DOI:
10.1039/C8RA02417K
(Paper)
RSC Adv., 2018,
8, 14760-14764
Persistent radical anion polymers based on naphthalenediimide and a vinylene spacer†
Received
19th March 2018
, Accepted 12th April 2018
First published on 18th April 2018
Abstract
Persistent n-doped conjugated polymers were achieved by doping the electron accepting PDNDIV and PFNDIV polymers with ionic (TBACN) or neutral (TDAE) dopants. The great electron affinities, as indicated by the low LUMO levels of PDNDIV (−4.09 eV) and PFNDIV (−4.27 eV), facilitated the chemical reduction from either TBACN or TDAE. The low-lying LUMOs of the neutral polymers PDNDIV and PFNDIV were achieved by incorporation of vinylene spacers between the electron poor NDI units to increase the conjugation length without the use of an electron donor, and this was lowered further by an electron-withdrawing fluorinated N-substituent on the NDI moiety. The polymer radical anions were found to persist for several days under ambient conditions by EPR spectroscopy. A distinguishing and noteworthy feature of these polymers is that they can be consecutively reduced by up to four electrons in acetonitrile. Conductivity measurements demonstrate the prospective impact of PDNDIV and PFNDIV for organic electronics.
Introduction
Doped conjugated polymers with excess electronic charge carriers are essential for many organic electronic applications such as organic thermoelectrics that require enhanced electrical conductivity. Applications for organic electronics require either p-doped (doping with positive charge carriers, holes) or n-doped (doping with negative charge carriers, electrons) polymers or both. Many conjugated polymers can be p-doped with ease using chemical or electrochemical methods. Several of these polymers retain their p-doping at ambient conditions making it easier to incorporate them into devices. On the other hand, obtaining stable n-doping in conjugated polymers is a challenge because many conjugated polymers have a low electron affinity. Thus, chemical n-doping requires very reactive dopants, and the doped polymers can easily de-dope by transferring the electrons to other molecules such as oxygen. Therefore, examples of extrinsically n-doped polymers1–5 are sparse and often require doping under dry nitrogen or argon. Recently, Katz and co-workers6 reported an n-doped polymer that they claimed to be the first demonstrated example of an air-stable n-doped polymer. Herein, we report a different class of naphthalenediimide-based polymers that can be n-doped under ambient conditions. We also demonstrate the important role of side chains in tuning and enhancing the air-stability of the doped polymers.
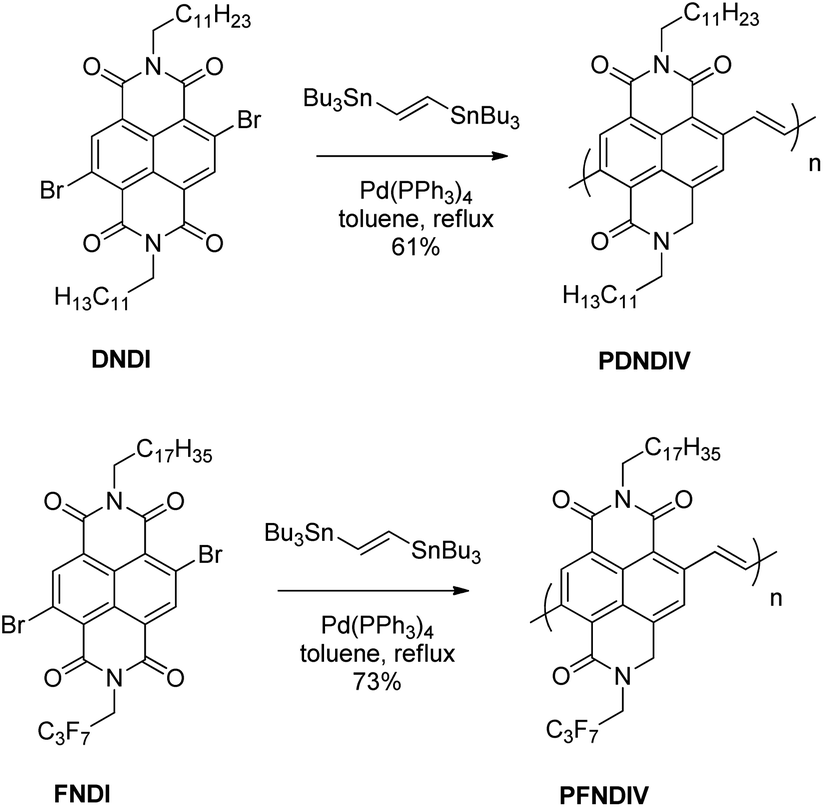
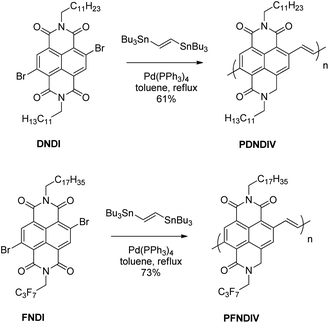
