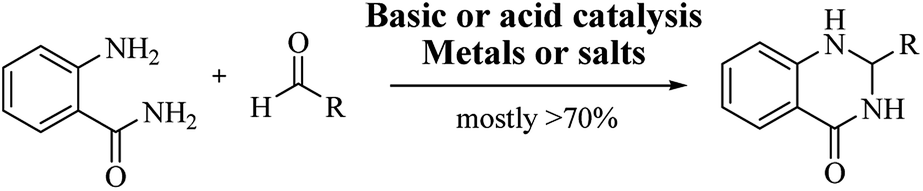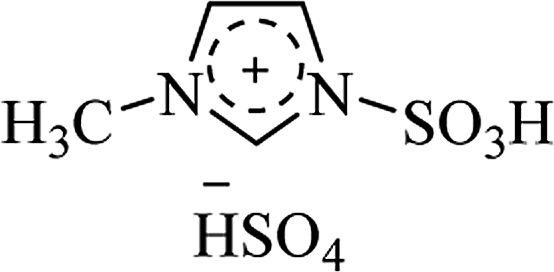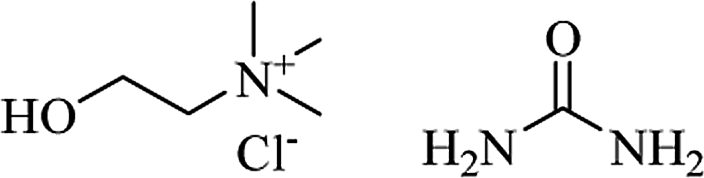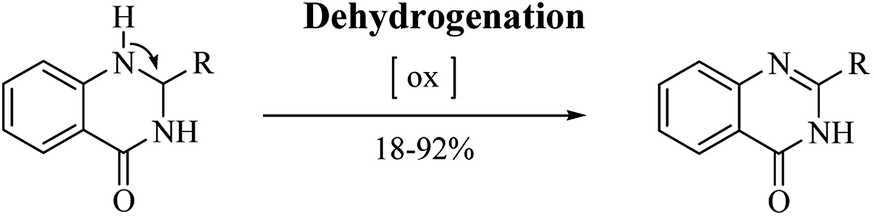DOI:
10.1039/C8RA02827C
(Review Article)
RSC Adv., 2018,
8, 20894-20921
2,3-Dihydroquinazolin-4(1H)-one as a privileged scaffold in drug design
Received
2nd April 2018
, Accepted 29th May 2018
First published on 7th June 2018
Abstract
2,3-Dihydroquinazolin-4-one (DHQ) belongs to the class of nitrogen-containing heterocyclic compounds representing a core structural component in various biologically active compounds. In the past decades, several methodologies have been developed for the synthesis of the DHQ framework, especially the 2-substituted derivatives. Unfortunately, multistep syntheses, harsh reaction conditions, and the use of toxic reagents and solvents have limited their full potential as a versatile fragment. Recently, use of green chemistry and alternative strategies are being explored to prepare diverse DHQ derivatives. This fragment is used as a synthon for the preparation of biologically active quinazolinones and as a functional substrate for the synthesis of modified DHQ derivatives exhibiting different biological properties. In this review, we provide a comprehensive assessment of the synthesis and biological evaluations of DHQ derivatives.
| Mariateresa Badolato is currently pursuing her PhD degree in Translational Medicine under the supervision of Dr Francesca Aiello and Professor Antonio Garofalo at the University of Calabria, Italy. She received her Master's degree in Pharmaceutical Technology and Chemistry at the University of Calabria in 2013. She worked for one year as a visiting scholar in Professor Neamati's laboratory at the Department of Medicinal Chemistry, College of Pharmacy, University of Michigan. With the project “RYGoldZip” guided by Dr Francesca Aiello, as a member of the team she won the Clinic Center Life Sciences award. Among various projects, she designed and synthesized several ligands for TRPV1 receptors. Her PhD dissertation focuses on the preclinical and mechanistic studies of small-molecule drugs targeting STAT3 and other signaling pathways for the treatment of various cancers. |
