
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSOMC grafting of vanadium oxytriisopropoxide (VO(OiPr)3) on dehydroxylated silica; analysis of surface complexes and thermal restructuring mechanism†
Manuel P. Högerl‡
a,
Li Min Serena Goh‡a,
Edy Abou-Hamada,
Samir Barmana,
Oliver Dachwaldb,
Farhan Ahmad Pashaa,
Jeremie Pelletiera,
Klaus Köhlerb,
Valerio D'Elia *ac,
Luigi Cavallo*a and
Jean-Marie Basset*a
*ac,
Luigi Cavallo*a and
Jean-Marie Basset*a
aKAUST Catalysis Center, King Abdullah University of Science and Technology, 23955-6900 Thuwal, Saudi-Arabia. E-mail: Jeanmarie.basset@kaust.edu.sa; luigi.cavallo@kaust.edu.sa
bDepartments of Chemistry and Catalysis Research Center, Technical University of Munich, Lichtenbergstrasse 4, Ernst-Otto-Fischer-Strasse 1, 85747 Garching, Germany
cDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), 21210, Payupnai, WangChan, Rayong, Thailand. E-mail: valerio.delia@vistec.ac.th
First published on 6th June 2018
Abstract
Vanadium oxytriisopropoxide (VO(OiPr)3), 1, was grafted on highly dehydroxylated silica (SiO2-700: aerosil silica treated at 700 °C under high vacuum) to generate compound 2 following the concepts and methodology of surface organometallic chemistry (SOMC). The resulting compound was analyzed by elemental analysis, FT-IR, 1H, 13C and 51V solid state (SS) NMR, Raman and EPR spectroscopies. The grafting reaction of 1 to generate 2 was found to lead to the formation of a monopodal surface complex [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O–)V(O)(OiPr)2], 2m, as well as bipodal [(Si–O–)2V(O)(OiPr)], 2b, formed along with (Si–O–iPr) moieties as an effect of the classical rearrangement of 2m with strained siloxane bridges. Upon controlled thermal treatment at 200 °C under high vacuum, 2m and 2b were found to mainly rearrange to tetrahedral VO4 moieties [(Si–O–)3V(O)] (3) with formation of propylene whereas the (Si–O–iPr) groups were preserved. The mechanism of the thermal rearrangement of the isopropoxide groups was investigated by a DFT approach revealing the occurrence of a concerted γ-H-transfer and olefin elimination mechanism.
Si–O–)V(O)(OiPr)2], 2m, as well as bipodal [(Si–O–)2V(O)(OiPr)], 2b, formed along with (Si–O–iPr) moieties as an effect of the classical rearrangement of 2m with strained siloxane bridges. Upon controlled thermal treatment at 200 °C under high vacuum, 2m and 2b were found to mainly rearrange to tetrahedral VO4 moieties [(Si–O–)3V(O)] (3) with formation of propylene whereas the (Si–O–iPr) groups were preserved. The mechanism of the thermal rearrangement of the isopropoxide groups was investigated by a DFT approach revealing the occurrence of a concerted γ-H-transfer and olefin elimination mechanism.
Introduction
Vanadium oxo complexes on oxide supports have attracted the attention of researchers in the fields of catalysis and inorganic chemistry for their role in promoting the oxidation of organic compounds1 and the non-oxidative2 and oxidative3 dehydrogenation of propane (ODHP) just to cite a few key applications. When employing traditional preparative techniques such as incipient wetness impregnation for the synthesis of supported vanadium oxo complexes, a distribution of monomeric and polymeric VOx species is generally formed on the surface irrespective of precursor and support.4–7 As an effect, it is difficult to characterize the individual surface domains and to evaluate their role in catalysis when using such materials.3,8–16 Among relevant vanadium oxo complexes, vanadium(IV) isopropoxide species have been often indicated as key intermediates within the mechanism of ODHP:17–19 the vanadium isopropoxide intermediate, formed after the first H-abstraction, should undergo a non-rate determining,17,19 H-transfer to an oxygen atom (V–O-support or V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the catalyst to generate propene before the catalytic cycle is completed by reoxidation of the V centers (Scheme 1).18–26
O) of the catalyst to generate propene before the catalytic cycle is completed by reoxidation of the V centers (Scheme 1).18–26
 | ||
| Scheme 1 Proton abstraction from V(IV) isopropoxide intermediates as proposed in several computational studies: (a) ref. 19, (b) ref. 18, (c) ref. 17, (d) ref. 20. | ||
The experimental observation and isolation of vanadium isopropoxide species under the reaction conditions of conventional ODHP catalysis is extremely challenging because they are labile intermediates formed after the rate determining step.19 Furthermore, it is extremely challenging to prepare vanadium(IV) complexes with uniform structure and oxidation state as well-defined and isolated species on the support surface. A viable strategy for the study of the transformation of supported vanadium isopropoxide species is by the grafting of more stable V(V) oxo isopropoxide complexes and the study of their stoichiometric transformations.27,28
In this context, a recent contribution to the elucidation of the structure and of the chemical reactivity of silica-supported vanadium oxo isopropoxide species has been reported by Hermans et al. by grafting 1 on highly dehydroxylated silica.28 The authors were able to show that, upon thermal treatment starting at 200 °C, the isolated [(Si–O–)V(O)(OiPr)2] complexes generated on the surface would undergo thermal restructuring leading to the formation of isolated tetrahedral VO4 sites along with two molecules of propene.
Surface organometallic chemistry (SOMC) allows the synthesis and investigation of well-defined and isolated surface complexes by advanced preparative and diagnostic tools.29,30 The grafting of suitable organometallic complexes by this strategy under rigorously controlled conditions is a useful tool for the preparation and the study of well-defined pre-catalytic complexes and supported reaction intermediates that could not be isolated by homogeneous organometallic chemistry.31–34 For instance, we have recently shown that by employing highly dehydroxylated silica supports,35 and by the careful application of SOMC-grafting techniques, it is possible to prepare truly isolated tetrahedral VO4 units that show a higher propylene productivity compared to that of traditional impregnated catalyst applied under identical conditions.36 This observation justifies the application of the advanced SOMC preparative tools for a more accurate study of vanadium oxo surface complexes related to the reaction network of ODHP.
Whereas, in previous studies, the grafting of 1 was carried out by the reaction of its vapors with silica,27,28 this work, analyzes the apparently different (and differently distributed) complexes formed by the solution-phase grafting of 1 on SiO2-700 by the SOMC protocol and the corresponding products of rearrangement after controlled thermolysis. Furthermore, given the lack of computational studies analyzing the reactivity of well-defined, supported vanadium(V) isopropoxide complexes, we applied density functional theory (DFT) calculations to gain deeper insights into the thermal restructuring of 2m. The occurrence of a concerted step of γ-H-transfer and olefin elimination, rather than a two-step radical process, is shown in this stoichiometric transformation.
Experimental section
All experiments were carried out using Schlenk equipment or in Argon filled gloveboxes (<0.1 ppm H2O, <0.1 ppm O2), and by using high-vacuum lines (∼10−5 mbar). Solvents were dried by use of a commercial solvent purification system (CuO catalysts/Alox) and degassed by two freeze–pump–thaw cycles. Molecular sieves were dried in vacuum 10−3 mbar for 12 h at 300 °C. Further information on analytical techniques and instrumentation is available in the ESI.†.Partially dehydroxylated silica (SiO2-700) was prepared by heat treatment of Evonik Aerosil 200 (10 g) under vacuum in a quartz tube. The starting silica material was pretreated with a minimal amount of water (∼10 mL) to ease its handling and was dehydroxylated at 700 °C for 18 h under vacuum (∼10−5 mbar).
