
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of wavy carbon nanowires via activation-enabled reconstruction and their applications towards nanoparticles separation and catalysis†
Fengting Lia,
Chenxue Yaob,
Yiqun Zheng *b and
Shifeng Houb
*b and
Shifeng Houb
aSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, Shandong 250100, P. R. China
bNational Engineering Research Center for Colloidal Materials, Shandong University, Jinan, Shandong 250100, P. R. China. E-mail: yiqunzheng@sdu.edu.cn; shifenghou@sdu.edu.cn; Tel: +86-531-88363180 Tel: +86-531-88362815
First published on 5th June 2018
Abstract
We report a facile synthesis of wavy carbon nanowires (WCNWs) derived from polyurethane via KOH activation. The success of this synthesis relies on a carefully designed activation procedure, which involved one pre-activation stage to form suitable precursor and one high-temperature activation stage to allow directional carbon reconstruction. In particular, PU was initially mixed with KOH and thermally treated sequentially at 400 °C and 800 °C for 1 hour, respectively. The resultant products exhibit high purity in the shape of wavy wire, together with a uniform diameter of 51 ± 5.2 nm and the length in the range of 2–8 μm. Systematic studies have been conducted to investigate the effect of reaction parameters in two activation stages on the morphology and structure of final products. It is worth noting that the as-prepared WCNWs could find promising use in the field of both nanoparticle separation and catalysis. For example, they exhibit outstanding separation abilities towards Au nanospheres with different sizes and enhanced catalytic performance when serving as the catalyst support for Pd towards ethanol oxidation reaction. Particularly, the peak current density of Pd/WCNWs catalysts can reach 2126 mA mgPd−1 and the value of its electrochemical active surface area is 60.5 m2 gPd−1.
Introduction
Carbon nanomaterials with at least one dimension between 1 and 100 nm can be classified as zero-dimensional (0D) (fullerenes, onion-like carbon, carbon/graphene dots and nanodiamonds),1–5 one-dimensional (1D) (carbon tubes/fibers, carbon nanohorns, and carbon wires),6–8 and two-dimensional (2D) nanomaterials (graphene, multilayer graphitic nanosheets and graphene nanoribbons).9–11 Among them, 1D carbon nanomaterials have attracted enormous research interest due to their unique properties and applications in biomedicine, adsorption, energy conversion and storage and fabrication nanoscale devices.12–14 This is owing to their excellent biocompatibility, large specific surface area and fabrication of junctions with different shapes and networks as conducting nanowires. As for their application in energy conversion, 1D carbon structures could provide both shortened pathways for electrolyte ion transport and large electrode/electrolyte interfaces to facilitate rapid charge-transfer reactions. To this end, it has become of great importance to explore feasible methods for synthesis of 1D carbons with tunable structure and properties.Thanks to the research efforts contributed by many groups, it is now possible to fabricate 1D carbon nanomaterials via a number of techniques, including arc-discharging,15 thermal evaporation,8 laser ablation,16 electrospinning technique,17 and chemical vapor deposition (CVD).18 For example, Lee and his colleagues first reported the synthesis of carbon nanowires (CNWs) by a thermal evaporation method in 1999.8 They obtained an amorphous CNW with the diameter of around 40 nm by heating a pressed tablet of graphite powder mixed with Ni catalyst in a quartz tube mounted inside a high-temperature tube furnace at 1200 °C. Azami and coworkers synthesized single-wall carbon nanohorn (SWCNH) with uniform and small diameter of about 50 nm, another type of 1D nano-carbon materials, via CO2 laser ablation of graphite in Ne-gas atmosphere.16 As for synthesis of carbon nanofibers, electrospinning and following carbonization technique was always involved. Yang and coworkers fabricated dimensionally thin, mechanically tough, electrically conductive nanofibers with diameters in the range of 200–300 nm by electrospinning of a polymer solution (PAN) and subsequent thermal treatment.19 In addition, the chemical vapor deposition (CVD) method, including catalytic chemical vapor deposition (C-CVD) and catalytic plasma-enhanced chemical vapor deposition (C-PECVD), has been extensively investigated to prepare 1D carbon nanomaterials. Dai and his co-workers synthesized kilogram scale of high quality single-walled carbon nanotubes (SWNTs) by the simple chemical vapor deposition of methane and they found that the success relied on the catalysts materials.20 Ostrikov and his colleagues fabricated metallic SWNTs and semiconducting carbon nanowires (CNWs) networks via slightly changing the process conditions in a simple CVD process using the same Fe/Al2O3 catalyst.21 SWCNT was obtained when the catalysts were dewetted at 900 °C under a flow of Ar (200 sccm) for 5 min. While the catalysts were pretreated at 1000 °C under a co-flow of Ar (200 sccm) and H2 (500 sccm) for 30 min, CNWs were synthesized. All these successful demonstrations have contributed to development in the synthesis of 1D carbon nanomaterials with distinct shapes and tunable structures.
It is worth noting that in addition to those as-mentioned approaches, the formation of 1D carbon nanomaterials should also be possibly realized via carbon reconstruction under appropriate conditions. Similar to the interesting case of semiconductor nanowires formed via oriented attachment of quantum dots,22 it is not unreasonable to assume that carbon atoms and/or aggregates can also be attached or connected along one direction to promote the formation of carbon nanomaterials with 1D structure. However, this concept has been rarely put into practice and reported. Recently, the carbon reconstruction allowed by KOH activation has caused research attention. In particular, carbon reconstruction was triggered by broken carbon species and dangling bonds which were generated via KOH activation at high temperature. For example, carbon reconstruction has occurred during graphene oxide activation. A three dimensional (3D) porous carbon, named MEGO, could be obtained via a simple activation with KOH of microwave exfoliated two dimensional (2D) graphite oxide (MEGO) at 800 °C.23 Later, Zhu and his colleagues found that the MEGO could remain quasi-2D morphology when KOH activation at relative low temperature of 450–500 °C. While the activation temperature was above 500 °C, a transition of quasi-2D MEGO to 3D porous structure could be observed.24 In short, it would be helpful to take advantage of this process in the synthesis of 1D carbon nanomaterials.
Herein, we present a niche and facile method for the synthesis of 1D carbon nanomaterials with the shape of wavy nanowire via a carefully-designed KOH activation. In particular, polyurethane (PU) was mixed with KOH and pre-activated at 400 °C, then followed by activation at 800 °C. The morphology and structure of resultant products were characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD), Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). Unlike 1D polymeric materials reported in previous studies, the atomic percentage of carbon in current product could reach up to 94.43% (Table S1†). To this end, the current product could be considered as one niche type of 1D carbon nanomaterial with the wavy shape. Moreover, several reaction parameters involved in both activation stages have been systematically studied to investigate their effect on the wavy carbon nanowires formation (WCNWs). Furthermore, the as-prepared WCNWs were found useful in both nanoparticle separation and catalysis. In particular, WCNWs exhibits excellent separation abilities for Au nanoparticles with the size of 10 and 50 nm. In addition, Pd/WCNWs catalyst displays outstanding catalytic activities and stabilities for ethanol oxidation reaction (EOR). The mass peak current density of Pd/WCNWs catalyst can reach 2126 mA mgPd−1 and stay at 459 mA mgPd−1 after 1000 cycles.