| Dr Francesca Aiello is an Assistant Professor of Medicinal Chemistry at the Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, Italy. She obtained her PhD degree in Pharmaceutical and Technologies Chemistry from the University of Siena in 1998. In 1999, she obtained her qualification as a Pharmacist. She completed a Postdoctoral Research Fellowship under the supervision of Professor Neamati at the Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, CA. She was a lecturer in Qualitative Analytical Medicinal Chemistry at the University of Calabria. During the past decade she received numerous grants from the University of Salerno, University of Calabria, and University of Siena to develop new anticancer chemotherapeutics, HIV-1 integrase inhibitors, and to synthesize new D3-receptor ligands. Dr Aiello has published 38 peer-reviewed manuscripts, 4 book chapters, and is an inventor in two patents in the area of Medicinal Chemistry. As leader of the project “RYGoldZip”, she discovered and formulated a topical product with wound healing properties for diabetic ulcers and she won the Clinic Center Life Sciences award. |
| Dr Nouri Neamati is the John G. Searle Professor of Medicinal Chemistry at the College of Pharmacy, University of Michigan and a member of the UM Translational Oncology Program. He obtained his PhD degree in Biomedical Sciences from the University of Texas Graduate School of Biomedical Sciences and MD Anderson Cancer Center, Houston, Texas in 1995. From 1995 to 2000, he was a postdoctoral and a research fellow at the National Institutes of Health (NIH) in Bethesda, MD. In September 2000, he joined the faculty at the School of Pharmacy, University of Southern