Synthesis of 2
A mixture of [VO(OiPr)3], (purchased from Sigma-Aldrich as a clear, pale yellow solution; used as received because the analysis of the sample by liquid NMR gave no indication of decomposition or impurities) (0.080 mL) and SiO2-700 (1 g) in pentane (10 mL) was stirred at r.t. overnight. After filtration, the solid (2) was washed 3 times with pentane and the liquid was transferred to a round bottom flask to quantify the isopropanol evolved during grafting by quantitative NMR using hexamethylbenzene C6Me6 as an internal standard. Analysis by NMR indicated the formation of 0.9 ± 0.1 isopropanol/V during grafting. The resulting white powder was dried under vacuum (∼10−5 mbar).Anal. found: V, 1.42; C, 2.02; H, 0.42 (%). 1H MAS NMR (600 MHz): δ 5.5, 4.3, 1.4, and 1.2 ppm. 13C CP-MAS NMR (600 MHz): δ 85, 67, and 23 ppm.
Thermolysis of 2 to 3
The thermolysis of 2 was carried out as a self-supporting silica pellet (∼30–50 mg) in a custom made vacuum IR cell. Experiments were carried out under static or dynamic vacuum to continuously remove any gaseous product (∼10−5 mbar). After the reaction time, the formed gases were (a) analyzed by transmission IR spectroscopy, and (b) sampled by gas-syringe from the reaction vessel and subjected to gas-chromatography, or (c) transferred to another vessel by trapping them with liquid N2 and then sampled, or (d) evacuated.Results and discussion
1 was reacted with silica (Aerosil® 200 from Evonik) partially dehydroxylated at 700 °C (SiO2-700) in pentane to generate 2. Elemental analysis of 2 revealed a vanadium loading of 1.42 ± 0.1 wt% suggesting an approximate average of 0.9 V-atoms per nm2 (0.28 mmol V g−1), which compares well with the number of available surface silanol groups (∼ 1.0 per nm2, 0.23–0.3 mmol silanol/g).35 The C/V molar ratio of 6.0 (expected C/V ratio for 1: 6) is in agreement with the presence of two isopropyl fragments per atom of vanadium.Controlled thermolysis reaction of 2 (2 °C min−1 from ∼23 to 200 °C, 1 h at 200 °C) as a self-supporting pellet under static vacuum conditions (∼10−5 mbar) to generate 3 led to the formation of nearly stoichiometric amounts of propene in the gas phase, as observed by IR and GC/FID/MS (0.95 equiv. propene C3H6 per V, see the ESI (Fig. S1†)). Oxygenates, such as isopropanol and acetone, possible products of ligand dissociation and dehydrogenation, were observed only in trace amounts.
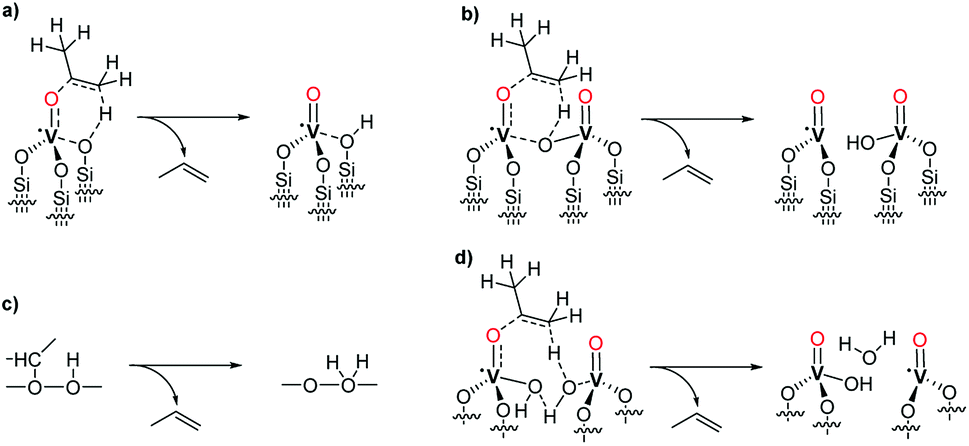
Grafting of 1 on SiO2-700 led to the appearance of the ν(C–H) bands (at 2970–2878 cm−1) and of the δ(C–H) bands (1467, 1453, 1384, 1371, 1326 cm−1) of 1 in the FTIR spectrum (Fig. 1) of 2 with the concomitant nearly complete disappearance of the ν(O–H) at 3741 cm−1 relative to the isolated silanols of the pristine support (see Fig. S2† for a comparison of 2 and SiO2-700).37 IR analysis before and after (3) the thermolysis reaction at 200 °C for 1 h under static vacuum showed a clear decrease of the intensity of the aliphatic bands present in 2, with the concomitant formation of a new sharp band at 3660 cm−1 and the reappearance of the silanol band at 3742 cm−1 (Fig. 1). The band at 3660 cm−1 can be assigned to a ν(VO–H) stretching mode.36,38
 | ||
| Fig. 1 IR spectra comparing the product of grafting of 1 (2, bottom trace), the material after thermolysis at 200 °C for 1 h (3, middle trace), and the difference spectrum of 2 and 3 (C, top trace). | ||
Along with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 V/initial silanols ratio obtained from the elemental analysis of 2, the nearly complete disappearance of the band relative to the isolated silanols, suggests that each precursor complex reacts stoichiometrically with a single silanol. To note, this result is different from what obtained by Hermans et al. when vapors of 1 were reacted with SiO2-700. In that case, an excess of precursor reacted with the support leading to a vanadium loading of about 2.9 wt% (about the double as obtained in our case). The authors attributed this observation to the reaction of the precursor also with the strained siloxane bridges of SiO2-700.28
:1 V/initial silanols ratio obtained from the elemental analysis of 2, the nearly complete disappearance of the band relative to the isolated silanols, suggests that each precursor complex reacts stoichiometrically with a single silanol. To note, this result is different from what obtained by Hermans et al. when vapors of 1 were reacted with SiO2-700. In that case, an excess of precursor reacted with the support leading to a vanadium loading of about 2.9 wt% (about the double as obtained in our case). The authors attributed this observation to the reaction of the precursor also with the strained siloxane bridges of SiO2-700.28
Furthermore, the IR spectra in Fig. 1 show that upon thermal treatment of 2 at 200 °C, only part of the organic isopropyl groups, responsible for the signal at 2970–2878 cm−1, are removed from the surface.
