
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater-soluble cyclometalated platinum(II) and iridium(III) complexes: synthesis, tuning of the photophysical properties, and in vitro and in vivo phosphorescence lifetime imaging†
Anastasia I. Solomatina a,
Shih-Hao Sub,
Maria M. Lukinac,
Varvara V. Dudenkovac,
Vladislav I. Shcheslavskiyd,
Cheng-Ham Wub,
Pavel S. Chelushkina,
Pi-Tai Chou*b,
Igor O. Koshevoye and
Sergey P. Tunik*a
a,
Shih-Hao Sub,
Maria M. Lukinac,
Varvara V. Dudenkovac,
Vladislav I. Shcheslavskiyd,
Cheng-Ham Wub,
Pavel S. Chelushkina,
Pi-Tai Chou*b,
Igor O. Koshevoye and
Sergey P. Tunik*a
aSt. Petersburg State University, Institute of Chemistry, Universitetskii pr. 26, 198504, St. Petersburg, Russia
bDepartment of Chemistry, National Taiwan University, Taipei, 10617 Taiwan, Republic of China
cInstitute of Biomedical Technologies, Privolzhskiy Research Medical University, Minin and Pozharsky Square, 10/1, Nizhny Novgorod 603005, Russia
dBecker & Hickl GmbH, Nahmitzer Damm 30, Berlin 12277, Germany
eDepartment of Chemistry, University of Eastern Finland, 80101 Joensuu, Finland
First published on 10th May 2018
Abstract
This paper presents synthesis and photophysical investigation of cyclometalated water-soluble Pt(II) and Ir(III) complexes containing auxiliary sulfonated diphosphine (bis(diphenylphosphino)benzene (dppb), P^P*) ligand. The complexes demonstrate considerable variations in excitation (extending up to 450 nm) and emission bands (with maxima ranging from ca. 450 to ca. 650 nm), as well as in the sensitivity of excited state lifetimes to molecular oxygen (from almost negligible to more than 4-fold increase in degassed solution). Moreover, all the complexes possess high two-photon absorption cross sections (400–500 GM for Pt complexes, and 600–700 GM for Ir complexes). Despite their negative net charge, all the complexes demonstrate good uptake by HeLa cells and low cytotoxicity within the concentration and time ranges suitable for two-photon phosphorescence lifetime (PLIM) microscopy. The most promising complex, [(ppy)2Ir(sulfo-dppb)] (Ir1*), upon incubation in HeLa cells demonstrates two-fold lifetime variations under normal and nitrogen atmosphere, correspondingly. Moreover, its in vivo evaluation in athymic nude mice bearing HeLa tumors did not reveal acute toxicity upon both intravenous and topical injections. Finally, Ir1* demonstrated statistically significant difference in lifetimes between normal tissue (muscle) and tumor in macroscopic in vivo PLIM imaging.
Introduction
In the last decade phosphorescent probes based on transition metal complexes attracted considerable interest for application in biosensing and bioimaging because their emissions demonstrate unique characteristics such as long lifetime and strong dependence on the oxygen concentration in the microenvironment, easily tunable emission energy and high photostability.1–10 Two of these features distinguish phosphorescent emitters from commonly used fluorescent probes and pave the way for their advanced application in imaging experiments. The lifetime in microsecond domain allows discrimination of the probe emission from background fluorescence and other short-time luminescent signals using time-resolved techniques. This considerably increases image resolution in luminescence microscopy and lowers detection threshold in sensing experiments.1,4,11,12 As a result, advanced sensing platforms were developed to recognize various biologically relevant analytes, including (but not limited to) detection of cyanide ions,13 biothiols14 (including thiourea15), β-galactosidase,16 dopamine receptor,17 or inhibitor of tumor necrosis factor-α.18 Moreover, the sensitivity of emission parameters (intensity and lifetime) to oxygen concentration makes possible mapping of oxygen concentration in in vitro and in vivo experiments,19,20 including quantitative measurements with the use not only emission intensity but also phosphorescence lifetime imaging (PLIM) techniques,21–25 which provides considerably higher precision in oxygen biodistribution mapping. This turns traditional luminescence imaging into the instrument, which allows for functional analysis of physiological status of the targeted objects. For example, it is well known that growing tumors intensively consume oxygen and hence induce hypoxia in these tissues.26,27 Application of suitable phosphorescent probes for oxygen content analysis gives valuable information, which is helpful for tumor localization and tracking its development and heterogeneity28 and even control of drug effect onto the tumor state.19 Oxygen is one of the major exogenous compounds in aerobic biological systems and any deviation from “normoxia state” indicates crucial changes in physiological status of the cells and organs studied. The oxygenation mapping with phosphorescent probes has found diverse utilization in various medical research such as oncology (see above), determination of local O2 concentration in bone marrow,22 retina,29 and live brain,30,31 tomographic oxygen mapping at the macroscopic level32 as well as monitoring of in situ oxygenation of respiring cells and their responses to metabolic stimulation.33However, for successful application of the probes, in particular, in in vivo experiments, it is highly desirable to use the compounds having specific properties, such as (i) solubility and stability in aqueous media accompanied with internalization into living cells, (ii) emission and excitation shifted as close as possible to the “window of transparency” of biological tissues (650–900 nm),34,35 and (iii) appreciable response of emission parameters to the changes in oxygen concentration. The first group of characteristics determines the presence of the probes in living cells and intrinsically implies their amphiphilic nature. Indeed, on the one hand, the probe has to be water (incubation media)-soluble (i.e., to display hydrophilicity) to reach the target from the blood flow; on the other hand, it also has to be hydrophobic to cross phospholipidic cell membranes. Consequently, the design of suitable molecules has to take into account both features, which is a challenging synthetic task.
In general, organometallic complexes are relatively hydrophobic because of the presence in their coordination sphere the ligands based on developed aromatic systems, which are extremely hydrophobic and normally give complete insolubility of these compounds in aqueous media. Thus, it is necessary to incorporate into ligands environment hydrophilic moieties to make the complex molecule amphiphilic. A few approaches can be employed to attain the aim. Encapsulation of luminescent complexes into a cavity of relatively large water-soluble molecules,36 covalent,37–41 and non-covalent conjugation42–44 with biomolecules substantially increase emitters compatibility with aqueous media to give internalizable luminescent species. In the case of cationic metal complexes a choice of suitable counterions (halides, acetate, etc.)45,46 may be helpful in conversion of initially water insoluble entities into readily soluble organometallic emitters. Attachment of hydrophilic substituents (e.g. amino-, amido–carboxy-, imidazo-groups, monosaccharides and peptides)25,33,43,47 and water soluble polymeric fragments (polyethylene glycol,24,31,48,49 polyvinylpirrolidone,19) to aromatic chromophores and ancillary ligands makes the luminescent probes soluble in physiological media thus extending the area of their application to in vivo experiments.19,21 Another group of ligands, which ensure water-solubility of the corresponding complexes and found a very wide application in homogeneous catalysis,50–52 are sulfonated N-heterocyclic carbenes, polypyridines and phosphines. Based on this type of ligands, a rather large variety of water-soluble luminescent Ru(II),53 Au(I),51 Au(III),54 Pt(II)55 and Ir(III)56 complexes were prepared and characterized. However, these complexes were rarely used in bioimaging55 and their suitability for advanced imaging experiments needs to be further explored.
In this paper we present systematic synthesis of cyclometalated water-soluble Pt(II) and Ir(III) complexes containing auxiliary sulfonated diphosphine (bis(diphenylphosphino)benzene (dppb), P^P*) and investigation of their structure and photophysical characteristics, which display considerable variations in emission gap, lifetime and sensitivity to molecular oxygen. The complexes obtained were applied as phosphorescent probes to study their internalization into cancer cells and to image molecular oxygen distribution in in vitro and in vivo tumor models using PLIM technique with one- and two-photon laser excitation.
Experimental section
General comments
Cyclometalated precursors [(C^N)Pt(DMSO)Cl]57, [(C^N)2IrCl2]58, were prepared according to the published procedures. Other reagents were used as received. The solution 1H, 31P{1H} NMR and 1H–1H COSY spectra were recorded on Bruker Avance 400 and Avance 500 spectrometers with chemical shifts referenced to residual solvent resonances or with respect to 85% H3PO4, δ = 0 ppm. Electrospray ionization (ESI) mass spectra were recorded using a maXis II ESI-QTOF instrument (Bruker, Germany) in the ESI+ and ESI− mode. Microanalyses were carried out at the analytical laboratory of the University of Eastern Finland.Photophysical measurements
The photophysical measurements in solution were carried out using distilled 1,2-dichloroethane, water was purified with a Simplicity Water Purification System, Merck Millipore (type 1 water). The solutions for lifetime and quantum yield measurements in the absence of oxygen were carefully deoxygenated with three freeze–pump cycles or bubbling solution with argon. UV/Vis spectra were recorded with a Shimadzu UV-1800 spectrophotometer at concentrations of ca. 1 × 10−5 M (1 cm cuvettes). Emission and excitation spectra in solution were recorded on a FluoMax-4 (JY Horiba Inc., Japan) spectrofluorimeter using concentration of ca. 4 × 10−5 M. The emission quantum yield in solutions was determined by the comparative method59 using coumarin 102 in distilled ethanol (Φr = 0.764) as the standard60 with the refraction indexes of 1,2-dichloroethane, water, and ethanol equal to 1.44, 1.33, and 1.36, respectively. Lifetimes in solution were determined by the Time-Correlated Single Photon Counting (TCSPC) method. The lifetime data were fitted using the Jobin-Yvon software package and the Origin 9.0 program. Oxygen-dependent phosphorescence lifetime studies were performed using an Edinburgh (FS920) fluorimeter, Edinburgh FL 900 photon-counting system with a hydrogen-filled lamp as the excitation source (Edinburgh Instruments, UK). Oxygen concentration was adjusted using gas dilution system DS-02 (Peak technology, Taiwan) equipped with mass flow controller EL-FLOW Base (Bronkhorst, Netherlands, 1500 sccm) with nitrogen as the dilution gas. The gas system was connected to thermostatic cuvette holder GS21530 (Specac, UK) with temperature controller Eurotherm 2216e (Specac, UK).Before phosphorescence decay measurements each sample was bubbled with pre-mixed gas at a defined oxygen concentration for 30 minutes, the temperature of the holder was fixed at 37 °C. The emission decays were fitted using Edinburg Instruments software by nonlinear least square fitting method with the deconvolution of instrument response function.