Experimental section
Materials
Polyurethane (PU, 8670AU) was purchased from Bayer. Commercial Pd/C catalyst was obtained from Shanghai River Sen Electric Co., Ltd. Potassium hydroxide (KOH, >85%), tetrachloroauric(III) acid hydrate (AuCl3 HCl·4H2O, >47.8%), sodium borohydride (NaBH4, 98%), L-ascorbic acid (AA, ≥99.7%), trisodium citrate dehydrate (TCD, C6H5Na3O7·2H2O, ≥99.0%), and isopropanol (C3H8O, ≥99.7%) were all obtained from Sinopharm Chemical Reagent Co., Ltd. Hexadecyltrimethyl ammonium bromide (CTAB, 99%), hexadecyltrimethyl ammonium chloride (CTAC, 97%) and sodium tetrachloropalladate(II) (Na2PdCl4, 98%) were all purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd. Poly(ethyleneimine) solution (PEI, average Mw ∼2000 by LS, 50 wt% in H2O) was purchased from Sigma-Aldrich Co., Ltd. Alcohol (CH3CH2OH, 99.5%) and hydrochloric acid (HCl, 12 M) were both obtained Tianjin Fuyu Fine Chemical Co., Ltd. All the chemicals were used as received without further purification. The water used in all experiments was ultrapure water prepared by an ultrapure water system (Ulupure, Zhengzhou, China) with a resistivity of 18.25 MΩ cm at room temperature. The quartz tube furnace (OTF-1200X) was purchased from Hefei Crystal Materials Technology Co., Ltd.Synthesis of wavy carbon nanowires (WCNWs)
In a standard procedure for preparation of WCNWs, 0.15 g of PU was mixed with 0.6 g of KOH in a 30 mL agate mortar by dry milling for 10 min at room temperature to obtain a solid mixture of PU/KOH. The as-prepared solid mixture was heated in a tube furnace at the heating rate of 10 °C min−1 under flowing argon at a flow rate of 100 mL min−1, pre-activated at 400 °C for 60 min, and then activated at 800 °C for another 60 min. After activation, the obtained black powder was neutralized with HCl (1 M) and washed with water until neutral pH was achieved. The resultant products were dried at 60 °C in the open air for overnight.Fabrication of WCNWs filter membrane
Filter membrane of the as-synthesized WCNWs was fabricated by a wet forming method using PEI as binder. In a typical experiment, 6 mg of WCNWs was dispersed in 2 mL of water with ultra-sonication (ultrasonic power: 120 W) for 60 min to form a 3 mg mL−1 suspension of WCNWs. Subsequently, 200 μL of 0.5 wt% PEI was injected into the suspension in one shot using a pipette, and the resultant mixture was ultra-sonicated for another 120 min. After that, the mixture was poured into a vacuum filter equipped with a 0.22 μm porous PTFE membrane and filtered under low pressure (−0.01 MPa) to produce a filter membrane followed by drying naturally.Preparation of Pd/WCNWs catalyst
15 mg of the as-prepared WCNWs was dispersed in 30 mL of isopropanol with ultra-sonication (ultrasonic power: 120 W) for 60 min to form a 0.5 mg mL−1 suspension of WCNWs. Next, 40 mL of aqueous TCD (4.7 mM) solution was poured into the as-prepared suspension. The mixture solution was heated at 85 °C in an oil bath under magnetic stirring for 5 min. Subsequently, 4.7 mL of aqueous solution containing 13.82 mg of Na2PdCl4 was injected into the preheated solution in one shot using a pipette. The reaction solution was kept at 85 °C under magnetic stirring. After 6 h, the products were collected by centrifugation, washed several times with water to remove excess TCD, and then dried at 60 °C in the open air for 6 h.Synthesis of Au nanospheres with the diameter of 10 nm and 50 nm
The synthesis of Au nanospheres were conducted using seeded growth. The Au seeds were prepared according to a reported protocol.25 In a typical experiment, 5 mL of aqueous HAuCl4 (0.5 mM) solution and 5 mL of aqueous CTAB (200 mM) solution were mixed in a 20 mL glass vial, then 0.6 mL of aqueous NaBH4 solution (10 mM) was added rapidly using a pipette. A brown solution immediately emerged after the introduction of NaBH4 solution. The brown solution was remained at room temperature for 10 min to ensure complete decomposition of NaBH4 remaining in the mixed solution. For synthesis of 10 nm Au nanospheres, 100 μL of the Au seeds solution was added into a mixed 5.5 mL of aqueous solution containing HAuCl4 (0.18 mM), CTAC (72.72 mM) and AA (27.27 mM) in one shot using a pipette. The reaction was allowed to continue at room temperature for 10 min. For synthesis of 50 nm Au nanospheres, 2 mL of aqueous CTAC (100 mM) solution, 150 μL of aqueous AA (10 mM) solution and 10 μL of 10 nm Au nanospheres aqueous solution were mixed in a 20 mL glass vital. Then 2 mL of aqueous HAuCl4 (0.5 mM) solution was dropped using a syringe pump at an injection rate of 2 mL h−1. The reaction was also allowed to proceed at room temperature for 10 min after the injection had been accomplished.Characterizations
The morphology of as-prepared materials was observed using a field emission scanning electron microscope (FESEM, SU8010, HITACHI, Japan) operated at 5 kV and transmission electron microscopy (TEM, JEM-1011, JEOL, Japan) at 120 kV. High-resolution TEM images were obtained using a field-emission microscope (HRTEM, JEM-2100F, JEOL, Japan) operated at 200 kV. Raman spectra were recorded via a high-resolution confocal-Raman system (LabRam HR800, HORIBA Jobin Yvon, Japan) with 633 nm lasers. X-ray diffraction (XRD) patterns were recorded by Bruker D8-Advance diffractometer (Bruker Ltd, Germany) with filtered Cu-Kα radiation (40 kV, 40 mA, 10° min−1 from 10 to 80°). X-ray photo electron spectroscopy (XPS) spectra were recorded by a VG ESCALAB MKII X-ray photoelectron spectrometer with an MgKα excitation source (1253.6 eV). The analysis of elements and their proportions in the products were performed with an elemental analyzer (vario EL cube, Elementar, Germany). The porous textures of the samples were analyzed by N2 adsorption/desorption at 77 K (ASAP 2020, Micromeritics, USA). The specific surface area was calculated using the BET method accorded to six points of relative pressure (P/P0) ranging from 0.05 to 0.08. The total pore volume was measured from the amount of nitrogen adsorbed at a relative pressure (P/P0) of 0.99. The pore size distribution was determined via density functional theory (DFT) method using nitrogen adsorption data. The tap density was calculated by the ratio of the mass to its tap volume, which was measured after the products were tapped 100 times in a vial. UV/vis extinction spectra were recorded on a U-4100 UV/VIS/NIR spectrometer (HITACHI, Japan). All the electrochemical experiments were carried out on a CHI-760E electrochemical workstation (Chenhua, Shanghai, China).Electrochemical measurement
The catalytic performance and stability of the as-synthesized Pd/WCNWs catalyst for EOR under alkaline conditions were examined in a three-electrode system by cyclic voltammetry (CV) and chronoamperometry using a CHI-760E electrochemical workstation. In the test, a platinum foil, a saturated calomel electrode (SCE), and a glassy carbon electrode (GCE, diameter: 3.0 mm) were used as the counter, reference, and working electrodes, respectively. For measurement, 4 mg of as-prepared Pd/WCNWs catalyst was dispersed in a mixture solution of water (2.0 mL), isopropanol (2.0 mL) and Nafion solution (10 μL, 5 wt%) with ultra-sonication (ultrasonic power: 120 W) for 30 min to form homogeneous suspension. 10 μL of the as-prepared homogeneous suspension was dropped on the surface of GCE and dried in the open air at 60 °C. As control, the catalytic performance of commercial Pd/C catalyst on the GCE was also tested by similar process.Results and discussion
Morphology and structure
As shown in Scheme 1, PU was first mixed with KOH by dry milling and thermally pre-activated at 400 °C and then activated at high temperature of 800 °C to form WCNWs. The weight yield of WCNWs was 20% versus the weight of raw PU. In addition, the tap density of WCNWs is 0.09 g cm−3. The morphology of WCNWs (see Experimental section for synthetic details) was characterized via typically scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Fig. 1a and b, exhibit SEM images of WCNWs in different magnifications, from which we can see clearly that WCNWs were obtained in high purity, together with an average diameter of 51 ± 5.2 nm and the length in the range of 2–8 μm (Fig. S1, ESI†). Unlike carbon fibers which are straight and smooth,26 these WCNWs are curvy and rough, indicating their different internal structures and a great potential for applications in metal nanoparticles carrier. The curvy and rough structure can be observed clearly with TEM image (Fig. 1c). The inset in Fig. 1d shows the selected-area electron diffraction (SAED) pattern obtained from an individual WCNW, indicating that the WCNWs are mainly amorphous or full of defects, probably due to the KOH activation and/or relative low growth temperature. | ||
| Scheme 1 Schematic illustration of the formation of WCNWs. | ||
 | ||
| Fig. 1 (a) Low- and (b) high-magnification SEM images of WCNWs. (c) TEM and (d) high-resolution TEM images of WCNWs. The scale bar in the inset of part (c) is 100 nm, and the inset of part (d) is the selected-area electron diffraction (SAED) pattern of the WCNWs. | ||
XRD and Raman spectra were obtained to further investigate the structure and crystallinity of the WCNWs (Fig. 2). XRD pattern in Fig. 2a reveals two broad peak at about 2θ = 23° and 43°, which are ascribed to the (002) and (100) reflections typically observed for microcrystalline, semi-graphitic carbons.27 The high intensity in the low angle region probably results from KOH activation, demonstrating the amorphous structure of the WCNWs. Raman spectrum in Fig. 2b further reveals the amorphous structure of WCNWs. The peaks appeared at 1320 and 1580 cm−1 can be attributed to the D (defects and disorder) and G (graphitic) bands of carbon materials, respectively.28 Generally, the D band corresponds to the breathing mode of k-point phonons of A1g symmetry, while the G band corresponds to the E2g phonons of sp2 carbon atoms.29 Furthermore, the value of D/G intense ratio of WCNWs is 1.10, indicating the structure of WCNWs is more amorphous, which is agreement with the diffuse diffraction ring in the inset of Fig. 1d. We can conclude that the as-obtained WCNWs have amorphous structure together with a wavy wire-like shape. Furthermore, N2 adsorption/desorption has been performed to investigate the specific surface area and pore size of the WCNWs. Indeed, the specific surface area of WCNWs is up to 845 m2 g−1. As shown in Fig. 2c, the isotherms is identified as type IV, illustrating that WCNWs is typical of micro- and mesoporous materials, which is confirmed by pore size distribution in Fig. 2d.30
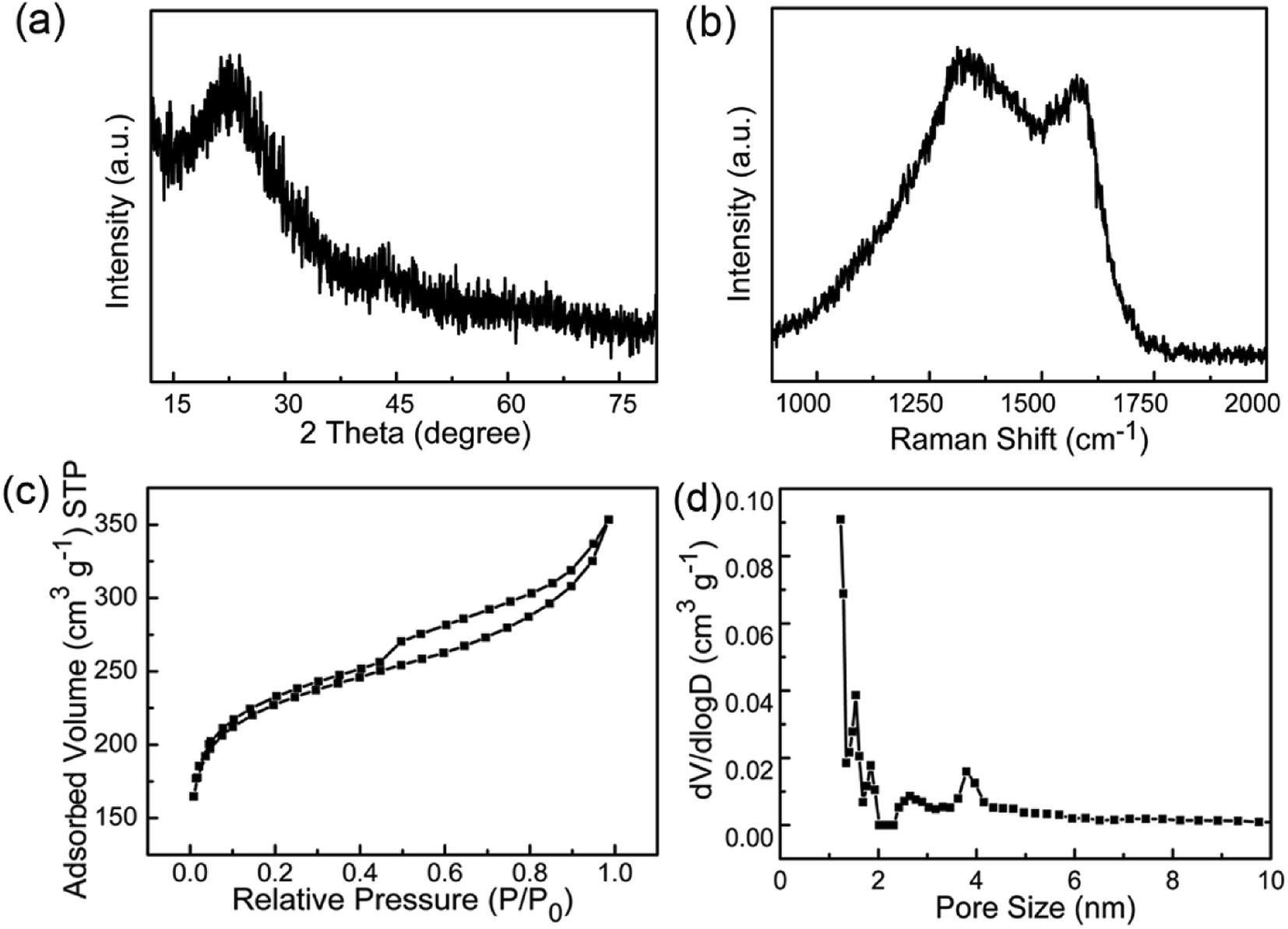 | ||
| Fig. 2 (a) XRD pattern and (b) Raman spectrum of WCNWs. (c) N2 adsorption/desorption isotherm and (d) pore size distribution curve of WCNWs. | ||
Fig. 3a shows the XPS survey spectra of WCNWs, where two primary peaks centered at 285.8 and 534.4 eV are observed, corresponding to C 1s and O 1s, respectively. The spectra of C 1s in Fig. 3b can be fitted to three line shapes with binding energies at 284.5, 285.2 and 286.1 eV, corresponding to C–C, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. It can be deduced that two functional groups exist on the surface of WCNWs besides the C–C peak, which is located at 284.5 eV.31 The spectra of O 1s in Fig. 3c exhibit peaks at 532.3 (CO) and 533.8 eV (C–O), revealing the existence of carbonyl and other oxygen groups, which is consistent with the results shown in Fig. 3b. Taken together, the surface of as-prepared WCNWs contains numerous oxygen-containing functional groups.
O, respectively. It can be deduced that two functional groups exist on the surface of WCNWs besides the C–C peak, which is located at 284.5 eV.31 The spectra of O 1s in Fig. 3c exhibit peaks at 532.3 (CO) and 533.8 eV (C–O), revealing the existence of carbonyl and other oxygen groups, which is consistent with the results shown in Fig. 3b. Taken together, the surface of as-prepared WCNWs contains numerous oxygen-containing functional groups.
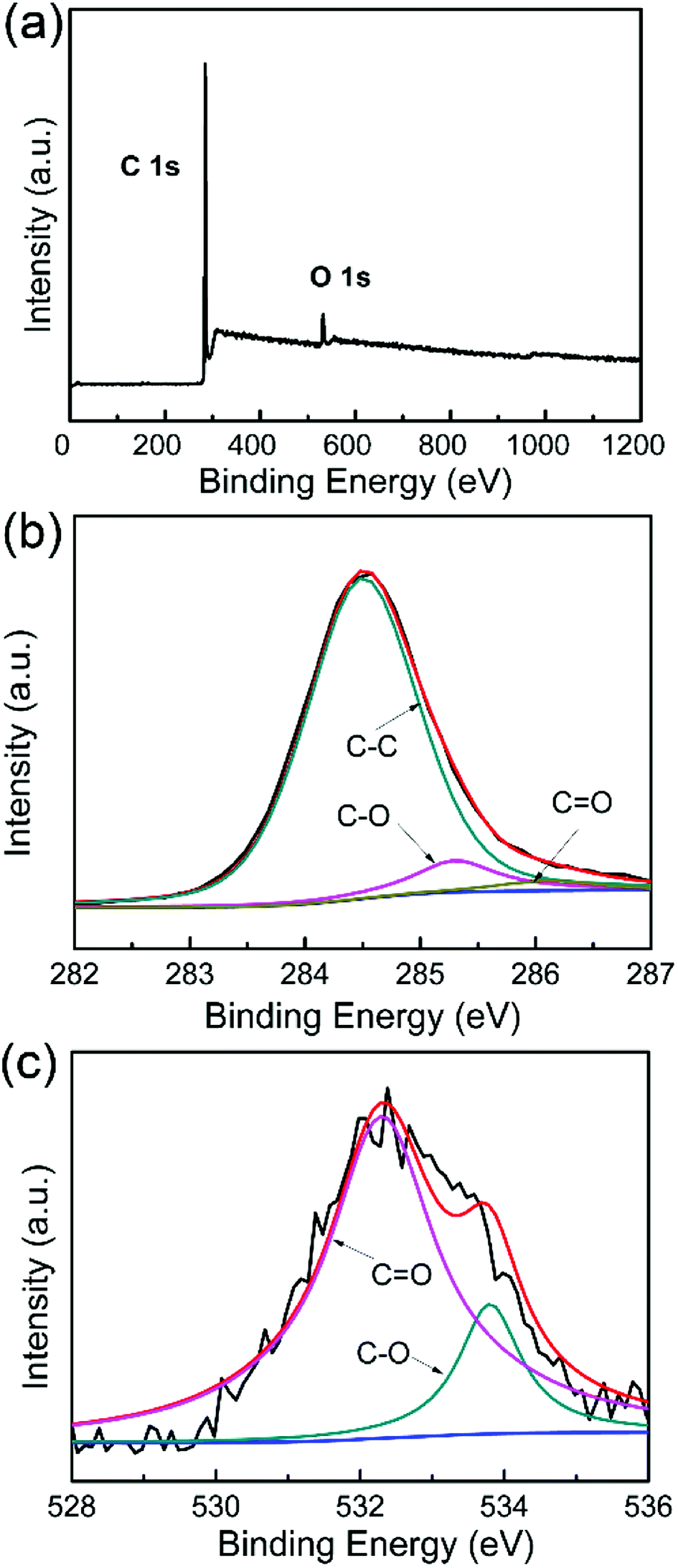 | ||
| Fig. 3 (a) XPS survey spectra of WCNWs. (b and c) High resolution XPS spectra of (b) C 1s and (c) O 1s. | ||
In addition, the surface chemical composition of WCNWs was analyzed by X-ray photoelectron spectrometer. As shown in Table S1,† the atomic percentage of carbon and oxygen in WCNWs is 94.43% and 5.57%, respectively. We can also enhance the carbon content in the current products by calcination under Ar/H2 (95/5) flow at 1000 °C for 1 h. In particular, the atomic percentage of carbon in the product reached 96.53% after calcination (Table S1†). Moreover, the elements and their proportions in the WCNWs were analyzed using an elemental analyzer. In addition to carbon and oxygen, WCNWs contained a small amount of hydrogen and nitrogen (Table S1†). It is worth noting that the calcination process caused unnoticeable change to the shape of the products and the wavy shape was still maintained (Fig. S2†).
Discussion on the formation mechanism
Effect of the amounts of KOH: to investigate the mechanism involved in the formation of WCNWs, we conducted a set of control experiments. We first examined the effect of the amounts of KOH, which had a direct correlation with the activation degree. When KOH was absent, only carbon monoliths was obtained (Fig. S3a†), indicating the necessary presence of KOH in the current procedure. When the mass ratio of PU to KOH was set to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:3, carbon fragments were obtained (Fig. S3b and c†). As more KOH was introduced (the mass ratio of 1:5, Fig. S3d†), products with the uniformity in both shape and size were obtained, which was similar to the results exhibited in Fig. 1. However, when the mass ratio was increased to 1:6, the product yield was pretty low and no solid residue was left after the activation. These results demonstrated that the amount of KOH played an important role in synthesis of WCNWs. Consequently, the optimum mass ratio of PU:KOH for synthesis of WCWNs was chosen at 1:4.