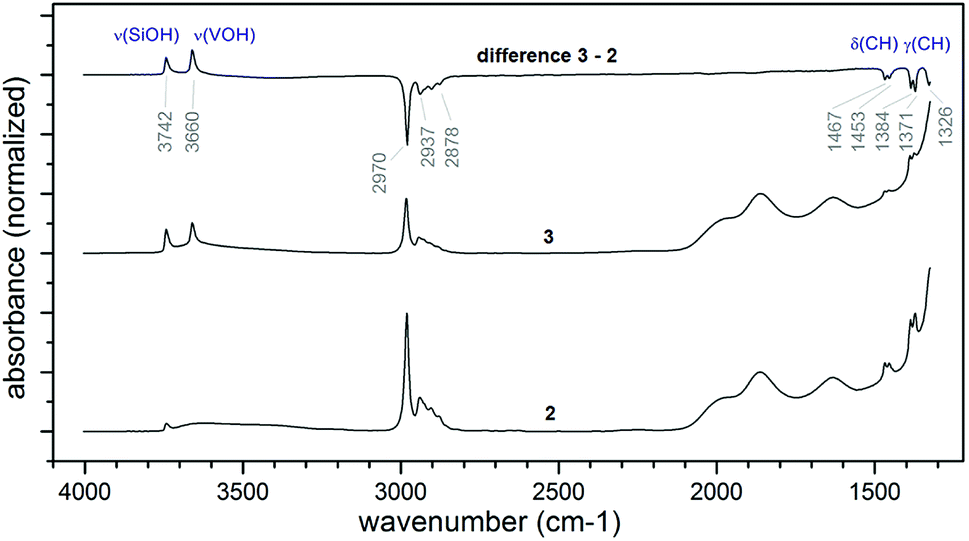
Analysis of 2 and 3 by Raman spectroscopy (Fig. 2) shows that either before and after thermal treatment the vanadyl group is present in the samples as supported by the intense absorption band at 1030–1035 cm−1 attributable to the isolated vanadyls of 2 and 3. The absence of bands in the 980–1000 cm−1 region excludes the formation of crystalline vanadia.3,6
 | ||
| Fig. 2 Raman spectra of SiO2-700, 2 and 3. | ||
To assess the oxidation state of the vanadium atoms before and after thermal treatment, 2 and 3 have been investigated by in situ electron paramagnetic resonance spectroscopy (EPR). In all cases, well-resolved EPR spectra of V(IV) were obtained with well resolved hyperfine structure (Fig. S3†). However, after careful quantitative calibration with standard V(IV) samples, it was found that the percentage of V(IV) relative to V(V) did not exceed 1.5% for 2. Also, after treatment of 2 to generate 3, the relation between V(IV) to V(V) did not exceed 2%. In addition, no indication of V(III) species was obtained at any stage. Therefore, the formation of paramagnetic V(IV) species, as observed by in situ EPR studies, was limited to a very minor component of 2 and 3.
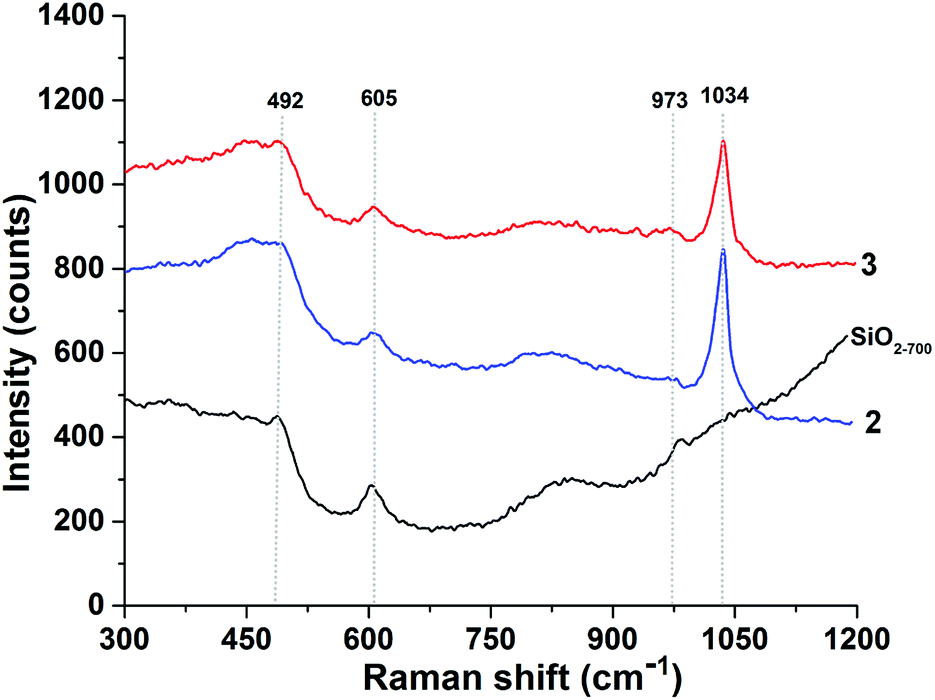
To assess the structure of 2 and 3 the 1H and 13C SS NMR spectra of these compounds were measured. The 1H spin-echo MAS NMR spectrum of 2 shows four signals at 1.2, 1.4, 4.3, and 5.5 ppm (Fig. 3A). The signals at 1.2 and 1.4 ppm auto correlate in 2D Double-Quantum (DQ) and Triple-Quantum (TQ) 1H–1H homonuclear dipolar correlation spectra (Fig. 3B and C respectively) and were therefore assigned to the methyl groups of the two isopropoxide ligands of 2 in two slightly different chemical environments.
 | ||
| Fig. 3 Solid-state NMR spectra of 2 (A) 1D 1H spin-echo MAS (20 kHz MAS, number of scans = 32, and repetition delay = 5 s) (B) 2D 1H–1H DQ (C) 2D 1H–1H TQ (22 kHz MAS, with a back-to-back recoupling sequence, number of scans = 32, repetition delay = 5 s, and number of t1 increments = 128, with the increment set equal to one rotor period of 45.45 μs) (D) 13C CP MAS NMR spectrum (10 kHz MAS, number of scans = 7000, repetition delay = 5 s, contact time = 2 ms, and line broadening = 80 Hz) (E) 2D CP MAS 1H–13C HETCOR NMR spectrum acquired with short contact times of 0.2 ms under 8.5 kHz MAS, number of scans per increment = 1500, repetition delay = 5 s, number of t1 increments = 32, and line broadening = 80 Hz. | ||
The proton resonances at 4.3 and 5.5 ppm display no autocorrelation in the DQ or TQ spectra, and were therefore assigned to C–H methine groups of isopropoxide ligands of 2 in two different chemical environments.
The 13C CP MAS NMR of 2 displays three signals at 23, 67, and 85 ppm (Fig. 3D). Additionally, the 2D 1H–13C HETCOR NMR spectrum (Fig. 3E, short contact time of 0.2 ms) shows a correlation between the methyl protons at 1.2 and 1.4 ppm and the carbon atoms at 23 ppm, which allows the assignment of the carbon–proton pairs to the methyl groups of the two isopropoxide ligands of 2. Furthermore, correlations are evident between the two CH methine protons at 4.3 and 5.5 ppm and the two carbon atoms at 67 and 85 ppm, respectively.
Additional off-diagonal correlations in the 1H DQ and TQ NMR spectra (Fig. 3B and C) between the first spin system of CH3 and CH (1.2 and 4.3 ppm in the single-quantum frequency; 5.5 and 6.7 ppm in indirect dimensions of the DQ and TQ spectra, respectively) and the second spin system of CH3 and CH (1.4 and 5.5 ppm in the single-quantum frequency; 6.9 and 8.3 ppm in indirect dimensions of the DQ and TQ spectra, respectively) indicating spatial proximity of the two chemical environments.
The 1H and 13C CP MAS NMR spectra of 3 (Fig. 4) as the product of the gradual thermolysis of 2, clearly show the nearly complete disappearance of the peaks at 5.5 ppm (slightly shifted to 5.8 ppm) and 85 ppm, respectively, which were present in 2 (Fig. 3A and B). Consistent with the results obtained from IR spectroscopy, and with the formation of propylene during thermal treatment, this observation suggests the removal of one kind of isopropoxide ligand from 2 upon thermolysis.
