
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUHPLC-ESI-Q-TOF-MS/MS analysis, antioxidant activity combined fingerprints for quality consistency evaluation of compound liquorice tablets†
Yujing Zhanga,
Chao Wangb,
Fangliang Yanga,
Zhe Yanga,
Fangren Wanga and
Guoxiang Sun *a
*a
aSchool of Pharmacy, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenyang, Liaoning 110016, P. R. China. E-mail: gxswmwys@163.com
bSchool of Pharmaceutical Engineering, Shenyang Pharmaceutical University, Shenyang, Liaoning, P. R. China
First published on 3rd August 2018
Abstract
Traditional Chinese medicines (TCM)/herbal medicines (HM) are too complicated to comprehensively investigate their quality consistency effectively with a single detection technique. Hence, finding an effective, rapid, and comprehensive quality control (QC) method is of great importance for guaranteeing the safety and efficacy of TCM/HM in clinical applications. In our current research, a novel strategy of multi-wavelength fusion HPLC fingerprints and ultraviolet (UV) spectroscopic fingerprinting was proposed and successfully applied to monitor the quality consistency of compound liquorice tablets (CLT). The quality grades of 35 CLT samples from two manufacturers were successfully discriminated and evaluated by the averaged linear quantified fingerprint method (ALQFM) from a qualitative and quantitative perspective. The results showed that the UV spectroscopic fingerprints agreed well with the multi-wavelength fusion HPLC fingerprints. In addition, ultra-high-performance liquid chromatography coupled with electrospray ionization quadrupole time-of-flight mass spectrometry (UHPLC-ESI-Q-TOF-MS) was applied to investigate the chemical constituents in CLT samples, providing an important chemical structural foundation for further QC and bioactivity studies. Additionally, a simple flow injection analysis (FIA) was developed to investigate the antioxidant capacity in CLT, which was based on the scavenging of 2,2-diphenyl-1-picrylhydrazyl radicals by antioxidants. Furthermore, the fingerprint–efficacy relationship between high-performance liquid chromatography (HPLC) fingerprints and the antioxidant activities of CLT samples was established utilizing orthogonal projection to latent structures (OPLS). In conclusion, this study indicated that integrating UHPLC-ESI-Q-TOF-MS/MS, UV spectroscopic fingerprints, and multi-wavelength fusion HPLC fingerprints coupled with the antioxidant activities reported could give important clues for further pharmacological and clinical studies of CLT. Meanwhile, it provides a practical strategy for the rapid screening and identifying of TCM/HM quality consistency.
1. Introduction
TCM/HM, as alternative medicines, are becoming increasingly popular worldwide. However, the therapeutic effects of most TCMs/HMs are combined with others in a single prescription based on the synergistic efficacies of multi-constituents toward multi-targets.1,2 The compatibility mechanisms of herb–drug interactions have attracted much attention, which could be attributed to the content variation of the chemical compounds in vitro.3 Clearly, it is not sufficient to just monitor the TCM/HM quality by quantifying a limited number of marker substances. Fingerprinting techniques, as rational and powerful methods for systemically characterizing complicated TCMs/HMs, have been widely accepted and adopted for TCM/HM QC in recent years.4 Nowadays, commonly used fingerprints involve single wavelength acquisition; however, this cannot reflect comprehensive chemical information of TCMs/HMs. Therefore, a multi-wavelength fusion HPLC fingerprints method5,6 has increasingly been adopted to realize the overall evaluation of the TCM/HM quality based on combining computer technology and macro fingerprint characteristics.In fact, various approaches for chromatographic fingerprinting based on various detection techniques, such as HPLC,7–9 gas chromatography (GC),10,11 and capillary electrophoresis (CE),12 are frequently used for TCM/HM QC; however, it should be noted that these tend to be time-consuming and laborious; moreover, in most cases they cannot offer information about all the chemical components in the TCM/HM, which restricts the overall QC of the multi-constituents. In this situation, flow-based methods, such as flow injection analysis (FIA), multisyringe FIA, and sequential injection analysis, have been proposed and even successfully applied in automated analysis for rapid screening purposes.13–15 On the basis of FIA, UV spectroscopic fingerprinting is preferred to monitor the TCM/HM quality, owing to its apparent analytical advantages, such as low experimental expenses, simple sample preparation, and short measurement time. It is common knowledge that UV spectra can display the features of unsaturated bonds information, such as for n–σ*, n–π*, and π–π*, qualitatively and quantitatively;16 therefore, UV spectroscopic fingerprinting in combination with chemometric methods can be used for monitoring the TCM/HM quality in a straightforward, rapid, and effective manner.17,18 In the past few years, UHPLC–ESI-Q-TOF-MS/MS, as a high resolution, excellent sensitivity, accurate mass measurement and high throughput technology, has been applied to integrate multi-constituent determination and fingerprint analysis for TCM/HM QC,19 and could be of great importance for facilitating further investigation of the compatibility mechanisms.
On the other hand, conventional chromatographic fingerprint analysis is typically used for authenticity and identification, but this only indicates the qualitative similarity and ignores the quantitative assessment.20,21 Although, the quantitative evaluation of chromatographic fingerprinting in the QC of CHM has been verified;22–24 nevertheless, multi-component quantification is not credible in situations where certain medicinal ingredients are ignored. In this study, both the averaged linear qualitative and averaged quantitative similarities of the fingerprints of the CLT samples were assessed by the averaged linear quantified fingerprint method (ALQFM), which was developed and successfully applied to address the issue of performing a qualitative and quantitative comparison of the reference standards and PPCE samples.25
CLT is an important TCM prepared from five medicinal herbs, including licorice root (extract) concrete (LRC), powdered poppy capsule extractive (PPCE), camphor, oleum anisi stellati, and sodium benzoic (SMB), which have been widely used for anti-inflammatory, antitussive, expectorant, and antiasthmatic actions.26 The compositions of CLT mainly include flavonoids, saponins, alkaloids, and other compounds, as reported in previous reports,27–44 and these chemical components have a common pharmacological activity, that is, their antioxidant activity.45–47 Moreover, the published studies have shown that the antioxidant components, especially flavonoids, saponins, and alkaloids of herbal products, can decrease the risk of numerous diseases, such as inflammation, senescence cardiovascular disease, neurodegenerative disorders, diabetes, and cancer, closely related to the damaging effects of free radicals.48–51 Accordingly, to discover natural antioxidants and their antioxidant properties from food and TCMs/HMs,52 the development of rapid, qualitative, and quantitative analytical techniques are important tasks. Stable free radical species, such as 1,1-diphenyl-2-picrylhydrazyl (DPPH), are often used for evaluating the free radical scavenging capacity of various antioxidants based on the ability of an antioxidant to quench free radicals by hydrogen donation.53 This encourages us to explore the antioxidant activities to reflect the bioactivity of CLT and to correlate them with their fingerprints.
In the present work, multi-wavelength fusion HPLC fingerprinting and UV spectroscopic fingerprinting method were developed for assessing the quality consistency of 35 CLT samples, where the two methods perfectly complement each other. In fingerprint assessments, ALQFM was established for scientific CLT quality analysis from a qualitative and quantitative perspective. In addition, the antioxidant capacity of CLT was determined based on FIA; besides, the fingerprint efficacy between a fusion fingerprint and antioxidant capacity was performed utilizing an OPLS model. An efficient UHPLC-ESI-Q-TOF-MS/MS was exploited to elucidate the chemical components in CLT. It was demonstrated that integrating UV spectroscopic fingerprinting and multi-wavelength fusion HPLC fingerprinting coupled with UHPLC-ESI-Q-TOF-MS/MS and assessing the antioxidant activities offers a powerful and effective method for QC and can facilitate further investigation of the compatibility mechanisms of CLTs.
2. Theory of ALQFM54
For the modeling, we assumed that the sample fingerprint vector (SFPV) and the reference fingerprint vector (RFPV) were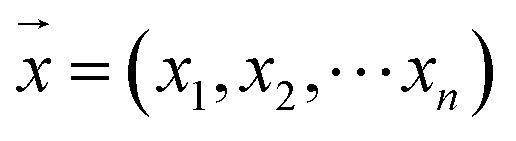 and
and 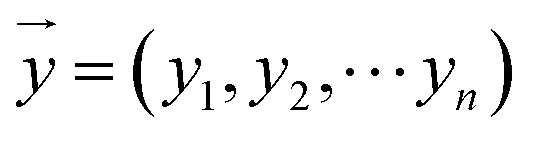 , respectively, where xi and yi are the ith peak area in the sample and reference fingerprints, respectively. The average linear qualitative similarity (SL), a qualitative parameter calculated by eqn (1), can accurately describe the resemblance in terms of the number and distribution of fingerprints between the RFPV and SFPV. On the other hand, the slope of the linear equation (b), as calculated by eqn (2), can quantitatively compare
, respectively, where xi and yi are the ith peak area in the sample and reference fingerprints, respectively. The average linear qualitative similarity (SL), a qualitative parameter calculated by eqn (1), can accurately describe the resemblance in terms of the number and distribution of fingerprints between the RFPV and SFPV. On the other hand, the slope of the linear equation (b), as calculated by eqn (2), can quantitatively compare 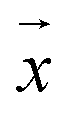 and
and 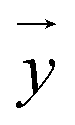 after being weight-corrected by mS and mR, where mS is the weight of each sample and the parameter mR is the average weight of the 35 CLT samples. Furthermore, the average linear quantitative similarity (PL), a quantitative parameter calculated by eqn (3), was revised by b to examine the total content of all the fingerprint components similarity for all ingredients between SFPV and RFPV. Finally, the apparent content similarity (R%, eqn (4)) and b were calculated for error evaluation of the fingerprints. A fingerprint variation coefficient (α), as defined in eqn (5), was determined as a statistical error that can reflect the fingerprint dissimilarity between SFPV and RFPV.
after being weight-corrected by mS and mR, where mS is the weight of each sample and the parameter mR is the average weight of the 35 CLT samples. Furthermore, the average linear quantitative similarity (PL), a quantitative parameter calculated by eqn (3), was revised by b to examine the total content of all the fingerprint components similarity for all ingredients between SFPV and RFPV. Finally, the apparent content similarity (R%, eqn (4)) and b were calculated for error evaluation of the fingerprints. A fingerprint variation coefficient (α), as defined in eqn (5), was determined as a statistical error that can reflect the fingerprint dissimilarity between SFPV and RFPV.
Accordingly, the quality evaluation method in terms of SL, PL, and α was named as ALQFM, by which the TCM quality could be classified into 8 grades (Table 1). In the evaluation system, the lower the grade values, the better the quality, where grades 1–5 are recognized as qualified.
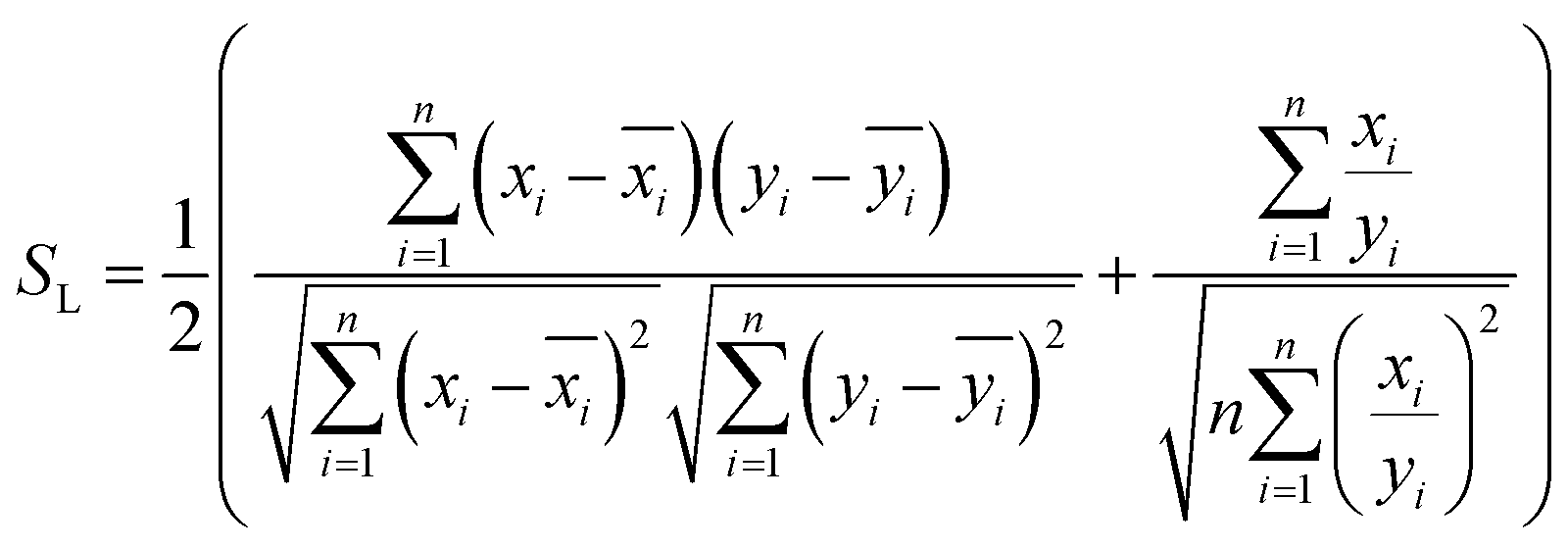 | (1) |
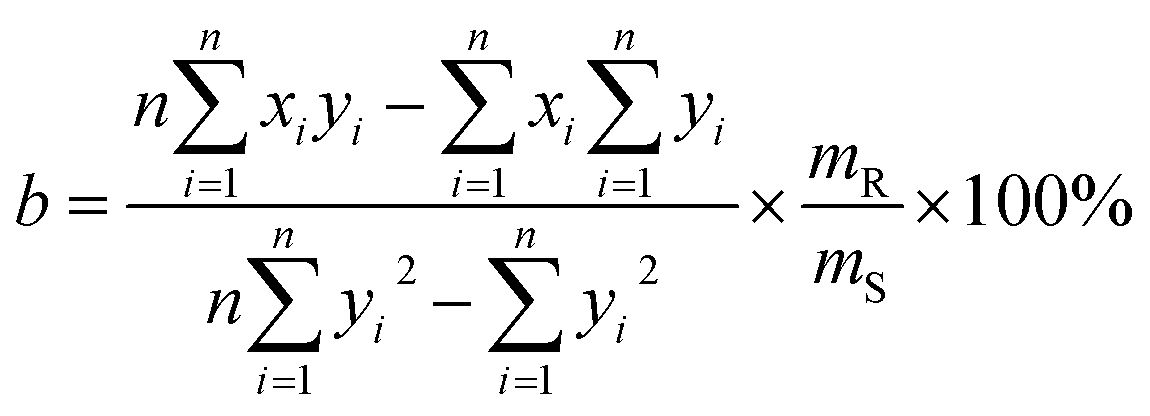 | (2) |
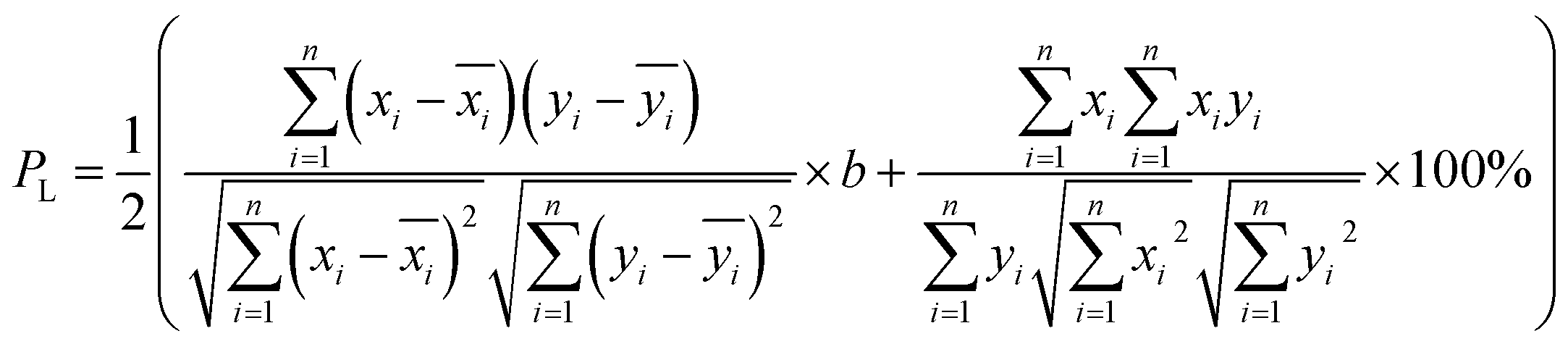 | (3) |
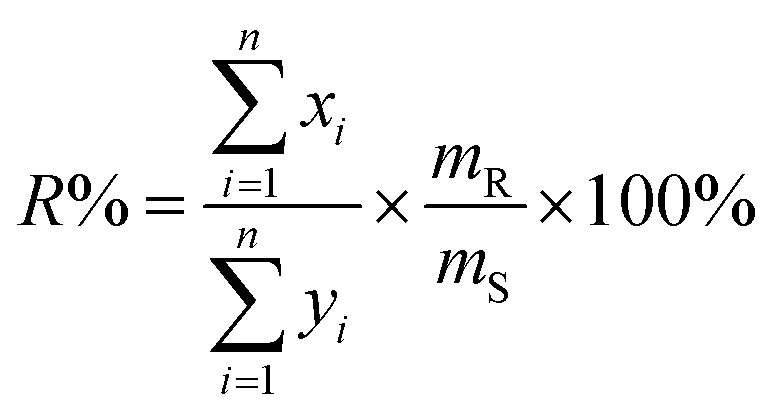 | (4) |
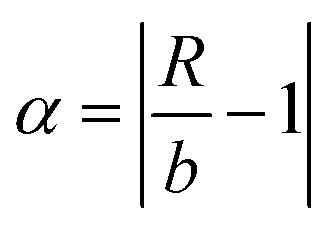 | (5) |
| Grade | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Sm≥ | 0.95 | 0.9 | 0.85 | 0.8 | 0.7 | 0.6 | 0.5 | Sm < 0.5 |
| Pm∈ | 95–105 | 90–110 | 80–120 | 75–125 | 70–130 | 60–140 | 50–150 | 0–∞ |
| α≤ | 0.05 | 0.1 | 0.15 | 0.2 | 0.3 | 0.4 | 0.5 | α > 0.05 |
| Quality | Best | Better | Good | Fine | Moderate | Common | Inferiors | Defective |
3. Materials and methods
3.1 Chemicals and reagents
A total of 35 batches of CLT samples (S1–S35) were supplied by Shandong Xinhua Pharmaceutical Co., Ltd. (Shandong, China; Manufacturer A, providing S1–S15) and China National Pharmaceutical Co., Ltd. (Hebei, China; Manufacturer B, providing S16–S35), respectively. Individual herbs, including glycyrrhiza extract and powdered poppy capsule extractive, were obtained from China National Pharmaceutical Co., Ltd. (Hebei, China). Nine reference standards, liquiritin (LQT), SMB, codeine phosphate (CON), and morphine (MPE), were acquired from the National Institutes for Food and Drug Control. Liquiritin apioside (LQA), liquiritigenin (LQG), isoliquiritigenin (ISG), isoliquiritoside (ISS), and glycyrrhizic acid (GLA) were provided by Shanghai Winherb Medical Technology Co., Ltd. (Shanghai, China). In the UHPLC-ESI-Q-TOF-MS/MS analysis, methanol (HPLC grade) and acetonitrile (HPLC grade) were provided from Merck (Darmstadt, Germany). Formic acid (HPLC grade) and ammonium acetate were purchased from Yuwang Chemical Industry Co., Ltd. (Shandong, China). In the HPLC and UV spectroscopic analysis, acetonitrile (HPLC grade), methanol (HPLC grade), and anhydrous ethyl alcohol (HPLC grade) were purchased from Yuwang Chemical Industry Co., Ltd. (Shandong, China). Sodium 1-heptanesulfonate was obtained by Zhongmei Chromatographic Co., Ltd (Shandong, China). Phosphoric acid (HPLC grade) was supplied from Kermel Chemical Reagent Co., Ltd (Tianjin, China). Deionized water was purified by a Milli-Q system (Bedford, MA, USA). All the other reagents were of analytical grade.3.2 Sample and standard preparation
The reference standards of LQT, SMB, CON, MPE, LQA, LQG, ISG, ISS, and GLA were accurately weighed separately and dissolved in methanol. To obtain CLT solutions, five tablets of CLTs were milled into powder and accurately weighed. The powder was then extracted with 50 mL methanol/water/phosphoric acid (160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40:1, v/v/v) solution in an ultrasonic bath for 20 min. All the solutions were filtered through 0.45 μm Millipore filters (Beijing Sunrise T&D Company, China) and stored at 4 °C prior to use.