California, with a joint appointment at the Norris Comprehensive Cancer Center. Professor Neamati is the recipient of numerous awards, including the NIH Technology Transfer, STOP CANCER, GlaxoSmithKline Drug Discovery Award, several Awards from the US Department of Defense, LUNGevity Discovery Award from the American Lung Association, and the Littlefield-AACR award in Metastatic Colon Cancer Research. He has published over 230 peer-reviewed manuscripts, 20 book chapters, and is an inventor in over 30 patents in the area of drug design and discovery. He was the founding Editor-in-Chief of Current Molecular Pharmacology, and is currently an Associate Editor of the Journal of Medicinal Chemistry as well as Current Anticancer Drug Targets. He has served as an Editorial Advisory Board member of several journals, including Expert Opinion on Drug Discovery, Expert Opinion on Investigational Drugs, and Hormones & Cancer. |
Introduction
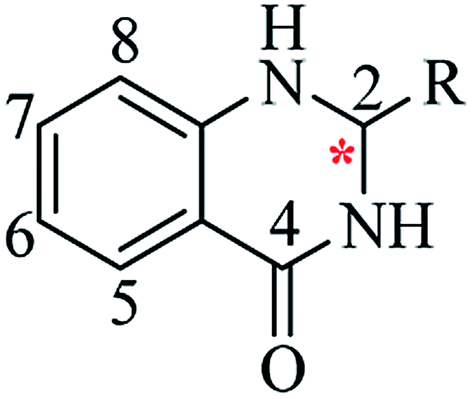
Nitrogen-containing heterocyclic scaffolds are quite common fragments in drugs and biologically active compounds.1,2 The 2,3-dihydroquinazolin-4(1H)-one (DHQ) is an important nitrogen-containing motif consisting of a phenyl ring condensed with a six-membered ring with two nitrogen atoms on positions 1 and 3, and a keto group on carbon 4 (Fig. 1). Most of the DHQ derivatives are substituted on the carbon 2 chiral center. Due to their attractive properties, 2-substituted DHQs are becoming a prominent synthetic intermediate for organic chemists and various methodologies are reported in the literature for their preparation as racemic mixtures. Although some asymmetric strategies have been attempted, the aminal chiral center is sensitive to racemization, making it difficult to synthesize pure enantiomers.3 The aim of the present review is to discuss reported methods for the synthesis of DHQ derivatives, highlighting their evolution towards alternative approaches and enantioselective strategies, and to summarize their use as synthons in organic chemistry and their important biological activities.