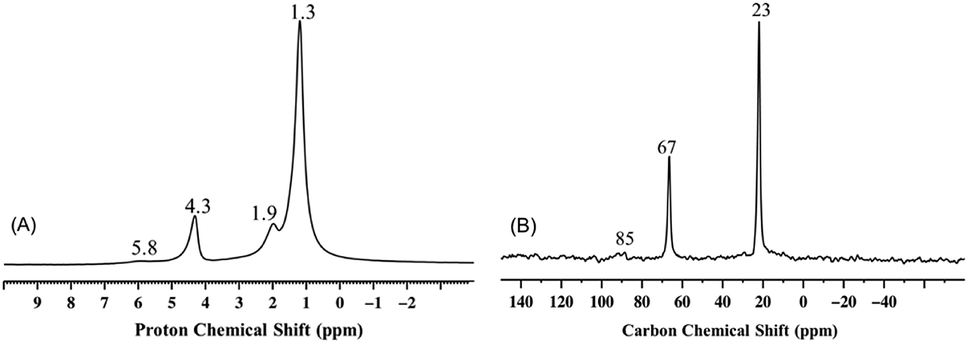 | ||
| Fig. 4 Solid-state NMR of 3 obtained from the gradual thermolysis of 2 at 200 °C for 1 h under static vacuum conditions. (A) 1D 1H spin-echo MAS spectra (acquired on 600 MHz NMR spectrometer under a 20 kHz MAS spinning frequency, number of scans = 32, and repetition delay = 5 s) and (B) 13C CP-MAS NMR spectrum (under 10 kHz MAS spinning frequency, number of scans 14000, repetition delay = 5 s, contact time = 2 ms, and line broadening = 80 Hz). | ||
It should be noted that the two isopropoxide signals of 2 display two significantly different sets of chemical shifts, 5.5 ppm for 1H NMR and 85 ppm for 13C NMR, versus 4.3 ppm for 1H NMR 67 ppm for 13C NMR. The chemical difference between the two isopropoxides is evident when, upon thermal treatment of 2, only the latter set of signals is preserved (Fig. 4). The first set of signals is compatible with a vanadium isopropoxide moiety, (V–O–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) (CH3)2) and (V–O–
(CH3)2) and (V–O–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H(CH3)2) respectively.27,28 The second set of signals is attributable to a non-metal bound isopropoxide (see, for instance, the spectrum of tetraisopropyl orthosilicate (Si–O–C(CH3)2): 4.2 ppm; (Si–O–H(CH3)2): 65.7 ppm. AIST database, Chart: 19429, accessed March 2018).
H(CH3)2) respectively.27,28 The second set of signals is attributable to a non-metal bound isopropoxide (see, for instance, the spectrum of tetraisopropyl orthosilicate (Si–O–C(CH3)2): 4.2 ppm; (Si–O–H(CH3)2): 65.7 ppm. AIST database, Chart: 19429, accessed March 2018).
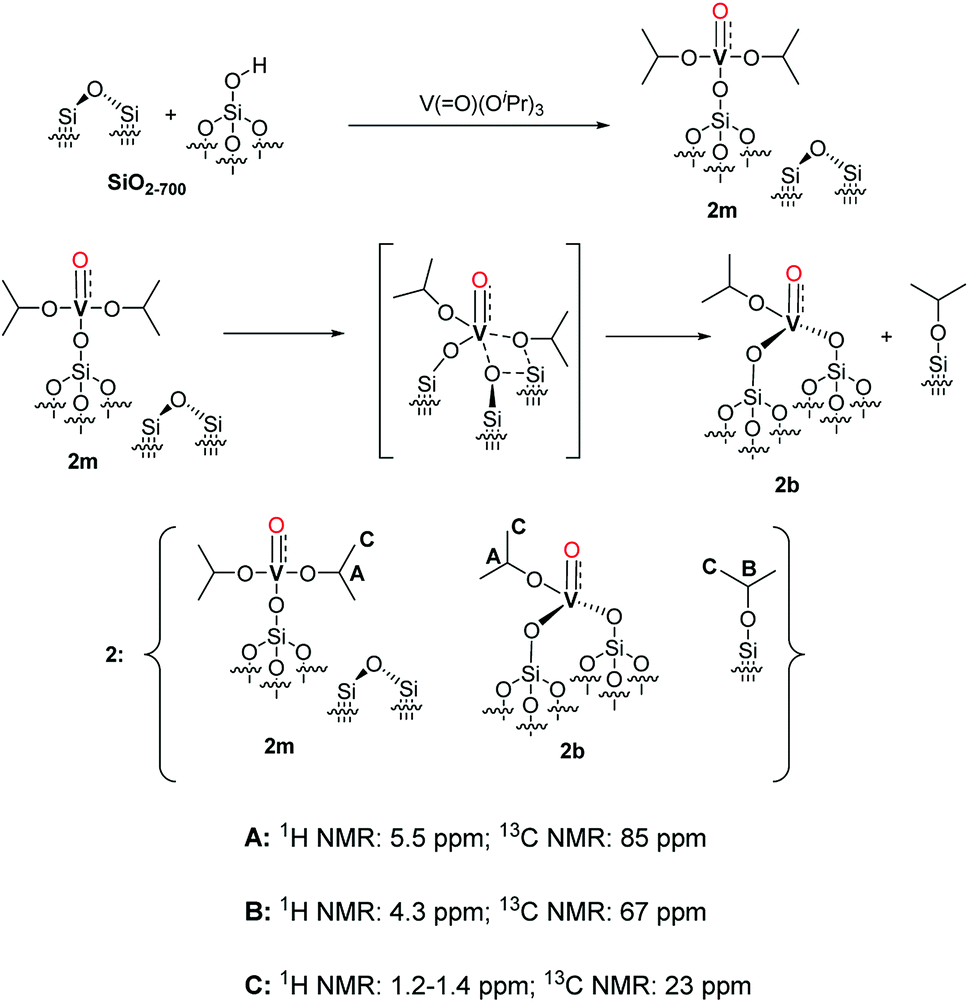 | ||
| Scheme 2 Proposed structure of 2 with a monopodal [(Si–O–)V(O)(OiPr)2] complex (2m), a bipodal [(Si–O)2V(O)(OiPr)] complex (2b) and alkylated silanol (Si–O–iPr) with the assignment of the respective NMR signals. The formation pathways of 2m and 2b are outlined in the first and second reaction lines of the Scheme respectively. The vanadyl bond of 2m, 2b is reported as a triple VO bond according to the NBO presented in the computational section and in Fig. S5.† | ||
Considering the grafting stoichiometry for 2 of a vanadium complex per silanol, as demonstrated by elemental analysis and FT-IR spectroscopy, an hypothesis on the identity and formation of the surface complexes in 2 can be drawn as in Scheme 2.
