
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning chain extender structure to prepare high-performance thermoplastic polyurethane elastomers†
Wei Juan Xu ,
Jian Jun Wang*,
Shi Yu Zhang,
Jun Sun,
Chuan Xiang Qin and
Li Xing Dai
,
Jian Jun Wang*,
Shi Yu Zhang,
Jun Sun,
Chuan Xiang Qin and
Li Xing Dai
College of Chemistry, Chemical Engineering and Materials, Science of Soochow University, Suzhou 215123, China. E-mail: wangjianjun@suda.edu.cn
First published on 6th June 2018
Abstract
In this work, a novel strategy is developed to solve the issue of mutually exclusive high mechanical robustness and thermo-stability for thermoplastic polyurethane (PU). A leaf-like and reticulate interfingering superstructure can be seen. The superstructure of polyurethanes can also be tuned by the polarity of chain extender molecular via changing the number for ferrocene redox centres, thus to further enhance the thermal stability and elasticity of PUs. As a result, by incorporating bisferrocene units into the main chain of PU, a high-performance PU elastomer can be synthesized with a highest initial degradation temperature of T5% of 345 °C, a highest tensile strength of 42.3 MPa with an elongation over 1000%, as well as a toughness of 19.6 GJ m−3. These results conclusively suggest that high-performance thermoplastic polyurethane elastomers had great promise for potential application in a wide range of practical fields.
1. Introduction
Thermoplastic polyurethane (PU) elastomers as the third generation rubbers combine the physical and mechanical properties of vulcanizates with the processability of thermoplastics,1 reflecting structural variability, performance controllability etc. PUs usually consist of alternating hard segments (HSs) and flexible diols structure of soft segments (SSs) to exhibit different mechanical and thermal properties. Therefore, there is a huge consumption of PUs in the global plastic application market at high growth rate.2–5 Recently it also shows interesting applications in self-healing materials,6–8 stimuli-responsive materials,9–12 and flexible conductive materials.13 Meanwhile, concerns about the poor heat resistance of PUs limit their further applications.14 The mechanical properties of PUs will be greatly reduced when the operating temperature is above 120 °C.15Generally, the initial thermal degradation of PUs starts from the breakdown of urethane groups in hard segments at the temperature range from 180 to 300 °C, and then the degradation of soft segments at higher temperatures.16 The first degradation step of hard segment is the most important one because it decides the highest operating temperature for PUs. Many efforts have been made to modify the HSs to be high heat-resistant to increase the thermo-stability of PUs. For example, Si–O,17–20 metallic organic polymers21,22 and heterocyclic23,24 have been shown to be effective to improve the thermo-stability, but still with a low stress modulus and strain behaviours. The high elasticity and high modulus of PUs were dependent on the aggregates of phase-separated.25 However, it is still difficult to rationally adjust the distribution of crystal and amorphous phases to avoid turning into obvious crystallization or clustering of polymers to result in a brittle superstructure.26,27 In other words, it seems to be mutually exclusive to achieve high mechanical robustness and thermo-stability. Therefore, it is still a great challenge to improve the heat resistance of PUs whilst achieving excellent mechanical properties.
Herein we are interested in adapting chemistry strategies to tune the chain extenders structure to solve above mutually exclusive issues. (1) Ferrocene derivatives were introduced as chain extenders into the backbone of polymers to improve the thermo-stability, due to their intrinsic heat-resistant and radical capture abilities.28–30 The thermo-stability and thermal decomposition of these PUs were characterized by simultaneous TGA-QMS analyzer and attenuated total reflection-fourier transform infrared (ATR-FTIR) spectroscopy. (2) Then those chain extenders were functionalized with ester groups to provide extra H-bonds sites to enhance modulus for polyurethanes. However, a large amount of H-bond will also induce unfavourable crystallization in polymer, thus decrease the mechanical performance obviously. (3) Finally, to avoid this situation, the superstructure of those polyurethanes can also be tuned by the polarity of chain extender molecular via changing the number for ferrocene redox centers, to provide more molecular spatial freedom to tune the crystallinities and phase separation of those polyurethanes. The crystallinities and morphology of these PUs were characterized by Differential Scanning Calorimeter (DSC), Small-Angle X-ray Scattering (SAXS), Wide-Angle X-ray Diffraction (WAXD), Polarizing Optical Microscope (POM) and Scanning Electron Microscopy (SEM). As a result, a high-performance thermoplastic polyurethane elastomer polymer have been synthesized by using 6,6′-bis [1-methyl-2-(β-hydroxyethyl)ester-2-methylpropyl]bisferrcene (bisFc) as chain extender. A highest initial degradation temperature of 345 °C and a highest tensile strength of 42.3 MPa with a strain over 1000% could be achieved, as well as a toughness of 19.6 GJ m−3.
2. Experimental
2.1 Materials
1,1′-Bis(hydroxymethyl)ferrocene (>99%, Energy Chemical, Shanghai), 1-methoxy-1-(trimethylsiloxy)-2-methyl-1-propene (>95%, Energy Chemical, Shanghai), BF3·OEt2 (98%, Energy Chemical, Shanghai), ethylene glycol (EG, AR, Sinopharm Chemical Reagent, Beijing), dibutyltin dilaurate (DBTDL, LR, Lingfeng Chemical Reagent, Shanghai), acetic anhydride (AR, Sinopharm Chemical Reagent, Beijing). Liquid reagents were further purified through distillation prior to use. Diphenyl methane-4,4′-diisocyanate (4,4′-MDI, >97%, Energy Chemical, Shanghai), polytetramethylene ether glycol (PTMEG, Mn is around 2000, Aladdin, Shanghai), it was dried under vacuum at 80 °C for 6 h prior to use. 1,4-Butanediol (BDO, AR, Sinopharm Chemical Reagent, Beijing).2.2 Synthesis of chain extenders
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) for 16.0 h. Then a solution of 3.48 g (12.70 mmol) 1,1′-bis(1-methoxy-methyl)ferrocene, 10.5 mL (50.80 mmol) 1-methoxy-1-(trimethylsiloxy)-2-methyl-1-propene and 3.5 mL (27.90 mmol) BF3·OEt2 in CH2Cl2 (180.0 mL) was stirred at −78 °C for 15 min. The reaction was quenched with a saturated solution of NaHCO3 and extracted with CH2Cl2. The organic phases were combined and dried to give a viscous yellow oil, which was chromatographed over a column of silica gel using ethyl acetate/petroleum ether (1:4 v/v) as the eluent. Yellow crystals of the title compound (4.74 g, 90%) were obtained by slow evaporation of a solution in dichloromethane/petroleum ether.
:1 v/v) for 16.0 h. Then a solution of 3.48 g (12.70 mmol) 1,1′-bis(1-methoxy-methyl)ferrocene, 10.5 mL (50.80 mmol) 1-methoxy-1-(trimethylsiloxy)-2-methyl-1-propene and 3.5 mL (27.90 mmol) BF3·OEt2 in CH2Cl2 (180.0 mL) was stirred at −78 °C for 15 min. The reaction was quenched with a saturated solution of NaHCO3 and extracted with CH2Cl2. The organic phases were combined and dried to give a viscous yellow oil, which was chromatographed over a column of silica gel using ethyl acetate/petroleum ether (1:4 v/v) as the eluent. Yellow crystals of the title compound (4.74 g, 90%) were obtained by slow evaporation of a solution in dichloromethane/petroleum ether.Then, 1,1′-bis[2-(β-hydroxyethyl)ester-2-methylpropyl]ferrocene was prepared by mixing 4.74 g (11.43 mmol) 1,1′-bis(1-methoxy-methyl)ferrocene, 3.9 mL (70.00 mmol) ethylene glycol and 0.1 mL (0.16 mmol) DBTDL at 110–120 °C under N2. After 3.0 h, the reaction was cooled to room temperature. The mixture was diluted with CH2Cl2, filtered and washed three times with deionized water, dried with magnesium sulfate to give viscous orange liquid (monoFc, 5.20 g, 96%). 1H NMR (600 Hz, CDCl3, ppm): δ = 4.07 (s, 2H, –CH2–OH), 4.00 (s, 4H, –CH2–OH), 3.88–3.84 (d, 8H, C5H5FeC5H4–), 3.61 (s, 4H, –CH2–CH2–OH), 2.48–2.47 (d, 4H, C5H5FeC5H4–CH2), 1.00–0.93 (s, 12H, –C(CH3)2–); 13C NMR (600 Hz, CDCl3, ppm): δ = 177.78 (–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–), 83.61, 70.36–68.22 (C5H5FeC5H4–), 65.74–60.46 (–CH2–CH2–OH), 43.64 (C5H5FeC5H4–CH2–), 40.87 (–C(CH3)2–), 24.64 (–C(CH3)2–).