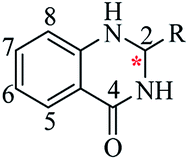 |
| | Fig. 1 2,3-Dihydroquinazolin-4(1H)-one framework. | |
2,3-Dihydroquinazolin-4(1H)-one: a privileged scaffold
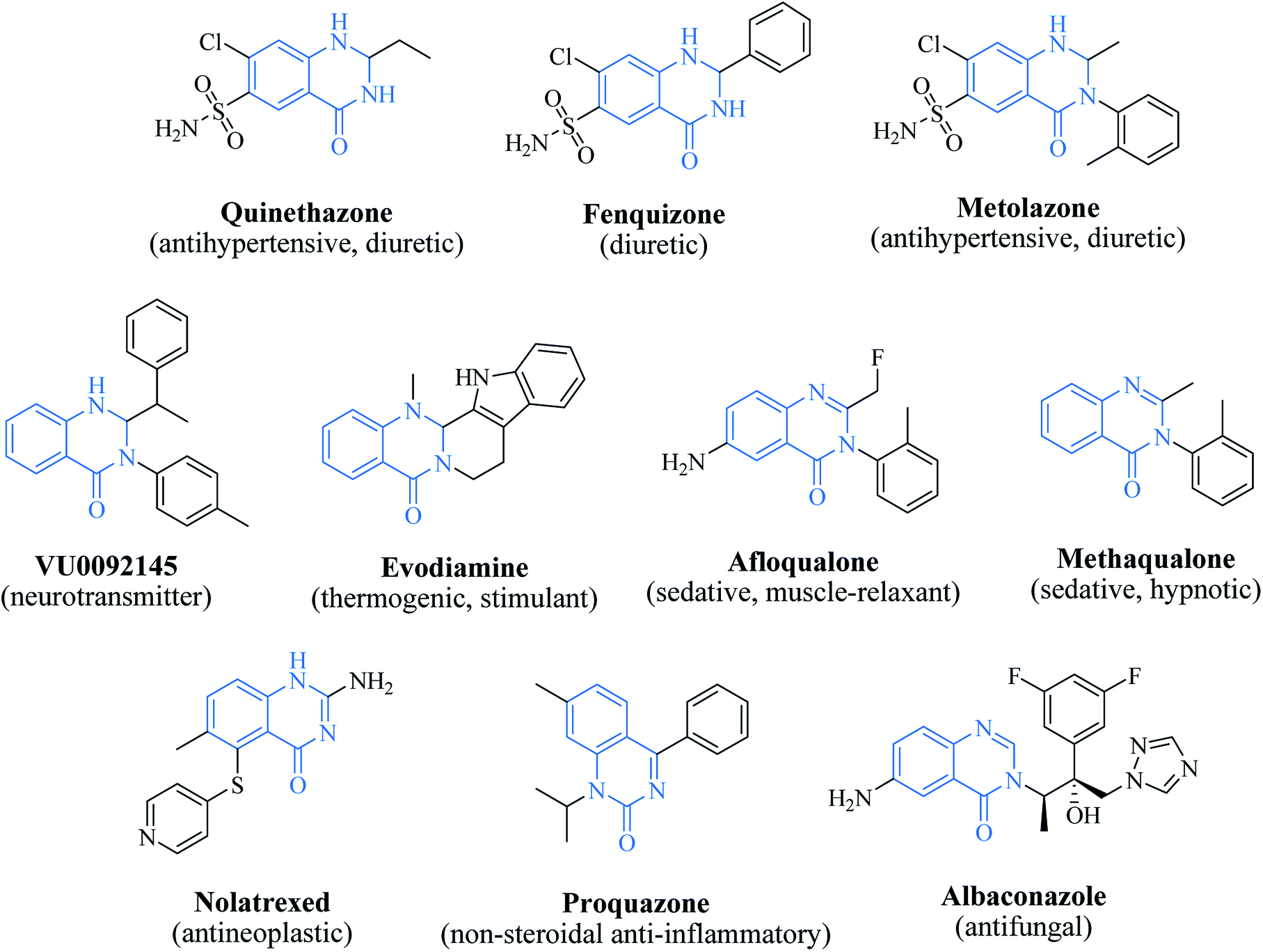
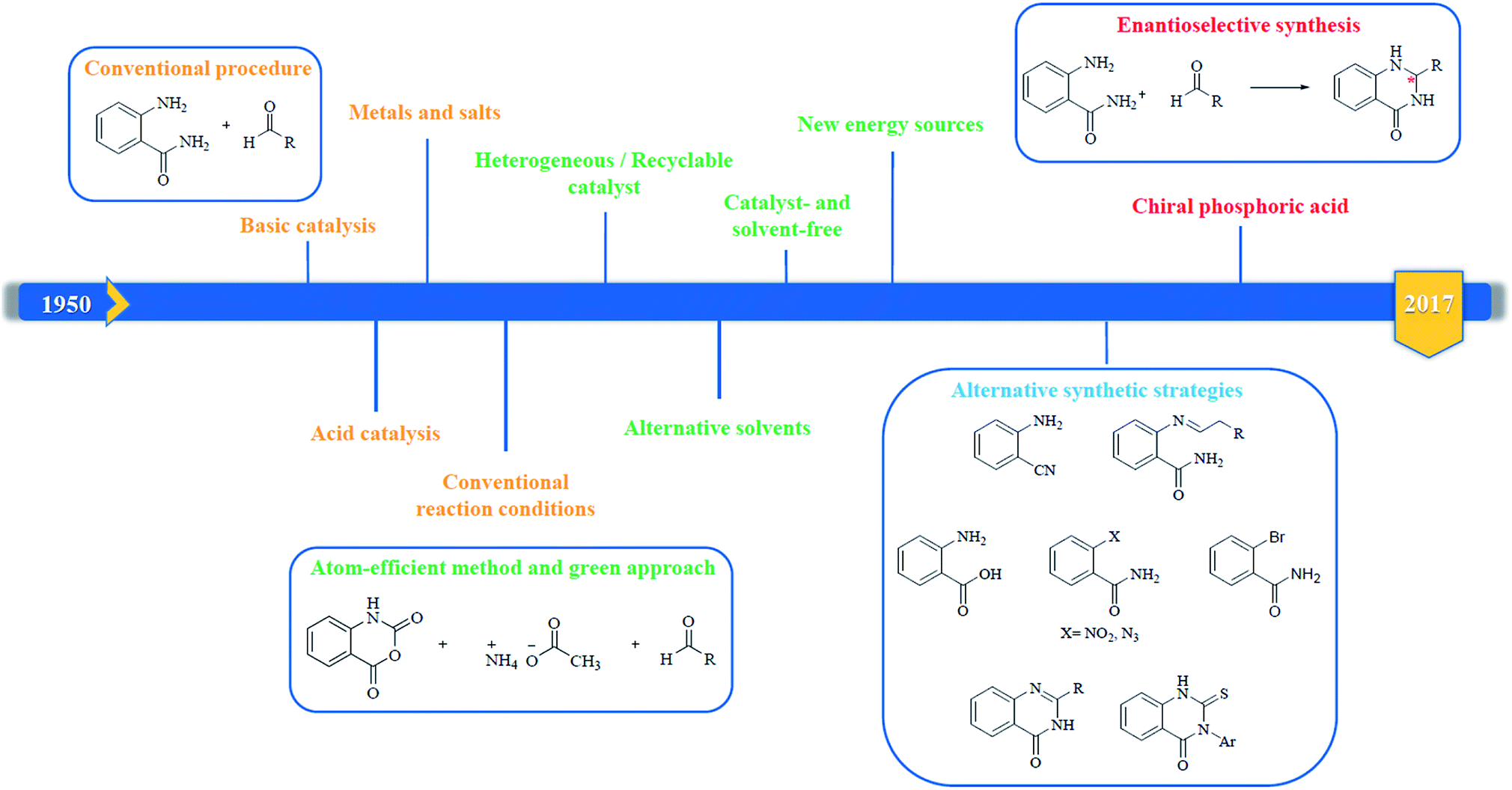
In 1988, Evans et al. introduced for the first time the concept of “privileged structures”. They are useful tools in the field of drug discovery since they represent suitable lead compounds for diverse receptors and the rational optimization of such structures could provide new receptor modulators and potential drugs.4 Medicinal chemists exploit the “privileged structures” to synthesize new libraries of compounds based on a central scaffold and screen them against various receptors implicated in different pathways, in some cases yielding biologically active compounds. In this regard, the DHQ core is emerging as a “privileged scaffold” and a variety of its derivatives, having diverse mechanism of action, are currently used for the treatment of various diseases.5–11 A panel of marketed drugs with the DHQ core is shown in Fig. 2. Besides these marketed drugs, a number of new DHQ derivatives have been designed that exhibit a wide range of pharmacological properties. Because of their importance, the synthesis of substituted DHQ derivatives has attracted much attention and different synthetic strategies have been developed. Since the classical protocols involved the use of toxic reagents and solvents in harsh reaction conditions, the evolution towards simple, clean, environmentally benign and high-yielding methods is gaining momentum (Fig. 3).
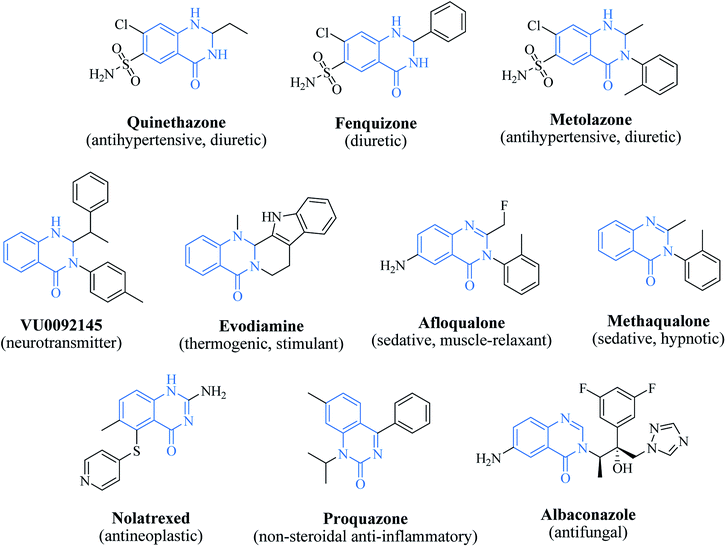 |
| | Fig. 2 The “privileged scaffold” DHQ in marketed drugs. | |
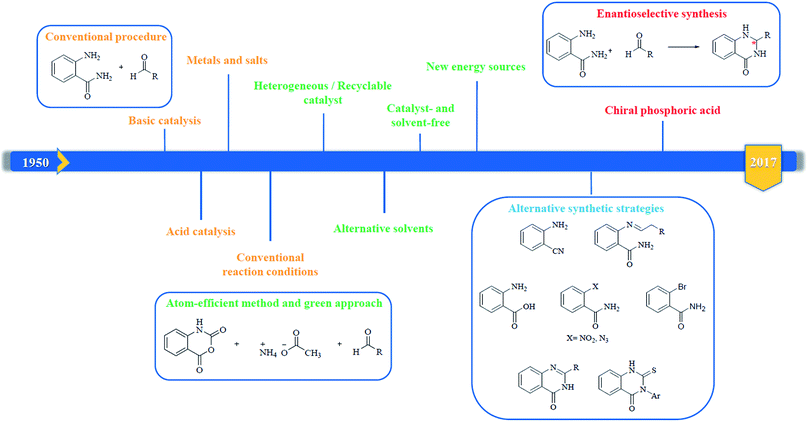 |
| | Fig. 3 Evolution of the synthetic strategies to prepare DHQ core. | |