The initial grafting of the precursor takes place in a monopodal fashion by reaction with the isolated silanols of SiO2-700 generating 2m. The latter complex can, in part, react with the strained siloxane bridges present on SiO2-700 by a metathesis reaction leading to 2b and (Si–O–iPr) moieties. The ability of the strained siloxane bridges of highly dehydroxylated silica to participate in grafting reactions and in the rearrangement of surface complexes by σ-bond metathesis is known.31,36,39 The rearrangement of 2m to 2b does not generate new silanol moieties in agreement with the lack of such functionalities in 2 as observed by FT-IR (Fig. 1). Further study of 2m by DFT calculations (see the computational section for further details) demonstrated the partial triple bond character (indicated as dashed bond in Scheme 2) of the vanadyl moiety of 2m with a bond order of 2.62 based on a natural bonding orbital (NBO) analysis of a model of 2m (see also Fig. S5 in the ESI†). The same observation is likely to hold also for 2b. When comparing our results to other studies on the grafting of vapors of 1 on dehydroxylated silica, Rice and Scott reported exclusively a signal at 84.3 ppm in the SS 13C NMR (not shown in the original manuscript) of the grafted complex.27 Hermans et al. observed a main signal at 85 ppm and a minor signal at 67 ppm. They attributed the former signal to monopodal [(Si–O–)V(O)(OiPr)2] and the latter to a monopodal complex (4, Scheme 3) formed by the reaction of 1 with a strained siloxane bridge.28
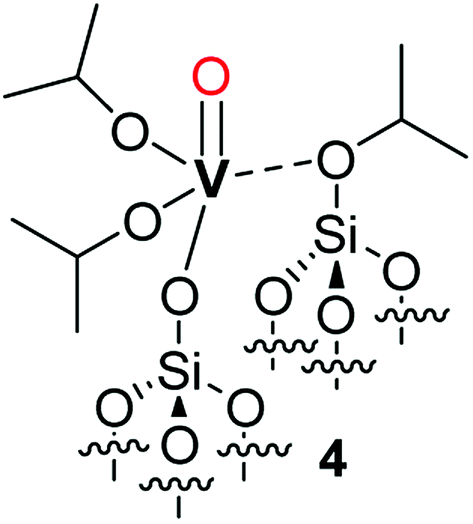 | ||
| Scheme 3 Representation of complex 4 proposed by Hermans et al.28 as the product of the reaction of 1 with a strained siloxane bridge. | ||
However, this attribution does not seem in agreement with the observation by the authors that only 4 is formed when using silica supports dehydroxylated at lower temperatures that present less strained and a lower density of siloxane bridges; the formation of bipodal species would be expected when less dehydroxylated silicas are employed.2
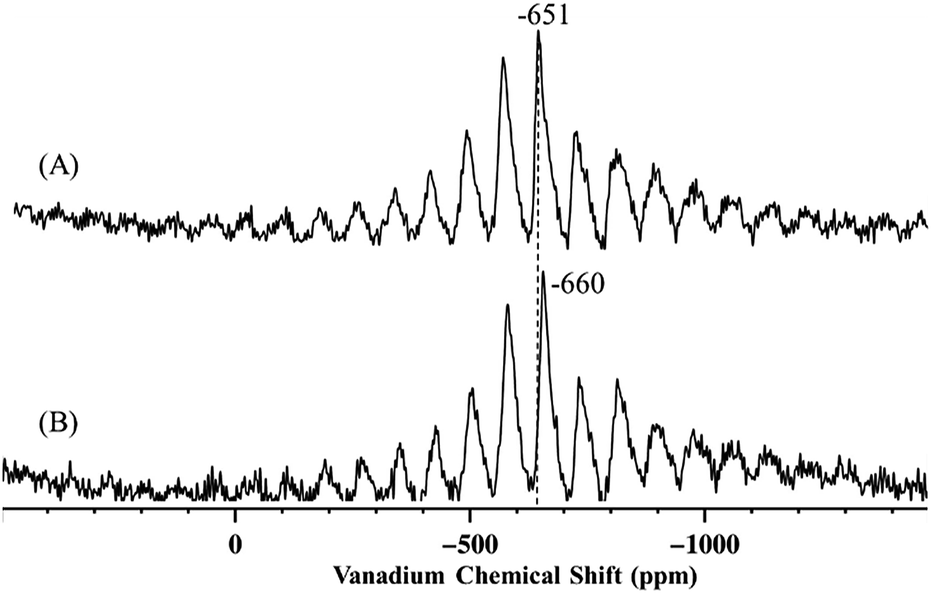
In any case, it appears that the bipodal complex 2b, to the formation of which we attribute the signal at 67 ppm (SiOH(CH3)2), is produced in significant amounts by solution phase grafting and in negligible amounts when 1 is grafted as a vapor. Further comparison between this work and previous literature was carried out by analysis of the 51V MAS NMR of the grafted complexes. The 51V MAS NMR of 2 and of 2-SiO2-300 (the complex prepared by grafting of 1 on silica dehydroxylated at 300 °C, SiO2-300) are displayed in Fig. 5A and B respectively.
 | ||
| Fig. 5 51V MAS NMR as determined by variation of the spinning frequency of (A) 2 and (B) 2-SiO2-300 (236.7 MHz, acquired on 900 MHz NMR spectrometer under a 20 KHz MAS spinning frequency, repetition delay = 0.25 s). | ||
The 51V MAS NMR spectra of 2 displays one resonance at −651 ppm, as determined by variation of the spinning frequency, slightly shifted from −630 ppm of precursor complex 1.27
The presence of a single band may appear in disagreement with the presence of two different vanadium complexes (2m and 2b) in 2, however, as proposed by Rice and Scott,27 the monopodal and bipodal complexes might have similar chemical shift and their resolution by 51V MAS NMR might be challenging.
Hermans et al. could resolve two signals, at −630 ppm and −655 ppm, and they attributed the first to monopodal [(Si–O–)V(O)(OiPr)2] and the second to less symmetrical complex 4.28
Based on these considerations, the resonance at −651 ppm for 2 could result from the coalescence of the signals relative to 2m and 2b. Indeed, a small shift to −660 ppm for 2-SiO2-300, that should contain predominantly bipodal species such as 2b,2,40 is observed compared to 2 suggesting that the bipodal species might display a slight upfield shift compared to the monopodal complexes.
Finally, when 2 was thermally treated to afford 3, the set of signals at 5.5 and 85 ppm (see Fig. 3 and 4) disappeared whereas that at 4.3 and 67 ppm was preserved. Based on the peak assignment in Scheme 2, this transformation should correspond to the loss of the isopropoxide moieties from 2m and 2b with the retention of the isopropoxide of (Si–O–iPr). Interestingly, a temperature of 200 °C is sufficient to entirely remove the isopropoxide groups bound to vanadium. This transformation is accompanied by the appearance of a small peak at 1.9 ppm in the 1H MAS NMR spectra of 3 (Fig. 4A), typically assigned to isolated SiOH groups, which are also present in the form of a band at 3741 cm−1 in the IR spectrum of 3 (Fig. 1).
The reconstruction and rearrangement of vanadium sites on silica and the formation of tripodal vanadium species under various conditions has been discussed previously.11,12,14,28,41
Based on the observed formation of propylene, the thermal restructuring of vanadium isopropoxide sites has been proposed to proceed via a γ-H transfer from the methyl group of the isopropoxide to the vanadyl oxygen producing V–OH moieties that can subsequently react with the siloxane bridges.28 Accordingly, the evolution of 2 upon thermal treatment can be described as in Scheme 4.