:1 and 1:3, carbon fragments were obtained (Fig. S3b and c†). As more KOH was introduced (the mass ratio of 1:5, Fig. S3d†), products with the uniformity in both shape and size were obtained, which was similar to the results exhibited in Fig. 1. However, when the mass ratio was increased to 1:6, the product yield was pretty low and no solid residue was left after the activation. These results demonstrated that the amount of KOH played an important role in synthesis of WCNWs. Consequently, the optimum mass ratio of PU:KOH for synthesis of WCWNs was chosen at 1:4.
Effect of activation temperature: in order to elucidate the effect of activation temperature on the formation of WCNWs, products obtained at different activation temperatures were collected for SEM characterization. As shown in Fig. 4a, at the activation temperature of 400 °C, only carbon monolithic, instead of WCNWs, was observed. As the activation temperature increases, e.g., to 600 °C, carbon chunks with a fraction of WCNWs were obtained, as exhibited in Fig. 4b, indicating that a restructuring to 1D carbon began to occur at this reaction condition. As the activation temperature raised up to 900 °C, many WCNWs were formed during the activation procedure (Fig. 4c). It was noticeable that these WCNWs obtained at 900 °C were shorter in length than those of synthesized via a standard experiment at 800 °C (Fig. 1), probably due to the enhanced intensity of KOH activation increased at higher temperature. When the activation temperature further increases to 1000 °C, no residue was collected after the reaction had been finished. Previously, it has been documented that 2D reduced graphene oxide could reconstruct to 3D porous carbons under KOH activation at 450 °C.24 This carbon reconstruction process was also observed in our case when activation temperature reached 600 °C.
 | ||
| Fig. 4 Effect of activation and pre-activation temperature on the morphology and structure of the resultant products. (a–c) SEM images of products obtained via the standard procedure, except that the activation temperature was varied from 800 °C to (a) 400 °C, (b) 600 °C, and (c) 900 °C, respectively; (d–f) SEM images of products obtained via the standard procedure, except that (d) no pre-activation process was involved and (e and f) pre-activation temperature was varied from 400 °C to (e) 300 °C and (f) 500 °C, respectively. | ||
Effect of pre-activation temperature: the current synthesis involved two stages of thermal treatment. In addition to the activation stage, we also investigated the effect of pre-activation temperature on the formation of WCNWs. Fig. 4d shows the typical SEM image of the resultant product obtained without a pre-activation procedure. We noted that only a few short and thin carbon nanowires are observed, illustrating a low yield of WCNWs under this reaction condition. It suggested the pre-activation stage was beneficial for an enhanced yield of wire-shaped product. As shown in Fig. 4e, when the pre-activation temperature was set to 300 °C, some quasi-spherical particles occurred. In contrast, as the pre-activation temperature further increases to 500 °C, only polyhedral carbons were observed (Fig. 4f). These results showed that the pre-activation temperature should be optimized at 400 °C for the best yield of WCNWs. To further elucidate the pre-activation temperature effect, we collected precursors obtained at different pre-activation temperature for SEM and/or TEM characterization. As exhibited in Fig. S4a and b,† PU was transformed into small particles via KOH activated at 400 °C for 1 h, which could be rearranged to 1D carbon of WCNWs at high activation temperature of 800 °C (Fig. 1). Furthermore, N2 adsorption/desorption has been performed to investigate the specific surface area and pore size of the precursor obtained via pre-activation at 400 °C. Compared with WCNWs, the precursor has larger specific surface area of 1616 m2 g−1 and higher tap density of 0.12 cm3 g−1, probably due to the presence of more micropores in the precursor (Fig. 2c and d and S5†). By contrast, the precursor obtained at 300 °C consisted of quasi-sphere particles and bulks (Fig. S4c†), which could be restructured to large carbon particles and/or quasi-wires during the activation procedure (Fig. 4e). For the samples collected at 500 °C, carbon mass with irregular shape was observed (Fig. S4d†), which could be reconstructed into polyhedral carbon via KOH assisted activation (Fig. 4f). Therefore, it is no unreasonable to assume that pre-activation temperature may affect the formation of WCNWs by varying the morphology and structure of its precursors.
Effect of activation time: to track the growth process, we terminated the reaction at 800 °C for different periods and collected the sample for SEM imaging. As shown in Fig. S6,† at t = 1 min, the sample was composed of large chunks, small particles aggregations, and thin WCNWs of around 25 nm. As reaction proceeded to 10 min, large chunks disappeared and only small particles aggregations and thin WCNWs existed. After another 20 min, the diameter of the WCNWs became about 45 nm. At t = 40 min, the product was dominated with WCNWs and the small particles aggregations disappeared. Based on these data, we believed that the precursor changed to large chunks and small particle aggregations in the initial stages, followed by the reconstruction of these particles along one direction to form 1D WCNWs.
In short, the growth mechanism is discussed as follows. KOH was firstly decomposed according to the reaction: 2KOH → K2O + H2O (g). Then carbon reacted with gaseous water to produce carbon monoxide and hydrogen (H2O + C → CO + H2).32,33 Similar to preparation of carbon nanotube/nanofiber via CVD, the CO formed in situ could mediate with H2 via hydrogenation and/or disproportionation to generate carbon atoms/clusters.34 These carbon species work as building blocks for the carbon nanoparticles and WCNWs observed in the initial stage of activation (Fig. S6†). It is worth noting that, in order to obtain WCNWs in high purity, the amount of KOH had to be more than 3 times of PU (Fig. S3†). It suggested that the concentration of CO/H2 affected by amount of KOH should reach a sufficiently high level to favor the generation of carbon atoms/clusters and thus allow the carbon reconstruction to occur. In contrast, when too much KOH was added (i.e., KOH/PU = 6:1), all the carbon precursor would be converted to gaseous product and no solid residue could be obtained. In a sense, the catalytic function of KOH could affect the reaction kinetics for this synthesis, which is crucial for the carbon reconstruction and thus the formation of WCNWs.