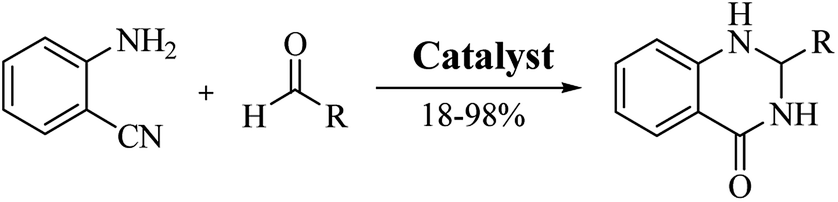
Conventional procedure: cyclocondensation of anthranilamide and an aldehyde
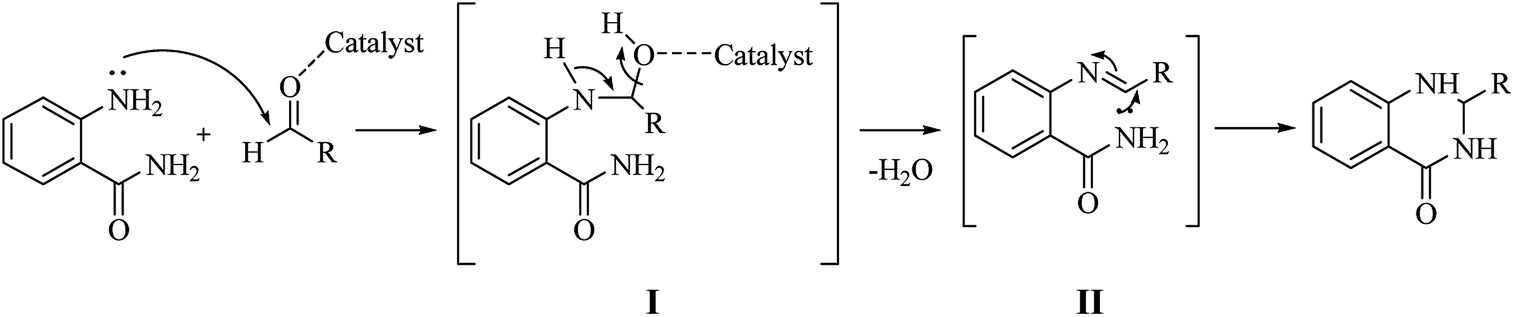
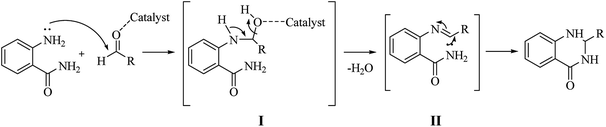
Although numerous strategies have been developed for the construction of the DHQ core, the most common and simple synthetic route for the preparation of DHQs is the direct cyclocondensation of anthranilamide and an aldehyde (Scheme 1). In the past decades, different catalysts and organic solvents have been used to speed up and improve the general yield of the reaction (Table 1). Regardless of the used catalyst and/or solvent, the most presumed mechanism of cyclocondensation is shown in Scheme 2.12 The first step involves the nucleophilic attack of the nitrogen of the amino group of the anthranilamide on the carbonyl carbon of the aldehyde, promoted by the catalyst, resulting in the formation of hydroxyl intermediate I. Next, the catalyst promotes the formation of the Schiff base (II) from I through the removal of a water molecule. Finally, the imine undergoes intramolecular cyclization by nucleophilic attack of the nitrogen of the amide group on the imine carbon, to furnish the corresponding DHQ derivative.
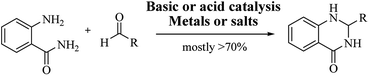 |
| | Scheme 1 Direct cyclocondensation of anthranilamide and aldehyde under conventional conditions. | |
Table 1 Conventional conditions for the cyclocondensation of anthranilamide and an aldehyde
| Nature catalyst |
Catalyst |
Conditions |
Time |
Yield |
Ref. |
| Strong base |
NaOH |
EtOH, reflux |
1 h |
60–86% |
13–15 |
| NaOEt |
EtOH, reflux |
3–4 h |
20–85% |
14 |
| Strong BrØnsted acid |
HCl |
EtOH, reflux |
4 h |
10% |
14 |
| Conc. HNO3/HCl |
Reflux → rt |
5 → 30 min |
>98% |
16 |
| H2SO4 |
Solvent-free, MWI |
Few min |
68–78% |
17 |
| Sulfonic acid functionality |
PTSA |
Chlorobenzene, reflux |
1 h |
74% |
13 |
| Benzene, reflux |
4 h |
70% |
18 |
| DMAC, rt/reflux |
1–2 h |
75–95% |