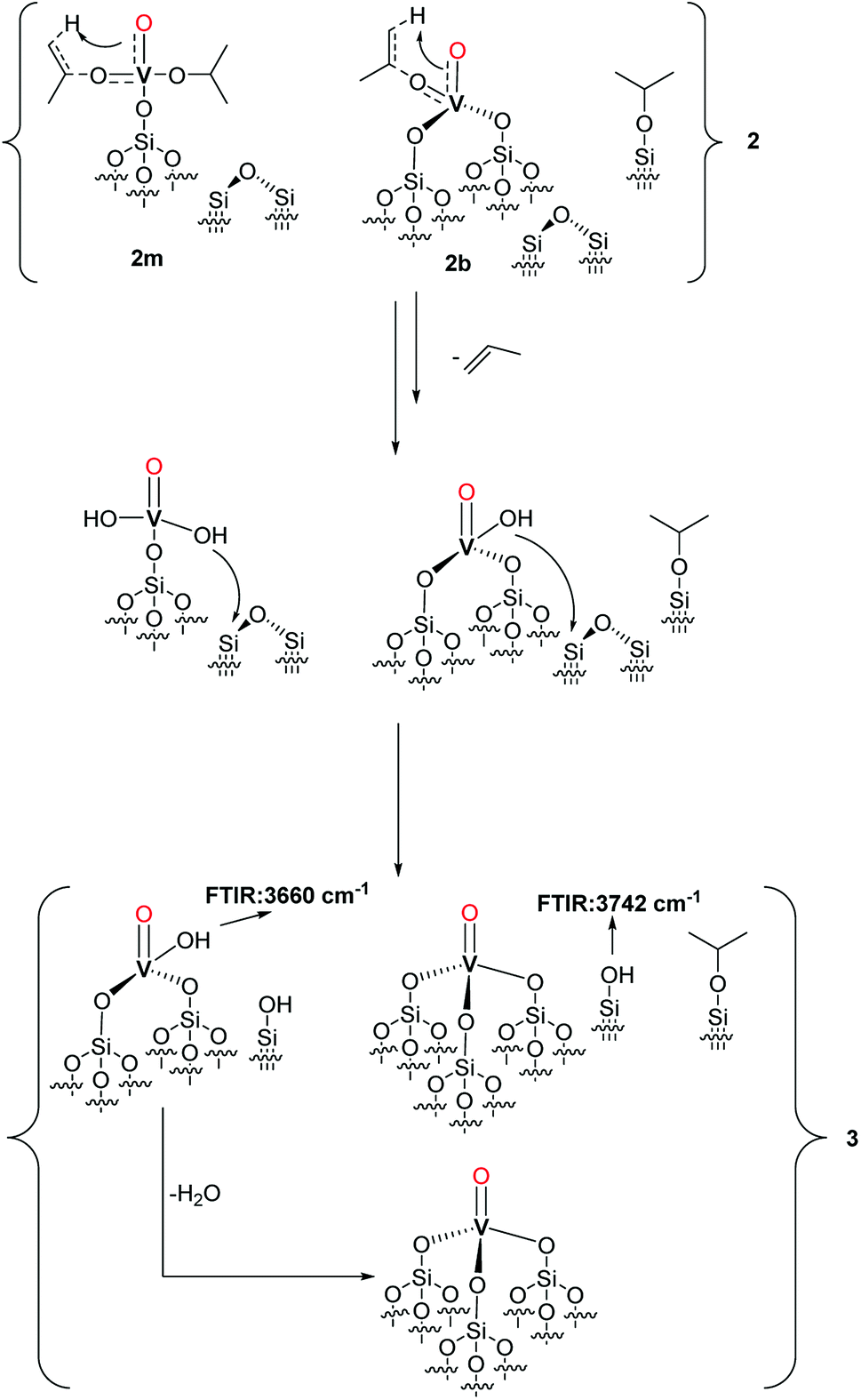 | ||
| Scheme 4 Proposed surface complexes and moieties in 3 as an effect of thermal rearrangement of 2. | ||
The produced V–OH groups, observed in 3 by the appearance of a band at 3660 cm−1 in the FT-IR spectrum (Fig. 1),36 can, in part, react with the strained siloxane bridges to generate multipodal species up to the formation of the tetrahedral VO4 complex. The latter complexes can also be generated by the reaction of the V–OH moieties with adjacent silanol groups. The (Si–O–iPr) groups are preserved in 3 likely because, in the absence of C–H activation by the vanadyl group, the treatment temperature of 200 °C is not sufficient to afford the complete decomposition of these species.
DFT mechanistic study
The key process for the transformation of complex 2 to 3 upon thermal treatment is the transfer of a proton of the methyl group of the isopropoxide to the vanadyl oxygen. In this section we provide deeper insight into this transformation by a DFT approach. Fig. 6 illustrates two possible reaction pathways for the abovementioned process: a concerted pathway (P1, green) and a two-step radical pathway (P2, purple).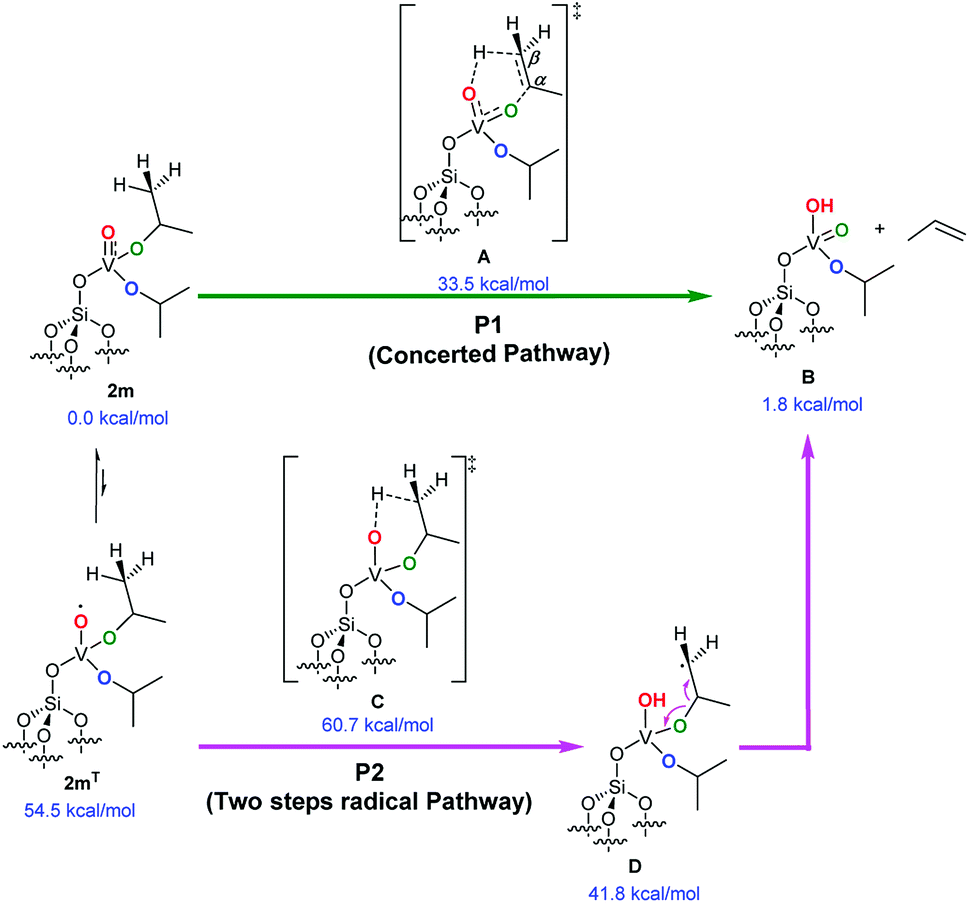 | ||
| Fig. 6 DFT-based reaction mechanisms for the γ-H-transfer and elimination of propene. The concerted pathway (P1, green) is energetically favorable over the two-step radical pathway (P2, purple; see the ESI for the energy diagram). | ||
Both pathways proceed via a six-membered arrangement where a methyl group from an isopropoxide ligand is placed in close vicinity to the vanadium-oxo functional group.