:40:1, v/v/v) solution in an ultrasonic bath for 20 min. All the solutions were filtered through 0.45 μm Millipore filters (Beijing Sunrise T&D Company, China) and stored at 4 °C prior to use.
3.3 Experimental conditions
The MS analysis was achieved on a Waters Xevo G2-S Q-TOF mass spectrometer (Waters Corporation, Manchester, UK) equipped with a Spray™ ESI source in both the positive and negative ion mode. The following operating parameters were set: ion spray voltage, +3.0 kV for the positive ion mode and −2.0 kV for the negative ion mode; cone voltage, 80 V for the positive ion mode and 40 V for the negative ion mode; source temperature, 80 °C for the positive ion mode and 120 °C for the negative ion mode; desolvation temperature, 200 °C for the positive ion mode and 150 °C for the negative ion mode; cone gas flow rate, 10 L h−1 for the positive ion mode and 50 L h−1 for the negative ion mode; and desolvation gas flow rate, 600 L h−1 for the positive ion mode and 700 L h−1 for the negative ion mode; collision energy range of 30–60 for the positive ion mode and 20–40 for the negative ion mode; scanned range of m/z 100–1200 for MS1 and 50–1200 for MS2.
:10:8, v/v/v). The separation was affected utilizing a linear gradient as follows: 6–18% B at 0–10 min, 18–33% B at 10–20 min, 33–46% B at 20–32 min, 46–60% B at 32–45 min, 60–78% B at 45–60 min, 78–80% B at 60–65 min. The injection volume and the flow rate were 10 μL and 1.0 mL min−1, respectively. The detection wavelength was set at 220 nm, 250 nm, 280 nm, and 344 nm.:50, v/v) was adopted as the carrier, with FIA being used as the analytical principle, as shown in Fig. 1A. The parameters for the separation were set as follows: temperature of the PTFE tube 35 °C, flow rate 0.5 mL min−1, CLT sample injection volume 1 μL, wavelength interval 1 nm, and slit width 1 nm, respectively.
 | ||
| Fig. 1 FIA analytical principle plot for the UV spectra (A), and 3D chromatogram plot (min × nm × absorbance) (B) for CLT samples, typical UV spectra (C) and four-wavelength fusion HPLC fingerprints (D) for 35 CLT samples. | ||
:50, v/v) was delivered at a flow rate of 0.4 mL min−1 and the other conditions were set as mentioned in Section 3.3.2. Finally, the absorbance of the mixture was measured by the decrease at 517 nm with a UV-vis DAD after reacting in the PTFE pipe. Then, the corrected sample elimination ratio (SER, eqn (8)) was calculated utilizing the elimination ratio (ER, eqn (6)) corrected by a correction factor (fi, eqn (7)) to evaluate the antioxidant activity of the sample.
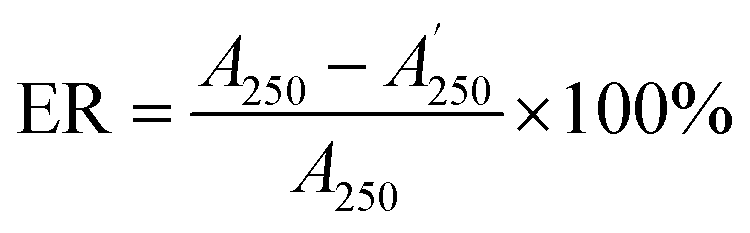 | (6) |
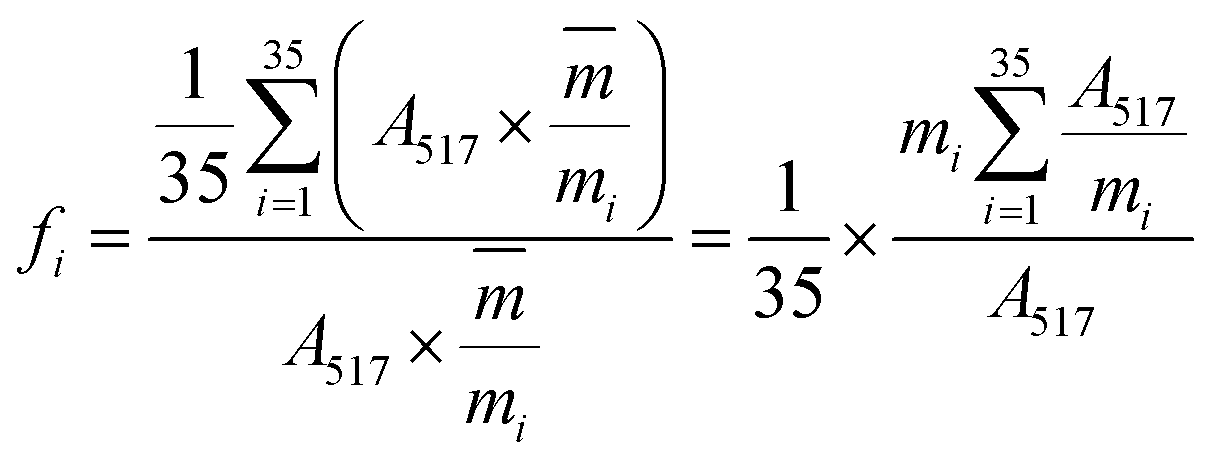 | (7) |
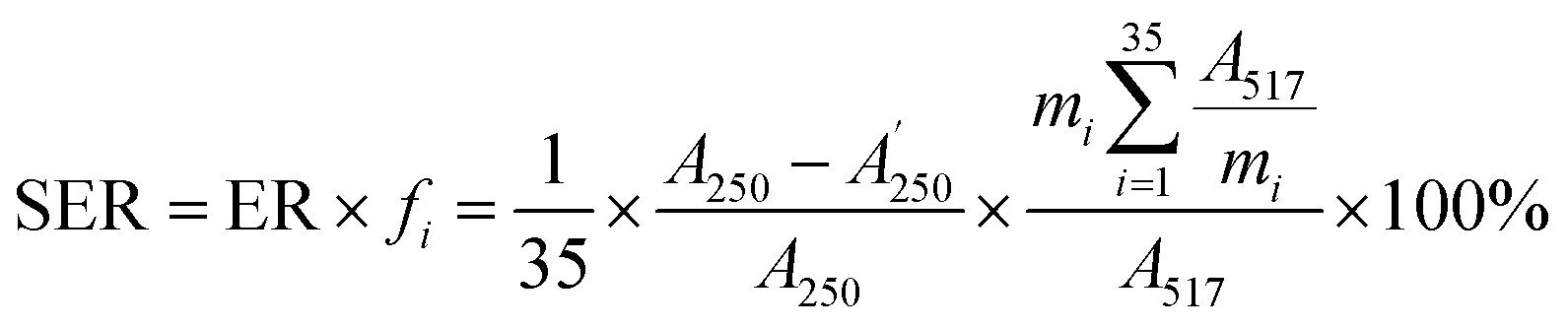 | (8) |
 were the chromatographic peak areas of the unseparated sample before and after reaction with DPPH at 250 nm, respectively. The negative peak area of DPPH after reaction with the sample at 517 nm was defined as A517. Also, fi was the ratio between the average of the 35 negative peak areas and each negative peak area, where the negative peak area was corrected by a mass correction factor. The higher the SER, the stronger the antioxidant activity of the sample.
were the chromatographic peak areas of the unseparated sample before and after reaction with DPPH at 250 nm, respectively. The negative peak area of DPPH after reaction with the sample at 517 nm was defined as A517. Also, fi was the ratio between the average of the 35 negative peak areas and each negative peak area, where the negative peak area was corrected by a mass correction factor. The higher the SER, the stronger the antioxidant activity of the sample.3.4 Data analysis
MassLynx 4.1 software (Waters, USA) was used for the UHPLC-ESI-Q-TOF-MS/MS data processing. Chromatographic fingerprints were assessed by laboratory-developed software (Digitized Evaluation System for Super-Information Characteristics of TCM Chromatographic Fingerprints 4.0; Software certificated no. 0407573, China). SIMCA 13.0 was applied for the data analysis.4 Results and discussion
4.1 UHPLC-ESI-Q-TOF-MS/MS analysis of the CLT sample
Before the MS data analysis, an in-house formula database involving the compound name, chemical structure, molecular formula, accurate mass, and related product ions of the compounds in the individual herbs of CLT was established by searching a number specialist of databases, such as Reaxys (https://www.reaxys.com/), Scifinder (https://scifinder.cas.org), PubMed (https://www.ncbi.nlm.nih.gov/pubmed) and Elsevier (https://www.sciencedirect.com/science/bookbshsrw). Both the positive and negative ion modes were employed in the chemical composition analysis of CLT (as shown in Fig. 2). However, to avoid repetition, we mainly list the negative ions in the ESI Table 1† unless some compounds could only be detected in the positive ion mode. In total, 41 compounds were identified (as depicted in Fig. 3), most of them originating from LRC (19 flavonoids and 19 saponins); only 2 alkaloids were from PPCE, the remaining ingredient was identified as SMB. Among the compounds identified, 9 compounds (peaks 1, 2, 6, 7, 8, 11, 15, 19, and 23) were unambiguously identified by comparing with the reference standards. The other compounds were tentatively characterized based on the mass accuracy of their precursor ions (within a 5 ppm error), tandem MS spectra, and fragmentation pathways, referring to the previous literature.27–44 After a thorough literature search, 2 compounds, comprising 1 flavonoid (isoliquiritigenin-4′-apiosyl(1→2)glucoside) and 1 saponin (apioglycyrrhizin or araboglycyrrhizin), were first identified in CLT in the present study.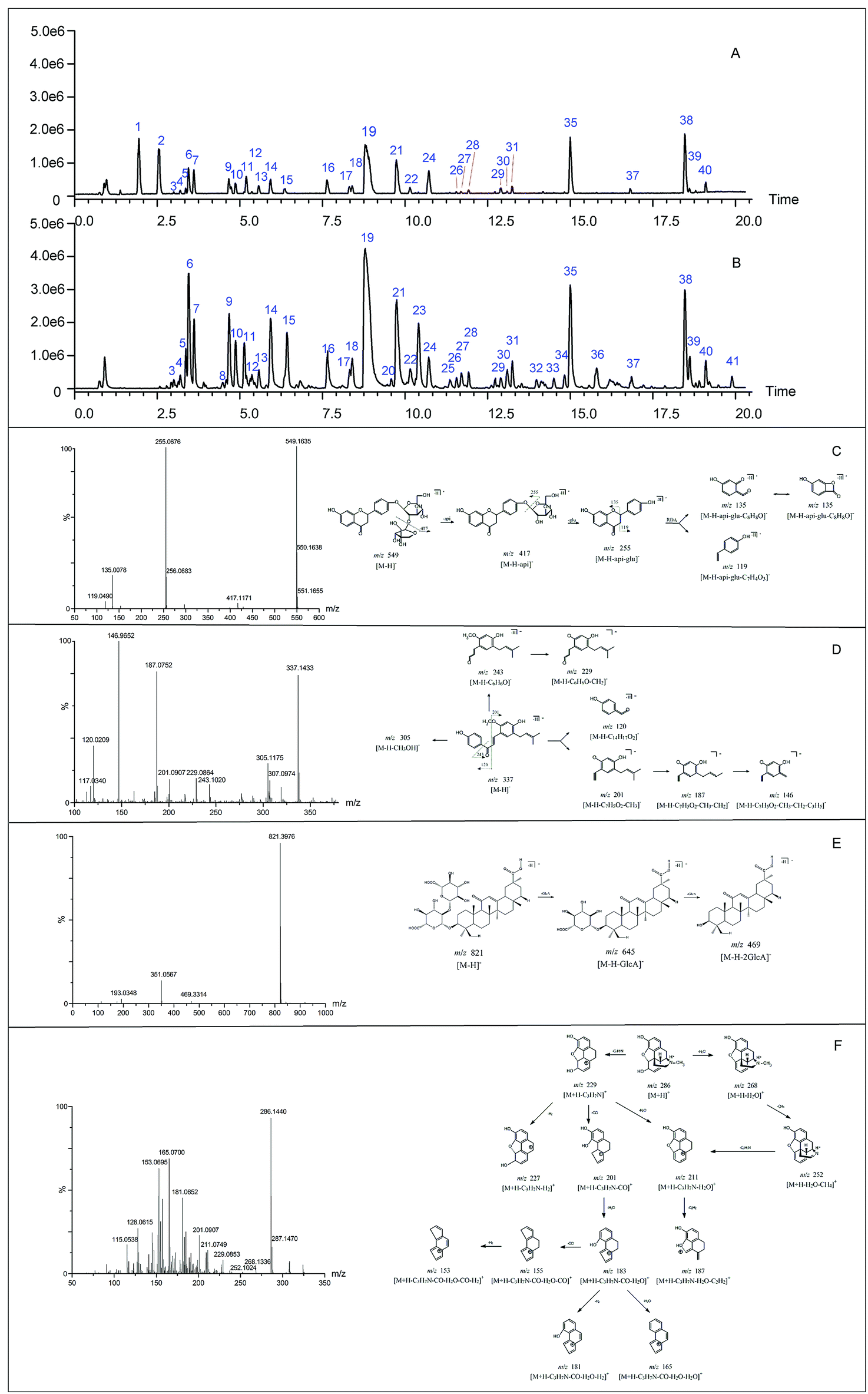 | ||
| Fig. 2 Total ion chromatogram of CLT in the positive (A) and negative ion mode (B), the ESI-MS/MS spectrum and proposed fragmentation pathways of liquiritin apioside (C), licochalcone C (D), glycyrrhizic acid (E) in the negative ion mode and morphine (F) in the positive ion mode. | ||
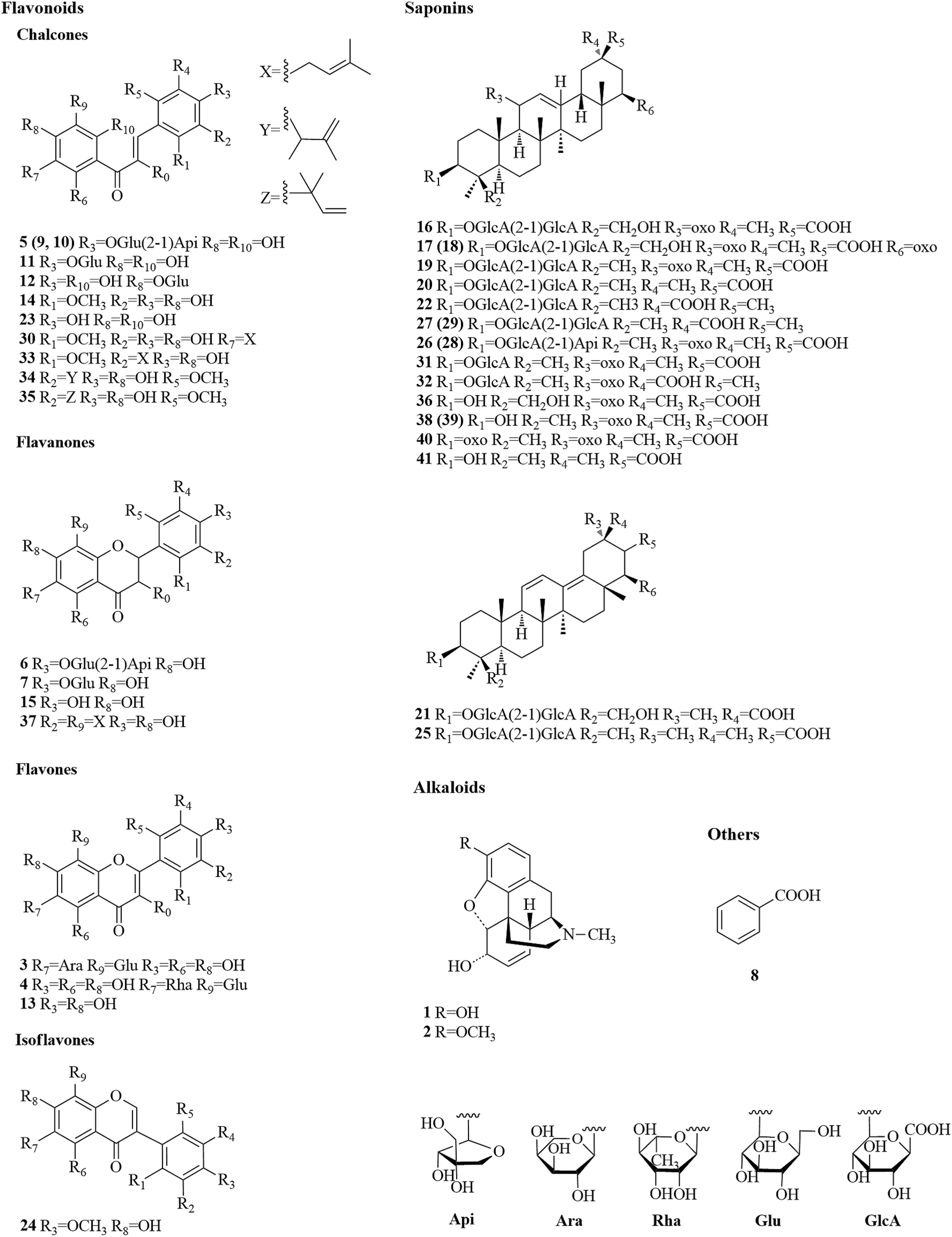 | ||
| Fig. 3 Chemical structures of 41 identified compounds in CLT (unless otherwise noted, the substituent groups (R) are H). | ||
Chalcones and flavanones. In light of the structural characteristics and typical fragment ions in the UHPLC-MS/MS data, chalcones and flavanones could be divided into two groups (as shown in Fig. 4): chalcones with R10 = OH could easily transform to the corresponding flavanone isomers. Thus, chalcones and flavanones showed similar fragmentation pathways to generate the characteristic ion at m/z 255 by elimination of an apiosyl group (Api, 132 Da), glucosyl group (Glu, 162 Da), or both of them, and yield fragment ions at m/z 135 and 119 via retro Diels–Alder (RDA) reaction in the negative ion mode. However, the UV spectrum could aid distinguishing them. Nevertheless, this only worked with a high purity and abundant intensity of chromatographic peaks. Liquiritigenin-4′-apiosyl(1→2)glucoside, a typical flavanone, was used to characterize the fragmentation pathway (Fig. 2C). Based on these fragmentation patterns, the reference compounds, and related literature, compounds 5, 9, 10, 11, 12, 23, 6, 7, and 15 were identified as isoliquiritigenin-4′-apiosyl(1→2)glucoside, isoliquiritigenin-4′-apiosyl(1→2)glucoside, isoliquiritigenin-4′-apiosyl(1→2)glucoside, isoliquiritoside, neoisoliquiritin, isoliquiritigenin, liquiritigenin-4′-apiosyl(1→2)glucoside, liquiritin, and liquiritigenin, respectively. In particular, since both A and B rings in glabrol (compounds 37) contained isopentenyl structures, two fragment ions at m/z 203.0701 and 187.1118 were produced via RDA reaction; in the case of no hydroxyl at R10, the fragmentation patterns were dramatically different. The cleavages mainly occurred around the carbonyl, and the ion abundance depended on the stabilities of the fragments. Generally, the number of phenolic hydroxyls determined the stability of the structure in the negative ion mode. Licochalcone B (compound 14), possessing three hydroxyl groups, gave a deprotonated molecule [M − H]− at m/z 285.0755 and produced predominant fragment ions at m/z 177.0181 [M − H–C6H5O–CH3]−, 150.0313 [M − H–C7H6O2–CH3]−, and 121.0283 [M − H–C9H10O3]− in the MS/MS spectra. Based on these cleavage patterns, peak 30 was identified as licochalcone D. Licochalcone C (compound 33), possessing two hydroxyl groups, gave the deprotonated molecule [M − H]− at m/z 337.1433 and produced fragment ions at m/z 305.1175 [M − H–CH3OH]−, 229.0864 [M − H–C6H5O–CH3]−, 201.0907 [M − H–C7H5O2–CH3]−, and 120.0209 [M − H–C14H17O2]− in the MS/MS spectra. Moreover, B rings, containing the isopentenyl structure, tended to strip off the neutral fragment of C4H7· (55 Da) and produced predominantly fragment ions at m/z 146.9652 (Fig. 2D). According to these fragmentation patterns, peaks 34 and 35 were identified as licochalcone E and licochalcone A, respectively.