19 and 20 |
| EtOH, reflux |
1 h |
70–90% |
21 |
| MES |
Aq. EtOH (50%), MWI |
5–20 min |
83–96% |
22 |
| Sulfanilic acid |
Aq. EtOH (50%), 70 °C |
|
|
23 |
| NaHSO4 |
EtOH, rt |
0.5–5.5 h |
91–97% |
24 |
| |
H2O, grinding rt → 60 °C |
0.5–7 h |
54–97% |
25 |
| SOCl2 |
EtOH, rt |
30–35 min |
93–95% |
26 |
| Weak BrØnsted acid |
Formic acid |
20 °C |
|
|
12 |
| Malonic acid |
Aq. EtOH (50%), rt |
5–37 min |
81–98% |
27 |
| Weak Lewis acid |
H3BO3 |
Solvent-free, 120 °C |
5 min |
82–90% |
28 |
| Organic |
T3P® |
AcCN, rt |
10–15 min |
85–94% |
29 |
| Lewis acid |
I2 |
ILs, 50 °C/80 °C |
0.5–10 h |
76–99% |
30 and 31 |
| EtOAc, hv |
1–15 h |
66–93% |
32 |
| Lugol's solution |
I2/KI |
H2O, rt |
2–12 h |
47–95% |
33 |
| Organic |
C3Cl3N3 |
AcCN, rt |
8–20 min |
60–96% |
40 |
| Lewis acid |
Mn(CH3COO)2 |
EtOH, reflux |
5 h |
|
34 |
| ZrCl2 |
EtOH, rt |
9–60 min |
80–97% |
35 |
| HgCl2 |
EtOH, 60 °C |
1–2 h |
88–94% |
36 |
| Cp2TiCl2 |
EtOH, rt |
7–9 min |
95–98% |
37 |
| InBr3 |
AcCN, rt |
10–60 min |
75–98% |
38 |
| BiBr3 |
AcCN, rt |
30 min |
80–95% |
39 |
| Sc(OTf)3 |
EtOH, 70 °C |
20–40 min |
85–92% |
42 |
| Dry DCM, rt |
4–7 h |
85–94% |
43 |
| PEG-400, 80 °C |
2 h |
78–90% |
44 |
| Yb(OTf)3 |
EtOH, 80 °C |
2–6 h |
>95% |
45 |
| IL, rt |
6–8 h |
85–96% |
46 |
| Y(OTf)3 |
EtOH, rt |
1.5 h |
88–99% |
47 |
| Ammonium salt |
NH4Cl |
EtOH, rt/reflux |
5–120 min |
78–98% |
48 and 49 |
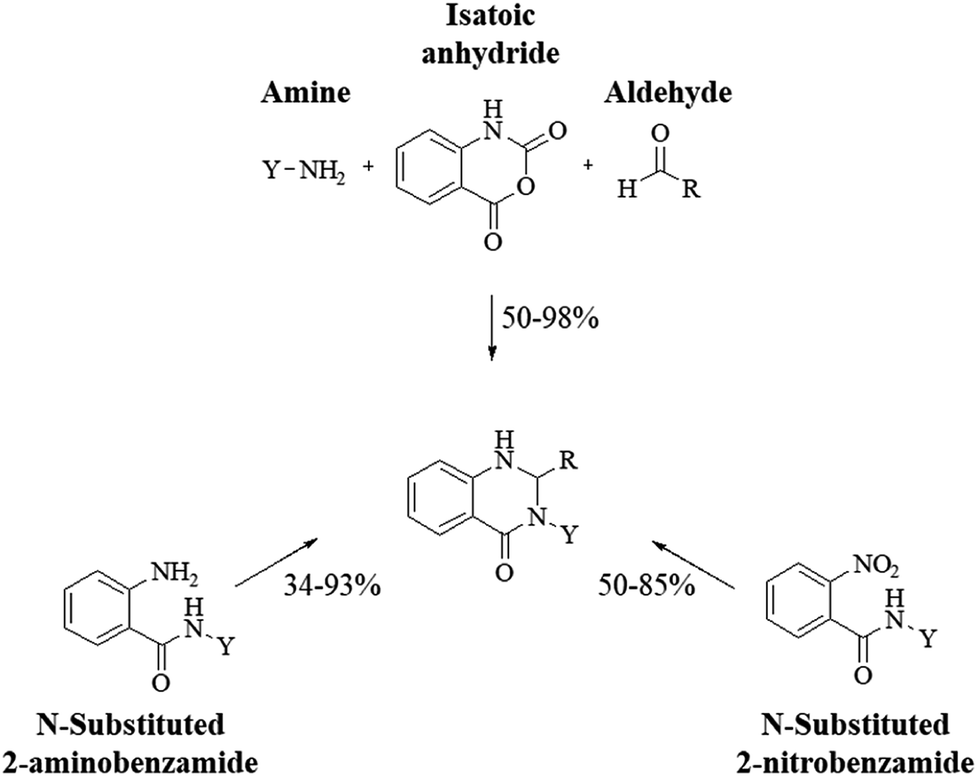
| CAN |
H2O, rt → 60 °C |
1–8 h |
62–97% |
50 |
| TBAHS |
MeOH, reflux |
2 h |
64–90% |
51 |
 |
| | Scheme 2 Presumed mechanism of the cyclocondensation of anthranilamide and an aldehyde. | |
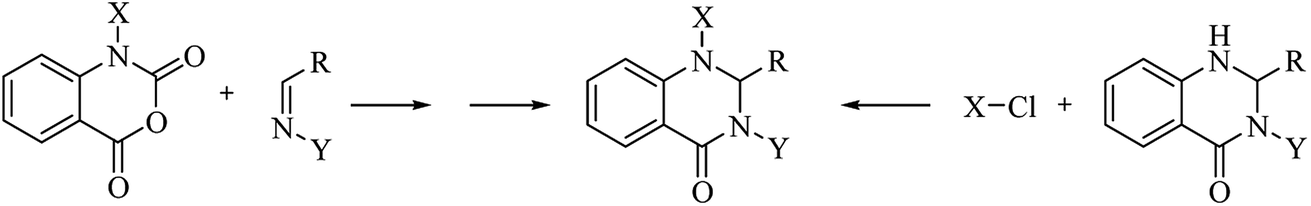
A variation of the traditional cyclocondensation of anthranilamide and an aldehyde is represented by the intramolecular cyclization of a Schiff base. As shown in Scheme 3, the nucleophilic attack of the nitrogen of the amide group on the imine carbon leads to the cyclic DHQ derivative.