The concerted pathway proceeds via a transition state A, in which both the C–H and C–O bonds of the involved isopropoxide ligand are cleaved.
The H atom is transferred to the oxygen of the vanadyl moiety, which is transformed into the V–OH group, whereas, cleavage of the O–C bond, converts the former vanadium isopropoxide moiety into a vanadium oxo moiety. At the same time, the Cα–Cβ bond of the reacting isopropoxide ligand assumes a double-bond character to form a molecule of propene that is eliminated at the end of the elementary reaction step.
In this mechanism, the vanadium center is always in a formal oxidation state of +5 (d0) and the overall reaction step occurs on the singlet spin state. The free energy barrier to access the A concerted transition state from the initial complex was calculated at 33.5 kcal mol−1. A natural population analysis (NPA) found a charge of 0.40 e on the transferred H atom that hints at a proton transfer step. Sauer et al. proposed a similar step earlier in their theoretical investigation into the mechanism of oxidative dehydrogenation of propane. They reported a barrier of 38 kcal mol−1 for the H-transfer from one of the methyl groups of a V-bound isopropoxide to the O atom of a Si–O–V moiety, with the V-metal center in the formal oxidation state of +4 (d1).19
Alternatively, the two-step radical pathway is initiated by a H-transfer from one of the methyl groups of the isopropoxide ligands to the vanadium-oxo group by the C transition state. This leads to intermediate D, which has a biradical character with one spare electron located on the isopropoxide group and the other on the V-metal center with a formal oxidation state of +4 (d1). Although the C transition state could exist in the triplet spin, and/or open-shell singlet states using a broken-symmetry approach, it was localized in the triplet spin state 60.7 kcal mol−1 above 2m. However, the main source of this large barrier is not to be found in the H-transfer but in the change of spin state, since 2m optimized in the triplet state, 2mT, is 54.5 kcal mol−1 above 2m in the singlet spin state. The C transition state collapses into intermediate D, shown in Fig. 6, located 41.8 kcal mol−1 above 2m. Intermediate D has its two excess electrons located at the V-metal center and at the isopropoxide Cβ atom (with a spin density of 1.03 e and 1.09 e, respectively). The energy difference between the C transition and the A transition states along the concerted pathway is 27.2 kcal mol−1; which is high enough to rule out the two-step pathway on the triplet spin state. Alternatively, we were unable to localize the open-shell singlet C transition state using a broken-symmetry approach, the system collapsed into a four-membered closed-shell singlet (V–C–C–O, D′) metallacycle (not shown). Because this metallacycle lies 45.1 kcal mol−1 above 2m on the singlet spin state, and because it is reached through a transition state of roughly 65 kcal mol−1 (C′, see Fig. S4† for an overview), this unlikely reaction pathway is not commented further. In conclusion, our analysis clearly indicates that propene elimination from 2m occurs on the singlet spin state via the concerted pathway A involving a single transition state.
The geometries of 2m and of transition states, A and C, are shown in Fig. 7. In 2m the V–O bond is calculated to be approximately 1.58 Å long suggesting a partial triple bond character42,43 as demonstrated by the NBO analysis (see Fig. S5† for an overview). In case of transition state A the geometry of the V-center is a distorted tetrahedron. The V–O bond that is formed by the cleavage of the C–O bond has a length of 1.64 Å and a partial triple bond character (bond order: 2.48). The V–O bond receiving a proton from the methyl group of the isopropyl moiety has a length of 1.67 Å and a bond order of 1.7. The two other V–O bonds have a length of about 1.8 Å.
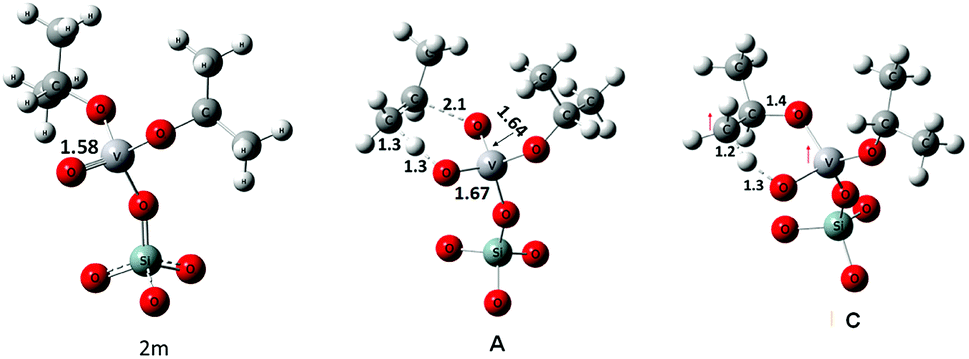 | ||
| Fig. 7 Geometries of the A and C transition states (selected distances in Å). | ||
The geometry of C shows that the cleaving C–H bond and the forming O–H bond are in the range of 1.2–1.3 Å, whereas the O–C bond, that must be cleaved to release propene, is still stable at 1.43 Å. Analysis of the spin density clearly indicates that one of the two excess electrons is located on the V-atom (spin density of 1.01 e) while the other is substantially shared between the O and C atoms involved in the H-transfer (with spin densities of 0.61 e and 0.38 e respectively).
Conclusions
The grafting of vanadium oxytriisopropoxide on the surface of dehydroxylated silica was studied by the surface organometallic chemistry approach. The complexes and moieties formed on the support upon grafting and after a controlled thermal treatment were carefully characterized by elemental analysis and FTIR, SS NMR, Raman and EPR spectroscopies. It was found that the reaction of 1 with the support surface generates monopodal (2m) and bipodal (2b) complexes, the latter, likely originated by the reaction of the monopodal species with a strained siloxane bridge. The complexes generated on the surface by the thermal rearrangement of 2m and 2b have been discussed. DFT analysis strongly supports a mechanism where a concerted intramolecular γ-H-atom transfer from an isopropoxide ligand's methyl group to the vanadyl oxygen takes place along with the cleavage of the O-isopropyl bond to liberate propene. Overall, we believe that this work contributes to a deeper understanding of the reactivity and wet-chemistry grafting patterns of vanadium alkoxide complexes. The latter compounds can be regarded as potential models to study hydrogen-transfer steps in reaction networks related to propane dehydrogenation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by King Abdullah University of Science and Technology, Thuwal, Saudi Arabia. The authors declare no competing financial interest. V. D. E. thanks the Thailand Research Fund (Grant No. RSA6080059) for funding this research.References
- M. Sutradhar, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2015, 301–302, 200–239 CrossRef.
- K. C. Szeto, B. Loges, N. Merle, N. Popoff, A. Quadrelli, H. Jia, E. Berrier, A. De Mallmann, L. Delevoye, R. M. Gauvin and M. Taoufik, Organometallics, 2013, 32, 6452–6460 CrossRef.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef.
- P. Gruene, T. Wolfram, K. Pelzer, R. Schlögl and A. Trunschke, Catal. Today, 2010, 157, 137–142 CrossRef.
- A. Dinse, T. Wolfram, C. Carrero, R. Schlögl, R. Schomäcker and K. P. Dinse, J. Phys. Chem. C, 2013, 117, 16921–16932 Search PubMed.