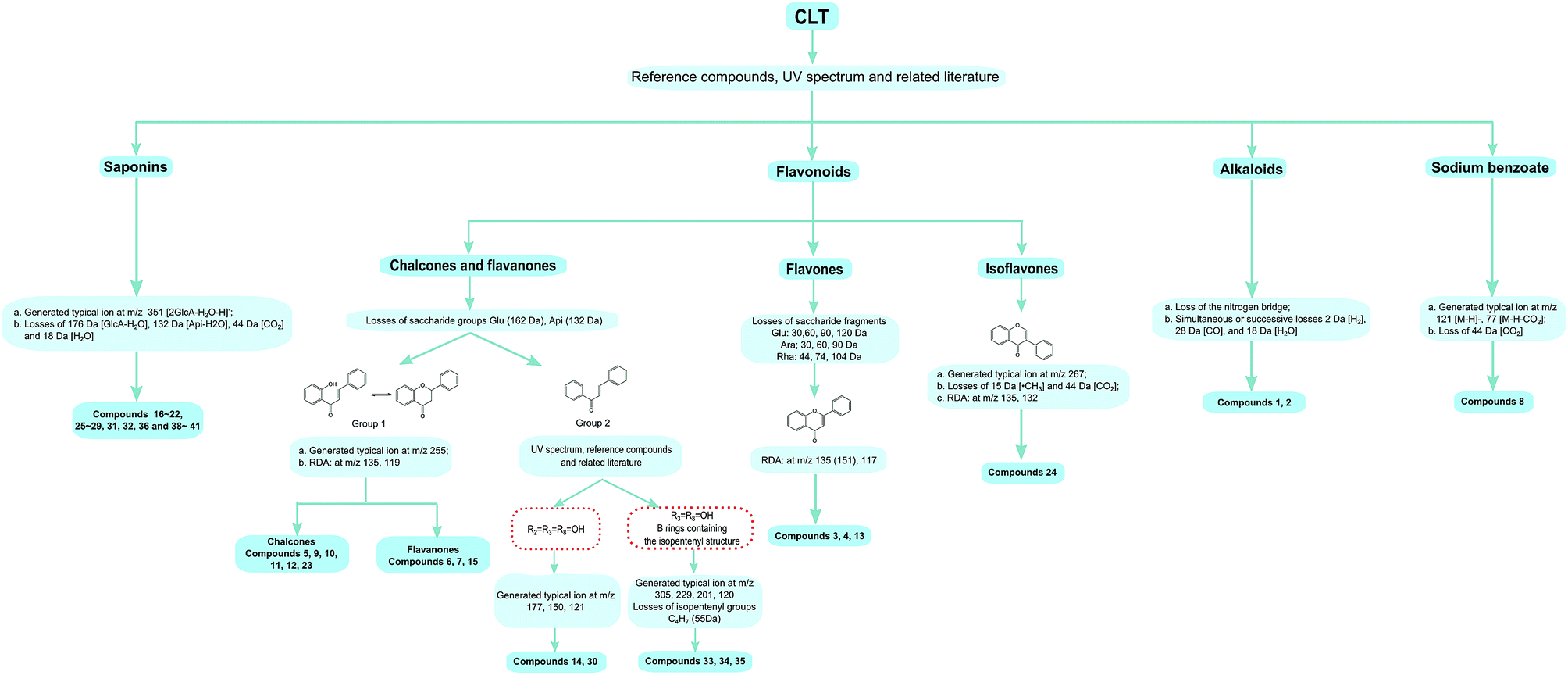 | ||
| Fig. 4 Strategy for rapid structural identification by MS/MS spectra in CLT. | ||
Flavones. Compound 13 (4′,7-dihydroxyflavone) represents the basic skeleton of flavones in licorice. The C-ring undergoes RDA fragmentation to generate fragments at m/z 135.0079 (1,3A−) and 117.0334 (1,3B−). Compounds 3 and 4 were characterized as flavone C-glucosides, which unlike flavone O-glycosides have that lost the whole glycosyl segment, which indicates a special cleavage pathway with successive or simultaneous losses of the glycosyl group. The flavone C-glucosides generated the product ions by losing 30, 60, 90, and 120 Da from the precursor ions. Similarly, the flavone C-arabinosides produced typical losses of 30, 60, and 90 Da, and flavone C-rhamnosides showed the characteristic elimination of 44, 74, and 104 Da. For example, isoviolanthin (compound 4, ESI Fig. S1A†) generated the product ions at m/z 457.1124, 383.0754, and 353.0657 from the precursor ions at m/z 577.1559, and then cleavage of the ion at m/z 253.0495 gave rise to fragments at m/z 135.0072 via RDA reaction. Based on these cleavage patterns, compound 3 was identified as an isoschaftoside.
Isoflavones. Formononetin (ESI Fig. S1B†) is a representative isoflavone in licorice. It shared a base peak at m/z 267.0653 in the ESI negative ion mode and was commonly observed with losses of ·CH3 (15 Da) and CO2 (44 Da). In the MS2 spectra, it produced fragment ions at m/z 252.0420 [M − H–CH3]−, m/z 223.0393 [M − H–CO2]−, and m/z 195.0444 [M − H–CO2–CO]−. The RDA reaction still occurred and generated fragments at m/z 135.0080 (1,3A−) and 132.0209 (1,3B−).
By comparison with the authentic compound, compound 19 was unambiguously attributed to glycyrrhizic acid, which was used to illuminate the fragmentation pathway as a typical triterpene saponin in LRC (Fig. 2E). The [M − H]− at m/z 821.3976 was fragmented into characteristic ions at m/z 645.3629, m/z 469.3314, and m/z 351.0567, corresponding to the [M − H–GlcA]−, [M − H–2GlcA]−, and [2GlcA–2H2O–H]−, respectively. By exploring the literature and these fragmentation patterns, compounds 16–18, 20–22, 25–29, 31, 32, 36, and 38–41 were identified as licorice saponin G2, yunganoside K2 or its isomer, yunganoside K3 or its isomer, an isomer of licorice saponin B2, licorice saponin K2, licorice saponin H2, licorice saponin C2, apioglycyrrhizin or araboglycyrrhizin, licorice saponin B2, apioglycyrrhizin or araboglycyrrhizin, an isomer of licorice saponin B2, glycyrrhetinic acid glucuronide, isomer of glycyrrhetinic acid glucuronide, glycyrrhetinic acid hydroxylate, 18-α-glycyrrhetic acid, 18-β-glycyrrhetic acid, 3-oxoglycyrrhetic acid, and 11-deoxoglycyrrhetic acid, respectively.
The loss of H2O produced m/z 268.1336, followed by a loss of CH4 or C2H3N to produce fragments m/z 252.1024 or 211.0749, respectively. The further fragmentation of ion m/z 211.0749 gave rise to other fragments. A key ion was m/z 229.0853 after the loss of the nitrogen bridge from the parent ion m/z 268.1336 and this was subsequently fragmented to ions m/z 227.0706, m/z 201.0907, and m/z 211.0749 after the losses of H2, CO, and H2O, respectively. The loss of H2O from m/z 201.0907 led to ion m/z 183.800, the other key ion, which was subsequently fragmented to ions m/z 181.0646, m/z 165.0700 (base peak with a highly conjugated frame), and m/z 153.0695 after the simultaneous or successive losses of H2, CO, and H2O.
According to above studies, it was concluded that the chemical components in the CLT samples mainly came from the principal individual herb, i.e., LRC (consisting of flavonoids and triterpene saponins), PPCE (consisted of alkaloids), and SMB. This information about the structures of the chemical components in CLT could help in developing research strategies for bioactivity and QC studies.