 |
| | Scheme 3 Intramolecular cyclization of a Schiff base | |
Basic catalysis
The base-catalyzed cyclocondensation of anthranilamide and an aldehyde was the first proposed strategy for the synthesis of DHQ derivatives. In 1967, Yale et al. prepared a new class of DHQs in the presence of 20% aqueous NaOH in refluxing EtOH. These compounds were tested for their inhibition of cell proliferation.13 Later, Bonola et al. used NaOH or NaOEt in absolute EtOH to prepare DHQs with antibacterial and antifungal activities.14 Ericsson et al. patented DHQ derivatives as anti-fertility agents; these compounds were prepared from anthranilamide and 1-naphthaldehyde in the presence of NaOH in refluxing absolute EtOH.15 The NaOH-catalyzed reactions gave moderate to high yields (60–85%) in 1 h compared to NaOEt, which took 3–4 h to complete the formation of DHQs (Table 1). Depending on the available aldehyde, the use of a strong base as catalyst was not always successful. Because of this limitation, different catalysts were commonly used for the synthesis of DHQs starting from anthranilamide and an aldehyde.
Acid catalysis
As shown in Table 1, more frequently, the cyclocondensation of anthranilamide and an aldehyde is performed in acid catalysis. HCl in EtOH14 or in association with concentrated HNO3 (ref. 16) was used as catalyst under reflux conditions. As reported, DHQ derivatives were often synthesized in the presence of a catalytic amount of H2SO4 (ref. 17) or by using catalysts bearing sulfonic acid functionality. Among them, p-toluenesulfonic acid was used in different solvents, including boiling chlorobenzene,13 benzene,18 N,N-dimethylacetamide19,20 and EtOH.21 2-(N-Morpholino)ethanesulfonic acid, commonly used as a buffering agent in biology, also stood out as a mild acid catalyst for the cyclocondensation of anthranilamide and an aldehyde.22 The use of sulfanilic acid,23 NaHSO4,24,25 and SOCl2,26 as a catalyst in EtOH has also been described. Moreover, formic acid12 as catalyst and solvent, as well as malonic acid27 in aqueous EtOH (50%), were used for the synthesis of DHQs. Furthermore, boric acid (H3BO3),28 and propylphosphonic anhydride,29 usually employed in the Fisher indole synthesis and the Pictet–Spengler reaction, efficiently catalyzed the cyclocondensation of anthranilamide and aldehydes.
Iodine and metal salts as catalysts
Molecular iodine (I2) emerged as a versatile, inexpensive and non-toxic catalyst, which serves as a Lewis acid. It is suitable in the cyclocondensation of anthranilamide and aldehydes, in ionic liquids,30,31 or in EtOAc.32 A synthesis in aqueous medium was also attempted. Due to poor solubility of I2 in water, 1 mol% of I2 as Lugol's solution (I2/KI) was used to prepare DHQs.33 In the presence of I2 as catalyst, DHQ derivatives were obtained with 66–95% yield, but the reactions required up to 15 h to complete.
Transition metal salts are excellent catalysts due to their kinetic stability, low toxicity and intrinsic metallic Lewis acidity. They were used in various organic transformations, including the cyclocondensation of anthranilamide and aldehydes.34,40 In particular, the use of transition metal salts reduced reaction time from hours to minutes without decreasing the yield. Compared to conventional Lewis acids, metal triflates are better catalysts in organic synthesis because of their chemical and physical properties, such as moisture and air-stability, recoverability, operational simplicity, and a strong tolerance to oxygen, nitrogen, phosphorus, and sulfur-containing reaction substrates and functional groups.41 Among them, scandium,42–44 ytterbium,45,46 and yttrium47 triflates were extensively used in various conditions to catalyze the cyclocondensation of anthranilamide and aldehydes, leading to DHQs with high yields (78–99%) in 0.3–8 h.
As shown in Table 1, even ammonium salts stood out as good catalysts for the preparation of DHQ derivatives.48–51 The reactions required short time and yields were up to 60%.