- M. Cavalleri, K. Hermann, A. Knop-Gericke, M. Hävecker, R. Herbert, C. Hess, A. Oestereich, J. Döbler and R. Schlögl, J. Catal., 2009, 262, 215–223 CrossRef.
- N. Magg, B. Immaraporn, J. B. Giorgi, T. Schroeder, M. Bäumer, J. Döbler, Z. Wu, E. Kondratenko, M. Cherian, M. Baerns, P. C. Stair, J. Sauer and H.-J. Freund, J. Catal., 2004, 226, 88–100 CrossRef.
- M. A. Bañares and I. E. Wachs, J. Raman Spectrosc., 2002, 33, 359–380 CrossRef.
- J. N. J. van Lingen, O. L. J. Gijzeman, R. W. A. Havenith and J. H. van Lenthe, J. Phys. Chem. C, 2007, 111, 7071–7077 Search PubMed.
- H. Kim, G. A. Ferguson, L. Cheng, S. A. Zygmunt, P. C. Stair and L. A. Curtiss, J. Phys. Chem. C, 2011, 116, 2927–2932 Search PubMed.
- L. Cheng, G. A. Ferguson, S. A. Zygmunt and L. A. Curtiss, J. Catal., 2013, 302, 31–36 CrossRef.
- I. E. Wachs, Dalton Trans., 2013, 42, 11762–11769 RSC.
- H. Tian, E. I. Ross and I. E. Wachs, J. Phys. Chem. B, 2006, 110, 9593–9600 CrossRef PubMed.
- S. L. Wegener, H. Kim, T. J. Marks and P. C. Stair, J. Phys. Chem. Lett., 2011, 2, 170–175 CrossRef.
- C. A. Carrero, C. J. Keturakis, A. Orrego, R. Schomäcker and I. E. Wachs, Dalton Trans., 2013, 42, 12644–12653 RSC.
- J. T. Grant, C. A. Carrero, A. M. Love, R. Verel and I. Hermans, ACS Catal., 2015, 5, 5787–5793 CrossRef.
- K. Alexopoulos, M.-F. Reyniers and G. B. Marin, J. Catal., 2012, 289, 127–139 CrossRef.
- X. Rozanska, R. Fortrie and J. Sauer, J. Am. Chem. Soc., 2014, 136, 7751–7761 CrossRef PubMed.
- X. Rozanska, R. Fortrie and J. Sauer, J. Phys. Chem. C, 2007, 111, 6041–6050 Search PubMed.
- F. Gilardoni, A. T. Bell, A. Chakraborty and P. Boulet, J. Phys. Chem. B, 2000, 104, 12250–12255 CrossRef.
- H. Fu, Z.-P. Liu, Z.-H. Li, W.-N. Wang and K.-N. Fan, J. Am. Chem. Soc., 2006, 128, 11114–11123 CrossRef PubMed.
- P. C. Redfern, P. Zapol, M. Sternberg, S. P. Adiga, S. A. Zygmunt and L. A. Curtiss, J. Phys. Chem. B, 2006, 110, 8363–8371 CrossRef PubMed.
- M.-J. Cheng, K. Chenoweth, J. Oxgaard, A. van Duin and W. A. Goddard, J. Phys. Chem. C, 2007, 111, 5115–5127 Search PubMed.
- J. Sauer, M. Pritzsche and J. Döbler, J. Phys. Chem. C, 2014, 118, 29159–29163 Search PubMed.
- J. Liu, F. Mohamed and J. Sauer, J. Catal., 2014, 317, 75–82 CrossRef.
- R. Schlögl, Concepts in Selective Oxidation of Small Alkane Molecules, in Modern Heterogeneous Oxidation Catalysis, ed. N. Mizuno, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 1–42 Search PubMed.
- G. L. Rice and S. L. Scott, Langmuir, 1997, 13, 1545–1551 CrossRef.
- A. M. Love, C. A. Carrero, A. Chieregato, J. T. Grant, S. Conrad, R. Verel and I. Hermans, Chem. Mater., 2016, 28, 5495–5504 CrossRef.
- J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- N. Maity, S. Barman, E. Callens, M. K. Samantaray, E. Abou-Hamad, Y. Minenkov, V. D'Elia, A. S. Hoffman, C. M. Widdifield, L. Cavallo, B. C. Gates and J.-M. Basset, Chem. Sci., 2016, 7, 1558–1568 RSC.
- Z. S. Qureshi, A. Hamieh, S. Barman, N. Maity, M. K. Samantaray, S. Ould-Chikh, E. Abou-hamad, L. Falivene, V. D'Elia, A. Rothenberger, I. Llorens, J.-L. Hazemann and J.-M. Basset, Inorg. Chem., 2017, 56, 861–871 CrossRef PubMed.
- Y. Bouhoute, A. Garron, D. Grekov, N. Merle, K. C. Szeto, A. De Mallmann, I. Del Rosal, L. Maron, G. Girard, R. M. Gauvin, L. Delevoye and M. Taoufik, ACS Catal., 2014, 4, 4232–4241 CrossRef.
- N. Maity, S. Barman, Y. Minenkov, S. Ould-Chikh, E. Abou-Hamad, T. Ma, Z. S. Qureshi, L. Cavallo, V. D'Elia, B. C. Gates and J.-M. Basset, ACS Catal., 2018, 8, 2715–2729 CrossRef.
- N. Maity, S. Barman, E. Abou-Hamad, V. D'Elia and J.-M. Basset, Dalton Trans., 2018, 47, 4301–4306 RSC.
- S. Barman, N. Maity, K. Bhatte, S. Ould-Chikh, O. Dachwald, C. Haeßner, Y. Saih, E. Abou-Hamad, I. Llorens, J.-L. Hazemann, K. Köhler, V. D'Elia and J.-M. Basset, ACS Catal., 2016, 6, 5908–5921 CrossRef.
- M. J. Kelly, A. Barthel, C. Maheu, O. Sodpiban, F.-B. Dega, S. V. C. Vummaleti, E. Abou-Hamad, J. D. A. Pelletier, L. Cavallo, V. D'Elia and J.-M. Basset, J. CO2 Util., 2017, 20, 243–252 CrossRef.
- C. Resini, T. Montanari, G. Busca, J.-M. Jehng and I. E. Wachs, Catal. Today, 2005, 99, 105–114 CrossRef.
- B. Werghi, A. Bendjeriou-Sedjerari, J. Sofack-Kreutzer, A. Jedidi, E. Abou-Hamad, L. Cavallo and J.-M. Basset, Chem. Sci., 2015, 6, 5456–5465 RSC.
- V. D'Elia, H. Dong, A. J. Rossini, C. M. Widdifield, S. V. C. Vummaleti, Y. Minenkov, A. Poater, E. Abou-Hamad, J. D. A. Pelletier, L. Cavallo, L. Emsley and J.-M. Basset, J. Am. Chem. Soc., 2015, 137, 7728–7739 CrossRef PubMed.
- Z. Wu, S. Dai and S. H. Overbury, J. Phys. Chem. C, 2009, 114, 412–422 Search PubMed.
- S. Pacigova, R. Gyepes, J. Tatiersky and M. Sivak, Dalton Trans., 2008, 121–130 RSC.
- S. DiBella, G. Lanza, A. Gulino and I. Fragala, Inorg. Chem., 1996, 35, 3885–3890 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods and details, additional IR spectra; computational coordinates and DFT energy diagram. See DOI: 10.1039/c8ra02419g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |