catalytic activity of ceria and ceria–cobalt mixed oxides in HAN decomposition.
Experimental
Materials
All the precursors required for the catalyst preparation were of analytical grade and were obtained from Sigma Aldrich.Preparation of catalyst
Cobalt doped ceria catalyst was prepared adopting co-precipitation method. Desired levels of the precursor ((NH4)2Ce(NO3)6) and doping agent (Co(NO3)2·6H2O) were dissolved in DM water. 1 M NaOH was used as the precipitating agent in the procedure adopted. The resulting precipitate was filtered, washed and dried overnight before subjecting to high temperature calcination. After subjecting the powder to calcination at 500 °C in air for 4 hours, forming of the powder was done with slip casting.27 This was followed by firing at 1000 °C (10 h soaking, 5°C min−1 ramp, air atmosphere) to achieve desired densification.27–30 Different ceria samples containing varying amounts of cobalt (10, 15, 20, 24, 26 and 28 mol%) were prepared and designated as CeCo y (y is the cobalt mole percentage initially added). In order to designate used catalyst after exposing to HAN, symbol x was suffixed to catalyst label.Iridium supported on γ-Al2O3 catalyst was prepared by multiple wet impregnation method. Iridium chloride was used as the precursor for iridium metal. The γ-Al2O3 pellets after impregnation and drying was calcined (400 °C for 2 h) and reduced in flowing hydrogen (500 °C for 12 h) atmosphere to get finely dispersed iridium coated alumina pellets.
HAN preparation
HAN was prepared by the neutralization of hydroxylamine with nitric acid.1 The concentration of the final solution was analysed by estimating the amount of water in the solution. The purity of the propellant prepared in-house was confirmed by various spectroscopic and analytical methods as reported elsewhere.1 A 75% concentrated solution of HAN was used for the study. The critical parameter being water content was double checked using Karl-Fisher titerator and TGA, with an apparent difference of 5% between the values. No ternary systems were examined in this work.Performance analysis
The efficiency of the catalyst prepared was assessed based on its ability to decompose HAN in thermal analysis studies. The temperature of onset of decomposition, rate of decomposition extracted from the rate of weight loss, and exothermicity during the decomposition were primarily utilized to characterize the catalyst's efficiency. A Perkin Elmer STA 6000 was used for simultaneous DTA-TG (differential thermal analysis-thermogravimetric analysis) studies. A heating rate of 10°C min−1 in nitrogen atmosphere was employed during the thermal analysis. A Perkin Elmer DSC 8000 was used for calorimetric experiments. A FTIR-DTA-TG hyphenated system was used for evolved gas analysis (EGA), where FTIR is Perkin Elmer make Frontier IR. All FTIR spectra are compared against open-source NIST Chemistry webbook (http://www.webbook.nist.gov).A schematic of the constant volume batch reactor used in the study for catalyst activity measurement, constructed as per the concept adopted by Kappenstein et al.,31 is shown in Fig. S1.† It essentially consists of a small enclosed chamber wherein the pressure and temperature rise arising from the decomposition of the monopropellant when injected onto a catalyst bed can be monitored as a function of time. In practice, around 200 μL of the monopropellant is dropped on to a heated catalytic bed packed with pellets. The pressure rise (overpressure in the range 0–3 bar) within the chamber of the reactor is monitored against time using electronic pressure transducers and data acquisition systems. The catalytic bed could be maintained up to 250 °C through a PID controlled band heater. The pressure rise data was acquired through a NI make USB based 14 bit 8 channel data card at 10 Hz.
Material characterization
Powder X-ray diffraction (XRD) of the catalyst was recorded with Rigaku Smartlab powder and thin film XR diffractometer having a Cu Kα source (1.5418 Å). The diffraction peaks of the samples examined were compared with Inorganic Crystal Structure Database (ICSD). The Ce to Co ratios were obtained using an Ultra 55 FE Karl Zeiss EDS. X-ray photoelectron spectroscopy (XPS) was performed on Axis Ultra XPS. High temperature thermal characterization (upto 1500 °C) of the catalyst was carried out using Netzsch STA 409 PC/PG at a heating rate of 10°C min−1.Results and discussion
Catalyst activity measurement
| Sample | Td (°C) | TG slope (mg min−1) | Specific decomposition enthalpy (J g−1) |
|---|---|---|---|
All values are average of atleast three measurements.a Catalyst to HAN ratio in every sample is kept 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 by weight. :10 by weight. |
|||
| Pure HAN | 199.5 ± 2.2 | 2 ± 0.003 | 551.4 ± 35.5 |
| CeO2 | 190.1 ± 9.2 | 3.6 ± 1.3 | 537.3 ± 23 |
| CeCo 10 | 139.3 ± 1.4 | 6.2 ± 1.1 | 589.7 ± 10.5 |
| CeCo 15 | 146.0 ± 3.1 | 10.5 ± 2 | 544.2 ± 35.2 |
| CeCo 20 | 130.3 ± 3.5 | 58.2 ± 7 | 589.9 ± 30.2 |
| CeCo 24 | 129.1 ± 2.4 | 71 ± 12.6 | 543.3 ± 28.1 |
| CeCo 26 | 130.2 ± 1.5 | 293.7 ± 55.9 | 643.1 ± 19 |
| CeCo 28 | 149.3 ± 3 | 29.1 ± 4.6 | 568.9 ± 13.5 |
| Ir/γ-Al2O3 | 119.7 ± 2 | 305.6 ± 36.7 | 377 ± 40.8 |
 | ||
| Fig. 2 Thermograms for HAN catalyst systems (a) TGA (inset: weight loss vs. time), (b) DSC curves and (c) images of catalyst after batch reactor decomposition studies (i) Ir/γ-Al2O3 before, (ii) Ir/γ-Al2O3 after 15 injections, (iii) CeCo 26 before and (iv) CeCo 26 after 50 injections. | ||
With the inclusion of cobalt, there appears to be a favourable shift in the decomposition temperature towards lower temperature. This is also accompanied by a significantly higher rate of decomposition (exhibited in the slope of TG curve) and relatively higher exothermicity. For ceria samples containing less cobalt, the reaction gets completed only at higher temperatures (180–200 °C) despite low temperature initiation.