Complex Pt1
[ppyPt(DMSO)Cl] (25 mg, 0.054 mmol) and 1,2-bis(diphenylphosphino)benzene (dppb, 24 mg, 0.054 mmol) were dissolved in degassed CH2Cl2 (5 mL) (Scheme 1). Then silver trifluoromethanesulfonate (AgOTf, 14 mg, 0.055 mmol) was added. The reaction mixture was stirred for 1 h under a nitrogen atmosphere, and then it was evaporated to dryness. Recrystallization by slow evaporation of a dichloromethane/hexane solution of Pt1 at +4 °C gave pale green crystalline material (54 mg, 98%). 1H NMR (400 MHz, CD3OD, 298 K): δ 8.42 (m with broad 195Pt satellites, 1H1), 8.17 (d, 3JH–H = 8.2 Hz, 1H4), 8.06 (t, 3JH–H = 7.8 Hz, 1H3), 7.89–7.48 (m, 25H5,9–13), 7.24–7.15 (m, 2H6,8), 7.02 (t, J = 6.6 Hz, 1H2), 6.84 (t, 3JH–H = 7.5 Hz, 1H7) ppm. 31P NMR (162 MHz, CD3OD, 298 K): δ 48.44 (d, 1JP–Pt = 1814 Hz, 1P), 36.69 (d, 1JP–Pt = 3760 Hz, 1P) ppm. ES MS (m/z): [M − CF3SO3]+ 795.174 (calc. 795.166). Anal. calc. for C48H37F3NO3P2PtS·CH2Cl2 (%): C 53.17; H 3.55; N 1.27; S 2.90. Found: C 53.32; H 3.40; N 1.48; S 3.24.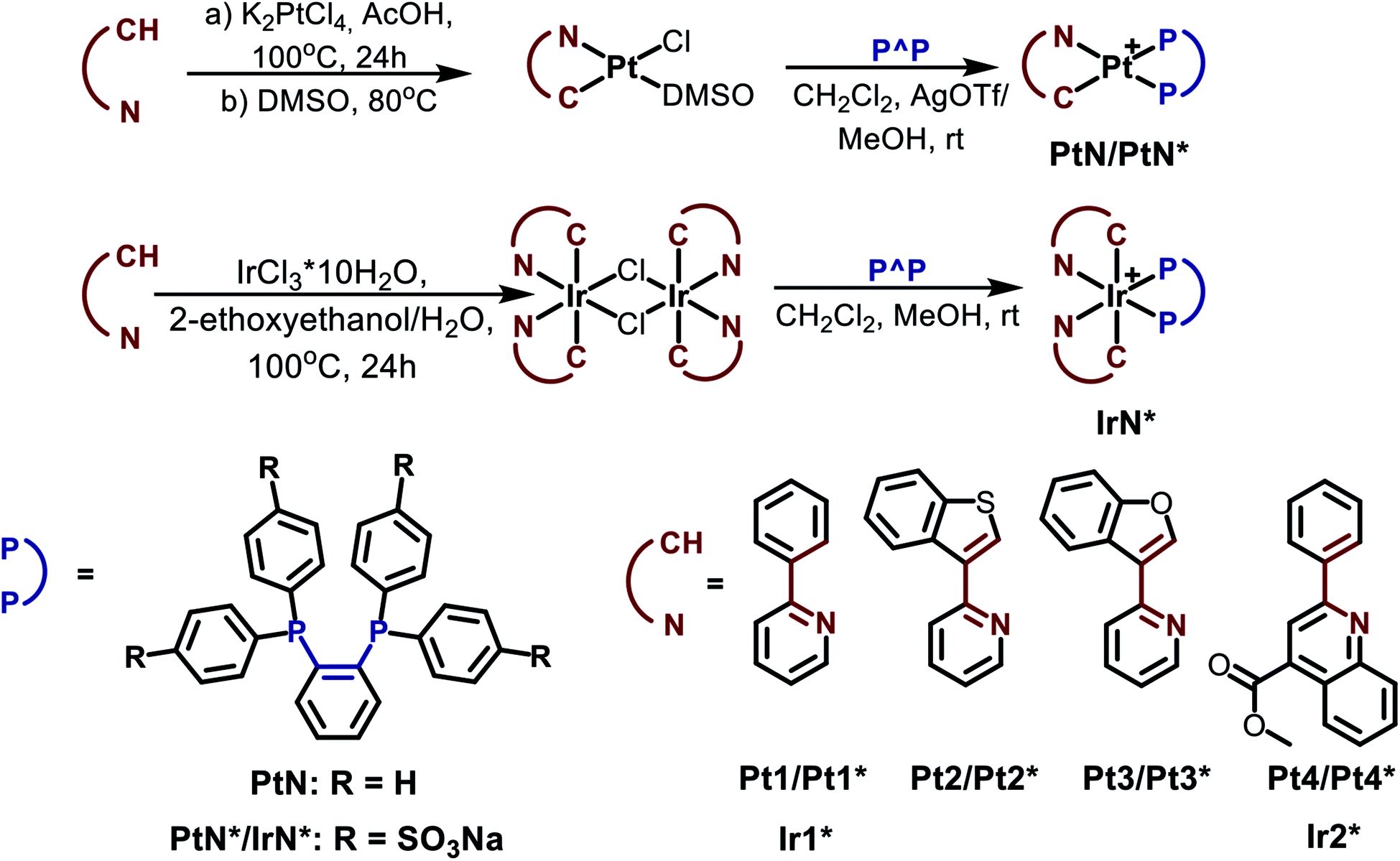 | ||
| Scheme 1 Preparation schemes and chemical structures of the Pt and Ir complexes used in the study. Water-soluble complexes are denoted by asterisks. | ||
Complex Pt2
Prepared analogously to Pt1 using [btpyPt(DMSO)Cl] as starting material (orange crystalline solid, 92%). 1H NMR (400 MHz, CD3OD, 298 K): δ 8.35–8.31 (m, 2H1,4), 8.28 (d, 3JH–H = 8.0 Hz, 1H5), 8.11 (t, 3JH–H = 8.2 Hz, 1H3), 7.86–7.74 (m, 11H), 7.66–7.60 (m, 6H), 7.59–7.51 (m, 8H), 7.45 (t, 3JH–H = 7.7 Hz, 1H6), 7.28 (t, 3JH–H = 7.6 Hz, 1H7), 6.95 (t, 3JH–H = 6.6 Hz, 1H2) ppm. 31P NMR (162 MHz, CD3OD, 298 K): δ 48.33 (d, 1JP–Pt = 2163 Hz, 1P), 25.09 (d, 1JP–Pt = 3462 Hz, 1P) ppm. ES MS (m/z): [M − CF3SO3]+ 851.144 (calc. 851.138). Anal. calc. for C44H32F3NO3P2PtS2·CH2Cl2 (%): C 49.78; H 3.16; N 1.29; S 5.91. Found: C 49.77; H 3.32; N 1.29; S 5.77.Complex Pt3
Prepared analogously to Pt1 using [bfpyPtb(DMSO)Cl] as starting material (yellow crystalline solid, 90%). 1H NMR (400 MHz, CD3OD, 298 K): δ 8.19 (m with broad 195Pt satellites, 3JPt–H = 28 Hz, 1H1), 8.06 (td, 3JH–H = 7.8, 1.3 Hz, 1H3), 7.99 (d, 3JH–H = 7.8 Hz, 1H4), 7.93–7.89 (m, 1Hpp), 7.86–7.74 (m, 12Hpp,5), 7.69–7.58 (m, 8Hpp), 7.54–7.50 (m, 4Hpp), 7.29 (t, 3JH–H = 7.5 Hz, 1H6), 7.20 (t, 3JH–H = 7.7 Hz, 1H7), 7.11 (d, 3JH–H = 8.2 Hz, 1H8), 6.96 (m, 1H2) ppm. 31P NMR (162 MHz, CD3OD, 298 K): δ 48.64 (d, 1JP–Pt = 2161 Hz, 1P), 34.02 (d, 1JP–Pt = 3498 Hz, 1P) ppm. ES MS (m/z): [M − CF3SO3]+ 835.168 (calc. 835.161). Anal. calc. for C44H32F3NO4P2PtS (%): C 53.66; H 3.28; N 1.42; S 3.26. Found: C 53.39; H 3.36; N 1.38; S 3.26.Complex Pt4
Prepared analogously to Pt1 using [qpyPt (DMSO)Cl] as starting material (red crystalline solid, 95%). 1H NMR (400 MHz, CD3OD, 298 K): δ 8.44 (s, 1H5), 8.36 (d, 3JH–H = 8.5 Hz, 1H9), 8.29 (d, 3JH–H = 8.5 Hz, 1H6), 7.84 (d, 3JH–H = 7.9 Hz, 1H4), 7.76–7.71 (m, 4Hpp), 7.67 (m, 3JH–H = 1.9, 7.4 Hz, 2Hpp), 7.61–7.54 (m, 7Hpp), 7.50–7.38 (m, 8Hpp,8), 7.25 (m, 3JH–H = 13.1, 6.6 Hz, 4Hpp), 7.21–7.11 (m, 2H1,3), 6.93 (t, 3JH–H = 7.7, 1.2 Hz, 1H7), 6.78 (t, 3JH–H = 7.5 Hz, 1H2), 4.11 (s, 3H6) ppm. 31P NMR (162 MHz, CD3OD, 298 K): δ 42.96 (d, 1JP–Pt = 1784 Hz), 35.95 (d, 1JP–Pt = 3872 Hz) ppm. ES MS (m/z): [M − CF3SO3]+ 903.194 (calc. 903.188). Anal. calc. for C48H36F3NO5P2PtS (%): C 54.76; H 3.45; N 1.33; S 3.05. Found: C 54.67; H 3.39; N 1.31; S 3.31.Complex Ir1*
Complex was obtained as compound Pt1*. Yield 85%, pale green amorphous solid. 1H NMR (400 MHz, CD3OD, 298 K): δ 8.21 (m, 1H9), 8.09–8.03 (m, 1H10), 7.87 (m, 2H11), 7.73 (m, 2H12), 7.66 (d, 3JH–H = 7.7 Hz, 1H4), 7.61 (d, 3JH–H = 7.9 Hz, 1H5), 7.56 (t, 3JH–H = 7.5 Hz, 1H3), 7.33 (d, 3JH–H = 5.8 Hz, 1H1), 7.20 (m, 2H11), 7.03 (t, 3JH–H = 7.4 Hz, 1H6), 6.92 (t, 3JH–H = 7.4 Hz, 1H7), 6.45–6.39 (m, 3H12,2), 6.16 (dd, 3JH–H = 7.4, 3.2 Hz, 1H8) ppm. 31P NMR (162 MHz, CD3OD, 298 K): δ 21.76 (s) ppm. ES MS (m/z): [M − 3Na]3− 421.026 (calc. 421.009), [M − 3Na + H]2− 632.042 (calc. 632.017), [M − 2Na]2− 643.035 (calc. 643.008), [2M − 3Na]3− 865.041 (calc. 865.007).Complex Ir2*
Complex was obtained analogous to compound Pt2*. The synthesis procedures of Pt1*–Pt4* complexes are given in ESI.† Yield 65%, orange amorphous solid. 1H NMR (400 MHz, CD3OD, 298 K): δ 8.58 (m, 1H10), 8.42 (s, 1H5), 8.36 (dd, 3JH–H = 8.5, 4JH–H = 1.2 Hz, 1H4), 8.18 (m, 1H11), 7.99 (d, 3JH–H = 8.1 Hz, 1H6), 7.60 (m, 4H12,13), 7.53 (dd, 3JH–H = 8.8, 4JH–H = 3.5 Hz, 1H1), 7.27 (t, 3JH–H = 8.1 Hz, 1H3), 7.17 (t, 3JH–H = 7.5 Hz, 1H7), 6.88 (t, 3JH–H = 7.5 Hz, 1H8), 6.76 (m, 2H12), 6.41 (t, 3JH–H = 8.5 Hz, 1H2), 6.32 (m, 2H13), 5.88 (dd, 3JH–H = 7.7, 4JH–H = 3.5 Hz, 1H9), 4.04 (s, 3H14) ppm. 31P NMR (162 MHz, CD3OD, 298 K): δ 13.53 (s) ppm. ES MS (m/z): [M − 3Na]3− 493.023 (calc. 493.023), [M − 2Na]2− 751.031 (calc. 751.029), [2M − 3Na]3− 1009.036 (calc. 1009.036).X-ray structure determination
The crystals of Pt1, Pt2 and Pt4 were immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 150 K. The X-ray diffraction data were collected with Bruker SMART APEX II and Bruker Kappa Apex II Duo diffractometers using Mo Kα radiation (λ = 0.71073 Å). The APEX2 program package61 was used for cell refinements and data reductions. The structures were solved by direct methods using the SHELXS-2014 (ref. 62) program with the WinGX63 graphical user interface. A semiempirical absorption correction (SADABS)64 was applied to all data. Structural refinements were carried out using SHELXL-2014.62 One phenyl ring of the diphosphine ligand in Pt1 and triflate counterion were disordered between two orientations each and were refined with occupancies of 0.63/0.37 and 0.69/031, respectively; geometry and displacement restraints and constraints were applied to these moieties. The crystallization solvent in Pt2 was partially lost from the crystal and could not be resolved unambiguously. The contribution of the missing solvent to the calculated structure factors was taken into account by using a SQUEEZE routine of PLATON.65 The missing solvent was not taken into account in the unit cell content. The structure of 6 was refined as a 2-component inversion twin. All H atoms in 1–6 and 8–11 were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95–0.99 Å and Uiso = 1.2–1.5Ueq (parent atom). The crystallographic details are summarized in Table S1.†Two-photon image experiments in vitro
For cell imaging experiments, about 5 × 104 HeLa cells were seeded in a 35 mm glass-bottom dish well with 2 mL of DMEM included. Incubation condition is at 37 °C and CO2 to air ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95. After 24 h of incubation, each dish was washed three times with PBS buffer solution and then introduced with 50 μM of iridium complex (for Ir1* and Ir2*) or 75 μM of platinum complex (for Pt2*, Pt3*, Pt4*). After this 18 hours incubation of metal complex, HeLa cells were washed three times with PBS buffer solution and then added phenol red-free culture medium in each dish. Finally, HeLa cells were studied by a Zeiss LSM710 confocal laser-scanning microscope with 63× objective for observation (P-APO, 1.40 oil immersion). The 80 MHz, 100 fs mode-locked Ti:sapphire laser (Mai-Tai DeepSee, Spectra-Physics) was used as a two-photon excitation light source. The temperature was maintained at 37 °C during the experiments.
:95. After 24 h of incubation, each dish was washed three times with PBS buffer solution and then introduced with 50 μM of iridium complex (for Ir1* and Ir2*) or 75 μM of platinum complex (for Pt2*, Pt3*, Pt4*). After this 18 hours incubation of metal complex, HeLa cells were washed three times with PBS buffer solution and then added phenol red-free culture medium in each dish. Finally, HeLa cells were studied by a Zeiss LSM710 confocal laser-scanning microscope with 63× objective for observation (P-APO, 1.40 oil immersion). The 80 MHz, 100 fs mode-locked Ti:sapphire laser (Mai-Tai DeepSee, Spectra-Physics) was used as a two-photon excitation light source. The temperature was maintained at 37 °C during the experiments.
Phosphorescence lifetime imaging (PLIM) in vitro
Zeiss LSM 710 (Zeiss, Germany) equipped with 80 MHz fs mode-locked Ti:sapphire laser (Mai-Tai DeepSee, Spectra-Physics) was used for the experiments. All data were collected using Plan-Apochromat 63×/1.4 oil immersion objective lens (Zeiss, Germany). To make time-resolved luminescence imaging we used time-correlated single-photon counting board SPC-150, 16-channel spectral detector PML-16-1-C and programmable pulse generator DDG-210 (Becker&Hickl GmbH, Germany). The DDG-210 is triggered by the pixel clock of the LSM 710 and delivers a laser modulation signal of programmable width, which is fed back into the beam blanking system of the microscope. The modulation of the laser signal on the pixel dwell time allows to record fluorescence and phosphorescence lifetime images simultaneously.66 Basically, when the laser is on the fluorescence signal is recorded and phosphorescence emission is built up, and when it is switched off, only phosphorescence signal is collected. The detailed operation of the FLIM/PLIM with Zeiss LSM systems can be found elsewhere.67 The TCSPC data were analyzed by the SPCImage 6.0 software (Becker & Hickl GmbH, Germany).In vitro cytotoxicity – MTT assay
For determining the cytotoxicity of metal complexes to HeLa cell, MTT assay was conducted by using a common colorimetric assay agent, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Roche, Germany). At first, HeLa cells were seeded in a 96-well incubation plate with 5 × 103 cells per well in a 90% Dulbecco's Modified Eagle Medium (HyClone, GE Healthcare, USA) along with 10% heat-inactivated fetal bovine serum (HyClone, GE Healthcare, USA). To compare the cytotoxicity effect of different metal complex concentration, six different concentrations were prepared for each plate (0–70 μM for Ir1*, Ir2*; 0–150 μM for Pt2*, Pt3*, and Pt4*). After 24 hours of incubation, every well was washed twice with PBS buffer solution, followed by addition of 200 μL of the culture medium containing 10% MTT agent. After 3 hours of reaction time, the culture medium was removed and 200 μL of dimethyl-sulfoxide (Merck, USA) was added to dissolve the deposited purple formazan crystal in the cell. The absorbance of the complexes was recorded at 595 nm. All experimental results (average and standard deviation) were calculated from three replicates collected by an ELISA reader system (VersaMax™ Microplate Spectrophotometers; Molecular-Devices, USA).Photostability experiments
Photostability study of the metal complexes was compared with commonly used chromophores (coumarin 480 and rhodamine B), which possess similar absorption and emission bands. Samples were dissolved in PBS (10 mM, pH = 7.4). Absorbances are all fixed to 0.05 at 360 nm (For Ir1* and C480, λexc = 720 nm) or at 430 nm (For Ir2*, Pt2*, Pt3*, Pt4* and rhodamine B, λexc = 860 nm). The excitation power was 32 mW at the objective lens. Luminescence intensity at the emission maxima was recorded with continuous exposure to excitation with 50 scan times with time interval of 2.0 seconds. Data was collected by Zeiss LSM710 microscope equipped with 63× objective (P-APO, NA = 1.40, oil immersion).Tumor model
Experiments were performed on female athymic nude mice purchased from the Pushchino animal nursery (Pushchino, Russia). To generate tumors, mice of 20–22 g body weight were injected subcutaneously in the left flank with HeLa cells (2 × 106 in 200 μL PBS). Experiments were carried out in 14 days after the cell injection, when the tumors reached ∼10–12 mm in size. For imaging procedures the mice were anesthetized intramuscularly with a mixture of Zoletil (40 mg kg−1, 50 μL, Virbac SA, Carros, France) and 2% Rometar (10 mg kg−1, 10 μL, Spofa, Czech Republic), and a skin flap over the tumor was surgically opened. After image acquisition, the animals were sacrificed by cervical dislocation and the tumors were excised for histopathology. For animal study, Ir1* was dissolved in distilled water in a concentration 30 mg mL−1 and injected topically, directly into the tumor or muscle 20–30 min prior to the study (50 μL of solution in 4–6 injections, total dose 75 mg kg−1 body weight). Tumor-bearing mice that did not receive the injection of Ir1* served as control.Two-photon microscopy in vivo
An LSM 880 (Carl Zeiss, Germany) fluorescence confocal laser-scanning microscope equipped with a femtosecond Ti:Sapphire laser with a repetition rate of 80 MHz and pulse duration of 140 fs was used to obtain two-photon fluorescence images. The images were acquired through a water immersion objective C-Apochromat 40×/1.2 NA W Korr. For in vivo image acquisition, an anesthetized animal was placed on the glass cover slip, so that the objective was focused on the region of interest (tumor or muscle surface). Ir1* was excited at a wavelength of 750 nm, emission was detected in the range 430–600 nm. For spectral imaging, emitted light was collected in the spectral range of 400–662 nm under lambda mode.PLIM in vivo
A confocal scanning system for macroscopic objects DCS-120 MACRO (Becker & Hickl GmbH, Germany) equipped with a TCSPC-based module was used for one-photon PLIM of tumors in vivo. Ir1* was excited with a picosecond diode laser at 405 nm (BDL-SMN-405, Becker&Hickl GmbH, Germany). Long pass (HQ435LP, Chroma, USA) and dichroic 560 nm (Chroma, USA) filters have been used to suppress the emission from the laser. The PLIM data were processed using SPCImage software (Becker & Hickl GmbH, Germany). Phosphorescence lifetime analysis was performed for 5 tumor nodules and 3 areas of muscles in 2 mice by selecting specific regions of interest. Data were represented as mean ± SD.This study was performed in strict accordance with the Russian national guidelines for the use of laboratory animals (the USSR Ministry of Higher and Middle Professional Education, Decreet No. 742, dated 13.11.1984) and was approved by the Ethical Committee of the Privolzhsky Research Medical University (Nizhny Novgorod, Russia).
Results and discussion
Synthesis and photophysical study of Pt(II) and Ir(III) complexes
Cyclometalated platinum(II) and iridium (III) complexes containing hydrophobic and water soluble diphosphines were obtained in good yield using slightly modified conventional procedures (Scheme 1). The complexes based on sulfonated 1,2-bis(diphenylphosphino)benzene (dppb) display excellent solubility and stability in aqueous media, good cellular uptake and very low cytotoxicity that provide a background for their application in imaging experiments (vide infra). For details of synthetic procedures and structural characterization (XRD, mass-spectrometry, NMR spectroscopy) of the platinum and iridium compounds see ESI, Section 1.†All complexes obtained excluding platinum phenylpyridine derivatives Pt1 and Pt1* are luminescent in organic solvents (Pt2–Pt4) and in aqueous solution (Pt2*–Pt4* and Ir1*, Ir2*). Their photophysical characteristics are summarized in Table 1; excitation and emission spectra are shown in Fig. 1. Absorption spectra and additional discussion of the spectroscopic properties are given in ESI file (Section 2).†
| No. | Abs, nm (ε × 10−3, M−1 cm−1) | λex, nm | λem, nm | τobs (aer/deg), μs | Φ (aer/deg), % |
|---|---|---|---|---|---|
| a In aqueous solution.b In 1,2-dichloroethane.c Observed emission lifetime are calculated from the data on double-exponential decay – τobs = (A1τ12 + A2τ22)/(A1τ1 + A2τ2). | |||||
| Pt1*a | 232 (80), 280 sh (21), 315 (6.5), 325 sh (6.0), 350 sh (2.5) | Non luminescent in solution | |||
| Pt2*a | 237 (85), 270 sh (36), 280 sh (29), 330 sh (6.6), 415 (3.1) | 330, 415 | 525 sh, 577 | 0.53/0.91c | 0.64/2.61 |
| Pt3*a | 232 (68), 280 sh (21), 305 sh (12), 415 (2.4) | 415 | 530 sh, 567 | 0.37/0.71c | 0.57/0.78 |
| Pt4*a | 225 sh (78), 245 sh (72), 275 sh (31), 358 (8.3), 410 sh (2.6) | 360, 410 sh | 620 sh, 648 | 0.18/0.38c | 0.11/0.22 |
| Ir1*a | 230 (85), 280 sh (22), 300 sh (13), 353 (5.2) | 308, 355 | 455, 485, 510 sh, 550 sh, 570 sh | 1.03/4.30 | 1.91/7.77 |
| Ir2*a | 255 sh (86), 284 (31), 361 (14), 420 (4.9) | 360, 420 sh | 620 | 0.38/0.43 | 1.63/2.12 |
| Pt1b | 265 sh (34), 320 (7.5), 332 (7.5), 358 (3.2), 375 sh (2.2) | Non luminescent in solution | |||
| Pt2b | 269 (33), 305 sh (12), 337 (7.1), 426 (3.5) | 270, 300 sh, 340, 426 | 510, 565 sh, 610 sh | 0.20/0.35 | 0.64/0.91 |
| Pt3b | 264 (35), 275 sh (25), 285 sh (20), 305 sh (13), 425 sh (2.7) | 285, 307 sh, 425 | 557, 600 sh | 0.10/0.14 | 0.39/0.48 |
| Pt4b | 270 sh (29), 278 (28), 285 (28), 363 (10), 420 sh (3.2) | 363, 420 sh | 620 sh, 655 | 0.09/0.10 | 0.05/0.08 |
 | ||
| Fig. 1 Normalized excitation (dashed) and emission (solid) spectra: (a) Pt2, Pt3 and Pt4 in 1,2-dichloroethane (λex = 420 nm for Pt2, 425 nm for Pt3, 440 nm for Pt4), (b) Pt2*, Pt3* and Pt4* in aqueous solution (λex = 415 nm for Pt2* and Pt3*, 425 nm for Pt4*), (c) Ir1* and Ir2* in aqueous solution, (λex = 355 and 425 nm for Ir1* and Ir2*, respectively). | ||
It can be easily seen that emission profiles of the hydrophobic and water-soluble platinum congeners (PtN–PtN*) are essentially similar, which is indicative of negligible effect of the phosphine ligands onto emission characteristics. The platinum complexes display a rather weak luminescence in fluid media with the broaden emission bands containing ill-defined vibronic structure that is in agreement with the predominant MLCT nature of emissive excited state associated mainly with metalating ligands. Large stokes shifts and lifetimes in the microsecond domain clearly show that emission occurs from the lowest triplet manifold, i.e. phosphorescence. Expansion of aromatic system in the C^N ligand and introduction of electron accepting substituent into quinoline fragment in the Pt4/Pt4* complexes results in a considerable bathochromic shift of the emission maxima in accord with the well-known trend revealed for metalated platinum complexes.68 It has to be noted that the effect of emission quenching with molecular oxygen is very minor for these platinum complexes, which makes difficult their application as oxygen sensors, with the only exclusion of Pt2* where quantum yields in degassed aqueous solution is four times higher than that in the aerated solution.
The photophysics of the [Ir(C^N)2(X^X)]1+/0 iridium complexes (X = N, P, O) have been extensively studied during the last decade69 because of attractive luminescent characteristics of these molecular emitters, in particular, for application in bioimaging.2,10 It has to be mentioned that a complete analogue of Ir1* with non-sulfonated diphosphine (Ir1) was earlier prepared and studied experimentally and theoretically.70,71 Similar to the platinum relatives, the peak wavelengths and profiles of the absorption and emission bands for Ir1* and its Ir1 analogue70,71 are nearly identical. The result points to similarity of their electronic structure and likeness of the nature of emissive transitions. Consequently, the emission of the Ir1 and Ir1* compounds may be assigned to a combination of metal perturbed IL and MLCT excited states with substantial contribution of the former that is also indicated by vibronically structured emission band. Ir2* containing substituted quinoline fragment in the metalating ligand displays a substantial red shift of emission and transformation of the emission profile into structureless band that points to domination of MLCT process in generation of the emissive excited state in this complex. Sensitivity of emission parameters to molecular oxygen for Ir1* and Ir2* is rather different; the higher degree of emission quenching with O2 has been found for Ir1*, which may be related to lower shielding of the chromophoric center in the phenyl pyridine complex and provides a background for the complex application in phosphorescence lifetime imaging (PLIM) experiments aimed at oxygen mapping in biological samples (vide infra).
To further evaluate applicability of the water-soluble iridium complexes for the bioimaging applications, we measured their two-photon absorption (TPA) cross sections (σ2) in water via a Z-scan method (see Experimental section in ESI, and Fig. S36†). Using a femtosecond laser (λem 800 nm, 180 fs, 1 kHz), σ2 values of Ir1* and Ir2* in water were measured to be 627 ± 9 GM and 655 ± 46 GM, correspondingly (1 GM = 10−50 cm4 s per photon).
These values are comparable to the highest TPA cross section values among the iridium complexes reported in the literature.72–75 As for the titled platinum complexes, the same excitation wavelength (800 nm) is applied at Pt2*, Pt3* and Pt4* in water, and their TPA cross sections were measured to be 416 ± 29 GM, 400 ± 50 GM, and 496 ± 44 GM, respectively, which are also comparable with that of potential Pt complexes for two-photon imaging.55,76–80
Analyses of the whole set of the photophysical data including measurements of TPA cross-sections show that Ir1* is the most suitable species for oxygen concentration mapping using PLIM technique since it shows the largest dynamic range of lifetimes (more than 4-fold increase in degassed state) and the highest Φdeg and σ2 values. To evaluate applicability of this complex for PLIM measurements we carried out a series of test experiments in solution, in vitro and in vivo: quantification of the lifetime characteristics on the oxygen concentration in solution, cytotoxicity assay and two photon imaging in PLIM mode on HeLa cells, as well as in vivo PLIM of nude mice bearing HeLa tumors.
Phosphorescence quenching
We performed exploration of Ir1* response to variations in oxygen concentration in attempts to monitor the cell hypoxia effect due to its potential correlation with tumor proliferation.26 In some cases, for example, the intracellular oxygen concentration in solid tumors can drop down to almost ∼0%.26,27,81There have been a number of oxygen-sensitive luminescent materials designed for monitoring intracellular oxygen level and tumor hypoxia,11,21,82–85 among which the phosphors based on transition-metal complexes have attracted considerable attention for their promising applications in imaging. The dynamic quenching of phosphorescence due to collisional energy transfer to the triplet ground state O2 molecules results in the radiationless deactivation of the phosphor lowest triplet state and lets phosphorescent emitters become suitable O2 sensors. Practically, O2 sensors based on this phenomenon increase their emission intensity and lifetime as the oxygen concentration decreases thus making detection of tumor hypoxia possible.
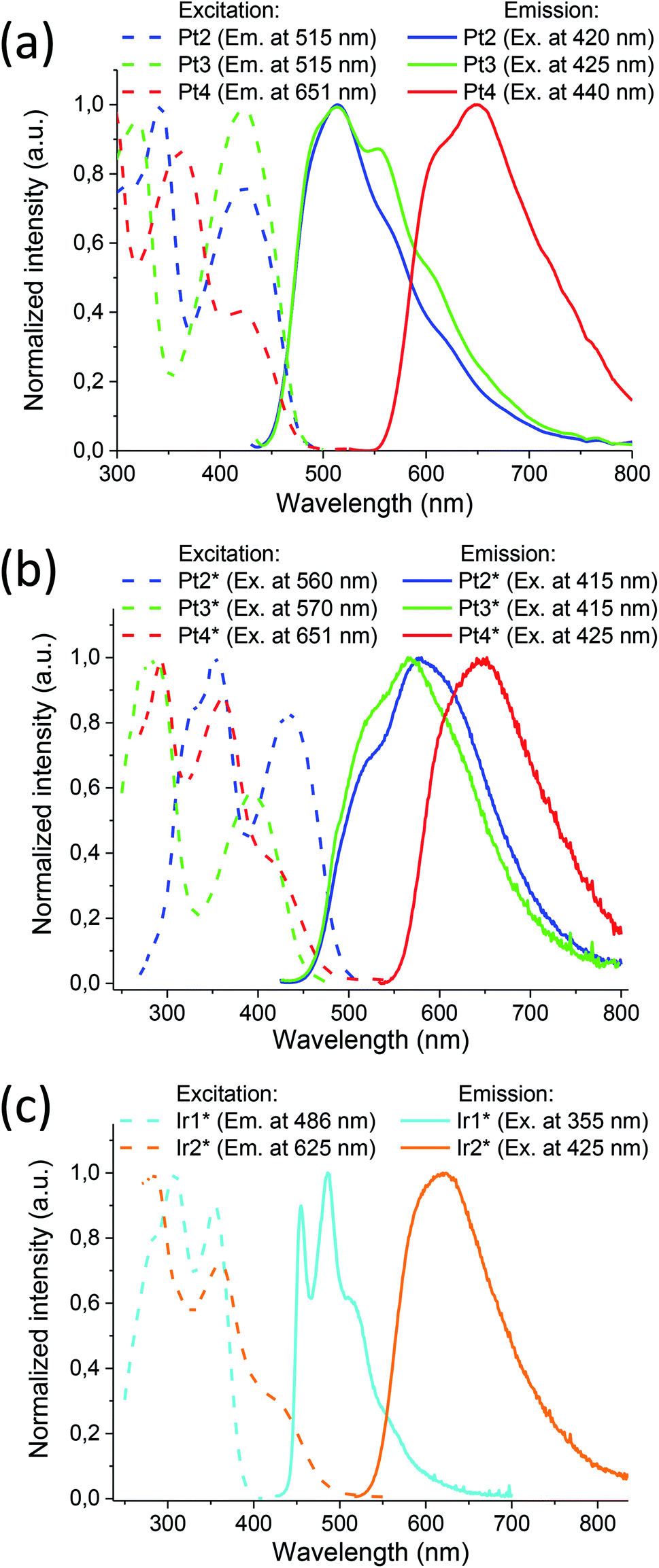
The ratio of phosphorescence lifetime is related to the concentration of existing quencher, i.e., oxygen, according to the well-known Stern–Volmer eqn (1),
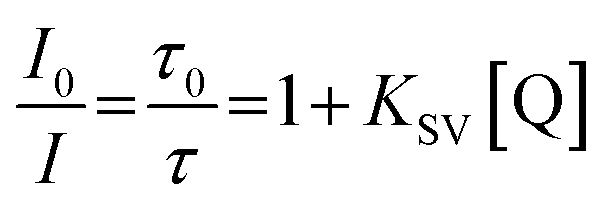 | (1) |
 | ||
| Fig. 2 (a) Phosphorescence decay curves (corresponding lifetimes are listed in the legend) of Ir1* in water under different oxygen pressures at 37 °C. (b) The resulting Stern–Volmer plot of Ir1* quenching by oxygen. | ||
Cellular uptake and cytotoxicity of Ir1* on HeLa cells
We applied laser-scanning microscopy (LSM) combined with two-photon excitation technique to study cellular uptake of water-soluble complexes developed in the present study. The two-photon excitation microscopy offers larger penetration depth and lower photodamage for intact tissue image in comparison with one-photon excitation.31,86 The LSM experiments indicated successful uptake of Ir1* by HeLa cells (18 hours incubation), and its primarily location within the cytoplasm (Fig. S37†). Note that similar behavior was observed for other water-soluble complexes (Fig. S38†) indicating that negatively charged complexes bearing sulfo groups can penetrate cells despite of electrostatic repulsion at cell membrane. This behavior can be attributed to amphiphilic nature of these complexes bearing not only hydrophilic phopshine ligands but also hydrophobic groups facilitating crossing cell membranes.Since cell-permeable transition metal complexes can potentially be highly cytotoxic, we have also performed MTT assay test to evaluate their cytotoxicity. The results (Fig. S39†) show that, upon incubation of HeLa cells with Ir1* and Ir2* up to 70 μM for 24 hours, more than 95% of HeLa cells has survived regardless of the complexes concentrations, affirming low cytotoxicity for both Ir1* and Ir2*. Analogously, the Pt complexes revealed no cytotoxic effect up to 70 μM for 24 hours, and rather high IC50 values ranging from 110 to 140 μM (Fig. S40†).
Two-photon in vitro PLIM on HeLa cells
For O2 sensing, the detection modalities can be categorized by emission intensity and lifetime measurements. The emission intensity assay can be attained either by monitoring at a single wavelength of O2-sensitive dye or by the ratiometric intensity referred to a O2-insensitive moiety.7 Unfortunately, this modality commonly encounters interferences from autofluorescence and dye delivery dynamics to the cell, which makes it difficult to quantify oxygen content.11 Compared with nanosecond-scale autofluorescence, fortunately, the phosphorescence originating from transition metal complexes possesses much longer (microsecond) lifetimes, so that the time-gated method is a superior technique benefitting from the above difference in the time scale.11 Accordingly, the phosphorescence lifetime imaging (PLIM) becomes a powerful tool in investigating lifetime distribution of phosphorescent materials in cell or tissue and hence in tracking of oxygen concentrations.12 Fig. 3 shows two-photon absorption PLIM of HeLa cells incubated with 50 μM Ir1* under standard oxygenated environment (Fig. 3a–c) and under N2 saturated atmosphere (Fig. 3d–f). In these images (panels b and e) the phosphorescence lifetime is symbolized by colors from blue (short decay) to red (long decay). The data obtained (Fig. 3c and f) indicate that the lifetime distribution of Ir1* in HeLa cells under the N2 saturated atmosphere is significantly longer than that in the standard atmosphere. The amplitude weighted mean lifetime was calculated to be 1.05 and 2.17 μs for standard and N2 saturated atmosphere, respectively (with relative uncertainty in lifetime calculations not exceeding 5%), affirming the potential of Ir1* for detecting intracellular oxygen level and hence monitoring hypoxia in vitro.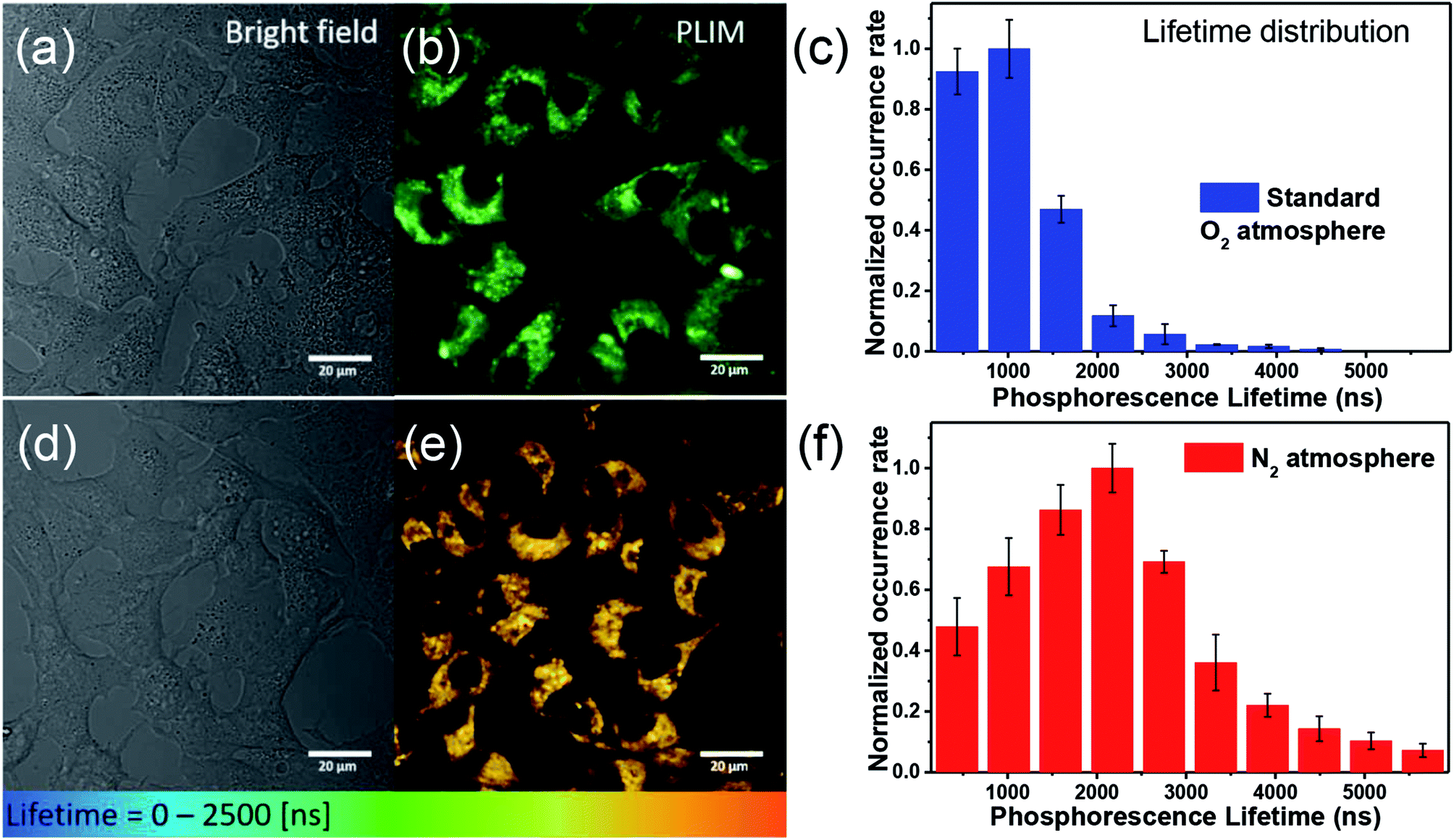 | ||
| Fig. 3 PLIM of HeLa cell incubated in 50 μM of Ir1* in the condition of Standard O2 atmosphere (a–c) and N2 atmosphere (d–f). Bright field images (a and d); PLIM mapping (b and e); the corresponding phosphorescence lifetime distribution (c and f). Excitation wavelength: 720 nm. The scale bar: 20 μm. | ||
The photostability of phosphorescence probes plays a key role in PLIM measurements. Under the high photon flux used in two photon excitation, photobleaching may take place during the data acquisition. This may cause a serious problem for PLIM since it requires a few minutes to collect sufficient data for lifetime analyses. For a fair photostability evaluation, we compared iridium complexes Ir1* and Ir2* with common luminophores coumarin 480 (C480) and rhodamine B.
Under the same power and excitation wavelength (λexc = 720 nm for Ir1* and C480, and λexc = 860 nm for Ir2*, Pt2*, Pt3*, Pt4* and rhodamine B), together with 50 scans (acquisition time of every scan is 1.94 second), each scan interval of 2 seconds, the emission intensity of C480 and rhodamine B dropped rapidly, whereas the emission intensity of Ir1* and Ir2* still exceeded 97% of the first scan emission intensity (see Fig. 4). In the similar way, photostabilities of Pt complexes were found to be higher than that of rhodamine B, though slightly lower than those of Ir complexes (Fig. S41†). We thus concluded that the titled water-soluble iridium complexes, especially Ir1*, are robust and have great photostability suitable for the PLIM measurements in vitro. In the next section, we have then evaluated its applicability for PLIM experiments in vivo.
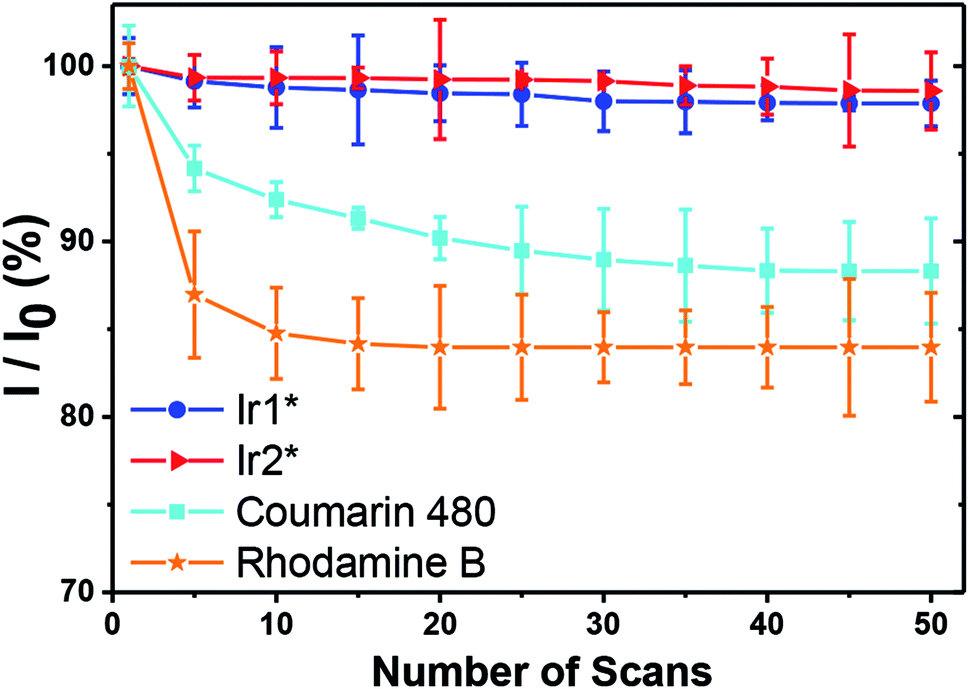 | ||
| Fig. 4 Photostability plot of Ir1* and Ir2* compared with common luminophores, coumarin 480 (C480) and rhodamine B. Experimental conditions: 10 mM PBS buffer (pH 7.4); all the absorbances are fixed to 0.05 at 360 nm (Ir1* and C480, λexc = 720 nm) or at 430 nm (Ir2* and rhodamine B, λexc = 860 nm). Excitation power was 32 mW. I is the emission intensity of each particular scan, and I0 is the emission intensity of the first scan. | ||
Evaluation of Ir1* applicability for in vivo PLIM imaging
In order to demonstrate the applicability of Ir1* to oxygen measurements in animal tissues, we performed in vivo experiments on mice with HeLa tumor xenografts.In this study we found that Ir1* shows detectable phosphorescence in tumor tissue upon local injection, whereas after intravenous administration of Ir1* in the same dose (75 mg kg−1) tumor uptake was insufficient for the phosphorescence detection using available technique. It is important to mention that no acute toxicity effects were observed in mice neither after local, nor after systemic administration of the probe. Examples of phosphorescence lifetime image of the tumor and the phosphorescence decay curve are shown in Fig. 5. The tumor areas had phosphorescence lifetimes 2590 ± 60 ns. In the muscle, the phosphorescence lifetime was 2410 ± 70 ns (Fig. S42†). The phosphorescence decay curves were fitted with a single-exponential decay mode with an average goodness of fit of less than 1.2. The average number of the analysed photons per curve were not less than 5000. Slightly longer phosphorescence lifetime in the tumor (p < 0.001) was, likely, due to reduced level of oxygenation as compared to muscle tissue. Reduced tumor oxygenation compared to surrounding normal tissue (muscle) was previously demonstrated in vivo using PLIM and O2-sensors for different subcutaneous mouse tumor models.23
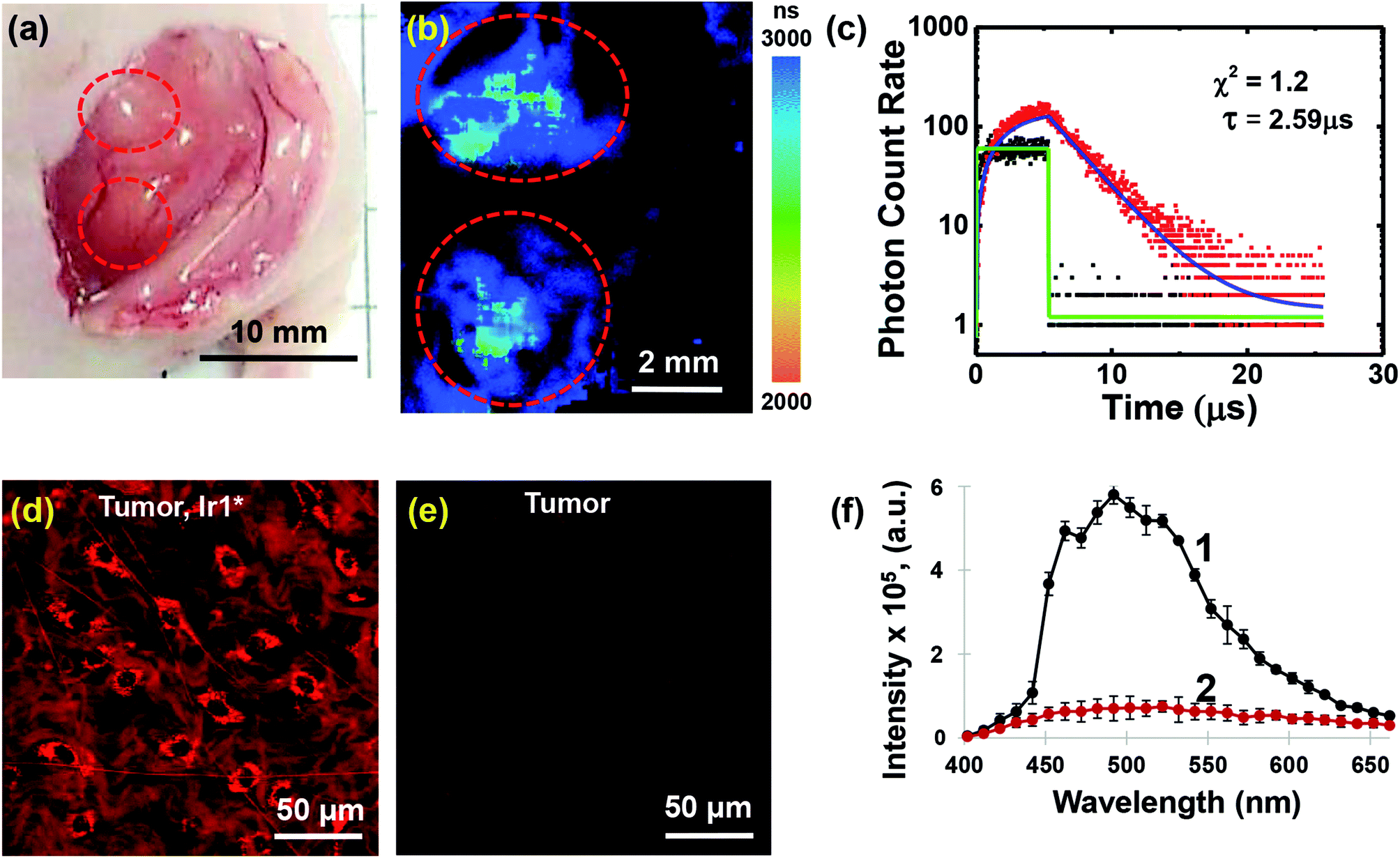 | ||
| Fig. 5 In vivo imaging of Ir1* phosphorescence in HeLa tumor xenograft. (a) Photograph of HeLa tumor in nu/nu mouse with surgically opened skin flap; (b) PLIM of tumor in vivo after local injection of Ir1*; (c) phosphorescence decay of Ir1* in tumor (red) and signal from control tumor without injection Ir1* in the selected spot in the tumor (black); (d) in vivo confocal laser-scanning microscopy of tumor with Ir1* (ex. 750 nm, reg. 430–600 nm); (e) in vivo microscopic image of the control tumor without injection of Ir1*; (f) emission spectra (ex. 750 nm, reg. 400–662 nm) of tumor with local injection Ir1* (1) and of control tumor without injection Ir1* (2), mean ± SD. Two tumor nodules that were injected with Ir1* are shown by the red dashed circles. Scale bars are indicated in each image. Blue curve on (c) is single exponential fit, green curve is instrument response function. | ||
After PLIM, the tumors were examined in vivo on confocal laser scanning microscope to analyze distribution of the probe at the microscopic level (Fig. 5d–f). It was revealed that intratumoral administration of Ir1* resulted in cellular internalization of the dye and staining of the cell cytoplasm along with connective tissue (collagen). A parallel detection of the emission spectra (the lambda scan) confirmed the accumulation of Ir1* in the tissue. Measurements made for control tumor without an injection of Ir1* did not show any phosphorescence signal.
Meanwhile, the phosphorescence lifetimes recorded in the tissue were generally longer than the values in solutions and in cultured HeLa cells. Besides oxygen concentration, some other factors are supposed to affect the phosphorescence lifetimes of oxygen sensitive probes, e.g. binding to proteins, temperature, pH. To estimate the effect of the protein binding, we performed additional PLIM measurements of Ir1* in distilled water and in the presence of 10% fetal bovine serum (Table S4†). We found that the presence of the serum proteins does not result in considerable change of the phosphorescence lifetimes. Consequently, interaction with proteins cannot be the reason for elongation of the phosphorescence lifetimes in living tissue. To convert the PLIM map into oxygen map, intracellular calibration of the lifetime vs. oxygen concentration is needed. Nevertheless, these pilot experiments on tumor-bearing mice illustrate the high potential of novel Ir1* complex for in vivo mapping of oxygen level in tissue.
Conclusions
In conclusion, we present synthesis and photophysical investigation of cyclometalated water-soluble Pt(II) and Ir(III) complexes containing auxiliary sulfonated diphosphine (bis(diphenylphosphino)benzene (dppb), P^P*) ligand. The complexes demonstrate considerable variations in excitation (extending up to 450 nm) and emission band wavelengths (maxima ranging from ca. 450 to ca. 650 nm), as well as in sensitivity of excited-state lifetimes to molecular oxygen, from almost negligible to more than 4-fold increase in degassed solution. Moreover, all the complexes possess high two-photon absorption cross sections (400–500 GM for Pt complexes, and 600–700 GM for Ir complexes).Despite their negative net charge, all these complexes demonstrate good uptake by HeLa cells and low cytotoxicity within the concentration and time ranges suitable for two-photon phosphorescence lifetime (PLIM) microscopy. The most promising complex, [(ppy)2Ir(sulfo-dppb)] (Ir1*), demonstrates two-fold lifetime changes in HeLa cell incubated under normal and nitrogen atmosphere, correspondingly. Further, in vivo evaluation of Ir1* in athymic nude mice bearing HeLa xenografts did not reveal acute toxicity upon both intravenous administration and topical injection. Finally, Ir1* demonstrated statistically significant difference in lifetimes between normal tissue (muscle) and tumor in whole-body in vivo PLIM imaging. Though the pilot experiments on tumor-bearing mice illustrate the high potential of novel Ir1* complex for in vivo mapping of oxygen level in tissue, improvement of the phosphorescence quantum yield and selectivity of the complex to tumor are desirable, and this work is now in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciate financial support from the Russian Science Foundation (grant 16-43-03003, synthesis, characterization, and spectroscopic studies; grant 14-15-00646-П, in vivo study) and Ministry of Science and Technology of Taiwan (MOST 106-2119-M-002-026-MY3, in vitro experiments). The work was carried out using the core facilities of St. Petersburg State University Research Park (Centre for Magnetic Resonance, Centre for Optical and Laser Materials Research, Centre for Chemical Analysis and Materials Research). The authors are thankful to Prof. Elena Zagaynova and Dr Marina Shirmanova (Institute of Biomedical Technologies, Privolzhskiy Research Medical University) for helpful discussions and valuable suggestions, and to Vasily V. Sivchik (Department of Chemistry, University of Eastern Finland) for the synthesis of 2-(benzofuran-3-yl)pyridine.Notes and references
- M. C. L. Yeung and V. W. W. Yam, Struct. Bonding, 2015, 165, 109–130 CrossRef CAS.
- K. Y. Zhang, S. Liu, Q. Zhao, F. Li and W. Huang, Struct. Bonding, 2015, 165, 131–180 CrossRef CAS.
- T. Zou, F. F. Hung, C. Yang and C. M. Che, Struct. Bonding, 2015, 165, 181–203 CrossRef CAS.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Struct. Bonding, 2015, 165, 205–256 CrossRef CAS.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43(12), 4144–4166 RSC.
- D. L. Ma, H. Z. He, K. H. Leung, D. S. H. Chan and C. H. Leung, Angew. Chem., Int. Ed., 2013, 7666–7682 CrossRef CAS PubMed.
- R. I. Dmitriev and D. B. Papkovsky, Cell. Mol. Life Sci., 2012, 69(12), 2025–2039 CrossRef CAS PubMed.
- D. L. Ma, V. P. Y. Ma, D. S. H. Chan, K. H. Leung, H. Z. He and C. H. Leung, Coord. Chem. Rev., 2012, 3087–3113 CrossRef CAS.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40(5), 2508–2524 RSC.
- K. K.-W. Lo, S. P.-Y. Li and K. Y. Zhang, New J. Chem., 2011, 35(2), 265–287 RSC.
- S. W. Botchway, M. Charnley, J. W. Haycock, A. W. Parker, D. L. Rochester, J. A. Weinstein and J. A. G. Williams, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(42), 16071–16076 CrossRef CAS PubMed.
- E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5(3), 879–886 RSC.
- J. Liu, Y. Liu, Q. Liu, C. Li, L. Sun and F. Li, J. Am. Chem. Soc., 2011, 133(39), 15276–15279 CrossRef CAS PubMed.
- G. Li, Y. Chen, J. Wu, L. Ji and H. Chao, Chem. Commun., 2013, 49(20), 2040 RSC.
- W. Wang, Z. Z. Dong, C. Yang, G. Li, Y. C. Tse, C. H. Leung and D. L. Ma, Sens. Actuators, B, 2017, 251, 374–379 CrossRef CAS.
- W. Wang, K. Vellaisamy, G. Li, C. Wu, C.-N. Ko, C.-H. Leung and D.-L. Ma, Anal. Chem., 2017, 89(21), 11679–11684 CrossRef CAS PubMed.
- K. Vellaisamy, G. Li, C.-N. Ko, H.-J. Zhong, S. Fatima, H.-Y. Kwan, C.-Y. Wong, W.-J. Kwong, W. Tan, C.-H. Leung and D.-L. Ma, Chem. Sci., 2018, 9(5), 1119–1125 RSC.
- C. H. Leung, H. J. Zhong, H. Yang, Z. Cheng, D. S. H. Chan, V. P. Y. Ma, R. Abagyan, C. Y. Wong and D. L. Ma, Angew. Chem., Int. Ed., 2012, 51(36), 9010–9014 CrossRef CAS PubMed.
- X. Zheng, X. Wang, H. Mao, W. Wu, B. Liu and X. Jiang, Nat. Commun., 2015, 6, 5834 CrossRef CAS PubMed.
- T. C. O'Riordan, K. Fitzgerald, G. V. Ponomarev, J. Mackrill, J. Hynes, C. Taylor and D. B. Papkovsky, Am. J. Physiol. Regul. Integr. Comp. Physiol., 2006, 292(4), R1613–R1620 CrossRef PubMed.
- T. Yoshihara, M. Hosaka, M. Terata, K. Ichikawa, S. Murayama, A. Tanaka, M. Mori, H. Itabashi, T. Takeuchi and S. Tobita, Anal. Chem., 2015, 87(5), 2710–2717 CrossRef CAS PubMed.
- J. A. Spencer, F. Ferraro, E. Roussakis, A. Klein, J. Wu, J. M. Runnels, W. Zaher, L. J. Mortensen, C. Alt, R. Turcotte, R. Yusuf, D. Côté, S. A. Vinogradov, D. T. Scadden and C. P. Lin, Nature, 2014, 508(7495), 269–273 CrossRef CAS PubMed.
- T. V. Esipova, A. Karagodov, J. Miller, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Anal. Chem., 2011, 83(22), 8756–8765 CrossRef CAS PubMed.
- O. S. Finikova, A. Y. Lebedev, A. Aprelev, T. Troxler, F. Gao, C. Garnacho, S. Muro, R. M. Hochstrasser and S. A. Vinogradov, ChemPhysChem, 2008, 9(12), 1673–1679 CrossRef CAS PubMed.
- R. I. Dmitriev, A. V. Kondrashina, K. Koren, I. Klimant, A. V. Zhdanov, J. M. P. Pakan, K. W. McDermott and D. B. Papkovsky, Biomater. Sci., 2014, 2(6), 853–866 RSC.
- A. L. Harris, Nat. Rev. Cancer, 2002, 2(1), 38–47 CrossRef CAS PubMed.
- P. Vaupel, K. Schlenger, C. Knoop and M. Hockel, Cancer Res., 1991, 51(12), 3316–3322 CAS.
- L. I. Cardenas-Navia, D. Mace, R. A. Richardson, D. F. Wilson, S. Shan and M. W. Dewhirst, Cancer Res., 2008, 68(14), 5812–5819 CrossRef CAS PubMed.
- R. D. Shonat and A. C. Kight, Ann. Biomed. Eng., 2003, 31(9), 1084–1096 CrossRef PubMed.
- J. Yu, A. Ramadeen, A. K. Y. Tsui, X. Hu, L. Zou, D. F. Wilson, T. V. Esipova, S. A. Vinogradov, H. Leong-Poi, N. Zamiri, C. D. Mazer, P. Dorian and G. M. T. Hare, Anaesthesia, 2013, 68(7), 723–735 CrossRef CAS PubMed.
- S. Sakadžić, E. Roussakis, M. A. Yaseen, E. T. Mandeville, V. J. Srinivasan, K. Arai, S. Ruvinskaya, A. Devor, E. H. Lo, S. A. Vinogradov and D. A. Boas, Nat. Methods, 2010, 7(9), 755–759 CrossRef PubMed.
- S. V. Apreleva, D. F. Wilson and S. A. Vinogradov, Appl. Opt., 2006, 45(33), 8547–8559 CrossRef CAS PubMed.
- R. I. Dmitriev, H. M. Ropiak, G. V. Ponomarev, D. V. Yashunsky and D. B. Papkovsky, Bioconjugate Chem., 2011, 22(12), 2507–2518 CrossRef CAS PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19(4), 316–317 CrossRef CAS PubMed.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4(11), 710–711 CrossRef CAS PubMed.
- L. R. Alrawashdeh, M. P. Cronin, C. E. Woodward, A. I. Day and L. Wallace, Inorg. Chem., 2016, 55(13), 6759–6769 CrossRef CAS PubMed.
- K. K. W. Lo, T. K. M. Lee, J. S. Y. Lau, W. L. Poon and S. H. Cheng, Inorg. Chem., 2008, 47(1), 200–208 CrossRef CAS PubMed.
- D. L. Ma, W. L. Wong, W. H. Chung, F. Y. Chan, P. K. So, T. S. Lai, Z. Y. Zhou, Y. C. Leung and K. Y. Wong, Angew. Chem., Int. Ed., 2008, 47(20), 3735–3739 CrossRef CAS PubMed.
- C. Li, M. Yu, Y. Sun, Y. Wu, C. Huang and F. Li, J. Am. Chem. Soc., 2011, 133(29), 11231–11239 CrossRef CAS PubMed.
- A. Baschieri, S. Muzzioli, V. Fiorini, E. Matteucci, M. Massi, L. Sambri and S. Stagni, Organometallics, 2014, 33(21), 6154–6164 CrossRef CAS.
- A. I. Solomatina, P. S. Chelushkin, D. V. Krupenya, I. S. Podkorytov, T. O. Artamonova, V. V. Sizov, A. S. Melnikov, V. V. Gurzhiy, E. I. Koshel, V. I. Shcheslavskiy and S. P. Tunik, Bioconjugate Chem., 2017, 28(2), 426–437 CrossRef CAS PubMed.
- P. K.-M. Siu, D.-L. Ma and C.-M. Che, Chem. Commun., 2005, 1(8), 1025–1027 Search PubMed.
- P. Wu, E. L. M. Wong, D. L. Ma, G. S. M. Tong, K. M. Ng and C. M. Che, Chem.–A Eur. J., 2009, 15(15), 3652–3656 CrossRef CAS PubMed.
- P. S. Chelushkin, D. V. Krupenya, Y.-J. Tseng, T.-Y. Kuo, P.-T. Chou, I. O. Koshevoy, S. V. Burov and S. P. Tunik, Chem. Commun., 2014, 50(7), 849–851 RSC.
- F. Scarpelli, A. Ionescu, L. Ricciardi, P. Plastina, I. Aiello, M. La Deda, A. Crispini, M. Ghedini and N. Godbert, Dalt. Trans., 2016, 45(43), 17264–17273 RSC.
- A. Ionescu, N. Godbert, L. Ricciardi, M. La Deda, I. Aiello, M. Ghedini, I. Rimoldi, E. Cesarotti, G. Facchetti, G. Mazzeo, G. Longhi, S. Abbate and M. Fusè, Inorg. Chim. Acta, 2017, 461, 267–274 CrossRef CAS.
- H.-J. Yin, Y.-J. Liu, J. Gao and K.-Z. Wang, Dalt. Trans., 2017, 46(10), 3325–3331 RSC.
- A. W.-T. Choi, M.-W. Louie, S. P.-Y. Li, H.-W. Liu, B. T.-N. Chan, T. C.-Y. Lam, A. C.-C. Lin, S.-H. Cheng and K. K.-W. Lo, Inorg. Chem., 2012, 51(24), 13289–13302 CrossRef CAS PubMed.
- Y. Zhu, C. Xu, Y. Wang, Y. Chen, X. Ding and B. Yu, RSC Adv., 2017, 7(52), 32632–32636 RSC.
- W. A. Herrmann and C. W. Kohlpaintner, Angew. Chem., Int. Ed. Engl., 1993, 1524–1544 CrossRef.
- Y. R. Hristova, B. Kemper and P. Besenius, Tetrahedron, 2013, 69(49), 10525–10533 CrossRef CAS.
- L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52(1), 270–289 CrossRef CAS PubMed.
- D. García-Fresnadillo and G. Orellana, Helv. Chim. Acta, 2001, 84(9), 2708–2730 CrossRef.
- F. F. Hung, W. P. To, J. J. Zhang, C. Ma, W. Y. Wong and C. M. Che, Chem. - A Eur. J., 2014, 20(28), 8604–8614 CrossRef CAS PubMed.
- C. K. Koo, K. L. Wong, C. W. Y. Man, H. L. Tam, S. W. Tsao, K. W. Cheah and M. H. W. Lam, Inorg. Chem., 2009, 48(16), 7501–7503 CrossRef CAS PubMed.
- S. Y. Yin, S. S. Sun, M. Pan, Y. Z. Fan, Y. X. Chen, H. P. Wang and Y. N. Fan, Inorg. Chem. Commun., 2017, 83, 81–83 CrossRef CAS.
- N. Godbert, T. Pugliese, I. Aiello, A. Bellusci, A. Crispini and M. Ghedini, Eur. J. Inorg. Chem., 2007, 32, 5105–5111 CrossRef.
- C.-L. Li, Y.-J. Su, Y.-T. Tao, P.-T. Chou, C.-H. Chien, C.-C. Cheng and R.-S. Liu, Adv. Funct. Mater., 2005, 15(3), 387–395 CrossRef CAS.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83(12), 2213–2228 CrossRef CAS.
- K. Rurack and M. Spieles, Anal. Chem., 2011, 83(4), 1232–1242 CrossRef CAS PubMed.
- Bruker, APEX II, Bruker AXS Inc., Madison, Wisconsin, USA, 2013 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45(4), 849–854 CrossRef CAS.
- Bruker, APEX II, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65(2), 148–155 CrossRef CAS PubMed.
- V. I. Shcheslavskiy, A. Neubauer, R. Bukowiecki, F. Dinter and W. Becker, Appl. Phys. Lett., 2016, 108(9), 091111 CrossRef.
- S. Kalinina, J. Breymayer, P. Schäfer, E. Calzia, V. Shcheslavskiy, W. Becker and A. Rück, J. Biophotonics, 2016, 9(8), 800–811 CrossRef CAS PubMed.
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, E. Schönhofer, J. Schönle and J. A. Zampese, Polyhedron, 2012, 35(1), 154–160 CrossRef CAS.
- S. X. Luo, L. Wei, X. H. Zhang, M. H. Lim, K. X. V. Lin, M. H. V. Yeo, W. H. Zhang, Z. P. Liu, D. J. Young and T. S. A. Hor, Organometallics, 2013, 32(10), 2908–2917 CrossRef CAS.
- L. He, C. P. Tan, R. R. Ye, Y. Z. Zhao, Y. H. Liu, Q. Zhao, L. N. Ji and Z. W. Mao, Angew. Chem., Int. Ed., 2014, 53(45), 12137–12141 CrossRef CAS PubMed.
- M. Sarma, T. Chatterjee, R. Bodapati, K. N. Krishnakanth, S. Hamad, S. Venugopal Rao and S. K. Das, Inorg. Chem., 2016, 55(7), 3530–3540 CrossRef CAS PubMed.
- R. M. Edkins, S. L. Bettington, A. E. Goeta and A. Beeby, Dalt. Trans., 2011, 40(47), 12765–12770 RSC.
- C.-L. Ho, K.-L. Wong, H.-K. Kong, Y.-M. Ho, C. T.-L. Chan, W.-M. Kwok, K. S.-Y. Leung, H.-L. Tam, M. H.-W. Lam, X.-F. Ren, A.-M. Ren, J.-K. Feng and W.-Y. Wong, Chem. Commun., 2012, 48(19), 2525–2527 RSC.
- C.-K. Koo, K.-L. Wong, C. W.-Y. Man, Y.-W. Lam, L. K.-Y. So, H.-L. Tam, S.-W. Tsao, K.-W. Cheah, K.-C. Lau, Y.-Y. Yang, J.-C. Chen and M. H.-W. Lam, Inorg. Chem., 2009, 48(3), 872–878 CrossRef CAS PubMed.
- M. G. Vivas, L. De Boni, T. M. Cooper and C. R. Mendonca, J. Phys. Chem. A, 2014, 118(30), 5608–5613 CrossRef CAS PubMed.
- Z. Ji, Y. Li, T. M. Pritchett, N. S. Makarov, J. E. Haley, Z. Li, M. Drobizhev, A. Rebane and W. Sun, Chemistry, 2011, 17(8), 2479–2491 CrossRef CAS PubMed.
- M. G. Vivas, L. De Boni, T. M. Cooper and C. R. Mendonca, ACS Photonics, 2014, 1(2), 106–113 CrossRef CAS.
- W. Sun, B. Zhang, Y. Li, T. M. Pritchett, Z. Li and J. E. Haley, Chem. Mater., 2010, 22(23), 6384–6392 CrossRef CAS.
- G. L. Semenza, Science, 2007, 318(5847), 62–64 CrossRef CAS PubMed.
- S. Liu, H. Liang, K. Y. Zhang, Q. Zhao, X. Zhou, W. Xu and W. Huang, Chem. Commun., 2015, 51(37), 7943–7946 RSC.
- L. Sun, G. Li, X. Chen, Y. Chen, C. Jin, L. Ji and H. Chao, Sci. Rep., 2015, 5, 14837 CrossRef CAS PubMed.
- A. Y. Lebedev, A. V. Cheprakov, S. Sakadzic, D. A. Boas, D. F. Wilson and S. A. Vinogradov, ACS Appl. Mater. Interfaces, 2009, 1(6), 1292–1304 CAS.
- M. Lepeltier, F. Appaix, Y. Y. Liao, F. Dumur, J. Marrot, T. Le Bahers, C. Andraud and C. Monnereau, Inorg. Chem., 2016, 55(19), 9586–9595 CrossRef CAS PubMed.
- C. Jin, J. Liu, Y. Chen, L. Zeng, R. Guan, C. Ouyang, L. Ji and H. Chao, Chem.–A Eur. J., 2015, 21(34), 12000–12010 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of platinum and iridium complexes (Section 1); photophysical and spectroscopic properties of platinum and iridium complexes (Section 2); two-photon luminescence imaging and cytotoxicities of platinum and iridium complexes in HeLa cells; photostability of Pt complexes; lifetimes of Ir1* complex in water and 10% fetal bovine serum (PDF). CCDC 1833001–1833003. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra02742k |
| This journal is © The Royal Society of Chemistry 2018 |