O)–O–), 83.61, 70.36–68.22 (C5H5FeC5H4–), 65.74–60.46 (–CH2–CH2–OH), 43.64 (C5H5FeC5H4–CH2–), 40.87 (–C(CH3)2–), 24.64 (–C(CH3)2–).
Then a solution of 5.08 g (10.24 mmol) 2,2′-(6,6′-diacetyl)bisferrocenylpropane in anhydrous diethyl ether (60.0 mL) was added into the mixed solution of 1.17 g (30.72 mmol) LiAlH4 and anhydrous diethyl ether (20.0 mL), stirring 5.0 h at room temperature. Cooling with ice water bath and adding water to decompose the remaining LiAlH4. The precipitate was filtered off and extracted with ether. The organic phases were combined and dried to give a viscous orange liquid, which was chromatographed over a column of silica gel using ether/CH2Cl2 (1:7 v/v) as the eluent to give 6,6′-bis(hydroxymethyl)bisferrocene (4.71 g, 92%).
Finally, 6,6′-bis[1-methyl-2-(β-hydroxyethyl)ester-2-methylpropyl]-bisferrocene was obtained through repeating the reaction steps of monoFc to prepare 4.71 g (9.42 mmol) 6,6′-bis(hydroxymethyl)-bisferrocene for the viscous bordeaux red liquid (bisFc, 5.90 g, 86%). 1H NMR (600 Hz, CDCl3, ppm): δ = 4.15 (s, 2H, –CH2–OH), 4.06 (s, 4H, –CH2–OH), 3.98–3.78 (d, 16H, –C5H4FeC5H4–), 3.65 (s, 4H, –CH2–CH2–OH), 2.86 (d, 2H, –C5H5FeC5H4–CH(CH3)–), 1.59–1.53 (d, 6H, –C5H4FeC5H4–C(CH3)2–C5H4FeC5H4–), 1.23 (d, 12H, –C5H4FeC5H4–CH(CH3)–C(CH3)2–), 0.96 (s, 6H, –C5H4FeC5H4–CH(CH3)–); 13C NMR (600 Hz, CDCl3, ppm): δ = 178.55 (–C(O)–O–), 90.29, 89.22, 70.03–67.74 (–C5H4FeC5H4–), 66.09–61.28 (–CH2–CH2–OH), 46.80 (–C5H4FeC5H4–CH(CH3)–), 40.45 (–C5H4FeC5H4–CH(CH3)–C(CH3)2–), 33.40–30.18 (–C5H4FeC5H4–C(CH3)2–C5H4FeC5H4–), 22.93–21.00 (–C5H4FeC5H4–CH(CH3)–C(CH3)2–), 22.93–21.00 (–C5H4FeC5H4–CH(CH3)–).
2.3 Preparation of polyurethane elastomers
The NCO prepolymer was prepared by reacting 4.05 g (16.80 mmol) 4,4′-MDI with 15.00 g (7.50 mmol) PTMEG in dry N,N-dimethylformamide (DMF, 5.0 mL) at 80 °C for 3.0 h. The reaction was carried out in a 250 mL three-neck round-bottom flask under N2 atmosphere with a mechanical stirrer. BDO, monoFc, and bisFc were dissolved in 60 mL DMF, respectively and then was added into the above prepolymer mixture with a molar ratio of 4,4′-MDI:PTMEG:diol = 2:1:1 at 80 °C. The polyurethane films were prepared in the polytetrahydrofuran mold to remove the solvent at 60 °C for 48.0 h, 70 °C for 12.0 h, and then 80 °C for 12.0 h.
2.4 Characterizations
The 1H NMR and 13C NMR spectra was recorded on Superconducting Pulse Fourier Transform Nuclear Magnetic Resonance spectrometry (Varian UNITY INOVA 600NB).Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy was recorded over the range from 4000 to 600 cm−1 by using a Nicolet 6700 spectrometer.
Gel permeation chromatograph (GPC) analysis of all the samples was performed on a Waters-1515 using DMF-LiBr as an eluent and polystyrene (PS) as standards. The sample concentration was 2–3 mg mL−1 and the flow rate was 0.800 mL min−1 at 40 °C.
The crystallization morphology of polyurethane films was observed through a Polarizing Optical Microscope (POM, Olympus OPTEC BK-POL) with 4× magnification.
The microstructures of the polyurethane films were observed on Scanning Electron Microscope (SEM, SU8010, Hitachi) with an accelerating voltage of 20 kV. Before the observation, all films were frozen in liquid nitrogen and then immediately snapped. The fractured surfaces of the sheets were sputtered with gold and then observed and photographed.
Thermogravimetric analysis (TGA) of all the samples was carried out on a TA TGA/DTG Q800 (Discovery) instrument. Simultaneous thermogravimetry-quadrupole mass spectroscopy analyzer (TGA-QMS, STA 449 F3 & QMS 403D) was used to detect the release of CO2 during the thermal decomposition of polymers. Around 2.5 mg sample was placed into an alumina pan at a heating rate of 10 °C min−1 from 50 to 700 °C under N2 atmosphere.
Thermal analyses of the polymers were per-formed under a N2 atmosphere through differential scanning calorimeter (DSC) using a TA Q200 operated at heating and cooling rates of 10 °C min−1, holding for 5 min at 240 and −80 °C, respectively.
Dynamic mechanical property of PU films were analysed in TA DMA Q800 by tension mode at a constant frequency of 1 Hz. The rectangular sample used in this testing was approximately 40 mm × 5.0 mm × 0.2 mm in dimensions. All the samples were first cooled to −100 °C by liquid nitrogen and then scanned within the temperature range −100 to 70 °C at a heating rate of 3 °C min−1.
Small-Angle X-ray Scattering (SAXS) and Wide-Angle X-ray Diffraction (WAXD) Measurements. SAXS and WAXD data for the PUs were acquired at the SAXSess mc2 facility in Austria. For SAXS measurements, the radiation wavelength of X-ray is 0.154 nm. The beam size is 1.2 × 0.6 mm (H × V). For WAXD measurements, the data were reduced from the 2D format to 1D by integrating with Fit2D. Both sets of data were expressed in terms of the wave vector, q, where q = 4πsinθ/λ.
The tensile properties of polyurethane films were investigated by Universal Testing machine (model BESTE KJ-1065, China) operated at a crosshead peed of 50 mm min−1 (2.0 min−1) with a load cell of 5 kN. Each sample was repeated for 5 times to get the average values. The dumbbell samples used in this testing were approximately 70.0 mm × 5.0 mm × 0.2 mm in dimensions. An initial length was set to be 25 mm. The cyclic testing was also performed using tensile tester to evaluate elastic property of the PUs. Tensile and retraction rates were set to be 2.0 min−1.
Activation energy (Ea) of non-isothermal kinetics of thermal decomposition was calculated based on Arrhenius equation (k(T) = Aexp(−Ea/RT)), as described elsewhere.16,31 The Doyle's approximation of the Flynn–Wall–Ozawa (FWO) method is used for temperature integration as shown in the following equation:
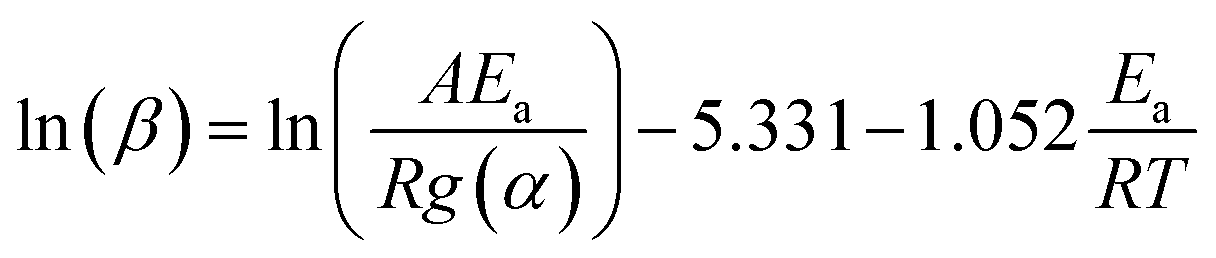 | (1) |
3. Results and discussion
3.1 Synthesis of PUs extended by ferrocenyl-diol
The polyurethanes were prepared via a 2-step reaction. As illustrated in Scheme 1, the prepolymer was prepared by the polymerization reaction between diols (–OH) group of polytetramethylene ether glycol (PTMEG) and isocyanate group (–NCO) of diphenyl-methane diisocyanate (4,4′-MDI) in the first step. Three different structure diols, 1,4-butanediol (BDO), 1,1′-bis[2-(β-hydroxyethyl)ester-2-methyl-propyl]ferrocene (monoFc), and 6,6′-bis[1-methyl-2-(β-hydroxyethyl)ester-2-methyl-propyl]bisferrocene(bisFc), were adapted as chain extenders in this work. BDO is an aliphatic structure, monoFc is ester-containing structure with only one aromatic ferrocene center, and aromatic bisFc is also an ester-containing structure but with 2 polar ferrocene centers. And then the prepolymers will undergo further polymerization reaction with chain extenders to prepare final products of polyurethanes. As shown in Fig. S10,† the δ shifts of 13C-NMR at 80.12 and 79.98 ppm corresponds to the cyclopentadienyl carbon which confirms the successful incorporation of ferrocenyl diol into the polymer via chain extension. Due to the steric and aromatic effect from the ferrocenyl molecular center, the δ for CO in urethane (–NH–COO–) shifted from 155.88 ppm in BDO-PU to lower δ shifts in monoFc-PU and bisFc-PU polymers. Meanwhile, in order to have a fair comparison for the final product polymers to understand the influence from these different chain extenders, these polyurethanes were prepared with a high average molecular weight (Mn) within the range from 2.9 × 104 to 3.0 × 104 g mol−1 and a narrow polydispersity index, respectively (Table 1 and Fig. S1†).
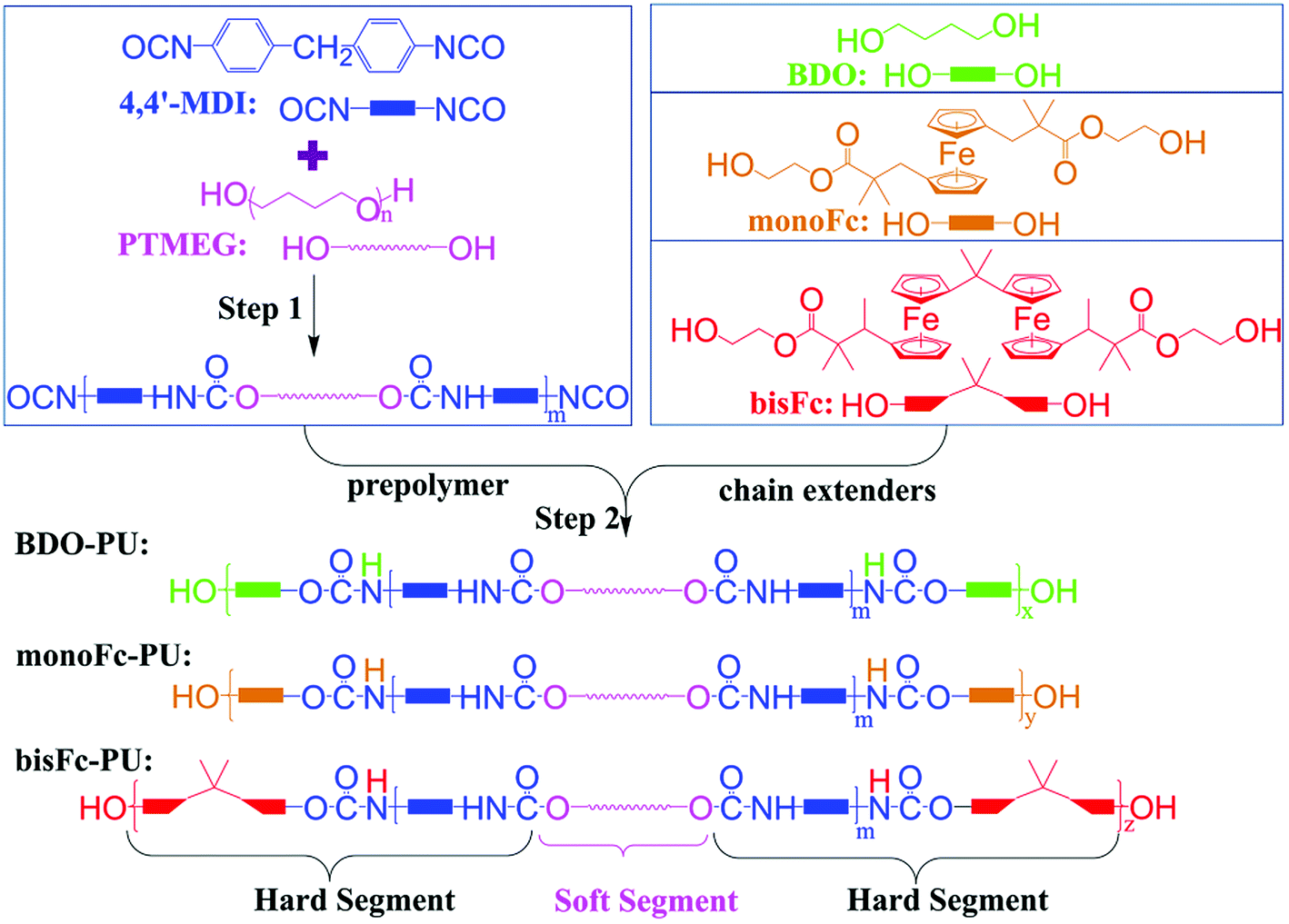 | ||
| Scheme 1 Synthetic routes for polyurethanes with different chain extenders: 1,4-butanediol (BDO), 1,1′-bis[2-(β-hydroxyethyl)ester-2-methylpropyl]ferrocene (monoFc), and 6,6′-bis[1-methyl-2-(β-hydroxyethyl)ester-2-methylpropyl]bisferrocene (bisFc). | ||
| PUs | 4,4′-MDI/PTMEG/extender | Hard segment content (%) | Mn × 104 (g mol−1) | PD | |
|---|---|---|---|---|---|
| Extender | Mass ratio (g) | ||||
| BDO-PU | 1,4-Butanediol | 4.05/15.00/1.91 | 28 | 3.6 | 2.0 |
| monoFc-PU | monoFc | 4.05/15.00/3.56 | 34 | 3.0 | 1.6 |
| bisFc-PU | bisFc | 4.05/15.00/5.46 | 39 | 2.9 | 1.5 |
3.2 Thermo-stability properties
The thermo-stability of the initial degradation temperature (T5% corresponds to 5% mass loss) of above polyurethanes were investigated by thermogravimetric analysis (TGA) method. As shown in Fig. 1a and S2,† BDO-PU shows a lowest T5% below 300 °C. BDO-PU is usually unstable at temperatures higher than 230 °C due to the decomposition of urethane bond and trans-urethane reaction, thus limits its further application.32 After introducing monoFc diols as the chain extenders into PU chains, T5% are increased to 320 °C. And it can further reach to a higher T5% of 345 °C with using the bisFc as the chain extender. Meanwhile, the decomposition temperature of Tmax for monoFc-PU and bisFc-PU were also increased by 50 °C and 70 °C than the one of BDO-PU, respectively.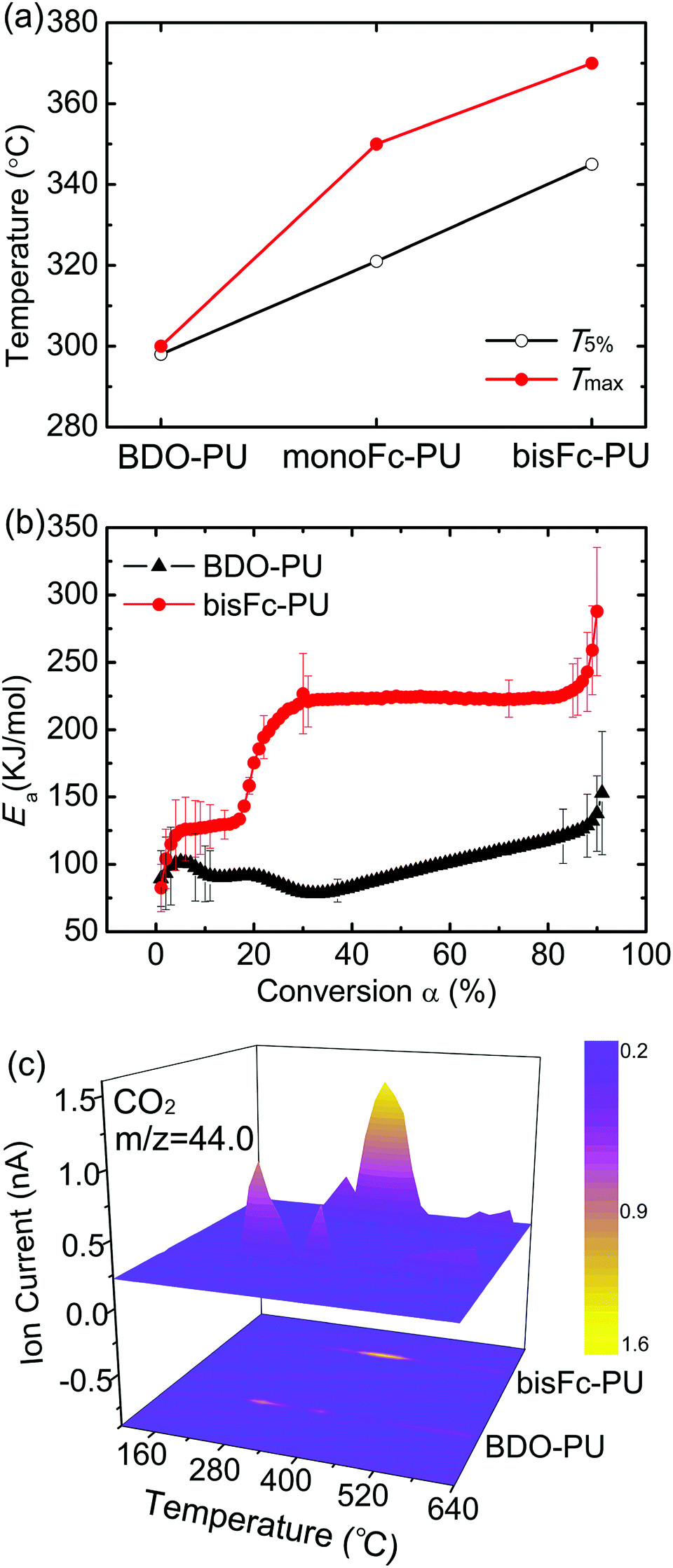 | ||
| Fig. 1 (a) The initial degradation temperature (T5%) and initial stage decomposition temperature (Tmax) of polyurethanes derived from the thermogravimetric analysis (TGA); (b) the comparison of activation energy (Ea) for BDO-PU and bisFc-PU versus conversion α; (c) the thermogravimetry-quadrupole mass spectroscopy (TG-QMS) shows the release of CO2 during BDO-PU and bisFc-PU degradation from 100 to 775 °C. | ||
Fig. S3† shows that there was no sharp melting peak for hard segments, because the crystallization degree was not enough to form one obvious melting peak when the hard segment content is low (23%–35%) or higher molecular weight of soft segment (>1000 g mol−1).33 We could still find a few small endothermic peak (Tm,H) at 154.9 °C for BDO-PU, and began to decompose when close to 200 °C. However, after the ferrocene units were introduced into the hard segments, Tm,H of monoFc-PU was increased to 171.6 °C and 204.1 °C, Tm,H of bisFc-PU could reach to 219.6 °C. It is shown that ferrocene in the hard segments is helpful to improve the thermal stability of polyurethane. In addition, the wider melting peak may be related to the organizational differences of crystal phases in the superstructure for PUs.
The influence of ferrocenyl diols was also further investigated by the comparison of activation energies (Ea) for BDO-PU and BisFc-PU (Fig. 1b and see details in Table S1†). The Ea for the degradation of BDO-PU was gradually increased to 100 kJ mol−1 for the reaction conversion of 0 < α < 5% due to the breakdown of urethane and the Ea was decreasing during the further degradation until α around 30%. This indicates that the breakdown of urethane of BDO-PU will be accelerated during the thermos-degradation process. However, the Ea for bisFc-PU was sharply increased to around 130 kJ mol−1 and levels until α around 20%. This indicates the thermo-degradation for bisFc-PU requires more energy to activate the breakdown of urethane. And the further degradation required a much higher Ea of 230 kJ mol−1 during the decomposition stage of 30% < α < 80%. As a result, the Ea required to degrade bisFc-PU was more than 2 times compared to the one for BDO-PU. Thus the presence of ferrocenyl molecular centers can effectively retard the degradation of urethane in polyurethanes.
The hard segments in PUs usually firstly degrade to form isocyanate, alcohol, and CO2, etc.14 Thus temperature-dependent attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy technique was also adapted to analyse the decomposition of urethane groups in these polyurethanes. As shown in Fig. S4,† the bonded N–H stretching band of BDO-PU were decreasing gradually, and the free N–H increased rapidly when the treating temperature increasing from 150 to 325 °C,34,35 while it had hardly changed in monoFc-PU and bisFc-PU. Until 375 °C, the N–H deformation band of them appeared to be weakened and split into two peaks. It implies that ferrocene units in the hard segments can obstruct the fracture and decomposition of N–H bonds. It also proves this point that the N–H in plane stretching band and C–N stretching band still exists in bisFc-PU until 375 °C. Therefore, although the CO of urethane stretching vibration peak intensities were decreasing greatly for BDO-TPU, it was still obvious in monoFc-PU and bisFc-PU. In addition, the CO stretching band of urea increased dramatically when the temperature was up to 325 °C. However, a wide peak band of 1651 cm−1 was generated in BDO-PU, which was different from monoFc-PU and bisFc-PU that tended to form narrow bands near 1640 cm−1. It illustrates that the thermal decomposition process of PU will be accompanied by the formation of urea structure, but ferrocene may induce the formation of urea structure with low wavenumber of CO bands.
Further investigation on the thermos-stability was confirmed by the Thermogravime-Quadrupole mass spectrometry (TG-QMS) coupling methodology result as shown in Fig. 1c. The emission of CO2 during the thermal decomposition started at a temperature of 325 °C for BDO-PU while it was retarded until 370 °C for bisFc-PU. As a result, the incorporation of ferrocenyl derivatives as the chain extenders are able to enhance the thermo-stability of urethane greatly so as to improve the thermo-stability of PUs. And the degradation temperature of polyurethanes will also be increased by the number for ferrocenyl molecular centers.
3.3 Morphology
The thermal behavior of the PUs was investigated by differential scanning calorimeter (DSC). Fig. S3† shows that Tm,H of PUs increased with an increase in the number of ferrocenyl molecular centers. An increase in Tm,H generally leads to a higher degree of phase separation.36 In addition, the thermal transition temperature of the hard segment (Tg,H) is not as easily observed as that of the soft segment phase. With the exception of BDO-PU, Tg,H of monoFc-PU and bisFc-PU occurred in 129 °C and 170 °C, respectively. Fig. S6† shows WAXD profiles for BDO-PU, monoFc-PU and bisFc-PU. In the WAXD profile for – (MDI-BD)n –, the crystalline peaks could be observed at q = 13.1, 13.7, 15.4, 16.8, and 18.0 nm−1.36 Some of weak crystalline peaks of hard segment chains were observed at the same positions for monoFc-PU (18.3 nm−1) and bisFc-PU (15.0, 16.1 and 18.3 nm−1). The intensity of crystalline peak of bisFc-PU was much higher than that of monoFc-PU, presumably due to larger cohesive force of bisferrocene and the formation of well-organized crystallized hard segment domains.36 Therefore, it is reasonable to prove the existence of the crystalline phase.Fig. 2 shows ATR-FTIR spectra in the ranges (a) N–H and (b) CO stretching band regions for BDO-PU, monoFc-PU and bisFc-PU. Because of the hydrogen bonding, the N–H group splits into two peaks in the expansion region. For all PUs, their hydrogen-bonded N–H groups with ether oxygen (ν(N–Hether)) and urethane carbonyl groups (ν(N–Hcarbonyl)) were observed at 3303 cm−1, and free one (ν(N–Hfree)) were at 3446 cm−1.34,35 The ratio of Iν(N–HH-bond) to Iν(N–Hfree) (Iν(N–HH-bond)/Iν(N–Hfree)) for BDO-PU was the largest. The peak width of monoFc-PU and bisFc-PU were much broader than for BDO-PU. Hence, it is likely to consider that N–H groups in monoFc-PU and bisFc-PU form many kinds of hydrogen bonds with ether and carbonyl groups,36,37 especially two kinds of novel chain extenders were functionalized with ester groups. Moreover, the hydrogen-bonded carbonyl stretching band (ν(COH-bond)) and the free one (ν(COfree)) were observed at 1705 and 1730 cm−1, respectively. The ratios of Iν(COH-bond) to Iν(COfree) (Iν(COH-bond)/Iν(COfree)) for monoFc-PU and bisFc-PU were close to each other and much smaller than the BDO-PU. These observations reveal that the hydrogen-bonded hard segment domains are formed well in BDO-PU, but those are not done in monoFc-PU and bisFc-PU.
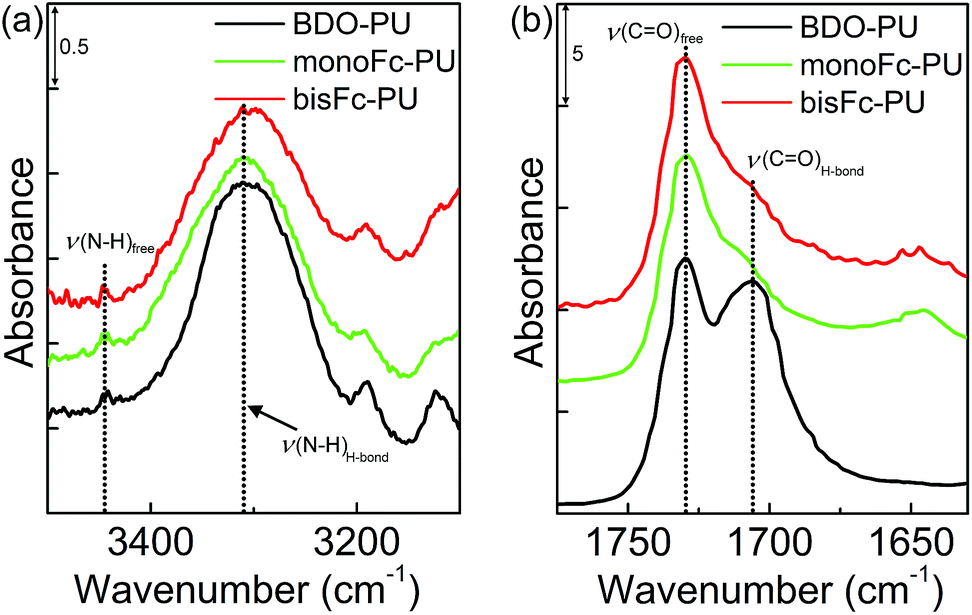 | ||
| Fig. 2 ATR-FTIR spectra of BDO-PU, monoFc-PU and bisFc-PU at room temperature. (a) N–H stretching and (b) CO stretching band regions. | ||
Furthermore, the glass transition temperatures (Tg,S) of the soft segment chains increased in the following order: BDO-PU → monoFc-PU → bisFc-PU (Fig. S5†). These observations suggest that compared with BDO-PU, more isolated hard segments of monoFc-PU and bisFc-PU enter the soft-segment phases because of higher Tg,S and ATR-FTIR data. However, there is π–π stacking interaction between Cp ring of ferrocene and benzene ring, which also affect crystallization and microphase separation.
Hu et al. pointed out that the relative crystallinity (Xc) of soft segments was a more sensitive criterion for distinguishing the degree of phase separation than Tg,S measurements in those highly segregated material.38 Obvious endothermic peaks were observed in the temperature range of 10 to 30 °C as shown in Fig. S5.† This is due to the melting of partially crystallized soft segment chains.36 Xc of the soft segments had increased from 12.5% to 17.5% or 13.0%, when the chain extender was changed from BDO to monoFc or bisFc. Combining with the Tm,H and Tg,H of the hard segments, it seems reasonable to conclude that the degree of microphase separation of the monoFc-PU and bisFc-PU is higher than BDO-PU. These two chain extenders containing ferrocene, as one part of the hard segments, which have a structure significantly different from that of the soft segments, will be less compatible with the soft-segment phases.38
SAXS measurements were also conducted on these samples to investigate the polymer morphology.39 As shown in Fig. S6,† weak and broad shoulder scattering peaks in the q range from 0.1 to 1.0 nm−1 were detected in the SAXS profiles for PUs. It seems that there is microphase separation for all PUs.40,41 Polyurethanes with hard-segment contents of 38% or lower (Table 1) have a discontinuous hard microdomain structure.41 Introducing ferrocene into the PU chains induced a peak shift to high q region. Based on Bragg's law, the interdomain spacing d = 2π/qmax, thus BDO-PU, monoFc-PU and bisFc-PU had one feature size of 16.2 nm, 13.0 nm and 13.4 nm, respectively. This phenomenon is likely to be caused by the higher cohesive energy of monoFc-PU and bisFc-PU, which was clarified by DSC, ATR-FTIR and WAXD.
The morphologies of the superstructure in those polymers were observed by polarized optical microscope (POM). The superstructure is formed based on the hard segment domains and a surrounding soft segment matrix. Bright-field images acquired in reflection mode shows that the polymeric film contained both an anisotropic crystalline phase mainly resulted from hard segments components and an isotropic amorphous phase at room temperature. As shown in Fig. 3a, BDO-PU exhibits a typical superstructure with the crystalline phases dispersed in continuous amorphous phases. Such morphology will enable it to exhibit an acceptable mechanical performance due to the noncovalent cross-linking among PU polymer chains.25,33,42–44 Taking ester-containing monoFc diols as chain extender, the polymers tend to form superstructure within size of 7–70 μm (Fig. 3b). This should be due to the crystallization of the polymers induced by the large amount of H-bonds from the esters groups and the π–π forces of the Cp rings. Interestingly, after replacing the chain extender with bisferrocenyl diol, the size of superstructure for bisFc-PU was only about 1 μm to 20 μm and closes to BDO-PU. Moreover, a coarse-textured and reticular superstructure for bisFc-PU was developed, which is more obvious than the one of BDO-PU (Fig. 3c). It is believed that a less ordered H-bonded geometrical structure will not induce crystallization and bisferrocene structure will also provide a high spatial freedom of structure conformation to regulate the phase transformation.26,45,46 The presence of the extra ester groups will enhance the noncovalent crosslinking interaction among the polymers network, while bisFc molecular is able to tune the polarity of the hard segments, thus a phase separated and superstructure of polyurethanes can be obtained with a relatively low crystallinity.
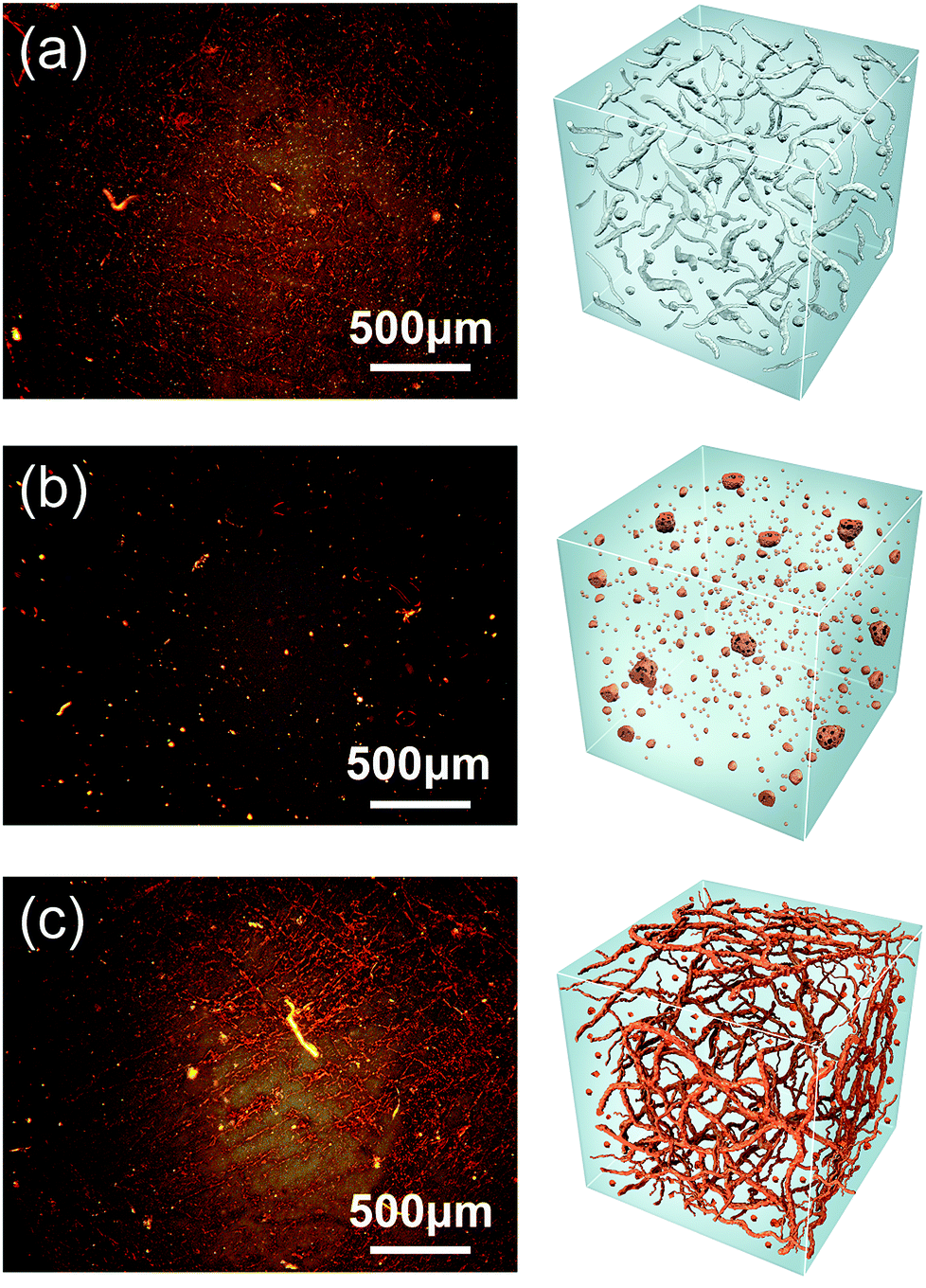 | ||
| Fig. 3 Polarized optical microscope (POM) images for (a) BDO-PU, (b) monoFc-PU, and (c) bisFc-PU with 4× magnifications at 35 °C, as well as the corresponding illustration for superstructure consisted of the crystalline phases and amorphous phases. | ||
Scanning electron microscopy (SEM) was used to study the fracture morphology and supramolecular structure of those PUs. As we can find in Fig. 4 and S7,† fracture appearance is almost the same except for some phase separation and crystal structure. From the Fig. 4a, morphology of BDO-PU was also relatively smooth. Phase separation occurs on a very small scale (Fig. S7†). By contrast, when introducing ester-containing monoFc or bisFc diols as chain extender, the phase separation was more obvious, which was consistent with the previous POM observations and crystallinity conditions measured by DSC (Fig. S5†). This indicates that changing the number of ferrocenyl molecular centers in the polymer chain will greatly influence the superstructure of PUs. Fig. 4b shows that the granular and wafer-shaped morphology, with inhomogeneous size, distributed in the fracture surface of monoFc-PU film. However, the fracture surface of the bisFc-PU is very rough with numerous grooves, as shown in Fig. 4c. Morphology of the sample belongs to the elastic fracture.47,48 Fig. 4d is a magnified image of the marked region in the bottom part in Fig. 4c. Morphology in this area is quite interesting. It can be clearly observed that the sample exhibits a profile of leaf-like micro/nanostructure.49 The size of each whole superstructure was about ca. 500 nm to 1 μm, which is consistent with the previous POM observations. Moreover, it can be found that all around those leaf-shaped morphology had numerous finer granular crystal group, the size of which were less than 40 nm, as shown in Fig. S7.†
 | ||
| Fig. 4 Fracture morphology of (a) BDO-PU, (b) monoFc-PU, and (c) bisFc-PU. (d) is partial enlargements (magnified by 12000 times) of the positions marked in (c). | ||
3.4 Mechanical properties
Furthermore, stress–strain behaviors of the above PUs films were shown in Fig. 5a. The results of their tensile strengths (σmax), elongations (εmax), Young's modules and toughness were also illustrated in Table S1.† The superior mechanical behaviors of bisFc-PU can be obviously distinguished from the ones of BDO-PU and monoFc-PU. The value of Young's modulus decreased in the following order: bisFc-PU > BDO-PU > monoFc-PU. According to the ATR-FTIR (Fig. 2), there is more isolated hard segment domains in monoFc-PU and bisFc-PU than of BDO-PU, but the strain-induced crystallization for bisFc-PU was more pronounced than in monoFc-PU and BDO-PU. This implies that the mechanical properties of PUs not only depend on hard segment domains. The tensile strength (σmax) of bisFc-PU was around 42.3 MPa with a strain over 1000% which was 1.54 times higher than BDO-PUs and 7.05 times higher than monoFc-PU. It can be found from SAXS that the size of microdomains for bisFc-PU was close to monoFc-PU, and was smaller than BDO-PU. According to POM and SEM observation, one reticular superstructure of bisFc-PU was formed with the amorphous region and the small size of crystalline region. This texture superstructure can give full play to the rigidity of hard segments and flexibility of soft segments to dispersing stress, and eventually to improve the tensile strength and toughness of PUs at the same time. The toughness of bisFc-PU was the highest compared with the others, which reach to 19.6 GJ m−3.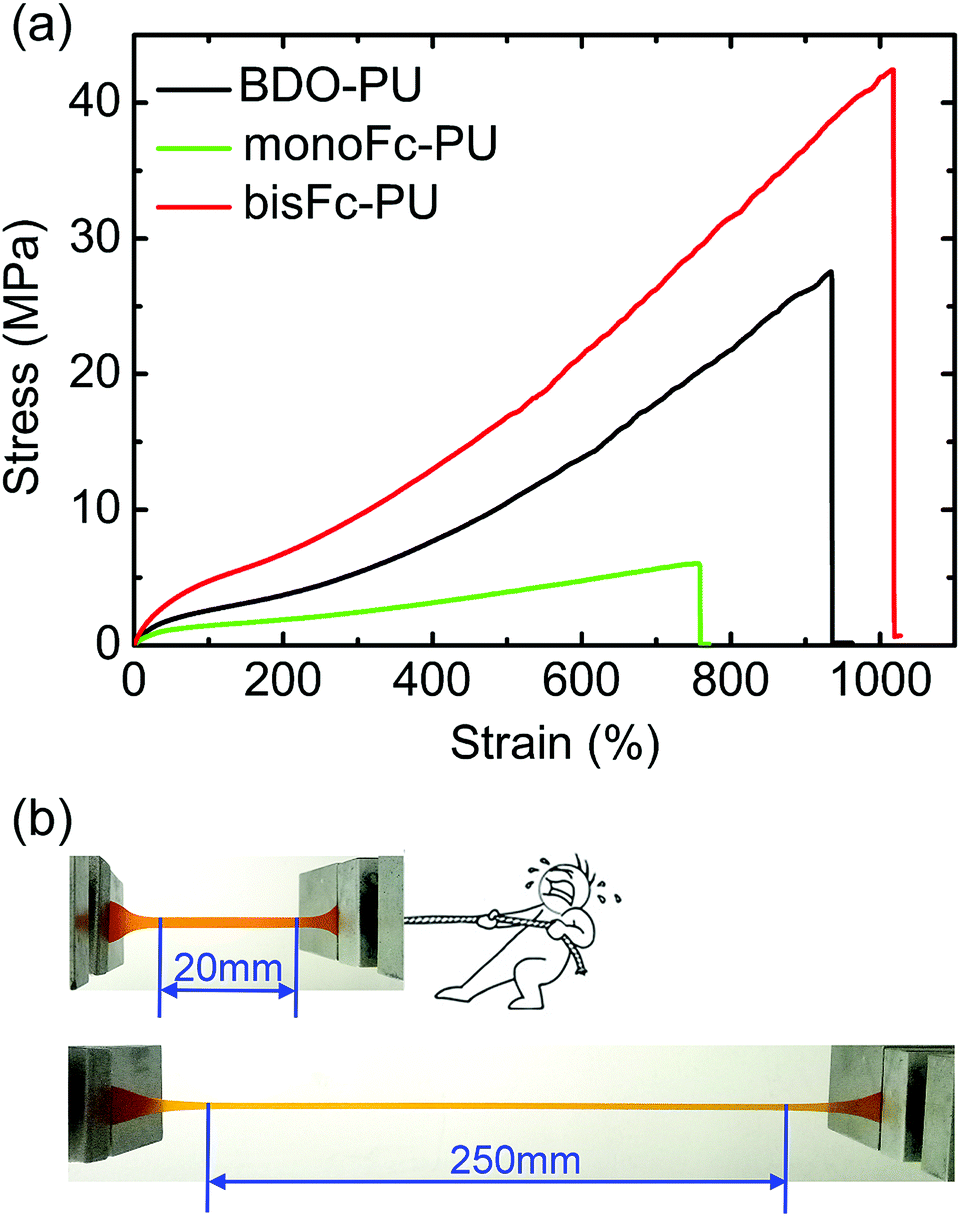 | ||
| Fig. 5 (a) Mechanical behaviors of BDO-PU, monoFc-PU, and bisFc-PU. (b) Digital photos showing bisFc-PU with an elongation of over 1000%. | ||
Fig. 6 shows cycle test data of BDO-TPU, monoFc-PU and bisFc-PU. The stress values of bisFc-PU at each cycle are all higher than of BDO-PU and monoFc-PU. These results are consistent with the data via stress–strain and strongly suggest higher mechanical properties of bisFc-PU. To investigate the elastic property, residual strain (εr) was normalized by maximum imposed strain (εm).36 The normalized residual strain (εr/εm) (Fig. 6b) is based on the data taken from Fig. 6a. As shown in Fig. 6b, the εr/εm of all PUs increased with εm, but with the exception of monoFc-PU (εr/εm > 0.2), the εr/εm was observed within 0 and 0.1 for BDO-PU and bisFc-PU. These results further prove that it is not the only factor to increase the mechanical properties of PUs through increasing microphase separation. The superstructure is the key to affecting the mechanical properties of PUs.
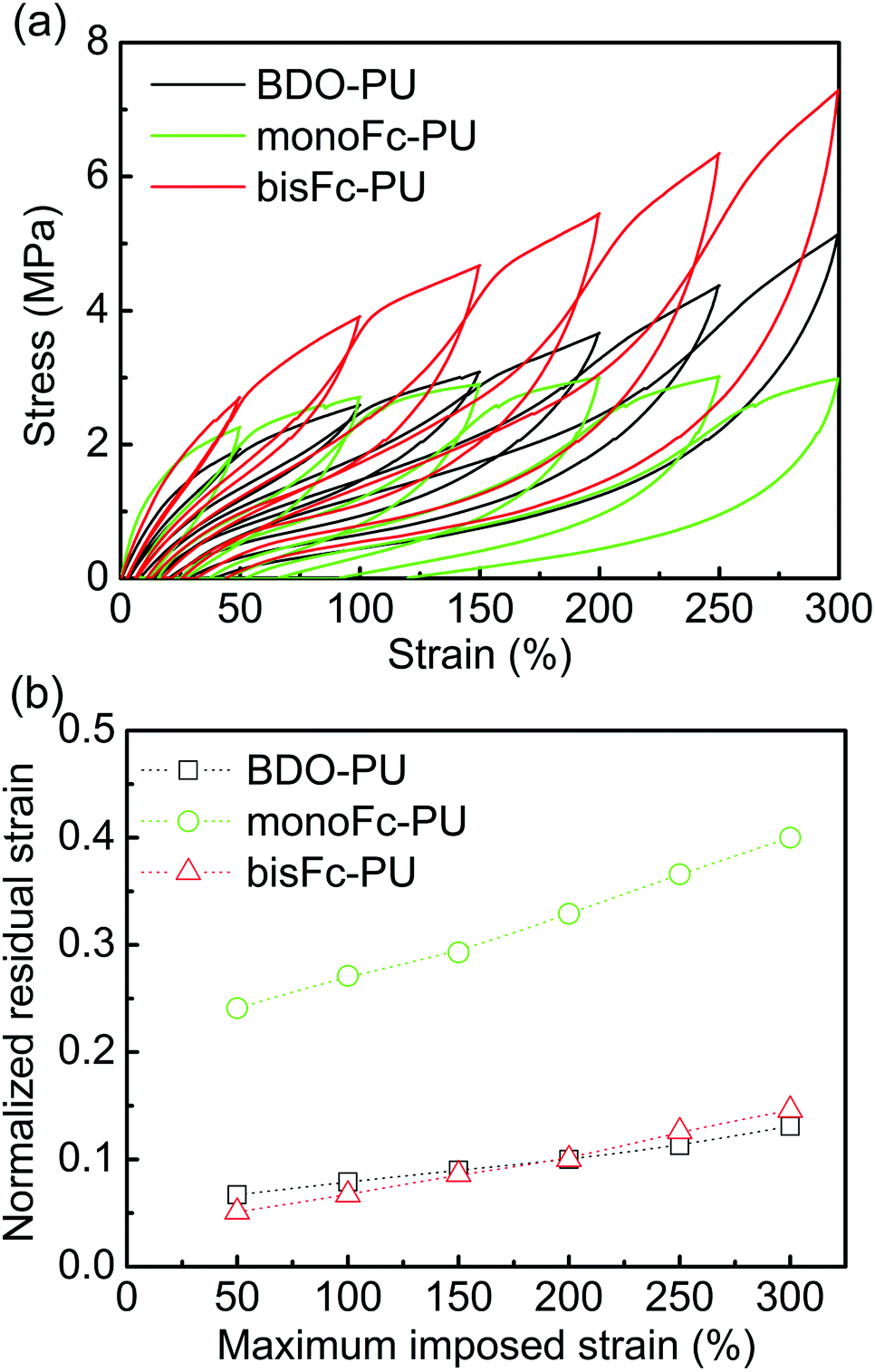 | ||
| Fig. 6 (a) Cycle test of BDO-PU, monoFc-PU, and bisFc-PU. (b) Normalized residual strain-maximum imposed strain relation obtained from (a). | ||
Introducing ferrocene and ester bond into the hard segments of PU chains, which will increase the cohesive force of the hard segments that mainly comes from the π–π force of the Cp rings, as well as more H-bonding interaction. These factors can lead to a greater difference about polarity and compatibility between the hard and soft segments. However, monoFc-PU did not show the same elastic property as good as bisFc-PU. This result can be ascribed to the unfavourable superstructure of monoFc-PU. Larger and inhomogenous size of superstructure are likely to cause more defects so as to arise local stress concentration (Fig. 3b) and thus turns into a brittle polymer structure. However, due to the high spatial freedom of polar bisferrocenyl diol, the H-bonded geometrical structure will be less ordered and thus texture superstructure of bisFc-PU could be obtained with a relatively low crystallinity. The leaf-shaped and micro-sized superstructure can also act as “dendrite topographies” to improve the mechanical properties. The presence of the extra ester groups will enhance the noncovalent crosslinking interaction among the PUs network and therefore brings bisFc-PU to exhibit a highest elongation at break (1018%) and toughness (19.6 GJ m−3). Moreover, bisFc-PU also delivers an excellent elastic shape memory performance. When the thin film (70 × 12 × 0.2 mm) was tested with a load of 4 kg, the shape recovery is up to 99.1% in 1 min while BDO-PU thin film is failure to perform in the same way (Fig. 7).
 | ||
| Fig. 7 The comparison test of shape recovery between BDO-PU and bisFc-PU with a load of 4 L (about 4 kg) in 1 min. | ||
Moreover, the thermo-stability of bisFc-PU was also increased greatly with a T5% of 345 °C by increasing the number of ferrocene centers into the main chain. This is the first thermoplastic polyurethane elastomers with such high thermo-stability and excellent mechanical properties, compared to the present reported PUs.7,12,19,50 Furthermore, the comprehensive performance of such pure polyurethane is even superior to many polyurethane-based composite polymers.51–56
4. Conclusions
In summary, a novel chemistry strategy was developed to solve the issue of mutually exclusive high mechanical robustness and thermo-stability for pure polyurethane. Ester-containing ferrocenyl diols were introduced as the chain extenders to tune the thermo-degradation temperatures and mechanical performance. A large number of H-bonds can enhance the noncovalent crosslinking inter-actions of polyurethanes while the unfavourable phase separation and superstructure of polyurethanes can also be modified by the polarity of ferrocenyl molecular structure. Thus a high-performance PU polymer can be synthesized with a high initial degradation temperature of 345 °C, a highest tensile strength of 42.3 MPa with a strain over 1000%, as well as a toughness of 19.6 GJ m−3. Ferrocene-containing polymers possess outstanding organometallic properties, including air-, heat- and photochemical stability, as well as interesting redox properties. These features will enable them to find more applications in smart materials and electrochemical areas.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Priority Academic Program Development of Jiangsu Higher Education Institutions and the State & Local Joint Engineering Laboratory for Novel Functional Polymeric Materials. We also thank Dr Jia Jia Chen for useful discussion.References
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed. Engl., 2013, 52, 9422–9441 CrossRef PubMed.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef PubMed.
- R. Klajn, L. Fang, A. Coskun, M. A. Olson, P. J. Wesson, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2009, 131, 4233–4235 CrossRef PubMed.
- T. Yuan, Y. Xu, C. Zhu, Z. Jiang, H.-J. Sue, L. Fang and M. A. Olson, Chem. Mater., 2017, 29, 9937–9945 CrossRef.
- W. Zhang, Y. Li, J. H. Sun, C. P. Tan, L. N. Ji and Z. W. Mao, Chem. Commun., 2015, 51, 1807–1810 RSC.
- Y. Yang and M. W. Urban, Angew. Chem., Int. Ed. Engl., 2014, 53, 12142–12147 CrossRef PubMed.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5261–5268 CrossRef.
- L. Huang, N. Yi, Y. Wu, Y. Zhang, Q. Zhang, Y. Huang, Y. Ma and Y. Chen, Adv. Mater., 2013, 25, 2224–2228 CrossRef PubMed.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef.
- G. A. Filonenko and J. R. Khusnutdinova, Adv. Mater., 2017, 29, 1700563 CrossRef PubMed.
- S. Ji, W. Cao, Y. Yu and H. Xu, Adv. Mater., 2015, 27, 7740–7745 CrossRef PubMed.
- A. Biswas, V. K. Aswal, P. U. Sastry, D. Rana and P. Maiti, Macromolecules, 2016, 49, 4889–4897 CrossRef.
- T. S. Hansen, K. West, O. Hassager and N. B. Larsen, Adv. Funct. Mater., 2007, 17, 3069–3073 CrossRef.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068–1133 CrossRef.
- X. Liu, J. Hao and S. Gaan, RSC Adv., 2016, 6, 74742–74756 RSC.
- H. Sui, X. Ju, X. Liu, K. Cheng, Y. Luo and F. Zhong, Polym. Degrad. Stab., 2014, 101, 109–113 CrossRef.
- M. Dascalu, V. E. Musteata, L. Vacareanu, C. Racles and M. Cazacu, RSC Adv., 2015, 5, 99193–99200 RSC.
- F. Askari, M. Barikani and M. Barmar, J. Appl. Polym. Sci., 2013, 130, 1743–1751 CrossRef.
- B. Hui and L. Ye, Eur. Polym. J., 2017, 91, 337–353 CrossRef.
- K. Wei, L. Wang and S. Zheng, Polym. Chem., 2013, 4, 1491–1501 RSC.
- N. Senthilkumar, A. Raghavan and A. S. Nasar, Macromol. Chem. Phys., 2005, 206, 2490–2500 CrossRef.
- N. Senthilkumar, T. Narasimhaswamy and I.-J. Kim, Mater. Sci. Eng. C, 2012, 32, 2258–2266 CrossRef.
- L. Y. Chiang, L. Y. Wang and C.-S. Kuo, Macromolecules, 1995, 28, 7574–7576 CrossRef.
- W. JianJun and S. TieJun, China Synth. Rubber Ind., 2001, 24, 347–349 Search PubMed.
- K. Kojio, M. Furukawa, S. Motokucho, M. Shimada and M. Sakai, Macromolecules, 2009, 42, 8322–8327 CrossRef.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 1–8 CrossRef PubMed.
- O. Lebel, T. Maris, M.-E. Perron, E. Demers and J. D. Wuest, J. Am. Chem. Soc., 2006, 128, 10372–10373 CrossRef PubMed.
- B. Lucio and J. L. de la Fuente, Eur. Polym. J., 2014, 50, 117–126 CrossRef.
- A. Neidlinger, T. Kienz and K. Heinze, Organometallics, 2015, 34, 5310–5320 CrossRef.
- J. Guasch, L. Grisanti, S. Jung, D. Morales, G. D'Avino, M. Souto, X. Fontrodona, A. Painelli, F. Renz, I. Ratera and J. Veciana, Chem. Mater., 2013, 25, 808–814 CrossRef.
- C.-D. Varganici, O. Ursache, C. Gaina, V. Gaina, D. Rosu and B. C. Simionescu, Ind. Eng. Chem. Res., 2013, 52, 5287–5295 CrossRef.
- M.-C. Kuo, R.-J. Jeng, W.-C. Su and S. A. Dai, Macromolecules, 2008, 41, 682–690 CrossRef.
- R. A. Nallicheri and M. F. Rubner, Macromolecules, 1990, 23, 1017–1025 CrossRef.
- C. M. Brunette, S. L. Hsu and W. J. MacKnight, Macromolecules, 1982, 15, 71–77 CrossRef.
- H. S. Lee, Y. K. Wang and S. L. Hsu, Macromolecules, 1987, 20, 2089–2095 CrossRef.
- S. Nozaki, S. Masuda, K. Kamitani, K. Kojio, A. Takahara, G. Kuwamura, D. Hasegawa, K. Moorthi, K. Mita and S. Yamasaki, Macromolecules, 2017, 50, 1008–1015 CrossRef.
- K. Kojio, T. Fukumaru and M. Furukawa, Macromolecules, 2004, 37, 3287–3291 CrossRef.
- C. B. Hu and R. S. Ward, J. Appl. Polym. Sci., 1982, 27, 2167–2177 CrossRef.
- Y. Xiao, H. Huang and X. Peng, RSC Adv., 2017, 7, 20093–20100 RSC.
- L. M. Leung and J. T. Koberstein, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 1883–1913 CrossRef.
- J. T. Koberstein, A. F. Galambos and L. M. Leung, Macromolecules, 1992, 25, 6195–6204 CrossRef.
- J. V. Duffy, G. F. Lee, J. D. Lee and B. Hartmann, ACS Symp. Ser., 1990, 424, 281–300 CrossRef.
- K. Kojio, M. Furukawa, S. Matsumura, S. Motokucho, T. Osajima and K. Yoshinaga, Polym. Chem., 2012, 3, 2287–2292 RSC.
- S. Nozaki, T. Hirai, Y. Higaki, K. Yoshinaga, K. Kojio and A. Takahara, Polymer, 2017, 116, 423–428 CrossRef.
- S. Barlow, A. L. Rohl and D. O'Hare, Chem. Commun., 1996, 257–260 RSC.
- S. Barlow, A. L. Rohl, S. Sh, C. M. Freeman and D. O'Hare, J. Am. Chem. Soc., 1996, 118, 7578–7592 CrossRef.
- X. Lv, Z. Huang, M. Shi, Y. Fan and G. Gao, RSC Adv., 2016, 6, 111688–111701 RSC.
- S. Zhang, Y. Cheng, W. Xu, J. Li, J. Sun, J. Wang, C. Qin and L. Dai, RSC Adv., 2017, 7, 56682–56690 RSC.
- X. Fu, J. Liu, Y. Wan, X. Zhang, F. Meng and J. Liu, J. Mater. Chem., 2012, 22, 17782–17791 RSC.
- M.-C. Kuo, S.-M. Shau, J.-M. Su, R.-J. Jeng, T.-Y. Juang and S. A. Dai, Macromolecules, 2012, 45, 5358–5370 CrossRef.
- Q. Zheng, Z. Ma and S. Gong, J. Mater. Chem. A, 2016, 4, 3324–3334 Search PubMed.
- X. Yao, X. Qi, Y. He, D. Tan, F. Chen and Q. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 2497–2507 Search PubMed.
- J. Li, G. Zhang, R. Sun and C.-P. Wong, J. Mater. Chem. C, 2017, 5, 220–228 RSC.
- M. Bera and P. K. Maji, Polymer, 2017, 119, 118–133 CrossRef.
- M. Kotal and S. K. Srivastava, J. Mater. Chem., 2011, 21, 18540–18551 RSC.
- J. Li, G. Zhang, L. Deng, S. Zhao, Y. Gao, K. Jiang, R. Sun and C. Wong, J. Mater. Chem. A, 2014, 2, 20642–20649 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data and supplementary figures. See DOI: 10.1039/c8ra02784f |
| This journal is © The Royal Society of Chemistry 2018 |