We had previously reported a straightforward route to electron-transporting polymers from readily available electron accepting monomers and vinylene spacers.7 Using this route, and monomers containing naphthalene diimide (NDI) units, we designed two new naphthalene diimide-vinylene (NDIV) copolymers. We chose to incorporate vinylene spacers instead of electron-rich donor units since vinylene groups impose planarity of the conjugated polymer while maintaining a high electron affinity. We chose to utilize two NDI monomers, one with alkyl hydrocarbon side chains on both imide nitrogens [N,N′-bis(n-dodecyl)-2,6-dibromo-1,4,5,8-naphthenedicarboximide (DNDI), Scheme 1] and the other with an alkyl fluorocarbon on one imide nitrogen and an alkyl hydrocarbon chain on the other imide nitrogen [N-(2,2,3,3,4,4,4-heptafluorobutyl)-N′-octadecyl-2,6-dibromo-1,4,5,8-naphthenedicarboximide (FNDI), Scheme 1]. We chose naphthalene diimide because it has been shown to form stable radical anions.8–10 Based on the work by Katz and co-workers,11 we hypothesized that the fluorocarbon sidechain will lower the lowest unoccupied molecular orbitals (LUMO), increase the electron affinity, and thus chemical stability of the n-doped polymer. To shed additional insight into the electronic properties of these systems, we carried out first-principles density functional theory (DFT) calculations on a series of oligomers ranging from 1 to 6 monomer units for both the FNDI and DNDI systems. Both the electron affinity and the spin density (see Fig. S1 in the ESI for details†) were computed at the ωB97XD/6-31g(d,p) level of theory,12 which has been previously benchmarked for a variety of charged, conjugated organic systems.13,14 Based on our DFT calculations, we found that oligomers with FNDI had an electron affinity that was 0.2 eV higher than oligomers with DNDI.
 |
| | Scheme 1 Synthesis of PDNDIV and PFNDIV by Stille coupling reaction. | |
The polymers, poly[N,N′-bis(n-dodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diylvinylene] (PDNDIV) and poly[N-(2,2,3,3,4,4,4-heptafluorobutyl)-N′-octadecyl-1,4,5,8-naphthalenedicarboximide-2,6-diylvinylene] (PFNDIV) were synthesized using previously reported methods.7,15,16 The polymers were doped by chemical reduction using either tetrabutylammonium cyanide (TBACN)17–19 or tetrakis(dimethylamino)ethylene (TDAE).5,20 Consistent with our hypothesis, we found that the incorporation of a fluorinated side chain in PFNDIV increased the electron affinity, as indicated by its decreased LUMO energy level (−4.27 eV), estimated from electrochemical measurements. Each polymer showed well-defined spectroscopic changes after gradual addition of either dopant due to the formation of radical anions, which were confirmed by EPR. Both of the doped polymers exhibit signatures of the radical anion for several days in THF under ambient conditions. We also found a distinguishing and noteworthy feature of these polymers: they can be consecutively reduced by up to four electrons in acetonitrile.
Results and discussion
Synthesis and characterization
The synthetic methods for DNDI, FNDI, PDNDIV, and PFNDIV are shown in Scheme 1, and the experimental procedures are described in the ESI.† The molecular weights of the polymers were measured by high temperature gel permeation chromatography (GPC) using 1,2,4-trichlorobenzene as the mobile phase and summarized in Table 1.
Table 1 Physical properties of the polymers
| Polymer |
Mn (g mol−1) |
| PDNDIV |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 622 622 |
| PFNDIV |
8304 |
The thermal stability of PDNDIV and PFNDIV was determined by using thermogravimetric analysis (TGA) with a heating rate of 10 °C min−1 under nitrogen atmosphere (Fig. S2a†). TGA curves showed that the decomposition temperatures (Td, at which 5% weight loss occurred) for PDNDIV and PFNDIV were found to be 239 and 279 °C, suggesting sufficient thermal stability of the polymers (Table 1). Differential scanning calorimetry (DSC, see Fig. S2b and S2c†) and powder X-ray diffraction patterns (Fig. S3†) of the polymers did not show any distinguishing peaks, which indicates that the solid-state structure of the polymers are amorphous.
Optoelectronic properties
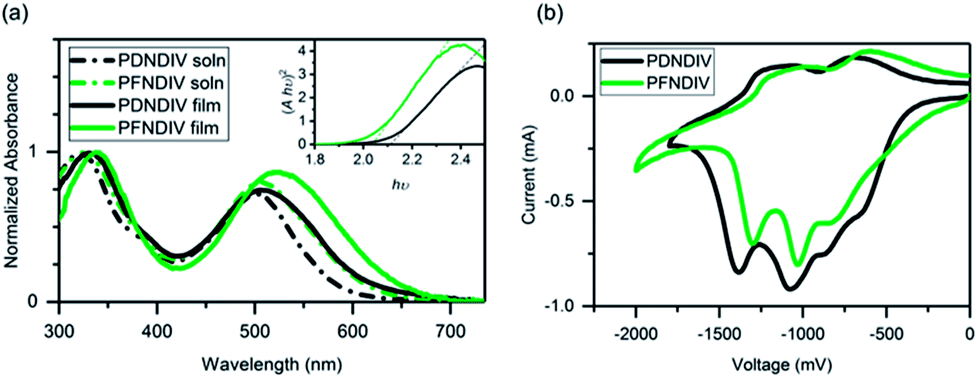
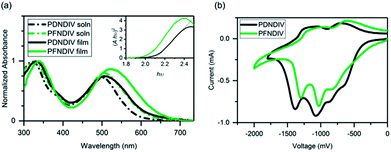
The UV-Vis absorption spectra of each polymer in dilute chloroform solution (10−5 M on the basis of repeating units) and of thin films on glass substrates coated with indium tin oxide (ITO) is shown in Fig. 1a. Both polymers showed two distinctive absorption bands, one in the blue region from 300–400 nm and the other in the red region 450–650 nm. We attribute these absorption to π–π* transitions of the NDI chromophore and electronic transitions between new delocalized bands in the polymer backbone,21 respectively (Table 2). A distinct broadening and red shift of the low energy transitions by 10–20 nm is observed in the polymer films, which indicates chromophore aggregation caused by interactions between the chains in the solid-state.22 The optical band gaps (Eg) obtained from the Tauc plot (see Fig. 1a inset) are 2.11 eV for PDNDIV and 2.04 eV for PFNDIV. The reduced Eg value of PFNDIV compared to that of PDNDIV was attributed to the presence of the electron-withdrawing fluorinated side chain, which can lower the LUMO energy level through inductive effects.11| | |
ELUMO = −4.8 eV − (Ereduction − 0.32 eV)
| (1) |
 |
| | Fig. 1 (a) UV-Vis spectra of the polymers in 10−5 M chloroform solution and as thin films on ITO coated glass. The Tauc plot of the film spectra, assuming r = 1/2 for a direct allowed transition, including the dashed trace used to determine the bandgap is inset. (b) Cyclic voltammograms of the polymer film dropcast onto a Pt disk (1.6 mm diameter) electrode as the working electrode, using Pt wire as the counter electrode, Ag/AgCl as the reference electrode, and 0.1 M tetrabutylammonium perchlorate as the supporting electrolyte at a scan rate of 50 mV s−1, reduction only scans. | |
Table 2 Optical and electrochemical properties of the polymers
| Polymer |
λmax solution (nm) |
λmax film (nm) |
Ereduction (eV) |
aEHOMO (eV) |
bELUMO (eV) |
cEg (eV) |
| Eqn (2). Eqn (1). Obtained from the Tauc plot. |
| PDNDIV |
322, 500 |
330, 509 |
−0.392 |
−6.20 |
−4.09 |
2.11 |
| PFNDIV |
325, 506 |
338, 524 |
−0.208 |
−6.31 |
−4.27 |
2.04 |
Cyclic voltammograms (CV) of the polymer films are shown in Fig. 1b. There were multiple reduction onsets in the CV, indicating that polymers can be consecutively reduced by up to 4 electrons in acetonitrile. LUMO energies of both the polymers were calculated from their first onset reduction potentials (eqn (1)) as −4.09 eV for PDNDIV and −4.27 eV for PFNDIV. The HOMO energy levels were estimated by subtracting the Eg from the above CV-determined LUMO energy levels and were found to be −6.20 eV for PDNDIV and −6.31 eV for PFNDIV (eqn (2)). CV data showed that both polymers are positioned at low-lying LUMO levels and the electron withdrawing N-substituents impart a significant impact on PFNDIV's superior π-acidity, which is evident from additional diminution in reduction potential.
Characterization and stability study of radical anion polymers
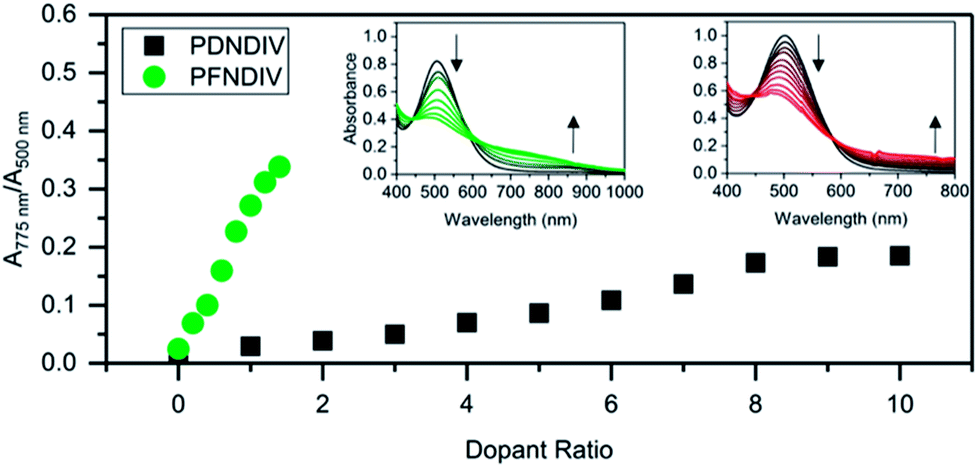
The polymers were doped using TDAE, a neutral electron donor, or TBACN, an anionic electron donor, under ambient conditions. TDAE17–19 and TBACN5,20 have been used as an electron donors for rylene diimides, and their proposed mechanism of doping is a single electron transfer. In the UV-Vis-NIR spectra, we found that the continuous addition of 0–1.4 equivalents (with respect to the polymer's repeating units) of TBACN with PFNDIV, progressively bleached the neutral polymer absorption band (450–650 nm) and simultaneously produced a new absorption band at 750 nm, with an isosbestic point near 600 nm (Fig. 2a). A similar behavior was observed for PDNDIV, but with larger numbers of equivalents (0–10) of TBACN (Fig. 2b) required to produce similar spectral changes.
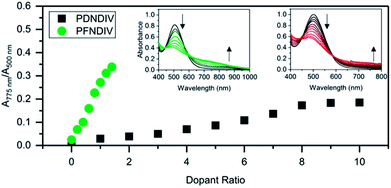 |
| | Fig. 2 Ratio of doped to undoped repeating units, estimated by UV-Vis-NIR spectroscopic changes, for PDNDIV and PFNDIV in THF with increasing molar equivalents of TBACN; insets are the UV-Vis-NIR spectra for PFNDIV (left) and PDNDIV (right). | |
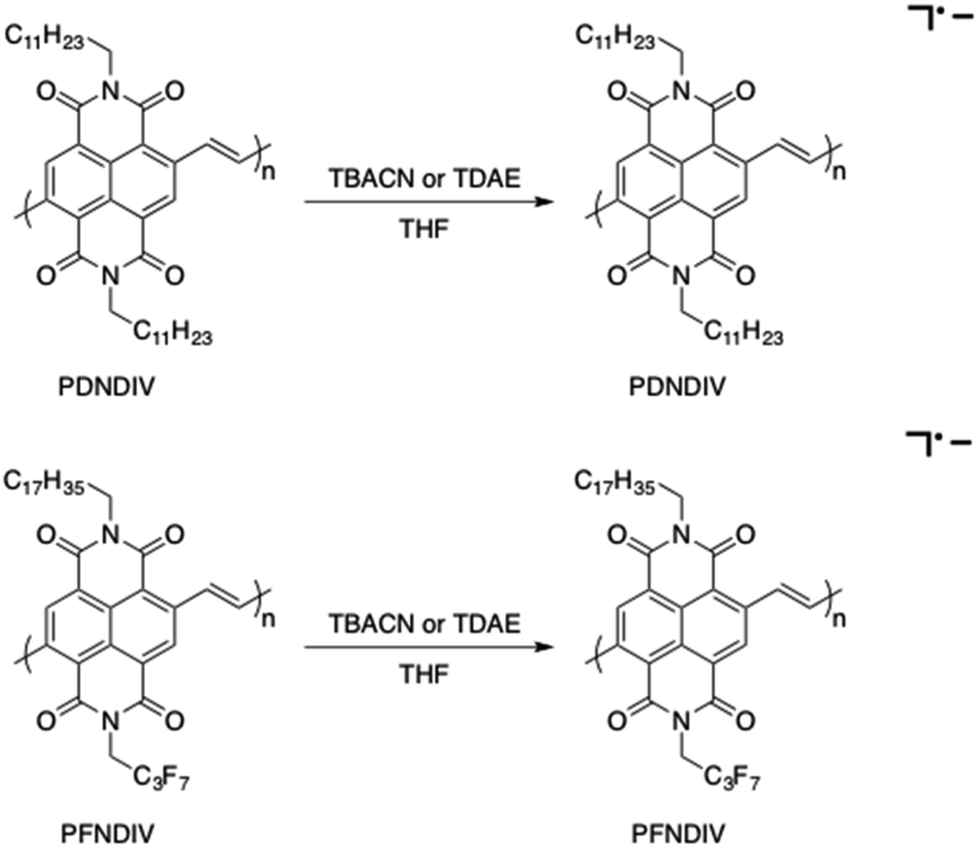
The formation of a weaker absorption band at ∼750 nm suggests the formation of the radical anion polymers [PDNDIV]˙− and [PFNDIV]˙− (Fig. 3), which was also confirmed by EPR spectroscopy (vide infra). The addition of TDAE also resulted in an instantaneous color change of the polymer solution (inset Fig. S5†), which is consistent with an n-doped polymer. TDAE-doped radical anion polymers were also characterized by UV-Vis-NIR absorption (Fig. S5†) and EPR spectroscopy (Fig. S6b†).
 |
| | Fig. 3 Synthesis of [PDNDIV]˙− and [PFNDIV]˙− under ambient condition in THF using TBACN or TDAE. | |
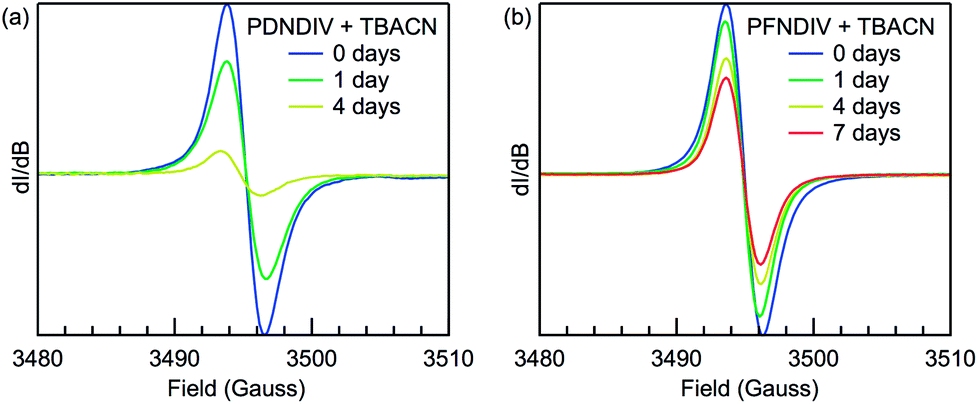
EPR spectroscopy was used to probe the formation of a radical anion and monitor the relative stability of the radical under ambient conditions over time. The EPR spectra of the polymers after addition of TBACN are shown in Fig. 4. Both PDNDIV and PFNDIV display a single, narrow resonance at g = 2.0035 upon mixing with TBACN, which indicates the formation of [PDNDIV]˙− and [PFNDIV]˙− radical anions. The absence of any hyperfine splitting indicates that the spin density of the radical anion is localized on the carbon of the aromatic ring. DFT calculations performed with the ωB97XD/6-31g(d,p) basis verify the nature of the SOMO as shown in the difference plots of the molecular orbital spin density (Fig. S1†). Both polymer radical anions are persistent as THF solutions stored in air, but the radical signal of [PFNDIV]˙− is more stable with an apparent half-life on the order of one week (Fig. 4b), comparable to the stability of the recently reported n-type polymer, ClBDPPV.6 The decay of the radical EPR signal is tentatively assigned to quenching by triplet oxygen. The difference in radical stabilities could possibly originate from variation in the reduction potentials of the LUMOs of [PDNDIV]˙− and [PFNDIV]˙−, with the more negative LUMO on PFNDIV. Interestingly, the EPR spectrum of the as-prepared PFNDIV displays the same radical signal and suggests PFNDIV can be partially doped under ambient conditions while PDNDIV displays no signal (Fig. S6a†).
