
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmino acid ionic liquids as catalysts in a solvent-free Morita–Baylis–Hillman reaction†
Mathias Prado Pereira ,
Rafaela de Souza Martins,
Marcone Augusto Leal de Oliveira and
Fernanda Irene Bombonato*
,
Rafaela de Souza Martins,
Marcone Augusto Leal de Oliveira and
Fernanda Irene Bombonato*
Group of Studies in Organic Synthesis and Catalysis, Department of Chemistry, University of Juiz de Fora, São Pedro 36036-900, Juiz de Fora, Minas Gerais, Brazil. E-mail: fernanda.bombonato@ufjf.edu.br; Fax: +55 32 2102-3310 ext. 30; Tel: +55 32 2102-3310 ext. 30
First published on 2nd July 2018
Abstract
In the present work, we describe the preparation of ten amino acid ionic liquids (AAILs) formed from ammonium salts as cations, derivatives of glycerol, and natural amino acids as anions. All of them are viscous oils, colorless or pale yellow, and hygroscopic at room temperature. They have appreciable solubility in many protic and aprotic polar solvents. The AAILs were used as catalysts in a Morita–Baylis–Hillman (MBH) reaction. The ionic liquids derivative from L-proline and L-histidine demonstrated the ability to catalyze the reaction between methyl vinyl ketone and aromatic aldehydes differently substituted in the absence of an additional co-catalyst under organic solvent-free conditions. The AAIL derivatives from L-valine, L-leucine, and L-tyrosine catalyzed the MBH reaction only in the presence of imidazole. The MBH adducts were obtained in moderate to good yields. Although the catalytic site in the ILs was in its enantiomerically pure form, all the MBH adducts were obtained in their racemic form.
Introduction
Ionic liquids (ILs) exclusively comprise an organic cation and an inorganic or organic anion. They are categorized as a green solvent due to their significant properties, such as good thermal stability, being liquid at/or close to room temperature, negligible vapor pressure, and nonflammability. In recent years, a new class of ILs has attracted the interest of several research groups. These ILs are known as task-specific ionic liquids (TSILs) and have a free functional group covalently tethered to the cation, or the anion. The presence of a free functional group can modify the physicochemical properties of an ionic liquid and also make it capable of acting as a catalyst in organic reactions.1 In this context, the amino acid ionic liquids (AAILs)2 have aroused interest due to the low cost of amino acids and the presence of a chiral carbon.There are two groups of AAILs: the first one has an amino acid as a cation or an anion in its natural form, and the other group has a modified amino acid.3–6 In the vast majority of cases, these AAILs are derivatives from L-proline and they can be used as an organocatalyst in Aldol, Mannich, Michael, and Morita–Baylis–Hillman reactions.7–10
The Morita–Baylis–Hillman (MBH) reaction is a powerful chemical transformation method, in which simple starting materials are converted into highly functionalized molecular synthons via a catalytic process.11 Historically, the most explored catalysts for this MBH reaction include tertiary amines or phosphines. The reaction is performed in polar protic solvents, such as water, and they increase the rate of the reaction. Common protocols involve a reaction between a substituted aldehyde and activated alkenes, such as vinyl sulfone, alkyl vinyl ketone, alkyl acrylate or acrylonitrile, in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) as a catalyst in a polar solvent.12–14
The mechanism proposed in the literature for MBH reactions catalyzed by a tertiary amine consists of a nucleophilic addition of 1,4 type to the α,β-unsaturated compound, generating the zwitterionic enolate (I) that acts as a nucleophile attacking the substituted aldehyde, creating a new carbon–carbon bond and a new zwitterionic species (II), which after a proton transfer followed by decomposition process that results in the MBH adduct (III) and regenerates the catalyst (Fig. 1).15
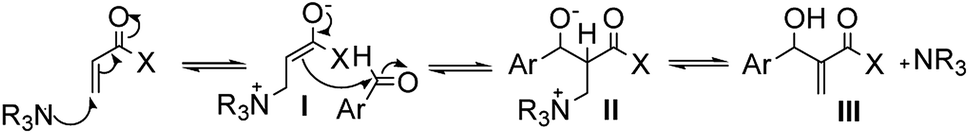 | ||
| Fig. 1 Simplified mechanism of the MBH reaction catalyzed by tertiary amine. | ||
Aggarwal16,17 and McQuade18,19 propose a different route the α hydrogen transfer in the intermediate II. This stage is considered the rate determinate step of the MBH reaction. MacQuade proposes that, in polar aprotic solvents, the alkoxide portion of the intermediate II reacts with another aldehyde molecule, resulting in a deprotonated hemiketal intermediate, and in the α hydrogen abstraction, the intermolecular form occurs due to the attack of the charged oxygen atom. After the hydrogen transfer, the hemiketal is hydrolyzed, resulting in the intermediate IV (Fig. 2(a)). After the elimination of the ammonium portion, the MBH adduct is formed. In Aggarwal's turn, the author suggests that the hydrogen transfer takes place between the intermolecular intermediate II and the protic polar solvent used in the reaction, and oxygen protonation and abstraction of the α hydrogen occur simultaneously (Fig. 2(b)).
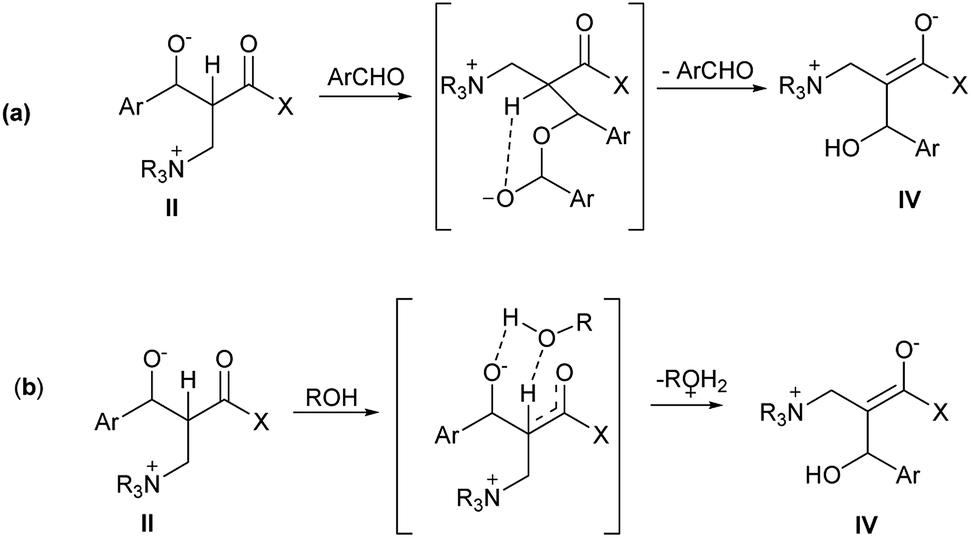 | ||
| Fig. 2 Intermediate from rate-limiting step proposed by Aggarwal (a) and MacQuade (b). | ||
In addition, there are several papers in the literature that describe the results obtained in the reactions of MBH catalyzed by L-prolines. In this case, the amine used as a catalyst is a secondary amine and the mechanism described in the works is based on the reaction of an α,β-unsaturated carbonyl compound with an L-proline by a nucleophilic addition of 1,2 type. It creates an α,β-unsaturated iminium ion (V), which, subsequently, suffers from a nucleophilic attack (the nucleophilic species at this stage depends on the reaction condition, such as excess of L-proline, presence of imidazole, base medium and L-proline) at the β position, resulting in a substituted β-enamine (VI). Enamine (VI) acts as a nucleophile and attacks the substituted aldehyde. It creates a condensation product (VII) which, after leaving the nucleophile (VIII) and hydrolyzing the imines, produces the MBH adduct (IX). Finally, it regenerates the catalyst (Fig. 3).20–27
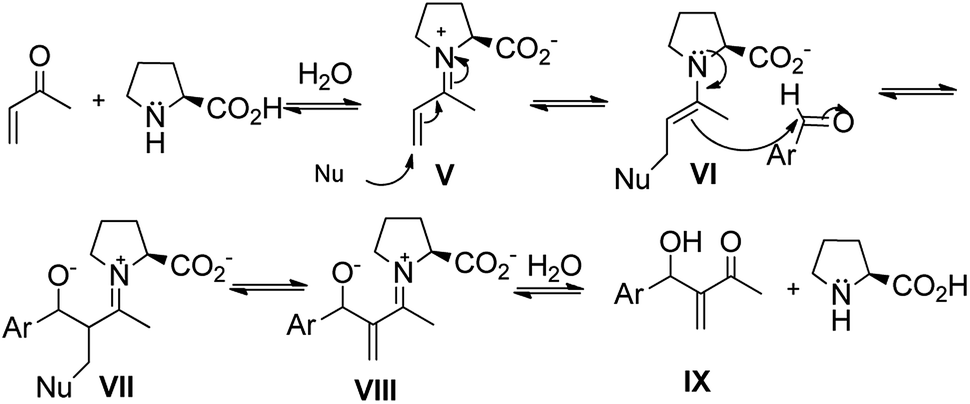 | ||
| Fig. 3 General mechanism proposed for the MBH reaction catalyzed by L-proline. | ||
Regardless of the nature of the amino-catalyst used (primary or tertiary amine), the reactional intermediates formed have a charge. In this sense, the use of aprotic polar, protic polar or even ionic polar solvents will influence the stabilization of these charged intermediates and, consequently, will affect the speed of the reaction.
In this sense, Santos et al.28 describe the influence of using a derivative imidazolium IL as a solvent in MBH reaction rate between methyl acrylate and benzaldehyde. The reaction performed 14.1 times faster in ILs than in acetonitriles. The authors suggest that a strong stabilization of zwitterionic intermediates occurs in ILs, affecting the reaction speed. Aggarwal et al. challenge the results described by Afonso et al. and question the experimental method used by the authors to determine the reaction speed. Afonso et al. determine the reaction rate by gas chromatography experiments, comparing the integration of the MBH adduct with the aldehyde integration. Aggarwal et al. go further and claim that benzaldehyde reacts with the ionic liquid derived from the imidazolium ion in a side reaction.
Coelho, Eberlin, and Neto28,29 go further into this investigation and study the origin of the stabilizing effect of imidazolium IL derivatives in the MBH by ESI-MS(/MS) and DFT calculations. The author identified an MBH intermediate associated with the ion pair of the ILs by ESI-MS(/MS). These experimental results evidenced the formation of an electrostatic intermediate complex between the ion pair of the IL and the cationic intermediate of the MBH reaction. Additionally, the analysis of theoretical calculations evidences the occurrence of ion-pairing and a larger supramolecular organization of the intermediates of the MBH reactions and the imidazolium IL used as a solvent.29
These two effects together, the ionic pairing and the supramolecular organization caused by imidazolium ionic liquids in the MBH reaction, changed this reaction faster when compared to reactions performed in molecular solvents.
No work in the literature describes the use of primary amines as catalysts in MBH reactions. Historically, primary amines are the least compounds used as catalysts due to the fact that in most aminocatalyzed reactions. The reaction mechanism undergoes a balance shift between the imine–enamine forms. This process is formed by the reaction of the catalyst with carbonyl compounds. It is disadvantageous when primary amines are used as catalysts, this phenomenon occurs the imine form is predominant in equilibrium.30,31 In this sense, the use of primary amines as catalysts requires special attention regarding the development of new and efficient catalytic systems.
In this context, in the present work, we investigate the synthesis of new AAIL's derivatives from glycerol and natural amino acids (L-valine, L-leucine, L-proline, L-histidine, and L-tyrosine). Further, we describe the use of AAILs prepared as catalysts in MBH reactions between MVK and differently substituted aldehydes.
Results and discussion
According to the Brazilian National Petroleum Agency (ANP), Brazil produced 3.94 million m3 of glycerol in 2015 due to the biodiesel production.32 The Brazilian market exports part of this production at a reduced cost, but still has a surplus of this raw material in the domestic market. With this in mind, we chose to use a derivative of glycerol as a cation in order to add value to this raw material that is abundant in our country.We started the synthesis of the ionic liquids with the preparation of quaternary ammonium salts 2 and 3 in four steps from glycerol. The intermediates 2 and 3 are described in the literature,33,34 but the characterization (1H and 13C NMR and IR) was incomplete or missing. Subsequently, salts 2 and 3 were submitted to an anion metathesis, which was mediated using Amberlyst A-26 OH, where iodide was substituted with a hydroxide ion. The hydroxide quaternary ammonium salts were reacted immediately with natural amino acids, L-valine, L-leucine, L-proline, L-histidine, and L-tyrosine, to result in an AAIL, as shown in 4a–e and 5a–e of Scheme 1. In this way, two series of AAILs with different chemical proprieties were prepared. AAILs 4a–e contained free hydroxyls and were capable of acting as donors and acceptors for hydrogen bondings. The AAILs 5a–e were capable of acting as acceptors for hydrogen bondings because the hydroxyls were protected as ketals.
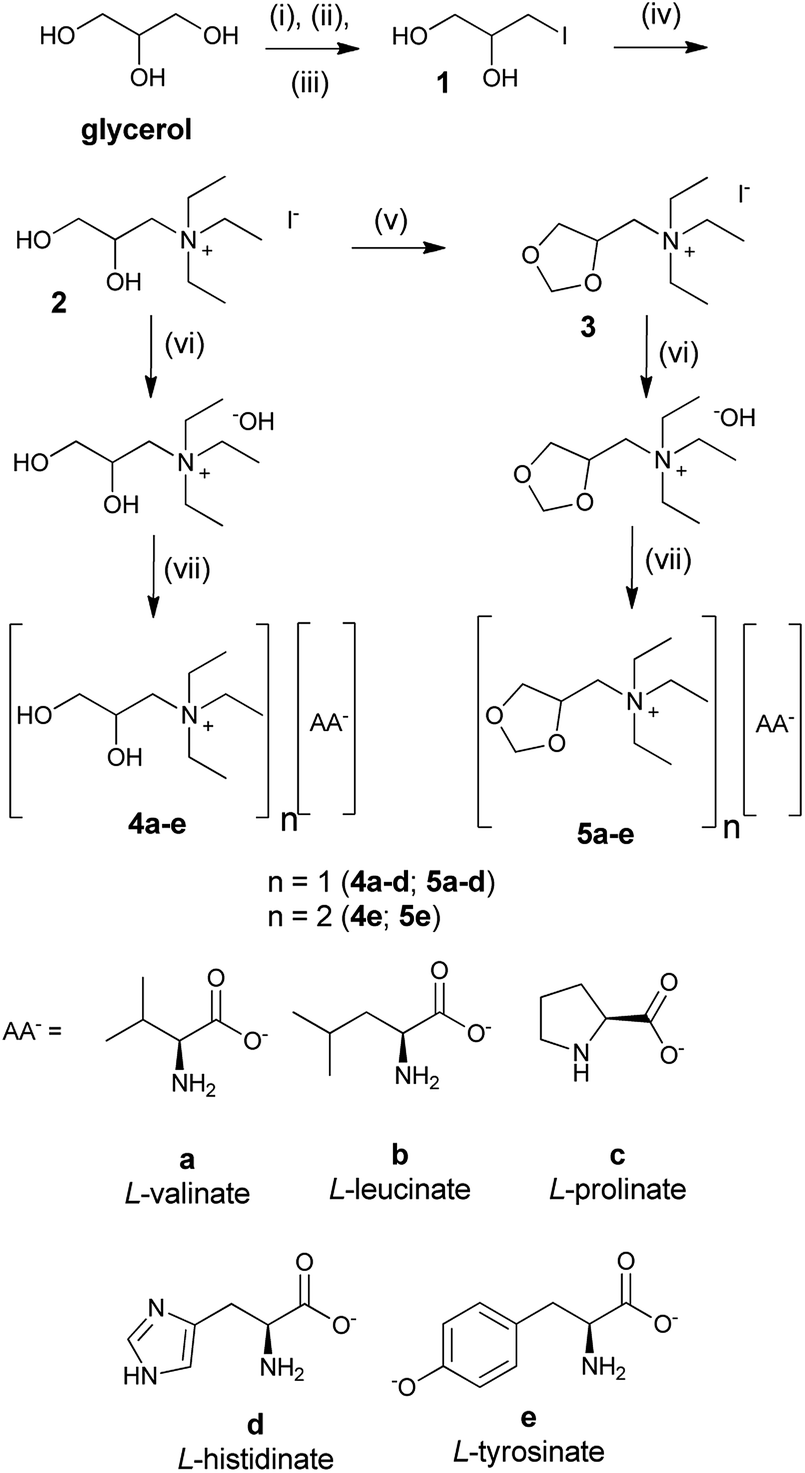 | ||
| Scheme 1 Synthetic route to AAILs 4a–e and 5a–e. Reagents and conditions: (i) acetone, TsOH, petroleum ether, Dean-Stark, 32 h, 60 °C, 80%; (ii) PPh3, I2, imidazole, toluene, 2 h, 90 °C, 81%; (iii) HCl 2 M, acetone, 4 h, 50 °C, 63%; (iv) triethylamine, ethanol, 16 h, 60 °C, 75%; (v) CH2O, TsOH, MeCN, 4 h, 90 °C, 77%; (vi) Amberlyst A-26 OH; (vii) L-amino acid, 48 h, r.t. 62–95%. | ||
The AAILs prepared as described above are viscous oils, colorless or pale yellow, and hygroscopic at room temperature. They have appreciable solubility in many protic and aprotic polar solvents, such as water, acetonitrile, dimethyl sulfoxide, N,N-dimethyl formamide, tetrahydrofuran, methanol, and chloroform, when compared with pure amino acids, and they are insoluble in ethyl acetate, diethyl ether, and hexane.
The AAILs structure was confirmed by 1H, 13C NMR, infrared spectroscopy (IR), and elemental analysis. The deprotonation of the acid group of amino acid was clearly evidenced by 13C NMR in the signal of carbonyl carbon that has a delocalization to high shift (Table 1).
Thermal gravimetric analysis (Table 2) showed that the ionic liquids had a decomposition temperature (Tdec) at ∼200 °C and that the ionic liquids 4a–e had a slightly higher temperature than 5a–e. This difference is due to the free hydroxyl group that increases the hydrogen bonds in AAIL 5a–e. A negative glass transition temperature (Tg) was observed via differential scanning calorimetric measurements.
| AAIL | Tdec/°C | Tg/°C | AAIL | Tdec/°C | Tg/°C |
|---|---|---|---|---|---|
| a *Not determined. | |||||
| 4a | 226 | * | 5a | 189 | −40 |
| 4b | 234 | −55 | 5b | 188 | −64 |
| 4c | 229 | −44 | 5c | 220 | −45 |
| 4d | 233 | * | 5d | 228 | * |
| 4e | 165 | * | 5e | 145 | −41 |
The AAILs were tested for catalysts in an MBH reaction between MVK and p-nitrobenzaldehydes. All the prepared ILs have the active catalytic site in an enantiomerically pure form, in this sense we expect to obtain the MBH adducts in their enantiomerically enriched form. Initially, the reactions were carried out with 20 mol% of the catalyst, in the absence of organic solvents, additives and at room temperature. The results of this sequence of experiments are summarized in Table 3 (entries from 1 to 17).
|
|||||
|---|---|---|---|---|---|
| Entry | AAIL (20 mol%) | Additive (20 mol%) | Solvent (1 mL) | Timeb (h) | Yieldc (%) |
| a All reactions were performed with 1.0 mmol MVK, 0.2 mmol p-nitrobenzaldehyde, 20 mol% of AAIL and 20 mol% of imidazole.b The reaction was monitorated by TLC until complete consumption of the aldehyde.c Isolated yield after column chromatographic purification.d Solvent 1 mL. | |||||
| 1d | Imidazole | CHCl3 | 72 | N.R. | |
| 2 | 4a | — | — | 72 | N.R. |
| 3 | 5a | — | — | 72 | N.R. |
| 4 | 4b | — | — | 72 | N.R. |
| 5 | 5b | — | — | 72 | N.R. |
| 6 | 4e | — | — | 72 | N.R. |
| 7 | 5e | — | — | 72 | N.R. |
| 8 | 4c | — | — | 72 | 82 |
| 9 | 5c | — | — | 72 | 51 |
| 10 | 4d | — | — | 4 | 68 |
| 11 | 5d | — | — | 4 | 66 |
| 12 | 4a | Imidazole | — | 5 | 44 |
| 13 | 5a | Imidazole | — | 5 | 35 |
| 14 | 4b | Imidazole | — | 5 | 41 |
| 15 | 5b | Imidazole | — | 5 | 38 |
| 16 | 4e | Imidazole | — | 5 | 30 |
| 17 | 5e | Imidazole | — | 5 | 26 |
| 18 | 4a | Imidazole | H2O | 5 | 29 |
| 19 | 5a | Imidazole | H2O | 5 | 23 |
| 20 | 4b | Imidazole | H2O | 5 | 34 |
| 21 | 5b | Imidazole | H2O | 5 | 24 |
| 22 | 4c | — | H2O | 72 | N.R. |
| 23 | 5c | — | H2O | 72 | N.R. |
| 24 | 4d | — | H2O | 4 | 40 |
| 25 | 5d | — | H2O | 4 | 32 |
| 26 | 4e | Imidazole | H2O | 5 | 20 |
| 27 | 5e | Imidazole | H2O | 5 | 18 |
Since we expect the MBH reaction to be catalyzed by the amine group and the prepared ionic liquids to have different amino acid residues, the amine function is classified differently (primary, secondary and heteroaromatic amines). To better understand the results, we will first discuss the MBH reactions catalyzed by the ILs containing primary amines (L-valine, L-leucine, and L-tyrosine) 4a, b and 5a, b and c, respectively. Later, we will discuss the results obtained when the ILs derived from the L-proline (4c and 5c) were used as catalysts. Finally, we will discuss the results in MBH reactions when the ILs derived L-histidine (4d and 5d) were used as catalysts.
The ionic liquids used also have cations with different characteristics. The series of ionic liquids 4 have cations capable of both donating and receiving hydrogen bonds. The ionic liquids of series 5 have cations capable of only receiving hydrogen bonds. In this work, we will compare our results with works described in the literature for MBH reactions where only amino acids were used as catalysts. In this sense, the reactions described up to the present moment in the literature report the use of protic or aprotic polar organic solvents in the presence of water as an additive. In this sense, we can affirm that the ILs used in this work as a solvent, and a catalyst, depending on the cation used have characteristics of protic or aprotic polar solvents and the residual water present in the ILs acts as an additive.
When the ILs 4a, b and e, and 5a, b and e (primary amines) were used as catalysts in the MBH reactions no formation of the desired MBH adduct was observed in the absence of solvents, organic additives and at room temperature. However, we can observe the consumption of both the MKV and the catalyst. By means of IR experiments, Calow et al. have observed that the MVK in the presence of primary amines rapidly undergo a nucleophilic addition reaction 1,4 type, generating a substituted β-ketone, which reacts again with another amine molecule and produces an imine, this species is inactive in the MBH reactions.36 In this context, the residues of the amino acids present in the ILs used in these reactions reacted with MVK inactivating the catalyst and disrupting the catalytic cycle, corroborating the results described by Calow and others.
Shi et al. described the MBH reaction between the MVK and the p-nitrobenzaldehyde, using the L-proline amino acid and the imidazole as the catalyst separately.20 The reactions were carried out using DMF as a solvent. Under these reaction conditions, it was not possible to observe the formation of the desired MBH adduct. However, when the two catalysts were used in combination, the MBH adduct was obtained in 91% after 24 hours of reaction at room temperature. The authors proposed that the reaction mechanism consists of the formation of an α,β-unsaturated imine formed by the reaction between the MVK and the L-proline. This imine undergoes a nucleophilic addition of the 1,4 type from the imidazole producing an enamine: the nucleophile from the reaction. It attacks the p-nitrobenzaldehyde forming the new carbon–carbon bond. Finally, this intermediate will consequently produce the MBH adduct.
Based on the results described by Shi et al., it was decided to cause MBH reactions between the MVK and the p-nitrobenzaldehyde, in the same reaction condition previously used; however, in the presence of 20 mol% of imidazole. In this new reaction condition, the MBH adducts were formed, after 5 hours, in yields ranging from 26 to 44%, Table 3 (entries, 12–17), and at significantly lower reaction times when compared to the reaction times described by Shi and others. As previously discussed, the ionic medium favors a supramolecular organization. The presence of imidazolium ion, formed during the reaction, favors the formation of ions pairing and both factors affect the velocity of the reaction positively. It should be observed that no variation of the reaction time was detected when there was an alteration of the cation type, hydrogen bonding donors/acceptors or only hydrogen bond acceptors.
In order to increase the yield of the MBH adduct and to decrease the reaction time, we decided to perform an MBH reaction between MVK and p-nitrobenzaldehyde, using the ILs 4a, b and e, and 5a, b, and e, in water. Several authors report an increase in yield and a decrease in reaction time when the imidazole was used as a catalyst in them MBH reaction.26,37–44 The results obtained from this sequence of reactions are shown in Table 3 (entries 18–21 and 26–27). The reaction times remained the same; however, the yield of the MBH adducts obtained was lower when compared to the reactions performed in the absence of water.
However, based on the results described by Calow, we believe that in the reaction condition used by us the mechanism of the reaction occurs in a different way to the one proposed by Shi. Since the addition reaction 1,4 occurs rapidly, it generates a species that contains an ammonium salt residue (which would be deprotonated by the imidazole) and an enolate (capable of acting as a nucleophile in the MBH reaction). It would be stabilized by forming an ion pair with the imidazole conjugate acid since the imidazole behaves better as a base than as a nucleophile.
When analyzing the results obtained using the ILs derived from the L-proline (4c and 5c), Table 3 (entries, 8–9), the MBH adducts were obtained without the need for additives in yields ranging from 51 to 72% after 72 hours of reaction. When water was used as a solvent, no reaction was observed (Table 3, entries 22–23). We analysed the ionic liquid by 1H NMR and ESI(−)-MS after the MBH reaction and observed that the residue of L-proline of IL 4c and 5c reacted slowly with the MVK excess present in the reaction medium, inactivating the catalyst which justifies the longer reaction time observed when the IL 4c and 5d were used.
Gruttadauria et al. and Inani et al. successfully described the MBH reaction catalyzed only by the amino acid L-proline in a mixture of polar aprotic and protic solvents (DMF: water, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and water, respectively).24,25 The two authors affirm that the L-proline is a bifunctional catalyst and that in the first stage of the reaction mechanism occurs with the reaction of L-proline with the MVK producing an iminium α,β-unsaturated ion and, then that a 1,4 type nucleophilic addition occurs. Gruttadauria et al. propose that the addition 1,4 is carried out by the carboxylate residue formed by the acid–base reaction between the carboxylic acid residue of L-proline with the NaHCO3 used in the reaction medium, to the β-carbon of the imine α,β-unsaturated. Inani et al. propose that the nucleophilic addition of the addition 1,4 type is carried out by another L-proline molecule that is used in excess in the reaction medium. The reaction time described in the two studies ranged from 13 to 120 hours. Based on these results, we can suggest that in the reaction of MBH, which was catalyzed by the ionic liquid derived from L-proline (4c and 5c), occurs via a mechanism similar to that described by Inani et al. Since the reaction was performed using only ILs derived from the L-proline, that is, we have an excess of L-proline residues in the reaction medium.
:1 and water, respectively).24,25 The two authors affirm that the L-proline is a bifunctional catalyst and that in the first stage of the reaction mechanism occurs with the reaction of L-proline with the MVK producing an iminium α,β-unsaturated ion and, then that a 1,4 type nucleophilic addition occurs. Gruttadauria et al. propose that the addition 1,4 is carried out by the carboxylate residue formed by the acid–base reaction between the carboxylic acid residue of L-proline with the NaHCO3 used in the reaction medium, to the β-carbon of the imine α,β-unsaturated. Inani et al. propose that the nucleophilic addition of the addition 1,4 type is carried out by another L-proline molecule that is used in excess in the reaction medium. The reaction time described in the two studies ranged from 13 to 120 hours. Based on these results, we can suggest that in the reaction of MBH, which was catalyzed by the ionic liquid derived from L-proline (4c and 5c), occurs via a mechanism similar to that described by Inani et al. Since the reaction was performed using only ILs derived from the L-proline, that is, we have an excess of L-proline residues in the reaction medium.
In addition to the work described by Shi and Gruttadauria and their collaborators, Guo et al. described the use of the amino acid L-proline combined with the L-histidine amino acid in MBH reactions, however, the formation of the MBH adduct was not observed when the two amino acids were used separately as catalysts. The authors highlighted that the success of the catalytic system used is based on the use of a mixture of protic polar solvents (methanol:water). MBH adducts were obtained in yields that range from moderate to good and long reaction times, from 7 to 24 days. Nevertheless, the authors do not describe a mechanistic proposal for the formation of MBH adducts. Surprisingly, when using ILs derived from the L-histidine 4d and 5d, MBH adducts were obtained with 4 hours of reaction in the absence of organic solvents or additives and in yields ranging from 66 to 68% (Table 3, entries 10–11).
Unfortunately, although all ILs have chiral active sites, no optical activity was observed in the MBH adducts obtained.
In order to understand more about the performance of the AAIL derived from the L-histidine in the MBH reaction, 1H NMR and IR experiments were performed and the results of in situ analyses suggested that the primary amine portion, present at the residue of L-histidine amino acid from the IL 4d, reacts with MVK by a nucleophilic addition of 1,4 type (as previously observed for the IL 4a, b and e and 5a, b, and e) generating a substituted β enolate. The enolate formed would be stabilized by the imidazole residue by the formation of an ionic pair, see the mechanistic proposal shown in Scheme 2.
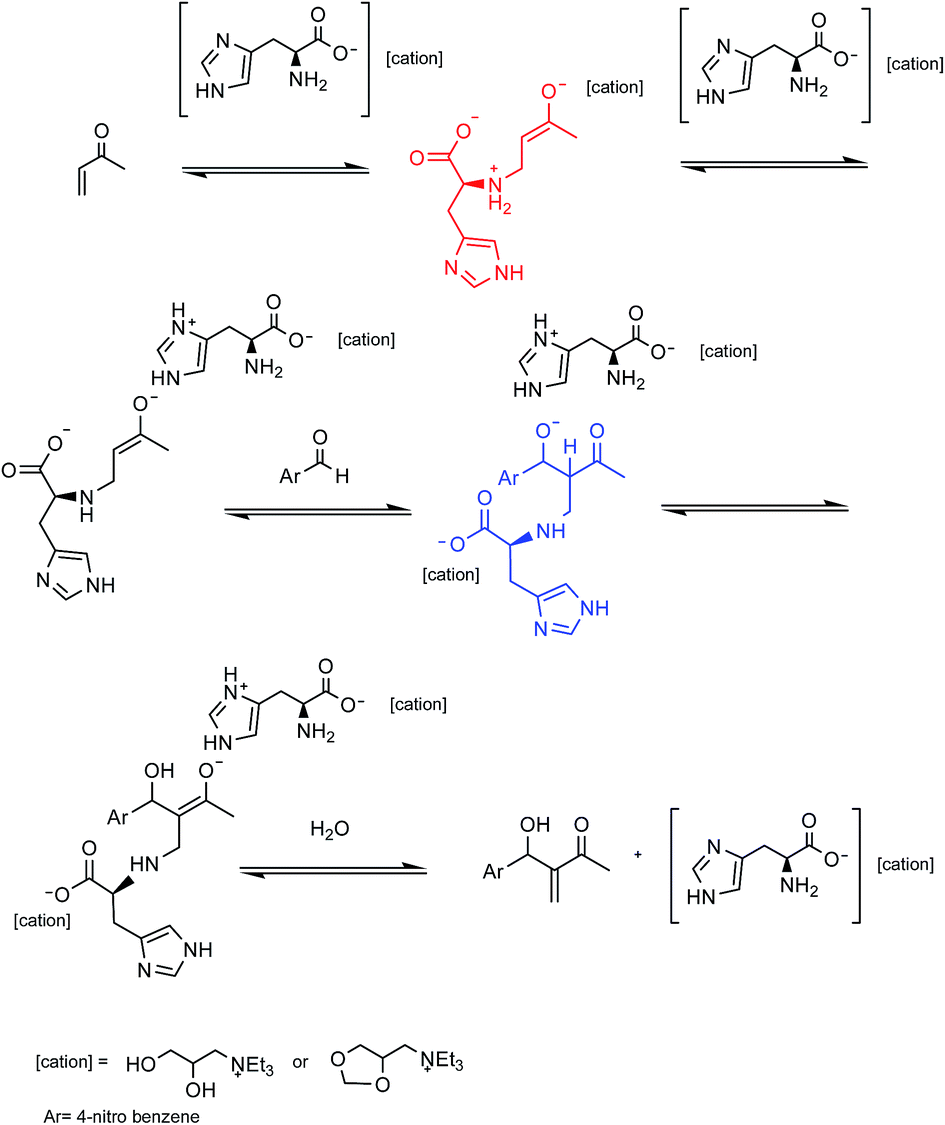 | ||
| Scheme 2 Mechanistic proposal for the reaction of MBH catalyzed by the ionic liquids 4d and 5d. | ||
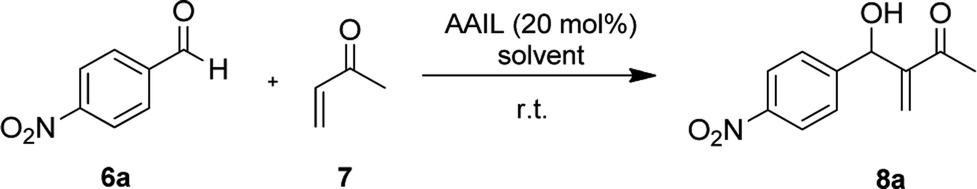
We also monitored the reaction across the mass-to-charge (m/z) characterization of the compounds which was made with 6120 Quadrupole LC/MS (Agilent Technologies, Singapore) equipped with an API-ES ion source, coupled to the Capillary Electrophoresis instrument, Agilent CE 7100 (Agilent Technologies, Singapore), used in compounds infusion into the MS by flush with acetonitrile. The sheath liquid (5 mM of ammonium acetate dissolved in 1:1 (v/v) methanol/water) was delivered at 0.001 mL min−1 flow using an LC isocratic pump (1260 Infinity, Agilent Technologies, Waldbronn, Germany). The MS was operated in the negative ionization mode applying electrospray voltage of 3.0 kV. Nitrogen was used as drying gas at 250 °C, with a flow rate of 6 L min−1; the pressure of the nebulizer gas was set at 15 psi. The m/z characterization of the MBH intermediate was made by the total ion current (TIC) mode. In order to perform the compounds infusion into the quadrupole MS by a flush with acetonitrile, the fused-silica capillary (Polymicro Technologies, Phoenix, USA) of 70 cm length and an internal diameter of 75 μm was used. The new capillary was conditioned by flushing with 1 M NaOH (10 min), water (10 min), and acetonitrile (15 min). Samples were injected hydrodynamically at 50 mbar for C10 s and cassette temperature was maintained at 250 °C. Among sample injections, a short preconditioning in the capillary was performed by flushing with 1 M NaOH (2 min), water (2 min) and acetonitrile (2 min). The data processing was performed with the Agilent ChemStation for CE-MS version software. The analysis was made within 15 min of reaction, and the spectra data were shown in Fig. 4. We observed the production of a nucleophilic 1,4 addition of amine residue of histidine to MVK, forming the MBH nucleophiles (intermediates A and B) and the product of the addition of MBH nucleophiles to the p-nitrobenzaldehyde (C, D, E, and F) (Fig. 4).
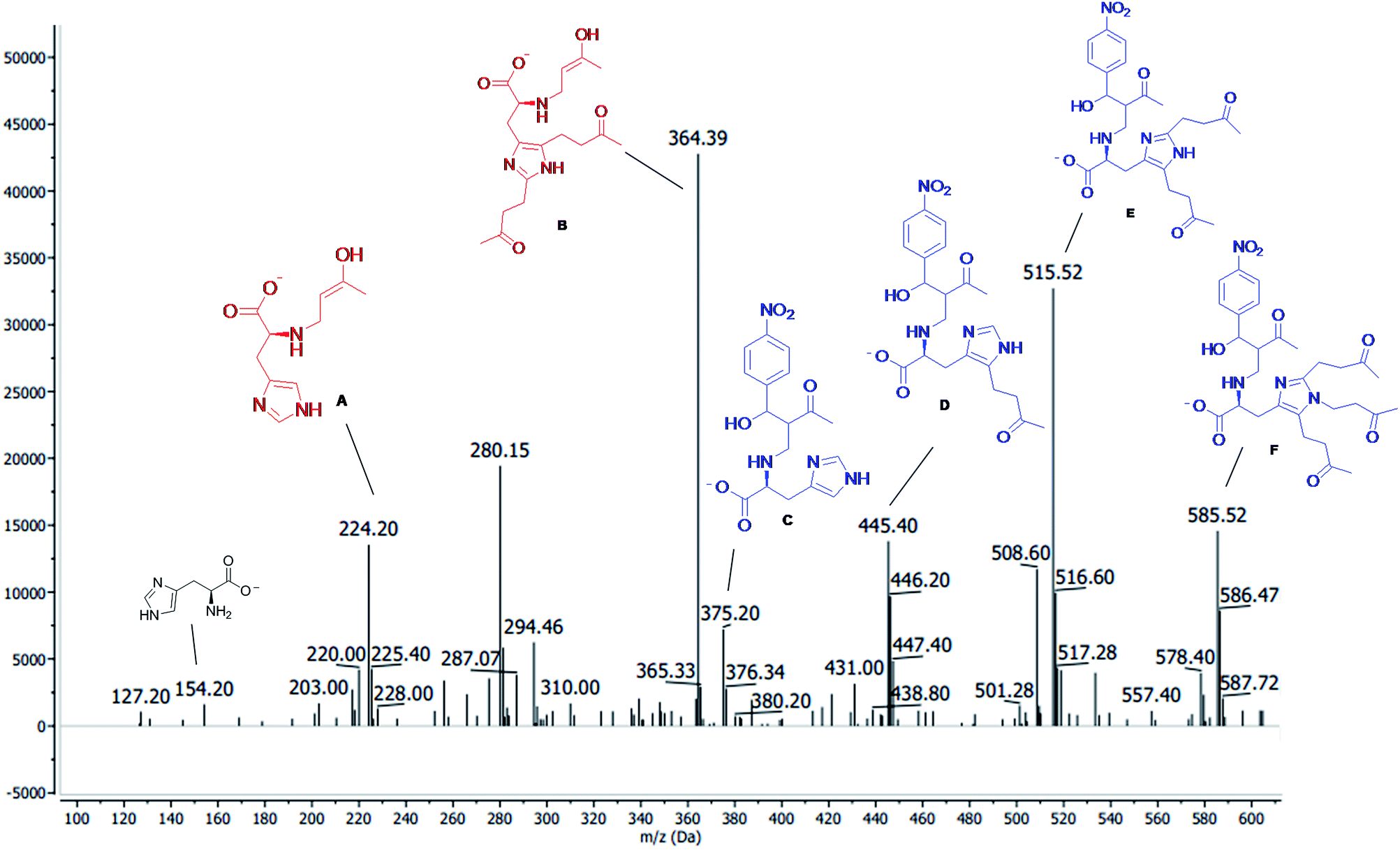 | ||
| Fig. 4 ESI(−)-MS after 15 min of reaction between MVK 1.0 mmol (7) and p-nitrobenzaldeyde 0.2 mmol (6a) and the AAIL 4d (20 mol%) as a catalyst. | ||
As described in the literature,45,46 we also observed the side reactions (electrophilic aromatic substitution, carbene reaction and N-alquilation) of imidazole residues of histidine with MVK and not with the aldehyde, as described by Aggarwal et al. This side reaction could be observed not only ESI(−)-MS (Fig. 4) but also in 1H NMR experiments (see ESI†). In view of these results, we decided to change the proportion between MVK and p-nitrobenzaldehyde. However, they do not assess the formation of side reactions in the imidazole residue.
Despite the occurrence of these lateral reactions in the imidazole residue, the active site of the catalyst (primary amine) remained free and the catalyst remained active even after five cycles of recycling. The yield of the adduct did not considerably change (68 to 60%). However, the lateral reactions suffered by the imidazole residue resulted from a considerable increase in the volume of the anion, making it difficult to ionic pairing, which can be observed in the increase in the reaction time when we reused the catalytic system more than once (Table 4). When water was used as a solvent (Table 3, entries 24–25), the reaction time was the same, but the yield decreased drastically, probably due to the decrease in the organization of the system generated by the presence of water used as solvents.
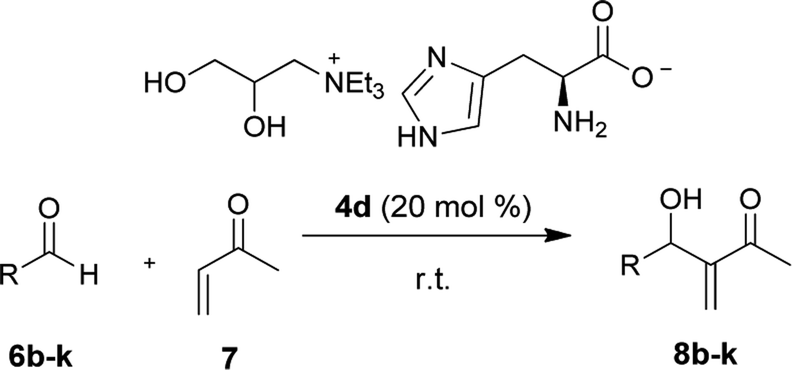
Although the lateral reactions occurred in the imidazole portion of ionic liquid 4d, this ILs were selected as the catalyst to study the scope of the reaction between the MVK and the aromatic aldehydes differently substituted. Similar to the results described in the literature, MBH adducts of aldehydes substituted by electron withdrawing groups were obtained at higher yields and at shorter reaction times, while the products of the aldehyde MBH addition, containing electron donating groups, were obtained with higher reaction times and lower yields (Table 5). All adducts described above are known compounds, no optical activity was observed, and their spectroscopic data was in accordance with the published literature.
|
|||
|---|---|---|---|
| Entry | R | Time (h) | Yieldb (%) |
| a All reactions were performed with 1.0 mmol MVK, 0.2 mmol of aromatic aldehydes and 20 mol% AAIL 4d.b Isolated yield after column chromatographic purification.c Reactions were performed with 1.5 mmol of MVK and 0.5 mmol of aromatic aldehyde and 20 mol% AAIL 4d | |||
| 1 | m-NO2C6H4 (8b) | 4 | 66 |
| 2 | o-NO2C6H4 (8c) | 4 | 62 |
| 3 | p-FC6H4 (8d) | 24 | 88 |
| 4 | p-ClC6H4 (8e) | 24 | 81 |
| 5 | o-ClC6H4 (8f) | 24 | 74 |
| 6 | p-Cf3C6H4 (8g) | 24 | 71 |
| 7 | 3,4,5-(OCH3)3C6H2 (8h) | 72 | 51 |
| 8 | 2-Furyl (8i) | 48 | 89 |
| 9c | Ph (8j) | 120 | 36 |
| 10c | p-OCH3Ph (8k) | 120 | 39 |
Conclusions
AAIL was prepared from glycerol via a simple method using a clearly defined protocol. All synthesized AAILs were viscous oils, colorless or pale yellow, and very hygroscopic at room temperature. Two series of AAILs with different chemical proprieties were prepared. AAILs 4a–e contained free hydroxyls and were capable of acting as donors and acceptors for hydrogen bondings. The AAILs 5a–e were capable of acting as acceptors for hydrogen bondings because the hydroxyls were protected as ketals.Both L-proline and L-histidine ionic liquids catalyzed the MBH reaction with no addition of additives or organic solvents. However, the ILs 4a, b and e and 5a, b and e catalyzed the MBH reaction only in the presence of imidazole. No previous work in the literature describes the use of primary amines (alone or associated with a co-catalyst) as a catalyst in the MBH reaction.
The ionic medium and the presence of imidazole (add or present in the amino acid structure) affect positively the rate of MBH reactions compared to molecular solvents.38,40,42–44,47,48 As described in the literature, these two effects added could affect the rate of the MBH reaction in an ionic medium: a supramolecular organization and a favoring of ion pairing.29
The reactions that the ILs 4c–d and 5d, used as catalysts, were monitored by ESI-(−)MS and 1H NMR. In that case, the IL 4d and 5d, derivatives from L-histidine produced by lateral reactions of imidazole portion, were observed (Fig. 4 B, D, E, and F), besides the MBH intermediates expect (Fig. 4 A and C and Scheme 2). This lateral reaction did not affect the catalytic site (primary amine) and the catalysts remained active. An increase in time reaction (4 to 96 hours) was observed at most when the catalysts 4d and 5d were used more than once. When the IL 4c and 5c were used, long-time reactions were observed due to the side reaction detected between the catalysts with the excess of the MVK used in the MBH reaction, that inactivated the catalyst site.
The best experimental condition was obtained with the IL 4d and it was employed to study the scope of the reaction between MVK and substituted aromatic aldehydes to result in different MBH adducts in good yields. Although all ILs has chiral active sites, no optical activity was observed in the MBH adducts obtained.
Experimental
Materials and methods
All the amino acids were purchased from Aldrich and the other chemicals and reagents were treated. The progress of reaction was monitored by using pre-coated TLC plates (E. Merck Kieselgel 60 F254). Melting points were determined by microquímica MQAPF-361. 13C NMR and 1H NMR were recorded on Bruker DPX-300 and DPX-500 spectrometer. Chemical shifts are reported in ppm. Peak multiplicities are designated by the following abbreviations: s, singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet; br, broad; and J, coupling constant in Hz. TMS and dioxane were used as an internal standard. IR spectra were recorded using Bruker FT-IR in the range of 4000–400 cm−1 and characteristics frequencies are expressed. Elemental analysis were recorded using a Perkin Elmer 2400 series II. Thermogravimetric analysis were performed under nitrogen atmosphere using an Shimadzu DTG-60 in the temperature range between 25 and 500 °C and a heating rate of 10 °C min−1. Differential scanning calorimeter were performed under nitrogen atmosphere using an Shimadzu DSC-60 in the temperature range between −90 and 100 °C and a heating rate of 10 °C min−1.Procedure for the preparation of intermediate compounds from glycerol
:1) to give the iodide compound as a colorless liquid (5.83 g, 81%). IR (plate, NaCl): 2986, 2935, 2877, 1058, 843 cm−1; 1H NMR (300 MHz, CDCl3): δ 4.30–4.22 (m, 1H); 4.14 (dd, J = 8.7 Hz and J = 6.0 Hz, 1H), 3.78 (dd, J = 8.7 Hz and J = 5.4 Hz, 1H), 3.23 (dd, J = 9.6 Hz and J = 4.5 Hz, 1H), 3.14 (dd, J = 9.9 Hz and J = 8.4 Hz, 1H), 1.44 (s, 3H), 1.33 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 110.6, 75.8, 69.8, 27.3, 25.8, 6.84.:8) to give the pure product 1 as a yellow solid (1.28 g, 63%). mp 46–48 °C; IR (film, KBr): 3356, 2925, 1054, 873 cm−1; 1H NMR (D2O, 300 MHz): δ 3.67–3.58 (m, 3H), 3.36 (dd, J = 10.8 Hz and J = 4.3 Hz, 1H), 3.27 (dd, J = 10.8 Hz and J = 6.0 Hz, 1H); 13C NMR (D2O, 75 MHz): δ 70.7, 64.8, 13.2.:0.5) to give the product 3 (2.44 g, 77%) as a yellow solid; mp 142–143 °C; IR (film, KBr): 3449, 2982, 2875, 1051, 1001 cm−1; 1H NMR (D2O, 300 MHz): δ 5.26 (s, 1H) 5.17 (s, 1H), 4.44–4.39 (m, 1H), 3.85–3.80 (m, 1H), 3.75–3.49 (m, 9H), 1.49–1.44 (t, J = 6.9 Hz, 9H); 13C NMR (D2O, 75 MHz): δ 96.1, 69.7, 68.2, 58.8, 54.6, 8.09.General procedure for the preparation of amino acid ionic liquids 4a–e, 5a–e
3-(Tri-ethylammonium)propan-1,2-diol hydroxide and N-((1,3-dioxolan-4-yl)methyl)-N,N,N-tri-ethylammonium hydroxide aqueous solution was prepared from ammonium salts (2) and (3), respectively, using anion exchange resin Amberlyst A-26 hydroxide. Hydroxide aqueous solution was added dropwise to a slightly excess equimolar of L-amino acid aqueous solution under cooling. The mixture was stirred under ambient temperature and protecting of light for 48 h. Then water was evaporated at 60 °C. To this reaction mixture were added a mixture of acetonitrile:methanol (9:1), and it was stirred vigorously. The mixture was then filtered to remove excess amino acid. Filtrate was evaporated to remove solvents. The product was dried in vacuum for 2 days at 60 °C.
General procedure for the Baylis–Hillman reaction
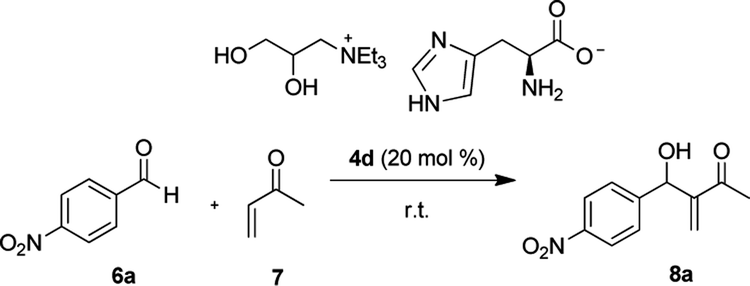
The p-nitrobenzaldehyde 6a (0.200 mmol) was added to catalyst 4d (0.0400 mmol) and methyl vinyl ketone 7 (1.00 mmol) in a 2.00 mL vial. The reaction mixture was stirred for 4 h at room temperature. The residue was purified using silica gel flash column chromatographic (ethyl acetate/hexane, 1:4) to give the product 8a 68% yield (30.0 mg).
General procedure for amino acid ionic liquid recycle experiments
Following extraction with ethyl acetate, the AAIL solution was subjected to vacuum to remove traces of ethyl acetate and water and charged with further portions of p-nitrobenzaldehyde 6a (0.200 mmol) and methyl vinyl ketone 7 (1.00 mmol) at room temperature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) for their financial support to the project research number CEX-APQ-03842-10, CEX-APQ-01283-14 and CEX–RED-00010-14. Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Rede Mineira de Química and MCT/FINEP/CT-INFRA 01/2013-REF 0633/13.Notes and references
- C. Yue, D. Fang, L. Liu and T.-F. Yi, J. Mol. Liq., 2011, 163, 99–121 CrossRef
.
- H. Ohno and K. Fukumoto, Acc. Chem. Res., 2007, 40, 1122–1129 CrossRef PubMed
- G. Tao, L. He, N. Sun and Y. Kou, Chem. Commun., 2005, 3562 RSC
- A. Safavi, S. Zeinali and M. Yazdani, Amino Acids, 2012, 43, 1323–1330 CrossRef PubMed
- D. Brégeon, J. Levillain, F. Guillen, J.-C. Plaquevent and A.-C. Gaumont, Amino Acids, 2008, 35, 175–184 CrossRef PubMed
- V. Gauchot and A. R. Schmitzer, J. Org. Chem., 2012, 77, 4917–4923 CrossRef PubMed
- Y. Qian, X. Zheng and Y. Wang, Eur. J. Org. Chem., 2010, 2010, 3672–3677 CrossRef
- X. Zheng, Y.-B. Qian and Y. Wang, Eur. J. Org. Chem., 2010, 2010, 515–522 CrossRef
- N. Morimoto, Y. Takeuchi and Y. Nishina, J. Mol. Catal. A: Chem., 2013, 368–369, 31–37 CrossRef
- N. Pothanagandhi, A. Sivaramakrishna and K. Vijayakrishna, Polym. Chem., 2017, 8, 918–925 RSC
- Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef PubMed
- J. Cai, Z. Zhou, G. Zhao and C. Tang, Org. Lett., 2002, 4, 4723–4725 CrossRef PubMed
- D. Roy and R. B. Sunoj, Chem.–Eur. J., 2008, 14, 10530–10534 CrossRef PubMed
- C. Faltin, E. M. Fleming and S. J. Connon, J. Org. Chem., 2004, 69, 6496–6499 CrossRef PubMed
- M. Shi, The chemistry of the Morita–Baylis–Hillman reaction, Royal Society of Chemistry, 2011 Search PubMed
- R. Robiette, V. K. Aggarwal and J. N. Harvey, J. Am. Chem. Soc., 2007, 129, 15513–15525 CrossRef PubMed
- V. K. Aggarwal, S. Y. Fulford and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2005, 44, 1706–1708 CrossRef PubMed
- K. E. Price, S. J. Broadwater, H. M. Jung and D. T. McQuade, Org. Lett., 2005, 7, 147–150 CrossRef PubMed
- K. E. Price, S. J. Broadwater, B. J. Walker and D. T. McQuade, J. Org. Chem., 2005, 70, 3980–3987 CrossRef PubMed
- M. Shi, J.-K. Jiang and C.-Q. Li, Tetrahedron Lett., 2002, 43, 127–130 CrossRef
- M. M. Vasbinder, J. E. Imbriglio and S. J. Miller, Tetrahedron, 2006, 62, 11450–11459 CrossRef
- P.-F. Guo, Q.-Y. Wei, H. Jiang and P.-F. Xie, Res. Chem. Intermed., 2012, 38, 639–644 CrossRef
- F. Giacalone, M. Gruttadauria, A. M. Marculescu, F. D'Anna and R. Noto, Catal. Commun., 2008, 9, 1477–1481 CrossRef
- M. Gruttadauria, F. Giacalone, P. Lo Meo, A. Mossuto Marculescu, S. Riela and R. Noto, Eur. J. Org. Chem., 2008, 2008, 1589–1596 CrossRef
- H. Inani, A. Jha and S. Easwar, Synlett, 2016, 28, 128–132 CrossRef
- K. Akagawa, S. Sakamoto and K. Kudo, Synlett, 2011, 817–820 Search PubMed
- S. H. Chen, B. C. Hong, C. F. Su and S. Sarshar, Tetrahedron Lett., 2005, 46, 8899–8903 CrossRef
- T. Regiani, V. G. Santos, M. N. Godoi, B. G. Vaz, M. N. Eberlin and F. Coelho, Chem. Commun., 2011, 47, 6593 RSC
- T. S. Rodrigues, V. H. C. Silva, P. M. Lalli, H. C. B. de Oliveira, W. A. da Silva, F. Coelho, M. N. Eberlin and B. A. D. Neto, J. Org. Chem., 2014, 79, 5239–5248 CrossRef PubMed
- F. Peng and Z. Shao, J. Mol. Catal. A: Chem., 2008, 285, 1–13 CrossRef
- L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC
- ANP divulga Anuário Estatístico 2016, http://www.anp.gov.br/wwwanp/noticias/3138-anp-divulga-anuario-estatistico-2016, (accessed 14 March 2018).
- M. A. Beckett, C. C. Bland and K. Sukumar Varma, Polyhedron, 2008, 27, 2226–2230 CrossRef
- D. J. Triggle and B. Belleau, Can. J. Chem., 1962, 40, 1201–1215 CrossRef
- E. Pretsch, P. Bühlmann and M. Badertscher, in Structure Determination of Organic Compounds, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009, pp. 1–88 Search PubMed
- A. D. J. Calow, J. J. Carbó, J. Cid, E. Fernández and A. Whiting, J. Org. Chem., 2014, 79, 5163–5172 CrossRef PubMed
- H. J. Davies, A. M. Ruda and N. C. O. Tomkinson, Tetrahedron Lett., 2007, 48, 1461–1464 CrossRef
- S. Luo, B. Zhang, J. He, A. Janczuk, P. G. Wang and J.-P. Cheng, Tetrahedron Lett., 2002, 43, 7369–7371 CrossRef
- R. Gatri and M. M. El Gaïed, Tetrahedron Lett., 2002, 43, 7835–7836 CrossRef
- S. Luo, P. George Wang and J.-P. Cheng, J. Org. Chem., 2004, 69, 555–558 CrossRef PubMed
- J. C. Gomes, J. Sirvent, A. Moyano, M. T. Rodrigues and F. Coelho, Org. Lett., 2013, 15, 5838–5841 CrossRef PubMed
- J. C. Gomes, M. T. Rodrigues Jr, A. Moyano and F. Coelho, Eur. J. Org. Chem., 2012, 2012, 6861–6866 CrossRef
- L.-R. Zhang, F.-P. Yi, X. Zhang and J.-Z. Zou, J. Chem. Res., 2012, 36, 418–420 CrossRef
- S. Zhao, H. Zhi, M. Zhang, Q. Yan, J. Fan and J. Guo, RSC Adv., 2016, 6, 62778–62784 RSC
- A. Richaud, N. Barba-Behrens and F. Méndez, Org. Lett., 2011, 13, 972–975 CrossRef PubMed
- V. K. Aggarwal, I. Emme and A. Mereu, Chem. Commun., 2002, 1612–1613 RSC
- Y. Wang, L. Liu, D. Wang and Y.-J. Chen, Can. J. Chem., 2012, 90, 85–91 CrossRef
- S. Zhao, M. He, Z. Guo, N. Zhou, D. Wang, J. Li and L. Zhang, RSC Adv., 2015, 5, 32839–32845 RSC
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR. See DOI: 10.1039/c8ra02409j |
| This journal is © The Royal Society of Chemistry 2018 |