Applications of WCNWs in nanoparticle separation and catalysis
It is well accepted that 1D carbon nanomaterials are highly promising for application in catalysis and separation membranes.35,36 Herein, we chose two models to explore their applications: (i) the separation ability towards gold (Au) nanoparticles when made into separation membrane; (ii) catalytic performance for EOR in alkaline medium when working as the catalyst support.Separation of Au nanospheres
Optical and electronic properties of metal nanoparticles are strongly dependent on their shape, size, and uniformity.37 To obtain samples with high purity, separation is necessary. So far, the main methodologies for separation of nanoparticles include ultracentrifugation, field flow fraction, electrophoretic, magnetic and membrane separation methods.35,38–42 We found that the as-prepared WCNWs could be fabricated into membrane to be used for separate Au nanospheres with different sizes of 10 and 50 nm from solution using the hand-made setup (Fig. S7,† 5a). As shown in Fig. 5a, 10 nm Au spheres passed through the membrane, while 50 nm spheres were trapped on the membrane. The corresponding experimental results were exhibited in Fig. 5c. Almost all the 10 nm Au nanospheres could freely pass through the membrane and about only 4.3% of them were remained inside the membrane or on the feed side, which was calculated from the adsorption intensities at 521 nm (Fig. 5d). In sharp contrast to the permeation of 10 nm Au spheres, all of the 50 nm Au spheres were been filtered out from solution. As for the mixed aqueous solution of 10 and 50 nm spheres, its color became slight red after filtering compared to the initial red solution (Fig. 5c), together with the weak intensity of the absorption peak after filtering (Fig. 5d). Besides, UV/vis extinction spectra for the mixture revealed a peak at near 527 nm, while the peak shifted to 521 nm after filtration, explaining that the filtrate only contained Au spheres of 10 nm. In addition, Fig. 5b shows the photographs of WCNWs membrane, from which we can see clearly that the membrane can be bent even without any cracking. Furthermore, the WCNWs membrane was characterized by SEM before (Fig. S8a†) and after filtration (Fig. S8b†). As shown in Fig. S8b,† Au nanospheres of 50 nm were captured on the membrane. The aggregation appeared on the membrane probably resulted from the removal surfactant of Au nanospheres and thus insufficient protection keep nanoparticles being attached to each other. In a word, these results illustrated the outstanding separation ability of WCNWs membrane in screening out metal nanoparticles with different sizes.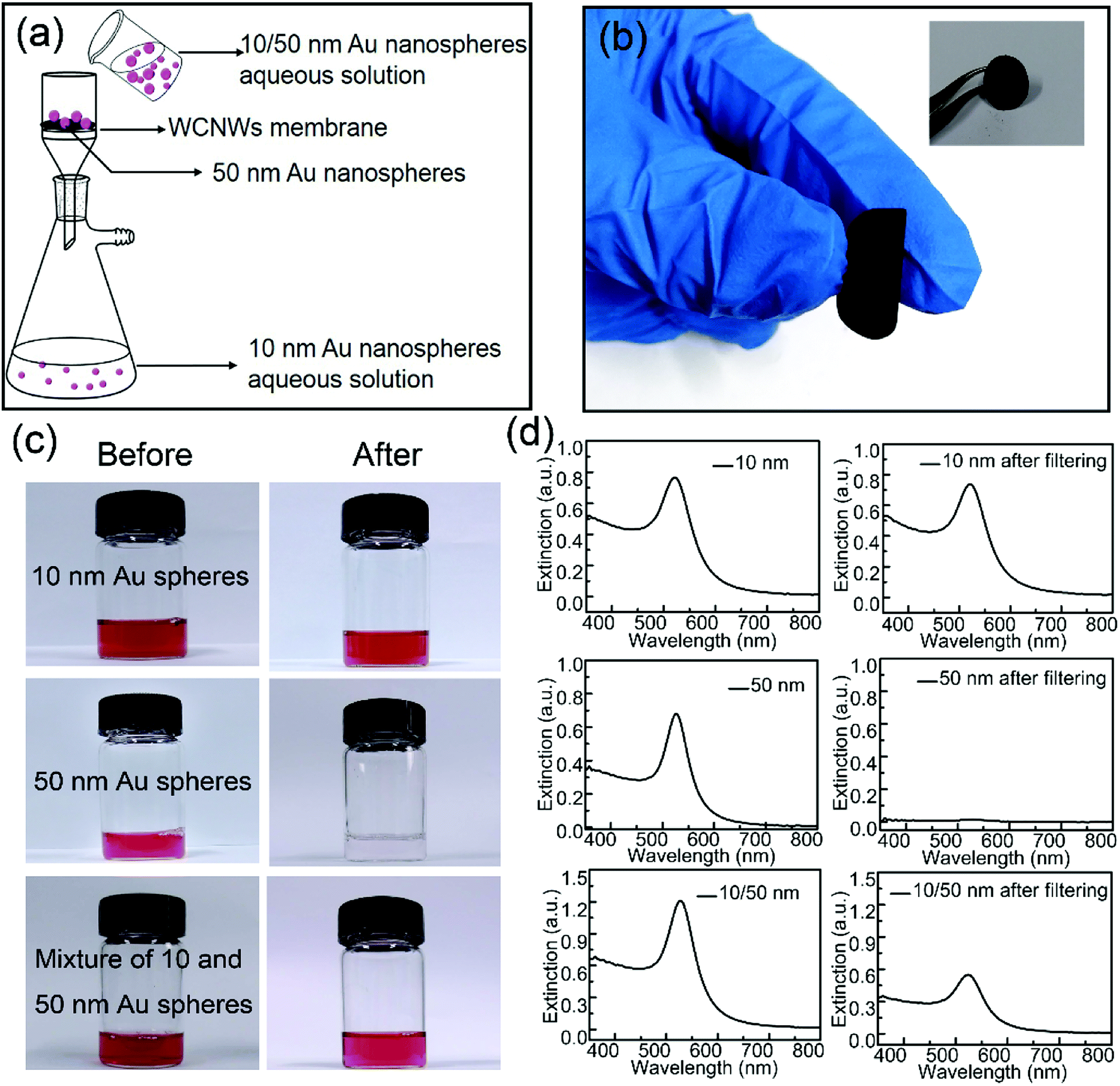 | ||
| Fig. 5 (a) Schematic illustration of the separation of Au nanospheres. (b) Digital photos of WCNWs membrane. (c) Digital photos and (d) corresponding UV/vis extinction spectra of aqueous suspensions containing Au nanospheres with different sizes before and after filtering. | ||
Pd/WCNWs catalyst for ethanol oxidation reaction
The as-prepared WCNWs was also found useful as catalyst support, which is probably due to their rough morphology and amorphous and/or full of defects structure, which could make it easier load Pd nanoparticles. The catalytic performance of Pd/WCNWs catalyst has been systematically investigated by CV experiments in a three-electrode system and the results were compared with that of commercial Pd/C catalyst. As shown in Fig. S9,† Pd nanoparticles with uniform diameter were well-distributed on the surface of WCNWs. The electrochemically active surface area (ECSA) is a primary parameter for the evaluation of active sites of catalysts and can be calculated from the area of the reduction peak of palladium oxide in the CV curves in 1.0 M of KOH.43 Fig. 6a exhibits the typical CV curves of Pd/WCNWs and commercial Pd/C catalysts in 1 M KOH solution at a scan rate of 50 mV s−1. The ECSA (m2 gPd−1) is reckoned in reference to the equation ECSA = Q/(0.405 × WPd), where Q is the coulombic charge by integrating peak area of the reduction of palladium oxide (mC) and WPd means the palladium loading (mg cm−2) on the work electrode, respectively. In addition, 0.405 is the charge required for the reduction of palladium oxide monolayer (mC cmPd−2).44 Therefore, the value of ECSA of Pd/WCNWs catalyst is 60.5 m2 gPd−1, which is nearly 2.67 times higher than that of commercial Pd/C catalyst (22.7 m2 gPd−1), elucidating that WCNWs used as catalysts support could enhance the mass transfer compared to the activated carbon carrier.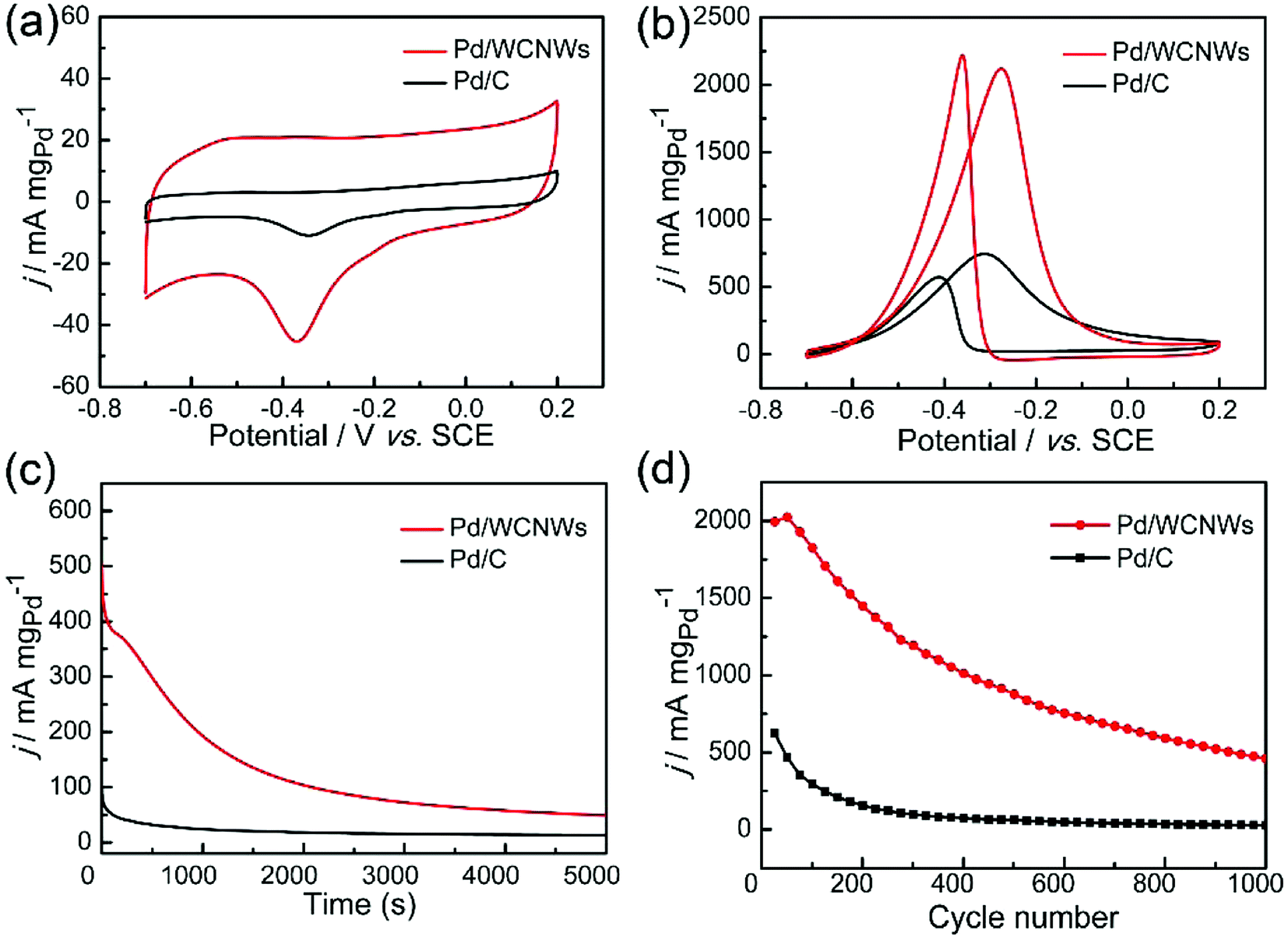 | ||
| Fig. 6 (a) CV curves of Pd/WCNWs and commercial Pd/C catalysts in 1.0 M KOH solution, scan rate: 50 mV s−1. (b) CV curves of Pd/WCNWs and commercial Pd/C catalysts in 1.0 M KOH + 1.0 M C2H5OH solution, scan rate: 50 mV s−1. (c) Chronoamperometric curves of Pd/WCNWs and commercial Pd/C catalysts in 1.0 M KOH + 1.0 M C2H5OH solution at −0.4 V. (d) The peak current density in the forward scan versus cycle number curves of Pd/WCNWs and commercial Pd/C catalysts in 1.0 M KOH + 1.0 M C2H5OH solution. | ||
In addition, catalytic performance of Pd/WCNWs and commercial Pd/C catalysts towards EOR was measured in the solution of 1.0 M KOH and 1.0 M C2H5OH at 50 mV s−1. As shown in Fig. 6b, the characteristic ethanol oxidation peaks of both samples are observed in the forward and backward scans, respectively. Clearly, the peak current density (the value is normalized to the mass of Pd) of Pd/WCNWs catalyst can reach 2126 mA mgpd−1, which is about 3.25 times higher than that of commercial Pd/C catalyst (654.2 mA mgPd−1), indicating that the specific catalytic ability of the Pd/WCNWs catalyst is much higher than that of commercial Pd/C catalyst. Furthermore, the Pd/WCNWs catalyst shows much slower current decay over time than commercial Pd/C catalyst (Fig. 6c). In particular, after 1000 s, the mass peak current density of Pd/WCNWs catalyst still remains of 192.7 mA mgPd−1, which is much higher than that of commercial Pd/C catalyst (24.2 mA mgPd−1). Moreover, the long-term stability of the Pd/WCNWs and commercial Pd/C catalysts, that is, 1000 cycles of CV curves were performed in 1.0 M KOH + 1.0 M C2H5OH solution at a scan rate of 50 mV s−1, was shown in Fig. 6d. After 1000 cycles, the peak current density of Pd/WCNWs catalyst maintains of 459 mA mgPd−1, which is 21.17 times higher than that of commercial Pd/C catalyst (21.68 mA mgPd−1), indicating the excellent stability of Pd/WCNWs catalyst. As result, all the above discussions suggest that Pd/WCNWs catalyst is more active and stable than commercial Pd/C catalyst for EOR, which may be mainly ascribed to the good electrochemical activity of Pd/WCNWs composites and the well-dispersed Pd nanoparticles.
Conclusion
In summary, we have developed a niche and facile method for synthesis of WCNWs derived from PU via KOH activation-enabled carbon reconstruction. The structural transformation and coalescence were observed during the synthetic process. The amount of KOH, pre-activation, and activation temperature have play important roles in the formation of WCNWs. Moreover, the as-prepared WCNWs exhibit outstanding separation ability towards Au nanospheres of 10 and 50 nm. In addition, the as-synthesized WCNWs are also served as carrier for Pd catalyst towards EOR in alkaline medium, and they exhibits high catalytic activities and stabilities. In particular, the forward peak current density reaches 2126 mA mgPd−1 and still keeps a high mass peak current density of 459 mA mgPd−1 after 1000 cycles. Due to their unique shape and structure, the as-synthesized WCNWs would hold widespread applications in the fields of wearable energy devices,45,46 solar cells,47 and water purification.48,49Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors appreciatively acknowledge financial support from the National Natural Science Foundation of China (Grant No. 21475076) and (Grant No. 21701100), and the International S&T collaboration Program of China (No. 2015DFA50060).Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brein, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef.
- D. Ugarte, Nature, 1992, 359, 707–709 CrossRef PubMed.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart and K. Raker, et al., J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef PubMed.
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef PubMed.
- O. A. Williams, Diamond Relat. Mater., 2011, 20, 621–640 CrossRef.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef.
- S. Iijima, M. Yudasaka, R. Yamada, S. Bandow, K. Suenaga and F. Kokai, et al., Chem. Phys. Lett., 1999, 309, 165–170 CrossRef.
- Y. H. Tang, N. Wang, Y. F. Zhang, C. S. Lee, I. Bello and S. T. Lee, Appl. Phys. Lett., 1999, 75, 2921–2923 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef PubMed.
- A. B. Bourlinos, T. A. Steriotis, R. Zboril, V. Georgakilas and A. Stubos, J. Mater. Sci., 2009, 44, 1407–1411 CrossRef.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef PubMed.
- Q. Sun, X.-Q. Zhang, F. Han, W.-C. Li and A.-H. Lu, J. Mater. Chem., 2012, 22, 17049 RSC.
- Ihsanullah, A. Abbas, A. M. Al-Amer, T. Laoui, M. J. Al-Marri, M. S. Nasser, M. Khraisheh and M. A. Atieh, Sep. Purif. Technol., 2016, 157, 141–161 CrossRef.
- O. Erol, I. Uyan, M. Hatip, C. Yilmaz, A. B. Tekinay and M. O. Guler, Nanomedicine, 2017 DOI:10.1016/j.nano.2017.1003.1021.
- N. Arora and N. N. Sharma, Diamond Relat. Mater., 2014, 50, 135–150 CrossRef.
- T. Azami, D. Kasuya, T. Yoshitake, Y. Kubo, M. Yudasaka, T. Ichihashi and S. Iijima, Carbon, 2007, 45, 1364–1369 CrossRef.
- M. Inagaki, Y. Yang and F. Kang, Adv. Mater., 2012, 24, 2547–2566 CrossRef PubMed.
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744–4822 CrossRef PubMed.
- C. Kim, K. S. Yang, M. Kojima, K. Yoshida, Y. J. Kim, Y. A. Kim and M. Endo, Adv. Funct. Mater., 2006, 16, 2393–2397 CrossRef.
- A. M. Cassell, J. A. Raymakers, J. Kong and H. Dai, J. Phys. Chem. B, 1999, 103, 6484–6492 CrossRef.
- Z. J. Han, S. Yick, I. Levchenko, E. Tam, M. M. Yajadda, S. Kumar, P. J. Martin, S. Furmana and K. Ostrikov, Nanoscale, 2011, 3, 3214–3220 RSC.
- N. Pradhan, H. Xu and X. Peng, Nano Lett., 2006, 6, 720–724 CrossRef PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef PubMed.
- S. Wu, G. Chen, N. Y. Kim, K. Ni, W. Zeng, Y. Zhao, Z. Tao, H. Ji, Z. Lee and Y. Zhu, Small, 2016, 12, 2376–2384 CrossRef PubMed.
- Y. Zheng, X. Zhong, Z. Li and Y. Xia, Part. Part. Syst. Charact., 2014, 31, 266–273 CrossRef.
- A. R. Postema, H. De Groot and A. J. Pennings, J. Mater. Sci., 1990, 25, 4216–4222 CrossRef.
- A. D. Roberts, J.-S. M. Lee, S. Y. Wong, X. Li and H. Zhang, J. Mater. Chem. A, 2017, 5, 2811–2820 Search PubMed.
- Y. Zhang, S. Liu, X. Zheng, X. Wang, Y. Xu, H. Tang, F. Kang, Q.-H. Yang and J. Luo, Adv. Funct. Mater., 2017, 27, 1604687 CrossRef.
- V. Wang, D. C. Alsmeyer and R. L. McCreery, Chem. Mater., 1990, 2, 557–563 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef.
- W. H. Lee, J. G. Lee and P. J. Reucroft, Appl. Surf. Sci., 2001, 171, 136–142 CrossRef.
- J. Laine and A. Calafat, Carbon, 1991, 29, 949–953 CrossRef.
- J. Laine, S. Simoni and R. Calles, Chem. Eng. Commun., 1991, 99, 15–23 CrossRef.
- B. Zheng, C. Lu, G. Gu, A. Makarovski, G. Finkelstein and J. Liu, Nano Lett., 2002, 2, 895–898 CrossRef.
- H. W. Liang, L. Wang, P. Y. Chen, H. T. Lin, L. F. Chen, D. He and S.-H. Yu, Adv. Mater., 2010, 22, 4691–4695 CrossRef PubMed.
- Y. Liang, M. G. Schwab, L. Zhi, E. Mugnaioli, U. Kolb, X. Feng and K. Müllen, J. Am. Chem. Soc., 2010, 132, 15030–15037 CrossRef PubMed.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- G. Chen, Y. Wang, L. H. Tan, M. Yang, L. S. Tan, Y. Chen and H. Chen, J. Am. Chem. Soc., 2009, 131, 4218–4219 CrossRef PubMed.
- T. K. Mudalige, H. Qu, G. Sanchez-Pomales, P. N. Sisco and S. W. Linder, Anal. Chem., 2015, 87, 1764–1772 CrossRef PubMed.
- N. Surugau and P. L. Urban, J. Sep. Sci., 2009, 32, 1889–1906 CrossRef PubMed.
- E. R. Essinger-Hileman, E. J. Popczun and R. E. Schaak, Chem. Commun., 2013, 49, 5471–5473 RSC.
- T. R. Gaborski, J. L. Snyder, C. C. Striemer, D. Z. Fang, M. Hoffman, P. M. Fauchet and J. L. McGrath, ACS Nano, 2010, 4, 6973–6981 CrossRef PubMed.
- J. W. Hong, Y. Kim, D. H. Wi, S. Lee, S.-U. Lee, Y. W. Lee, S.-I. Choi and S. WooHan, Angew. Chem., Int. Ed., 2016, 2753–2758 CrossRef PubMed.
- L. Chen, L. Lu, H. Zhu, Y. Chen, Y. Huang, Y. Li and L. Wang, Nat. Commun., 2017, 8, 14136 CrossRef PubMed.
- S. Hong, J. Lee, K. Do, M. Lee, J. H. Kim, S. Lee and D.-H. Kim, Adv. Funct. Mater., 2017, 27, 1704353 CrossRef.
- J. Zhong, Y. Zhang, Q. Zhong, Q. Hu, B. Hu, Z. L. Wang and J. Zhou, ACS Nano, 2014, 8, 6273–6280 CrossRef PubMed.
- V. T. Tiong, N. D. Pham, T. Wang, T. Zhu, X. Zhao, Y. Zhang, Q. Shen, J. Bell, L. Hu, S. Dai and H. Wang, Adv. Funct. Mater., 2018, 1705545 CrossRef.
- Y. Han, Z. Xu and C. Gao, Adv. Funct. Mater., 2013, 23, 3693–3700 CrossRef.
- J. Ge, D. Zong, Q. Jin, J. Yu and B. Ding, Adv. Funct. Mater., 2018, 1705051 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02639d |
| This journal is © The Royal Society of Chemistry 2018 |