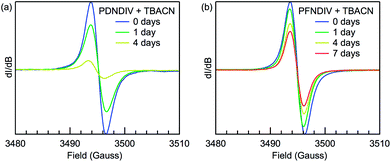 |
| | Fig. 4 Room-temperature X-band EPR spectra of (a) 2 mM (repeating units) PDNDIV with 10 mM TBACN and (b) 2 mM PFNDIV with 2 mM TBACN in THF yielding [PDNDIV]˙− and [PFNDIV]˙−, respectively. The 7 day point for [PDNDIV]˙− is omitted and unreliable due to polymer precipitation. | |
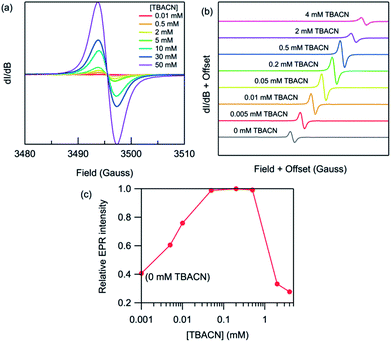 |
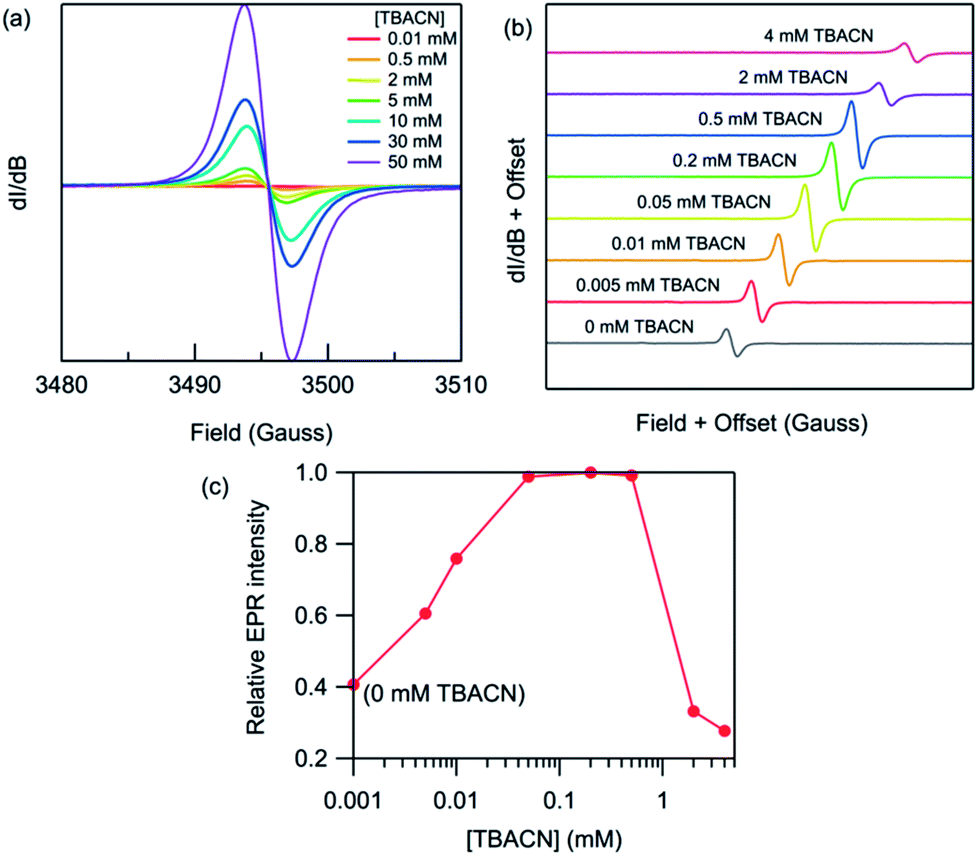
| | Fig. 5 Room-temperature X-band EPR spectra of (a) PDNDIV (2 mM) and (b) PFNDIV (0.5 mM) after mixing with increasing concentration of TBACN in THF. (c) Normalized EPR intensity of PFNDIV (0.5 mM) with increasing concentration of TBACN in THF. | |
We then studied the impact of the concentration of TBACN on the intensity of the EPR signal Fig. 5. For PDNDIV we found that 1 equivalent of TBACN with respect to polymer repeating units was insufficient to dope the polymer. At higher concentrations, we observed an EPR signal, which indicated the formation of the radical anion. The intensity of the signal increased steadily with increasing concentration of the dopant. This observation is consistent with the presence of an equilibrium, with a slight preference for the reactants. This is consistent with the expected LUMO energy levels of PDNDIV and the cyanide anion of TBACN. In the case of PFNDIV, we found a steady increase in the EPR signal up to 1 equivalent of the TBACN. Further increase of the cyanide concentration led to a decrease in the EPR signal. From CV studies, we know that the polymer can accept up to four electrons. Therefore, we tentatively attribute this decrease in [PFNDIV]˙− EPR signal with increasing cyanide concentration to the formation of an EPR-silent singlet or triplet [PFNDIV]2− species.
Conductivity studies
To investigate the solid-state conductivity of these radical anion polymers under ambient conditions, we fabricated two-terminal devices using vacuum depositing Au electrodes (Fig. S11,† inset) using neutral or doped PDNDIV or PFNDIV acting as the active layer. Either the neutral or doped polymers were drop-cast from THF on the fabricated channels, and current (I)–voltage (V) measurements were carried out under ambient conditions (Fig. S9 and S10†). The conductivity of each doped polymer, 6.98 × 10−7 S cm−1 for [PDNDIV]˙− and 3.28 × 10−7 S cm−1 for [PFNDIV]˙−, was at least an order of magnitude greater than the conductivity of the neutral polymers, 3.49 × 10−8 S cm−1 for PDNDIV and 2.61 × 10−8 S cm−1 for PFNDIV. While these conductivities are lower than those of other reported n-doped conjugated polymers and are too low for practical applications, the drastic increase in conductivity upon doping and the air stability of the doped polymers suggests that these and similar NDI-vinylene polymers may find potential use as n-type organic electronic materials. The low conductivities may be due, in part, to the amorphous nature of these polymers in the solid state.
Conclusions
In summary, two new NDI-vinylene based electron acceptor polymers have been successfully synthesized. Due to the low-lying LUMOs (−4.09 and −4.27 eV) both the polymers behave as strong π-acids. The electron-withdrawing effect of the fluorinated side chain reduces the LUMO level of PFNDIV significantly. Multistep reduction potentials indicate the presence of several electron accepting sites in the polymer backbone. Chemical reduction from electron donors to the polymer generates persistent radical anion polymers with ambient stability. Time-dependent EPR spectroscopy showed the stability of the radical anions exists beyond several days. Finally, conductivity measurements showed the positive impact of chemical doping on the polymer's electrical conductivity, making these materials promising for organic electronic applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the Department of Chemistry University of Massachusetts Amherst for research facilities. S. D. acknowledges IUSSTF for a research fellowship. B. M. W. acknowledges the National Science Foundation for the use of supercomputing resources through the Extreme Science and Engineering Discovery Environment (XSEDE), Project No. TG-CHE150040.
Notes and references
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Adv. Mater., 2014, 26, 2825–2830 CrossRef CAS PubMed.
- K. Shi, F. Zhang, C.-A. Di, T.-W. Yan, Y. Zou, X. Zhou, D. Zhu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 6979–6982 CrossRef CAS PubMed.
- B. D. Naab, S. Zhang, K. Vandewal, A. Salleo, S. Barlow, S. R. Marder and Z. Bao, Adv. Mater., 2014, 26, 4268–4272 CrossRef CAS PubMed.
- S. Hwang, W. J. Potscavage Jr., Y. S. Yang, I. S. Park, T. Matsushima and C. Adachi, Phys. Chem. Chem. Phys., 2016, 18, 29199–29207 RSC.
- S. Wang, H. Sun, U. Ail, M. Vagin, P. O. Å. Persson, J. W. Andreasen, W. Thiel, M. Berggren, X. Crispin, D. Fazzi and S. Fabiano, Adv. Mater., 2016, 28, 10764–10771 CrossRef CAS PubMed.
- X. Zhao, D. Madan, Y. Cheng, J. Zhou, H. Li, S. M. Thon, A. E. Bragg, M. E. DeCoster, P. E. Hopkins and H. E. Katz, Adv. Mater., 2017, 29, 1606928 CrossRef PubMed.
- G. Nagarjuna, A. Kokil, J. Kumar and D. Venkataraman, J. Mater. Chem., 2012, 22, 16091–16094 RSC.
- S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, J. Am. Chem. Soc., 2014, 136, 12004–12010 CrossRef CAS PubMed.
- Q. Song, F. Li, Z. Wang and X. Zhang, Chem. Sci., 2015, 6, 3342–3346 RSC.
- S. K. Keshri, S. Kumar, K. Mandal and P. Mukhopadhyay, Chem.–Eur. J., 2017, 23, 11802–11809 CrossRef CAS PubMed.
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. R. Golder, B. M. Wong and R. Jasti, Chem. Sci., 2013, 4, 4285–4291 RSC.
- P. Li, B. M. Wong, L. N. Zakharov and R. Jasti, Org. Lett., 2016, 18, 1574–1577 CrossRef CAS PubMed.
- X. Liang, L. Tan, Z. Liu, Y. Ma, G. Zhang, L. Wang, S. Li, L. Dong, J. Li and W. Chen, Chem. Commun., 2017, 53, 4934–4937 RSC.
- H. Zhang, Y. Xie, X. Chen, T. Jia, W. Huang, S. Luo, Q. Hou, R. Zeng and Z. Sun, J. Electrochem. Soc., 2017, 164, A290–A294 CrossRef CAS.
- Y. Kumar, S. Kumar, S. Kumar Keshri, J. Shukla, S. S. Singh, T. S. Thakur, M. Denti, A. Facchetti and P. Mukhopadhyay, Org. Lett., 2016, 18, 472–475 CrossRef CAS PubMed.
- M. R. Ajayakumar, D. Asthana and P. Mukhopadhyay, Org. Lett., 2012, 14, 4822–4825 CrossRef CAS PubMed.
- M. R. Ajayakumar, P. Mukhopadhyay, S. Yadav and S. Ghosh, Org. Lett., 2010, 12, 2646–2649 CrossRef CAS PubMed.
- M. Fujitsuka, S. S. Kim, C. Lu, S. Tojo and T. Majima, J. Phys. Chem. B, 2015, 119, 7275–7282 CrossRef CAS PubMed.
- P. M. Alvey and B. L. Iverson, Org. Lett., 2012, 14, 2706–2709 CrossRef CAS PubMed.
- L. Ye, X. Jiao, H. Zhang, S. Li, H. Yao, H. Ade and J. Hou, Macromolecules, 2015, 48, 7156–7163 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02417k |
| ‡ These authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Kevin R. Kittilstved
b,
Kevin R. Kittilstved