4.2 UV spectroscopic/HPLC fingerprint analyses
In the UV fingerprint analysis, unseparated chromatograms (single chromatographic peak collected within 1 min) at 250 nm and UV spectra of samples in the region 190–400 nm were recorded. A typical 3D chromatogram plot of the CLT sample (S1) is shown in Fig. 1B. The retention time (RT) and the peak area (RA) with an unseparated chromatogram were used to estimate the repeatability, precision, and stability. The obtained results showed that the RSDs of RT and RA were all less than 2.0% for the precision, repeatability, and stability validation.
In the HPLC fingerprint analysis, the average linear qualitative similarity (SL) and average linear quantitative similarity (PL) of the HPLC fingerprints was used to estimate the precision, repeatability, and stability. The obtained results showed that, for precision, the RSDs of SL and PL were less than 0.2% and 0.8%; for repeatability, the obtained value did not exceed 0.3% and 1.1%; for stability, the RSDs were less than 0.7% and 1.6%. Thus, these results demonstrated that the developed UV spectroscopic and HPLC methods met the fingerprint analysis requirements for the CLT samples.
| Sample | UV | Fusion | Pred (SCR) | Var (SCR) | REd(%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SL | PL% | α | Grade | SL | PL% | α | Grade | ||||
| a Used for the calibration model.b Used for the prediction model.c Outliers.d RE, relative error. | |||||||||||
| S1a | 0.997 | 112.9 | 0.008 | 3 | 0.959 | 118.7 | 0.037 | 4 | 42.9020 | 43.3005 | −0.92 |
| S2a | 0.992 | 104.8 | 0.000 | 1 | 0.961 | 112.8 | 0.053 | 3 | 44.5328 | 44.8081 | −0.61 |
| S3c | 0.997 | 104.1 | 0.009 | 1 | 0.934 | 110.5 | 0.037 | 3 | |||
| S4a | 0.997 | 111.0 | 0.008 | 3 | 0.956 | 120.1 | 0.049 | 5 | 41.3406 | 43.0298 | −3.93 |
| S5b | 0.997 | 106.7 | 0.004 | 2 | 0.962 | 114.9 | 0.048 | 3 | 41.6980 | 41.3589 | 0.82 |
| S6a | 0.995 | 106.0 | 0.002 | 2 | 0.953 | 114.8 | 0.038 | 3 | 40.8171 | 41.9347 | −2.67 |
| S7b | 0.996 | 105.3 | 0.000 | 2 | 0.951 | 116.7 | 0.049 | 4 | 41.1791 | 40.6103 | 1.40 |
| S8a | 0.994 | 111.0 | 0.004 | 3 | 0.960 | 116.5 | 0.048 | 4 | 41.7202 | 41.5415 | 0.43 |
| S9b | 0.996 | 106.1 | 0.001 | 2 | 0.950 | 120.8 | 0.052 | 5 | 41.3713 | 41.4627 | −0.22 |
| S10a | 0.986 | 107.6 | 0.006 | 2 | 0.949 | 119.1 | 0.031 | 4 | 41.4828 | 39.7119 | 4.46 |
| S11a | 0.996 | 106.5 | 0.005 | 2 | 0.956 | 119.0 | 0.040 | 4 | 41.3830 | 39.8654 | 3.81 |
| S12c | 0.985 | 110.8 | 0.001 | 3 | 0.884 | 127.1 | 0.011 | 5 | |||
| S13c | 0.991 | 111.8 | 0.015 | 3 | 0.856 | 124.9 | 0.021 | 5 | |||
| S14a | 0.990 | 108.0 | 0.021 | 2 | 0.899 | 118.4 | 0.080 | 4 | 41.4751 | 41.5693 | −0.23 |
| S15c | 0.991 | 105.6 | 0.012 | 2 | 0.843 | 128.8 | 0.021 | 5 | |||
| S16a | 0.996 | 90.3 | 0.005 | 2 | 0.886 | 66.8 | 0.061 | 6 | 33.9911 | 34.7101 | −2.07 |
| S17a | 0.997 | 95.8 | 0.007 | 1 | 0.881 | 67.8 | 0.056 | 6 | 33.8583 | 33.0766 | 2.36 |
| S18a | 0.996 | 93.0 | 0.006 | 2 | 0.891 | 66.8 | 0.088 | 6 | 34.4281 | 33.0232 | 4.25 |
| S19a | 0.996 | 87.2 | 0.001 | 3 | 0.907 | 67.1 | 0.071 | 6 | 34.4853 | 34.8612 | −1.08 |
| S20b | 0.996 | 94.0 | 0.007 | 2 | 0.906 | 67.4 | 0.074 | 6 | 34.0604 | 33.4956 | 1.69 |
| S21a | 0.996 | 91.4 | 0.007 | 2 | 0.924 | 74.6 | 0.029 | 5 | 36.3014 | 36.8928 | −1.60 |
| S22a | 0.996 | 87.8 | 0.009 | 3 | 0.902 | 75.8 | 0.012 | 5 | 36.5263 | 36.3241 | 0.56 |
| S23a | 0.979 | 84.6 | 0.014 | 4 | 0.922 | 70.8 | 0.040 | 5 | 36.6813 | 37.6926 | −2.68 |
| S24a | 0.989 | 90.2 | 0.011 | 2 | 0.924 | 75.2 | 0.024 | 5 | 36.8588 | 36.1093 | 2.08 |
| S25b | 0.996 | 92.4 | 0.007 | 2 | 0.892 | 76.1 | 0.031 | 5 | 36.6918 | 37.1300 | −1.18 |
| S26a | 0.996 | 89.6 | 0.020 | 3 | 0.921 | 82.9 | 0.004 | 4 | 38.4024 | 38.8688 | −1.20 |
| S27a | 0.996 | 94.5 | 0.015 | 2 | 0.918 | 80.9 | 0.010 | 4 | 38.6008 | 37.8988 | 1.85 |
| S28a | 0.995 | 94.1 | 0.014 | 2 | 0.930 | 79.9 | 0.017 | 5 | 37.3817 | 37.1949 | 0.50 |
| S29a | 0.994 | 94.1 | 0.016 | 2 | 0.943 | 83.2 | 0.011 | 4 | 37.5003 | 36.5895 | 2.49 |
| S30b | 0.991 | 88.7 | 0.017 | 3 | 0.934 | 83.2 | 0.013 | 4 | 38.1066 | 38.3294 | −0.58 |
| S31a | 0.987 | 101.3 | 0.001 | 1 | 0.970 | 95.7 | 0.060 | 2 | 35.4962 | 34.6755 | 2.37 |
| S32a | 0.999 | 104.8 | 0.005 | 1 | 0.973 | 95.2 | 0.052 | 2 | 35.5709 | 37.1779 | −4.32 |
| S33a | 0.998 | 101.2 | 0.002 | 1 | 0.971 | 93.0 | 0.034 | 2 | 35.1585 | 35.8153 | −1.83 |
| S34a | 0.998 | 99.7 | 0.010 | 1 | 0.969 | 95.8 | 0.052 | 2 | 35.3569 | 35.6429 | −0.80 |
| S35b | 0.976 | 102.8 | 0.001 | 1 | 0.965 | 100.7 | 0.059 | 2 | 35.5584 | 36.2390 | −1.88 |
For the UV fingerprints, the qualitative parameters SL and α values of the 35 CLT samples were, respectively, above 0.979 and below 0.021, illustrating that all the CLT samples had similar chemical compositions. Based on the qualitative parameters SL and α, the quality grades of the 35 CLT samples should have the highest quality. In fact, only 8 samples (S2, S3, S17, and S31∼S35) met the level of grade 1, while the remaining ones were in the range of grade 2–4 in combination with PL from a quantitative perspective. For example, 17 samples (S5∼S7, S9∼S11, S14∼S16, S18, S20, S21, S24, S25, and S27∼S29) were judged as grade 2 with PL in the range 94.1–106.7%; 9 samples (S1, S4, S8, S12, S13, S19, S22, S26, and S30) were judged as grade 3 with PL in the range 88.7–112.9%; 1 sample (S23) was judged as grade 4 with PL 84.6%, respectively. The above results demonstrated that, although the qualitative evaluation (SL and α) was important, the further quantitative assessment (PL) should not be ignored. As a quantitative parameter, PL describes the overall ingredient content in the samples. Therefore, PL has a great potential to be associated with the medicinal efficacy in clinical situations. In general, samples above grade 5 were recommended as the qualified ones. Accordingly, in this study, the qualities of the 35 CLT samples were all judged as qualified with the UV fingerprints.
For the fusion fingerprints, the acceptable SL and α values of the 35 CLT samples were above 0.843 and below 0.088, respectively, indicating that they were similar in the number and distribution of chemical components, and there was little variability between the samples. The acceptable PL values were set in a wider range (70.0–120.0%) and could exactly discriminate the CLT samples from every piece of content of the fingerprints, but actually SL and α were disabled for this function. For example, S2, S3, S5, and S6 should be the grade 2 based on the parameters SL and α. However, they were the higher grades in combination with PL. S16–S20 had unqualified integrated grades (grade 6) due to the much lower contents of components, while the remaining 30 samples had qualified ones (grades 1–5, where there was no judgment sample in grade 1; grade 2 with PL in the range 93.0–100.7%: S1–S35; grade 3 with PL in the range 110.5–114.9%: S2, S3, S5, and S6; grade 4 with PL in the range 80.9–119.1%: S1, S7, S8, S10, S11, S14, S26, S27, S29, and S30; grade 5 with PL in the range 70.8–128.8%: S4, S9, S12, S13, S15, S21–S25, and S28). ALQFM could balance the effects of the large and small peaks; therefore, the qualities of the 35 CLT samples could be more accurately distinguished with the HPLC fingerprints.
The PCA score plot in Fig. 5A and B revealed that most samples fall into one cluster except for S12, S13, and S15 in HPLC. The PCA results were in good agreement with the ALQFM analysis, providing strong evidence that the quality of S12, S13, and S15 might be different from the other samples. In fact, the interesting PL value of S12, S13, and S15 were the highest among the CLT samples, which just better states why these were considered outliers in the PCA score. Then, the 35 CLT samples in the PCA score plot for the UV and HPLC methods, excluding the outliers, could be clearly divided into two clusters marked as group 1 and 2, respectively. Group 1 (S1–S15, from manufacturer A) was obviously different from group 2 (S16–S35, from manufacturer B). It should be noted that the samples in each cluster were more concentrated in Fig. 5B than Fig. 5A, indicating that UV spectral fingerprinting could be rapidly detected and evaluated, and that HPLC fingerprint analysis was recommended for a more accurate assessment. Consequently, from the PCA results, the products from the same manufacturer had a relatively good quality consistency, while the products among different manufacturers exhibited large differentiation.
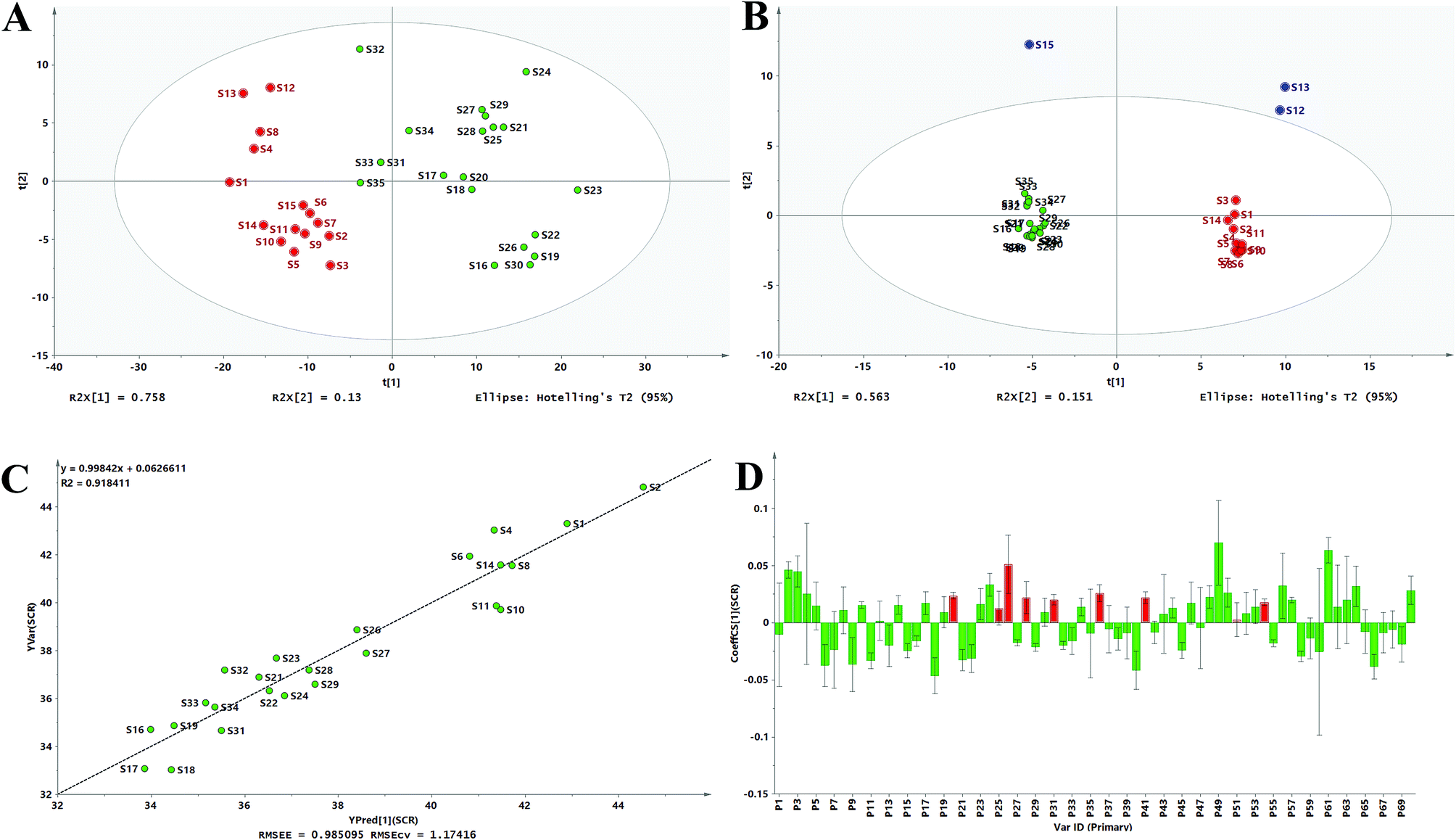 | ||
| Fig. 5 PCA scores scatter plot for the UV spectra (A) and fusion fingerprint (B), measured versus predicted values for the calibration model (C) and OPLS standardized regression coefficient plot (D). | ||
Compared with the quality grades assessed by the UV fingerprinting method, the fusion fingerprints results exhibited some fluctuations and even greater differences, and this could be attributed to two different analytical principles, which mainly reflected the features of the separation and unseparated chemical ingredients in the CLT samples, respectively. For example, S16, S17, S18, S19, and S20 had an unqualified separated quality (grade 6 in the HPLC method); however, they had better unseparated ones (grades 2, 1, 2, 3, 2 in the UV method, respectively). The fusion HPLC fingerprints combined with a UV fingerprints assessment strategy thus provided a feasible and reliable means to monitor the quality consistency of the CLT samples, which was very necessary to avoid a bias caused by the UV fingerprinting method.
4.3 Relationship between the fusion fingerprints and antioxidant activities in vitro
According to the existing literature, flavonoids, triterpene saponins, and alkaloids possess excellent antioxidant abilities.45–47 Simultaneously, combined with the chemical components in ESI Table 1,† the antioxidant abilities of the CLT samples might be due to the presence of chemical constituents, such as flavonoids, triterpene saponins, and alkaloids. This information encouraged us to investigate the fingerprint–efficacy relationship between the fusion fingerprints and the antioxidant activities. The fusion fingerprints and antioxidant activities in vitro were investigated under the conditions described in Sections 3.3.2 and 3.3.4, respectively. Typical fusion chromatograms with 68 co-possessing fingerprints for the 35 CLT samples are shown in Fig. 1D. Correlation analysis between the SER values (as the Y variables) and the HPLC fingerprints (as the X variables) was performed by the OPLS method.After omitting the four outliers (S3, S12, S13, and S15) based on the t[1]–t[2] score plot, the remaining CLT samples were randomly divided into two groups (Table 2) of the calibration set (24 samples) to establish a validation set (7 samples) to validate the OPLS model. The established calibration model (Fig. 5C) achieved an explained variance (R2) of 91.84%, a predictive ability (Q2) of 87.00%, and a root mean square error of estimation of 0.9851, respectively, indicating that the present model was excellent. The validation set was used to assess the activity prediction of the obtained model. A satisfactory result with an explained variance (R2) of 97.49% and a root mean square error of prediction (RMSEP) value of 0.4584 was obtained, indicating the calibration model possessed a well predictive ability. The predicted vs. measured SER values for both the validation and calibration models are shown in Table 2, where no significant difference could be observed for the CLT samples. As shown by the standardized regression coefficients plot (Fig. 5D) of the calibration model, 38 peaks (2–5, 8, 10, 12, 14, 17, 19, 20, 23–26, 28, 30, 31, 34, 36, 41, 43, 44, 46, 48–54, 56, 57, 61–64, and 70) out of 70 fingerprints in the fusion chromatogram were positively correlated, while the remaining peaks were negatively correlated with SER, indicating that the majority of the chemical components in the CLT samples possessed antioxidant abilities. Furthermore, the peaks 20, 25, 26, 28, 31, 36, 41, 51, and 54 in the fusion fingerprints were identified as MPE, LQA, LQT, CON, SMB, ISS, LQG, ISG, and GLA, respectively, by comparing the retention time and the online UV spectra with reference standards.
5 Conclusions
In this paper, a reliable and sensitive UHPLC-ESI-Q-TOF-MS/MS method was performed for identification of the chemical profiling of CLT samples. A total of 41 compounds, including 19 flavonoids, 19 saponins, and 2 alkaloids, were identified or tentatively deduced by comparing their retention times and the MS spectrometry data in the literature. Based on UHPLC-ESI-Q-TOF-MS/MS analysis and the UV absorption, the four main UV absorption bands (220 nm, 250 nm, 280 nm, and 344 nm) of the CLT samples could basically reflect the overall information of the CLT samples. Accordingly, the rapid UV spectroscopic fingerprints and the accurate multi-wavelength (220 nm, 250 nm, 280 nm, and 344 nm) fusion HPLC fingerprints were integrated in equal weight to reflect the overall characterizations of the CLT samples. In the fingerprint assessments, ALQFM with qualitative and quantitative assessment advantages was recommended and established for scientific CLT quality differentiation first, and it could overcome the defects in the quantitative criteria lacking in evaluation methods. According to the integrated UV spectroscopic fingerprints and multi-wavelength fusion HPLC fingerprints as well as ALQFM, the quality consistency of the 35 CLT samples from two manufacturers exhibited almost the same results, indicating that the combination of the two fingerprinting methods provided a reliable means to monitor the quality consistency of the CLT samples. Moreover, PCA was applied to explore the discriminating ability of the two fingerprinting methods, and the results demonstrated that samples from the same manufacturer had a relatively good quality consistency and that the UV spectra provided less information than from HPLC. To further investigate the antioxidant potential of CLT, the free radical scavenging capacity was rapidly assessed by FIA, which was suitable for batch samples in antioxidants screening. In addition, the fingerprint–efficacy relationship between multi-wavelength fusion HPLC fingerprints and antioxidant activities was conducted utilizing OPLS, providing important supplemental bioactivity information for CLT QC. This study reported important clues for the further pharmacological study of CLT and offered a rapid, holistic, and scientific analytical strategy for CLT/TCM QC, which could play an important role in CLT practical production.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundations of China (81573586).References
- R.-F. Hu and X.-B. Sun, Chin. J. Nat. Med., 2017, 15, 436–441 Search PubMed.
- X. Liu, W. Y. Wu, B. H. Jiang, M. Yang and D. A. Guo, Trends Pharmacol. Sci., 2013, 34, 620–628 CrossRef PubMed.
- B. C. Hogle, X. Guan, M. M. Folan and W. Xie, J. Food Drug Anal., 2017, 26, 26–31 CrossRef PubMed.
- J. Cheng, S. He, Q. Wan and P. Jing, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1077–1078, 22–27 CrossRef PubMed.
- J. R. Lucio-Gutierrez, J. Coello and S. Maspoch, Anal. Chim. Acta, 2012, 710, 40–49 CrossRef PubMed.
- Y. Ni, Y. Lai, S. Brandes and S. Kokot, Anal. Chim. Acta, 2009, 647, 149–158 CrossRef PubMed.
- L. Cui, Y. Zhang, W. Shao and D. Gao, Ind. Crops Prod., 2016, 85, 29–37 CrossRef.
- A. Sabir, M. Rafi and L. K. Darusman, Food Chem., 2017, 221, 1717–1722 CrossRef PubMed.
- Z. Yang, J. Zhu, H. Zhang and X. Fan, J. Ginseng Res., 2017, 42, 334–342 CrossRef PubMed.
- L. Bai, J. Smuts, J. Schenk, J. Cochran and K. A. Schug, Fuel, 2018, 214, 521–527 CrossRef.
- Y. Yang, W. Kong, H. Feng, X. Dou, L. Zhao, Q. Xiao and M. Yang, J. Pharm. Biomed. Anal., 2016, 121, 84–90 CrossRef PubMed.
- I. Garcia-Perez, M. Vallejo, A. Garcia, C. Legido-Quigley and C. Barbas, J. Chromatogr. A, 2008, 1204, 130–139 CrossRef PubMed.
- A. S. Arribas, M. Martinez-Fernandez, M. Moreno, E. Bermejo, A. Zapardiel and M. Chicharro, Food Chem., 2013, 136, 1183–1192 CrossRef PubMed.
- E. J. Llorent-Martinez, P. Ortega-Barrales, M. L. Fernandez-de Cordova and A. Ruiz-Medina, Anal. Chim. Acta, 2011, 684, 21–30 CrossRef PubMed.
- N. Mrazek, K. Watla-iad, S. Deachathai and S. Suteerapataranon, Food Chem., 2012, 132, 544–548 CrossRef PubMed.
- S. Chen, S. Li, C. Chen, J. Kong, Q. Wang, S. Ma and F. Zheng, Appl. Chem. Ind., 2017, 46, 2441–2446 Search PubMed.
- Y. Lu, A. Memon, P. Fuerst, A. Kizonas, C. Morris and D. Luthria, J. Food Compos. Anal., 2017, 60, 10–16 CrossRef.
- G. G. Shimamoto and M. Tubino, Fuel, 2016, 186, 199–203 CrossRef.
- H. Li, W. Yao, Q. Liu, J. Xu, B. Bao, M. Shan, Y. Cao, F. Cheng, A. Ding and L. Zhang, Molecules, 2017, 22, 1–14 CrossRef.
- M. Goodarzi, P. J. Russell and Y. Vander Heyden, Anal. Chim. Acta, 2013, 804, 16–28 CrossRef PubMed.
- L. Gong, X. Haiyu, L. Wang, Y. Xiaojie, Y. Huijun, W. Songsong, L. Cheng, X. Ma, S. Gao, R. Liang and H. Yang, J. Sep. Sci., 2016, 39, 611–622 CrossRef PubMed.
- L. Yang, G. Sun, Y. Guo, Z. Hou and S. Chen, PLoS One, 2016, 11, 1–19 Search PubMed.
- L. Yang, X. Xie, J. Zhang and G. Sun, PLoS One, 2016, 11, 1–19 Search PubMed.
- Y. Zhang, L. Yang, J. Zhang, M. Shi and G. Sun, J. Sep. Sci., 2017, 40, 2800–2809 CrossRef PubMed.
- Y. Zhang, G. Sun, Z. Hou, B. Yan and J. Zhang, J. Sep. Sci., 2017, 40, 4511–4520 CrossRef PubMed.
- G. Sun and X. Zhi, Cent. South Pharm., 2008, 6, 349–355 Search PubMed.
- S. Cheddah and T. Hang, J. Chin. Pharm. Sci., 2014, 23, 694–710 Search PubMed.
- M. A. Farag, A. Porzel and L. A. Wessjohann, Phytochemistry, 2012, 76, 60–72 CrossRef PubMed.
- M. He, H. Wu, J. Nie, P. Yan, T. B. Yang, Z. Y. Yang and R. Pei, J. Pharm. Biomed. Anal., 2017, 146, 37–47 CrossRef PubMed.
- Y. J. Li, J. Chen, Y. Li, Q. Li, Y. F. Zheng, Y. Fu and P. Li, J. Chromatogr. A, 2011, 1218, 8181–8191 CrossRef PubMed.
- L. Liang, J.-s. Yang, W.-h. Lin, S.-y. Xiao and H.-x. Liu, Chin. Herb. Med., 2015, 7, 62–68 CrossRef.
- C. Liu, Z. Hua and Y. Bai, Forensic Sci. Int., 2015, 257, 196–202 CrossRef PubMed.
- J. Liu, L. Luo, H. Zhang, B. Jia, J. Lu, P. Li and J. Chen, J. Funct. Foods, 2015, 16, 40–49 CrossRef.
- L. Luo, L. Shen, F. Sun and Z. Ma, Food Chem., 2013, 138, 315–320 CrossRef PubMed.
- P. Montoro, M. Maldini, M. Russo, S. Postorino, S. Piacente and C. Pizza, J. Pharm. Biomed. Anal., 2011, 54, 535–544 CrossRef PubMed.
- Y. Qi, S. Li, Z. Pi, F. Song, N. Lin, S. Liu and Z. Liu, Talanta, 2014, 118, 21–29 CrossRef PubMed.
- X. Qiao, W. Song, S. Ji, Q. Wang, D. A. Guo and M. Ye, J. Chromatogr. A, 2015, 1402, 36–45 CrossRef PubMed.
- X. Qiao, M. Ye, C. Xiang, T. Bo, W. Z. Yang, C. F. Liu, W. J. Miao and D. A. Guo, Steroids, 2012, 77, 745–755 CrossRef PubMed.
- S. Wang, L. Chen, J. Leng, P. Chen, X. Fan and Y. Cheng, J. Pharm. Biomed. Anal., 2014, 98, 22–35 CrossRef PubMed.
- Y. Wang, S. He, X. Cheng, Y. Lu, Y. Zou and Q. Zhang, J. Pharm. Biomed. Anal., 2013, 80, 24–33 CrossRef PubMed.
- T. Xu, M. Yang, Y. Li, X. Chen, Q. Wang, W. Deng, X. Pang, K. Yu, B. Jiang, S. Guan and D. A. Guo, Rapid Commun. Mass Spectrom., 2013, 27, 2297–2309 CrossRef PubMed.
- Y. Yang, X.-J. Yin, H.-M. Guo, R.-L. Wang, R. Song, Y. Tian and Z.-J. Zhang, Chin. J. Nat. Med., 2014, 12, 542–553 Search PubMed.
- L. Zhang, L. Zhu, Y. Wang, Z. Jiang, X. Chai, Y. Zhu, X. Gao and A. Qi, J. Pharm. Biomed. Anal., 2012, 62, 203–209 CrossRef PubMed.
- J. Zhu, X. Yi, J. Zhang, S. Chen and Y. Wu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1060, 262–271 CrossRef PubMed.
- H. H. Koolen, E. M. Pral, S. C. Alfieri, J. V. Marinho, A. F. Serain, A. J. Hernandez-Tasco, N. L. Andreazza and M. J. Salvador, Phytochemistry, 2017, 134, 106–113 CrossRef PubMed.
- C. A. Puente-Garza, C. Meza-Miranda, D. Ochoa-Martinez and S. Garcia-Lara, Plant Physiol. Biochem., 2017, 115, 400–407 CrossRef PubMed.
- A. Zahari, A. Ablat, Y. Sivasothy, J. Mohamad, M. I. Choudhary and K. Awang, Asian Pac. J. Trop. Med., 2016, 9, 328–332 CrossRef PubMed.
- J. M. Lorenzo, M. Pateiro, R. Domínguez, F. J. Barba, P. Putnik, D. B. Kovačević, A. Shpigelman, D. Granato and D. Franco, Food Res. Int., 2017, 106, 1095–1104 CrossRef PubMed.
- H. Cui, Y. Kong and H. Zhang, J. Signal Transduction, 2012, 2012, 646354 Search PubMed.
- S. Losada-Barreiro and C. Bravo-Diaz, Eur. J. Med. Chem., 2017, 133, 379–402 CrossRef PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef PubMed.
- B. Kirschweng, D. Tátraaljai, E. Földes and B. Pukánszky, Polym. Degrad. Stab., 2017, 145, 25–40 CrossRef.
- I. Hamlaoui, R. Bencheraiet, R. Bensegueni and M. Bencharif, J. Mol. Struct., 2018, 1156, 385–389 CrossRef.
- G. X. Sun, Y. Wu, Z. B. Liu, Y. F. Li and Y. Guo, Anal. Methods, 2014, 6, 838–849 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02431f |
| This journal is © The Royal Society of Chemistry 2018 |