The highest catalytic activity was observed for CeCo 26 composition. The decomposition reaction for this catalyst was found to have initiated at 120–130 °C with significantly higher exothermicity. Besides, slope of weight loss curve which corresponds to rate of reaction was also highest for CeCo 26 catalysed HAN decomposition which has a direct bearing on the momentum. However, further addition of cobalt was found to lower the high catalytic activity observed for CeCo 26. The lowering of decomposition temperature aids the thruster in saving on board power over longer duration by maintaining the catalyst bed at relatively lower temperatures. The higher exothermicity observed for this catalytic system can translate to higher specific impulse and possibly an altered reaction mechanism. The sharp DSC peaks shown in Fig. 2b shows the instantaneous nature of the energy release. The specific decomposition enthalpy given in Table 1 calculated from DSC data can be used for a comparison of catalytic activity. Though some of the catalyst composition show similar specific enthalpy values, CeCo 26 stands out due to its every value being highest. Apparently, the performance of the new Ce–Co based oxide catalyst appears extremely promising when compared to the benchmark catalyst based on Ir/γ-Al2O3. The parameters which decide Ce–Co catalyst's supremacy include low decomposition temperature, higher enthalpy, higher weight loss which represents higher mass generation rate and higher thrust. The only issue in the cerium oxide based catalyst is the slightly higher initiation temperature when compared to Ir/γ-Al2O3. However, in a practical situation decomposition temperature is least important among all other parameters as the thruster's catalyst bed is kept well above 300 °C which is way above the observed decomposition temperature to avoid cold start problems. In a continuous mode operation wherein, the temperature goes above 1000 °C, decomposition temperature plays possibly insignificant role as the bed is already heated to very high temperature levels.
The efficiency of the catalyst in a pulse mode operation can be more effectively examined in a batch reactor. Catalyst bed of the reactor was maintained at 120 °C for entire duration of injections. The pressure rise data obtained for CeCo 26 and Ir/γ-Al2O3 are given in Table 2. The pressure–time curves obtained for CeCo 26 is shown in Fig. S2.† The most significant observation is that the cerium based catalyst retained its activity for the 50 injections without any visible signs of deterioration in activity while iridium based catalyst showed decline in activity just after 15 injections either due to possible poisoning or thermal shock based attrition.
| Sample | ΔP (mbar) | dP/dt (mbar s−1) |
|---|---|---|
| CeCo 26-HAN | 373.7 | 81.33 (±6%) |
| Pure HAN | 370.46 | 70.66 (±6%) |
| Ir/γ-Al2O3-HAN | 335.35 | 72.43 (±6%) |
The cerium oxide based catalyst pellets remained remarkably intact even after the 50 trials while Ir/γ-Al2O3 catalyst was found to have broken into pieces and dust due to the highly exothermic reaction, as shown in Fig. 2c.
The higher dP/dt and ΔP values from batch reactor studies along with higher exothermicity obtained in DSC studies point to modified reaction routes for CeCo 26 catalyst based systems (discussed later along with product profile). These high dP/dt values matches with higher slopes observed in TGA studies except in pure HAN wherein thermal history appears to play a significant role in deciding the rate of reaction. The difference in the ΔP value between pure HAN and catalysed system is not significant. Similar observations have been made earlier also wherein thermal and catalytic decompositions were examined.9
Catalyst characterization
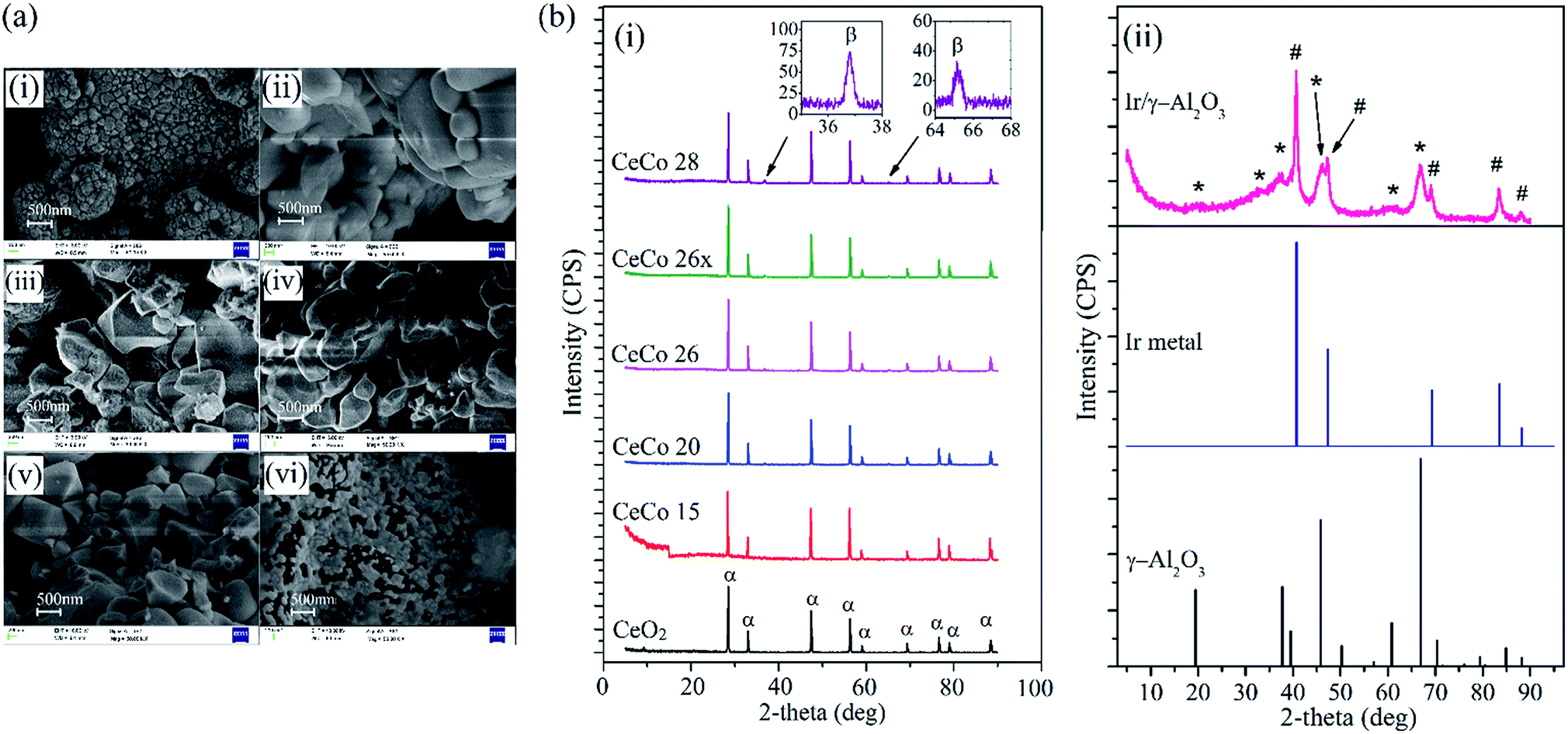 | ||
| Fig. 3 (a) SEM images of ceria catalyst with different cobalt doping in final pellet form (i) CeO2, (ii) CeCo 20 (iii) CeCo 26, (iv) CeCo 26x, (v) CeCo 28 and (vi) Ir/γ-Al2O3. (b) Stack plot of XRD curves for different catalysts (i) doped ceria catalysts (α for CeO2 and inset showing β for Co3O4 peaks) and (ii) Ir/γ-Al2O3 (* and # stands for γ-Al2O3 and Ir metal, respectively). | ||
The intact elemental compositional ratio observed in used CeCo 26x is also indicative of the efficiency of catalyst in resisting any form of poisoning during HAN decomposition.
The Co3O4 peak at 36.7° becomes visible only beyond 15 mol% loading of cobalt. Similar observation is reported elsewhere, wherein at low cobalt loading, diffraction peaks corresponding to neither CoO nor Co3O4 can be seen in cobalt oxide doped ceria matrix.17,18 The crystallite sizes calculated from the most intense XRD reflections of CeO2 show an initial rise (upto 15 mol%) and subsequent fall due to well dispersed cobalt oxide restricting growth of CeO2 crystallites.17,19 The crystallite size at highest loading of cobalt (i.e. 28 mol%) is lowest among doped ceria catalyst species which still remains higher than pure CeO2 crystallite size.
The crystallite size of active cobalt doped catalysts (CeCo 26) was found to be 51 nm. Earlier studies on cobalt doped ceria, report variations in dispersion of cobalt oxide where best dispersion and catalytic activity are reported when cobalt doping is around 20–30 mol%.17,18 Interestingly, a similar trend is observed for HAN decomposition also. The cell parameter of cobalt doped cerium oxide remain more or less identical, which again is consistent with observation made elsewhere.18,25 Since the catalytic activity of the composite oxide during HAN decomposition studies using thermal techniques showed decline in activity for higher loading of cobalt, we did not further explore characterization of Co doped ceria with higher cobalt content. XRD studies on Ir/γ-Al2O3 showed reflections corresponding to iridium metal (ICSD collection code-64992) and γ-Al2O3 (ICSD collection code-66558) with no sign of any of iridium oxide.
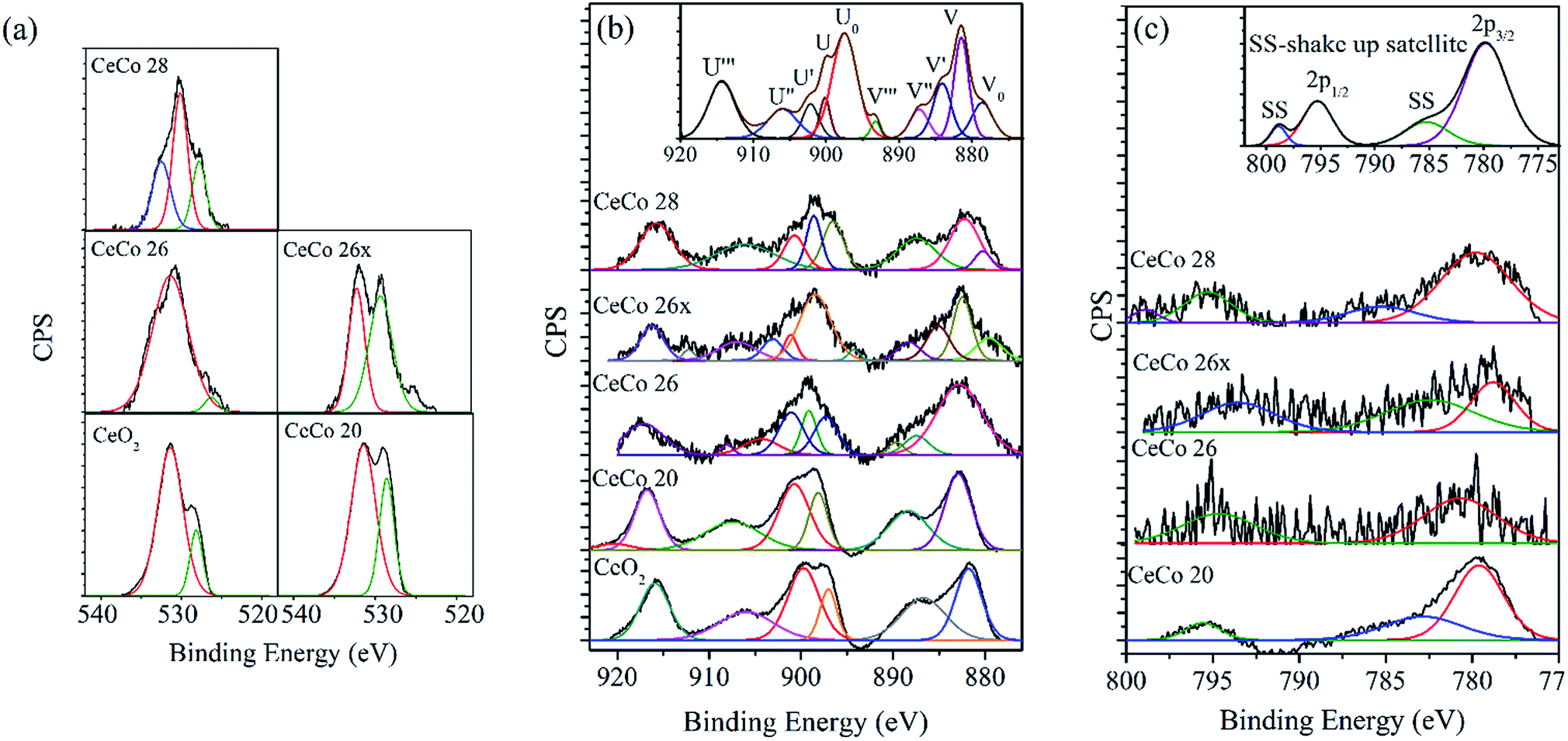 | ||
| Fig. 4 Comparative XPS for (a) O 1s orbital, (b) Ce 3d orbital, and (c) Co 2p orbital (insets are typical final state positions). | ||
The XPS spectra for O 1s obtained for various doped ceria catalysts are shown in Fig. 4a. It is reported that the O 1s peak is generally broad for multi component oxide samples due to the non-equivalence of the surface O ions.25,33 O 1s peak tends to be broad with multiple overlapping components depending on the nature of oxidation state. The peak at ∼529.8 eV is presumed to be due to lattice oxygen while the broad peak ranging from ∼530–534 eV could be attributed to chemically adsorbed oxygen, weakly bonded oxygen, adsorbed water or carbonates.17,25,33 The intensity of adsorbed oxygen peak appears to be more prominent than the lattice oxygen. The ratio of the intensity of lattice oxygen to adsorbed oxygen peaks appears to vary significantly with the cobalt doping with CeCo 26 showing large amount of the latter. Significantly high intensity of oxygen other than lattice oxygen in CeCo 26, the most active form of catalyst, is a favourable feature.18,25 Possibly, adsorbed oxygen is not the prime criteria for activity, though their propensity to favour oxidation is known.25 It could also be reflection of non-equivalence of various cationic and anionic species on the surface. A summary of the quantitative estimation of percentage lattice oxygen is given in Table 3. The pronounced effect of adsorbed oxygen maybe treated entirely as a surface feature.
| Co:Ce atomic ratios |
||||
|---|---|---|---|---|
| Sample | Precursor solution | Final solid sample | Ce3+/(Ce3+ + Ce4+) mol% | Olattice/(Olattice + Oadsorbed) mol% |
| Pure CeO2 | 0 | 0 | 0 | 18.7 |
| CeCo 20 | 20:80 |
15.4:84.6 |
0 | 31.8 |
| CeCo 26 | 26:74 |
14:86 |
18.5 | 3.7 |
| CeCo 26x | 26:74 |
13.9:86.1 |
22.3 | 54 |
| CeCo 28 | 28:72 |
20.7:79.8 |
0 | 17.8 |
| Ir wt% in final sample | |
|---|---|
| Ir/γ-Al2O3 | 31.67 |
The Ce 3d XP spectra hold information to identify and quantify all primary valence species of cerium but the spectral features get convoluted due to Ce 4f orbital hybridization with O 2p orbital and partial occupancy of 4f valence orbital as reported.29,32 Accordingly, there are five spin orbit doublets for two multiplets, classed as 3d5/2 and 3d3/2 in a Ce 3d spectrum.29,32 While defining Ce4+ 3d final state, peaks for 3d3/2 are labelled as U (901.1 eV), U′′ (907.6 eV), U′′′ (916.4 eV) and 3d5/2 as V (882.3 eV), V′′ (888.4 eV), and V′′′ (898.2 eV); similarly for Ce3+ 3d final states, U0 (899.2 eV), U′ (904.6 eV) and V0 (880.3 eV), V′ (886) are the corresponding peaks for 3d3/2 and 3d5/2 ionizations, respectively. Doublet designations U0/V0, U/V, U′/V′, U′′/V′′ and U′′′/V′′′ are assigned to represent hybridization states of Ce 3d94f2 O 2p5, Ce 3d94f2 O 2p4, Ce 3d94f1 O 2p6, Ce 3d94f1 O 2p5 and Ce 3d94f0 O 2p6, respectively.
According to Jampaiah et al.,25 the feature at 918 eV (U′′′, shake up satellite) can be attributed as signature Ce4+ state. This peak can very well be used to differentiate Ce4+ and Ce3+. So, reduction in its area fraction and it's creeping towards lower binding energy, is a definitive sign of Ce4+ concentration drop. The peak intensity profile and binding energy of U′′′ in our studies, does comply with literature, and is lowest for catalytically active CeCo 26 species in comparison to CeO2 indication of Ce3+ presence. Other characteristic highlight of the influence of cobalt dopant on Ce oxidation state is the increase in 886 and 904 eV peak intensities in CeCo 26 sample, a characteristic feature of Ce3+ state.17,25,29,32 In other words CeCo 26 has undisputable presence of Ce3+ which remains intact even in CeCo 26x sample also. It has been shown in earlier XPS studies on mixed oxides of cobalt and cerium that loading of cobalt promotes the production of Ce3+, which enhances Ce4+ ↔ Ce3+ redox process.17,18 In order to quantify Ce3+ percentage, an area based formula was used25 and values are reported in Table 3. The Ce3+/Cetot values achieved in current study (18.5 mol%; Table 3) seems pretty close to values obtained by Liotta et al. (14 mol%).18 Sustained catalytic activity of CeCo 26 even after 50 pulse mode decompositions and presence of Ce3+ even in used catalyst are promising features. Presence of even minute amounts of Ce3+ improves reducibility of ceria or generation of oxygen vacancies which increase redox capabilities of doped ceria.21–25 On the other hand, iridium based catalysts are known to get oxidized and get poisoned in the course of multiple pulse mode operation.7
Unlike other two XPS spectra, spectra of Co 2p were quite noisy and vague (Fig. 4c), possibly due to low and uneven concentration on the surface. The primary peaks are at 779.7 eV for Co 2p3/2 and 795 eV for Co 2p1/2.34 The cobalt oxide identified from XRD pattern of the samples was Co3O4 and was present in all the samples beyond 15 mol% loading of cobalt. The other possible oxide that could be present is CoO. XPS spectrum is quite handy in differentiating Co3O4 and CoO. The satellite peak for Co 2p3/2 (786 eV) and Co 2p1/2 (802 eV) become more prominent in CoO unlike Co3O4 wherein peak intensities are very low. While indication of CoO presence was evident in all the samples, CeCo 26, the most active catalyst appeared to contain only Co3O4. Co3O4 compound has cobalt in both oxidation state Co3+ and Co2+, which makes it difficult to differentiate completely from CoO. CeCo 26x on the other hand showed a complex spectra with prominence of CoO. Possibility of high temperature transition between different oxides during decomposition can't be ruled out. Co3O4 formation at a particular percentage of cobalt is reported earlier.17,18 It is assumed that, Co3O4 form would have been stabilized in the presence of ceria. The high reaction temperature and possible phase change around this temperature experienced by CeCo 26x would have been responsible for an altered XPS spectrum vis-à-vis to CeCo 26.
Catalyst thermal decay studies
To evaluate catalyst's ability and tolerance at high temperature normally experienced in HAN thruster chamber, CeCo 26 catalyst pellets were subjected to 1400 °C for 2 h in a tubular furnace in air. No apparent physical damage or breakage was observed after annealing at 1400 °C. Annealed samples were firstly analysed for its activity towards HAN in DTA-TG. As shown in thermogram in Fig. 5c, the activity is retained even at high temperature exposure for 2 h. This endorses the thermal robustness of the catalyst CeCo 26, a critical requirement for high temperature application. The TG slope was however reduced possibly due to some deterioration in activity. These thermally aged samples were further examined for possible modification of chemical composition and compared with pristine samples using XRD and XPS as illustrated in Fig. 5a and d, respectively. The HAN decomposition temperature (Td) and slope of TG curve along with details of elemental oxidation states for fresh and thermally aged samples are given in Table 4 and cell parameters in Table S2.† | ||
| Fig. 5 (a) Comparison of XRD curves for CeCo 26 samples (pristine and thermally aged) (α, β & δ stands for CeO2, Co3O4 & CoO, respectively); (b) high temperature STA plot for CeCo 26 sample along annealing temperature; (c) DTA-TG of annealed and pristine CeCo 26 with HAN for activity estimation (inset: weight loss with respect to time); (d) XP spectra for different elements in thermally aged CeCo 26 samples (i) O 1s orbital, (ii) Ce 3d orbital and (iii) Co 2p orbital. | ||
| Samples | Td (°C) | TG slope (mg min−1) | Ce3+/(Ce3+ + Ce4+) mol% | Olattice/(Olattice + Oadsorbed) mol% |
|---|---|---|---|---|
| CeCo 26 | 130.2 | 293.72 | 18.53 | 3.65 |
| CeCo 26 after annealing at 1400 °C for 2 h | 136.3 | 107.53 | 8.97 | 32.84 |
In XRD, reflections corresponding to both the pre-existing phases (CeO2 and Co3O4) along with peak for CoO (Fig. 5a, inset) were identified in the thermally aged samples. The crystallite size shows enlargement due to annealing but cell parameter contracts marginally. Co3O4 seems to have reduced in quantity since beyond 1000 °C Co3O4 conversion to CoO is obvious.
Ce 3d XPS analysis of thermally aged samples shows smaller features for Ce3+ compared to pristine CeCo 26. The Ce3+ which got probably stabilized in CeO2 matrix in presence of cobalt is known to undergo oxidation depending on oxygen availability. The annealing condition probably would have favoured a similar change causing the change in the ratio of cerium oxidation state. Examination of O 1s and Co 2p spectral characteristics of annealed samples show similar characteristic features as observed in CeCo 26x.
The high temperature thermal analysis of the pristine sample CeCo 26 carried out to examine any possible thermal degradation, further confirmed the thermal robustness of the samples. Fig. 5b do not indicate any thermal change other than minute weight loss (0.35%) at 1000 °C possibly due to conversion of Co3O4 to CoO evident from XRD, leading to release of oxygen from samples.
Evolved gas analysis and reaction mechanism
An evolved gas analysis was performed during HAN decomposition with and without catalysts (Ir/γ-Al2O3 and CeCo 26), employing a hyphenated DTA-TG-FTIR set up. Fig. 6a shows thermograms for catalysed and non-catalysed HAN decompositions along with FTIR spectrum (inset) of evolved gases at peak decomposition temperature. At peak decomposition temperature, products identified for HAN are HNO3, NO2, and N2O. N2 and H2O are other products which are not identified, thanks to the technique's constraints, but known as decomposition products as per available literature.14,16,35 H2O was separated from evolved gas stream due to an in-built desiccator of FTIR gas cell. In catalysed system the overall product profile remains similar but with differing proportions of the constituent gases. An approximate quantification of the product species identified in FTIR was carried out based on absorbance intensity of specific peaks of individual species. A relative quantitative analysis of products during whole duration of the decomposition is shown in Fig. 6b.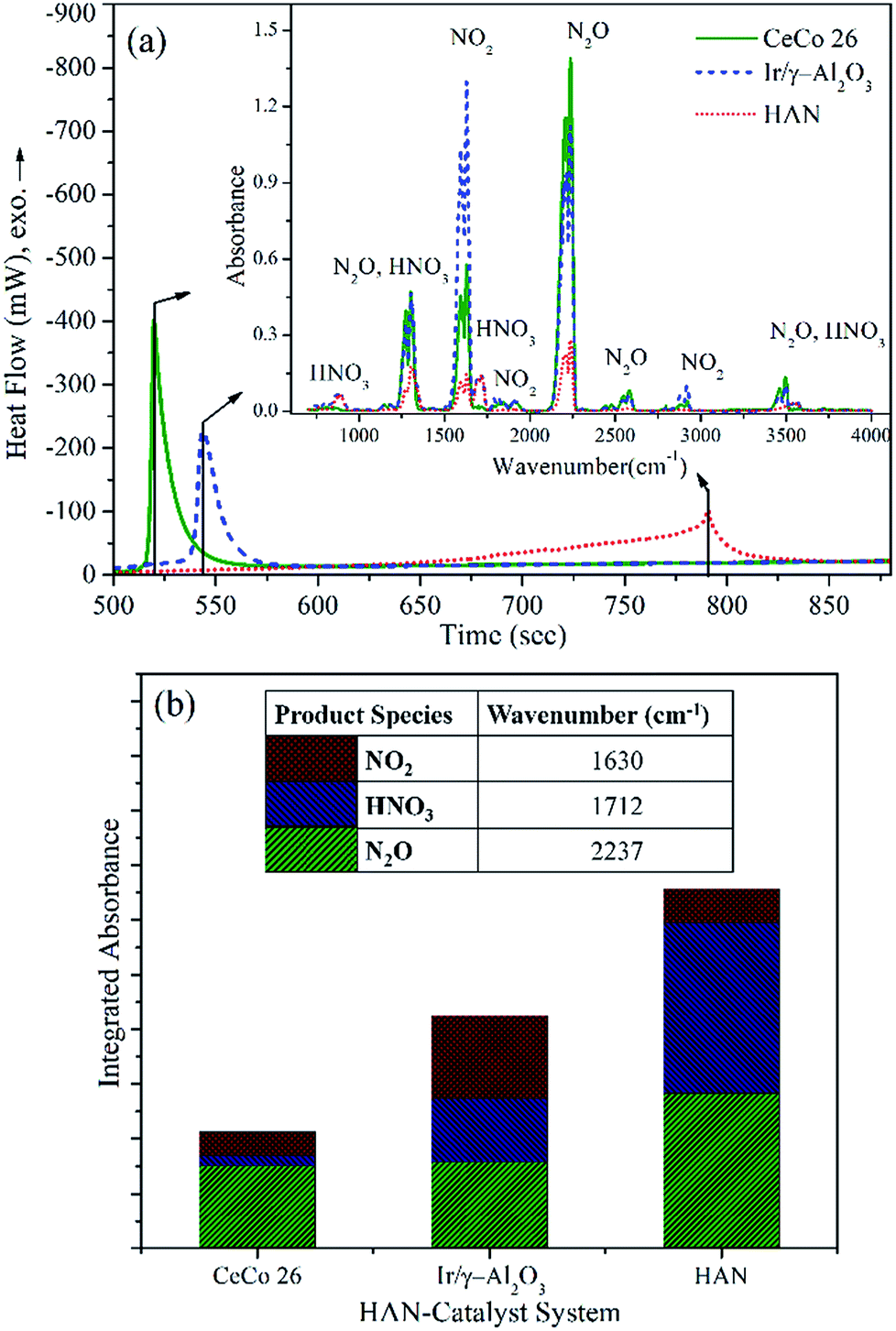 | ||
| Fig. 6 (a) DTA curve for catalyzed and non-catalyzed HAN decomposition; FTIR spectrum of product gas at peak temperature (inset); (b) absorbance for specific product species (inset table: wavenumber at which integration is performed). | ||
HNO3 and N2O are the major FTIR identifiable decomposition products during HAN thermal decomposition while NO2 was also observed in relatively small quantities. Both the nitrogen oxides in measurable quantities and HNO3 in relatively smaller proportion were identified during Ir/γ-Al2O3-HAN decomposition. Evidently, N2O and HNO3 concentrations were found to be lesser in the Ir/γ-Al2O3 catalysed decomposition when compared to thermal decomposition of HAN. NO2 concentration, on the other hand was found to be higher in Ir/γ-Al2O3 catalysed decomposition. This evidently, implies a modified reaction mechanism. Both the NOx species, namely N2O and NO2, particularly the latter, were found to be less when CeCo 26 catalyst was used. Interestingly, HNO3 was scarcely detected in this case. This further suggests distinctively different reaction route even for CeCo 26 catalysed system. Lowering of nitrogen oxide and acidic species during decomposition over CeCo 26 suggest enhancement of nitrogen concentration (IR insensitive) which also implies a lower average molecular weight of the product gases and higher thruster performance.
Yet another highlight is the reduction in the concentration of HNO3 when moving from thermal to catalytic, especially for CeCo 26. This is one of the prominent corrosive and undesirable product that renders catalytic pellet prone to poisoning and attrition. Besides, reduction of HNO3 as much as possible has been suggested as the best optimized condition for HAN monopropellant thruster.13,36
Presence of NO though has been reported in a few rapid thermal analysis studies, its concentration is rather negligible in our studies. Moreover, NO is not included in the final products in many proposed reduced reaction mechanisms.6,37 Nitric acid and its products with hydroxylamine like nitrous acid play a critical role as intermediate products in the overall decomposition mechanism of HAN. The relation between NO2 and HNO3 is quite subtle as per mechanisms reported in literature;16,37,38 The proposed dissociation is as follows
| HNO3 → NO2˙ + OH˙ | (1) |
Higher conversion of intermediate nitric acid and nitrous acid leads to higher percentages of nitrogen oxides in the product. The product profile observed for catalysed systems appears to follow this path. As discussed CeCo 26 catalysed systems show negligible nitric acid and substantial N2O in relation to NO2. Parallels can be drawn from the reported mechanism of HAN with plutonium or iron.5 Based on the reported reaction of cerium ions with hydroxylamine the following redox mechanism can be assumed to play an intermediary role in deciding the extreme reactivity of these catalysed systems.39–43
| Ce4+ + NH3OH+ → Ce3+ + ½N2 + 2H+ + H2O | (2) |
| Ce3+ + 3/2H+ + ½NO−3 → Ce4+ + ½HNO2 + ½H2O | (3) |
The product profiles obtained by these catalysed systems appear to follow the reduced reaction mechanism proposed by Lee and Litzinger.37 The reaction appears to favour thermodynamic path with obvious change in specific reaction enthalpies shown in Table 1. The ratio of cerium ions present in differing oxidation states in the catalyst also will have an apparent influence on the reactivity.
Conclusions
Possibility of hydroxylammonium nitrate (HAN) emerging as a dependable and promising high performing liquid monopropellant depends on the development of high temperature tolerant catalysts. High temperature robustness, resistance to catalytic poisoning and thermal shock based attrition at high temperature are the critical challenges in developing a dependable catalyst for application in long duration space thrusters. A new class of high temperature tolerant active catalyst for HAN decomposition based on cerium oxide doped with cobalt has been developed from co-precipitation route and sintered to pellets to be used in a practical space thruster. The activity of the newly developed catalyst was found to be extremely promising and its performance levels in ensuing a low temperature, high exothermic decomposition was highly encouraging. Compared to exiting iridium based alumina supported catalyst, the new catalyst was found to be more durable in terms of physical integrity and resistant to catalyst poisoning. The catalyst was characterized for its elemental composition and in situ phases formed at different doping levels of cobalt were established in XRD & XPS studies. The cobalt doped ceria catalyst was found to be extremely active at a particular cobalt loading. Presence of Ce3+ in ceria matrix was found to be critical for its extreme catalytic activity in the HAN decomposition studies. A possible redox reaction pathway involving cerium ions has also been proposed. The spent catalyst was also characterized for any possible chemical poisoning and was found to be robustly immune. Features like lack of attrition, resistance to poisoning, high temperature tolerance and tendency to initiate a low temperature decomposition reaction with high exothermicity etc. makes the new class of cerium oxide catalysts highly promising. The real test of durability and activity of the catalyst in a high temperature and pressure thruster is however a prerequisite for its real time application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank ARDB for funding this work and ICER-NCCRD for permitting its characterization facilities.References
- S. Hoyani, R. Patel, C. Oommen and R. Rajeev, J. Therm. Anal. Calorim., 2017, 129, 1083–1093 CrossRef.
- R. Spores, R. Masse, S. Kimbrel and C. H. McLean, GPIM AF-M315 Propulsion system, presented in part at 49th AIAA/ASME/SAE/ASEE Joint propulsion conference & exhibit, San Jose, CA, USA, 15-17 July, 2013, Paper: AIAA pp. 2013–3849 Search PubMed.
- F. B. Apollo, N. Sukae, M. Haruki and A. Muneo, IHI Eng. Rev., 2010, 43(1), 22–28 Search PubMed.
- T. Katsumi, T. Inoue, J. Nakatsuka, K. Hasegawa, K. Kobayashi, S. Sawai and K. Hori, Combust., Explos. Shock Waves, 2012, 48(5), 536–543 CrossRef.
- G. S. Barney and P. B. Duval, J. Loss Prev. Process Ind., 2011, 24, 76–84 CrossRef.
- J. R. Pembridge and G. Stedman, J. Chem. Soc., Dalton Trans., 1979, 1657–1663 RSC.
- R. Amrousse, T. Katsumi, Y. Niboshi, N. Azuma, A. Bachar and K. Hori, Appl. Catal., A, 2013, 452, 64–68 CrossRef.
- S. Kang, J. Lee and S. Kwon, J. Propul. Power, 2014, 30(1), 249–251 CrossRef.
- L. Courthéoux, D. Amariei, S. Sylvie Rossignol and C. Kappenstein, Appl. Catal., B, 2006, 62, 217–225 CrossRef.
- A. J. Fortini, J. R. Babcock and M. J. Wright, US Pat. 20080064913, Ultramet, 2008.
- M. D. Fokema and J. E. Torkelson, US Pat., 20080064914A1, 2008.
- E. Savrun and S. J. Sawhill, US Pat., 20120297779A1, Sienna Technologies Inc., 2012.
- S. Kang and S. Kwon, J. Spacecr. Rockets, 2015, 52(5), 1508–1509 CrossRef.
- R. Amrousse, T. Katsumi, N. Itouyama, N. Azuma, H. Kagawa, K. Hatai, H. Ikeda and K. Hori, Combust. Flame, 2015, 162, 2686–2692 CrossRef.
- E. W. Schmidt and D. F. Gavin, US Pat. 005485722A, 1996.
- R. Amrousse, T. Katsumi, N. Azuma and K. Hori, Combust. Flame, 2017, 176, 334–348 CrossRef.
- J. Liu, Z. Zhao, J. Wang, C. Xu, A. Duan, G. Jiang and Q. Yang, Appl. Catal., B, 2008, 84, 185–195 CrossRef.
- L. F. Liotta, G. Di Carlo, G. Pantaleo, A. M. Venezia and G. Deganello, Appl. Catal., B, 2006, 66, 217–227 CrossRef.
- J. Xu, G. Lu, Y. Guo, Y. Guo and X.-Q. Gong, Appl. Catal., A, 2017, 535, 1–8 CrossRef.
- S. Roy, M. S. Hegde and G. Madras, Appl. Energy, 2009, 86, 2283–2297 CrossRef.
- P. Singh, N. Mahadevaiah, S. K. Parida and M. S. Hegde, J. Chem. Sci., 2011, 123(5), 577–592 CrossRef.
- G. Xiao, S. Li, H. Li and L. Chen, Microporous Mesoporous Mater., 2009, 120, 426–431 CrossRef.
- D. E. P. Vanpoucke, P. Bultinck, S. Cottenier, V. Van Speybroeck and I. Van Driessche, J. Mater. Chem., 2014, A2, 13723–13737 RSC.
- D. E. P. Vanpoucke, S. Cottenier, V. Van Speybroeck, I. Van Driessche and P. Bultinck, J. Am. Ceram. Soc., 2014, 97(1), 258–266 CrossRef.
- D. Jampaiah, K. M. Tur, S. J. Ippolito, Y. M. Sabri, J. Tardio, S. K. Bhargava and B. M. Reddy, RSC Adv., 2013, 3, 12963–12974 RSC.
- M. Mori, Z. Wang and T. Itoh, J. Fuel Cell Sci. Technol., 2011, 8, 01100711–01100716 Search PubMed.
- E. Jud and L. Gauckler, J. Electroceram., 2005, 14, 247–253 CrossRef.
- G. Bernard-Granger and C. Guizard, J. Am. Ceram. Soc., 2008, 91(5), 1703–1706 CrossRef.
- C. B. Carter and M. G. Norton, Ceramic Materials Science and Engineering, Springer Science+Business Media, New York, USA, 2007 Search PubMed.
- J. Svoboda, H. Riedel and R. Gaebel, Acta Mater., 1996, 44(8), 3215–3226 CrossRef.
- D. Amariei, L. Courthéoux, S. Rossignol and C. Kappenstein, Chem. Eng. Process., 2007, 46, 161–174 CrossRef.
- A. V. Thorat, T. Ghoshal, P. Carolan, J. D. Holmes and M. A. Morris, J. Phys. Chem. C, 2014, 118, 10700–10710 CrossRef.
- X. Tang, Y. Li, X. Huang, Y. Xu, H. Zhu, J. Wang and W. Shen, Appl. Catal., B, 2006, 62, 265–273 CrossRef.
- T. J. Chuang, C. R. Bridle and D. W. Rice, Surf. Sci., 1959, 59, 413–429 CrossRef.
- R. Amrousse, T. Katsumi, A. Bachar, R. Brahmi, M. Bensitel and K. Hori, React. Kinet., Mech. Catal., 2014, 111, 71–88 CrossRef.
- T. Katsumi, R. Amrousse, Y. Nibosh and K. Hori, Int. J. Energ. Mater. Chem. Propul., 2015, 14(4), 307–319 Search PubMed.
- H. S. Lee and T. Litzinger, Combust. Flame, 2003, 135, 151–169 CrossRef.
- S. D. Chambreau, D. M. Popolan-Vaida, G. L. Vaghjiani and S. R. Leone, J. Phys. Chem. Lett., 2017, 8, 2126–2130 CrossRef PubMed.
- A. M. M. Doherty, M. D. Radcliffe and G. Stedman, J. Chem. Soc., Dalton Trans., 1999, 3311–3316 RSC.
- R. W. Johnson and D. S. Martin Jr, J. Inorg. Nucl. Chem., 1959, 10, 94–102 CrossRef.
- M. Matheswaran, S. Balaji, S. J. Chung and I. S. Moon, Bull. Korean Chem. Soc., 2007, 29(8), 1329–1334 Search PubMed.
- Y. Wei, B. Fang, T. Arai and M. Kumagai, Journal of Applied Electrochemistry, 2005, 35, 561–566 CrossRef.
- T. A. Sedneva, Russ. J. Appl. Chem., 2005, 78(6), 907–911 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02368a |
| This journal is © The Royal Society of Chemistry 2018 |