Intramolecular cyclization of a Schiff base
The synthesis of DHQs through intramolecular cyclization of a Schiff base was performed under different conditions, providing DHQs with 42–99% yield in 1–16 h (Table 2). While strong basic conditions gave the final product in long time,52 the use of molecular nitrogen shortened the completion of the reaction.53 A rapid synthesis was obtained using metal oxide nanoparticles,54 and under catalyst-free conditions.55,56
Table 2 Reaction conditions for the intramolecular cyclization of a Schiff base
| Substrate |
Conditions |
Time |
% Yield |
Ref. |
| Schiff base |
NaH, THF, 0 °C → rt |
16 h |
42–91 |
52 |
| N2, EtOH, reflux |
6 h |
83 |
53 |
| Fe3O4 NPs, EtOH, reflux |
1–3.5 h |
94–98 |
54 |
| AcOH, reflux |
1.5 h |
80–92 |
55 and 56 |
An atom-efficient method: one-pot three-component synthesis
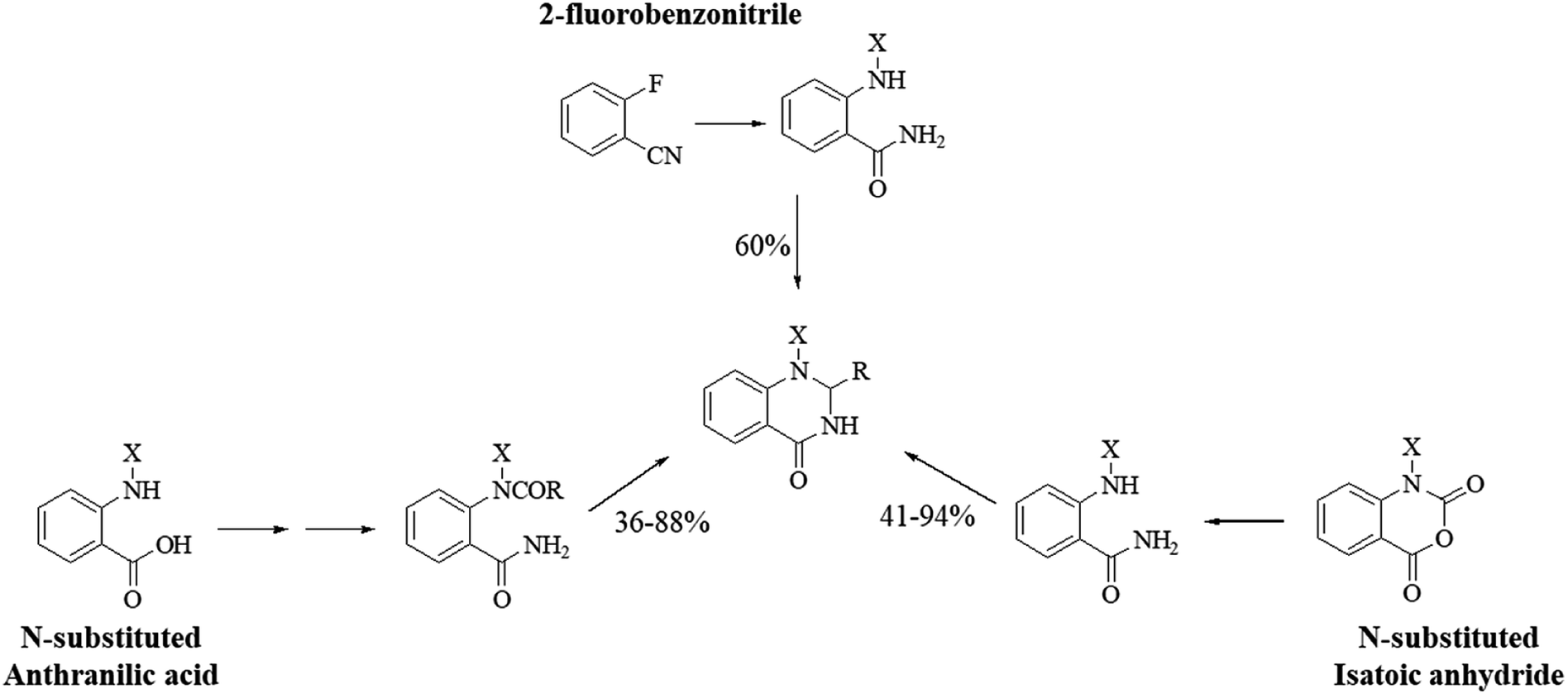
Although the cyclocondensation of anthranilamide and aldehydes is a facile and simple approach to obtain the DHQ core, more advantageous strategies to synthesize DHQ derivatives remain a desired goal in organic chemistry. The first evolution towards a more convenient approach was moving from a two-component reaction (anthranilamide and an aldehyde) to a one-pot three-component reaction (isatoic anhydride, ammonium acetate and an aldehyde) (Scheme 4). Initially, Staiger et al. suggested this strategy about half a century ago,57 and recently the advantages of multi-component reactions have been highlighted. Compared to conventional synthesis, the one-pot three-component reaction represents an attractive and atom-efficient method to efficiently prepare the DHQ framework.
 |
| | Scheme 4 Preparation of DHQ through one-pot three-component reaction. | |
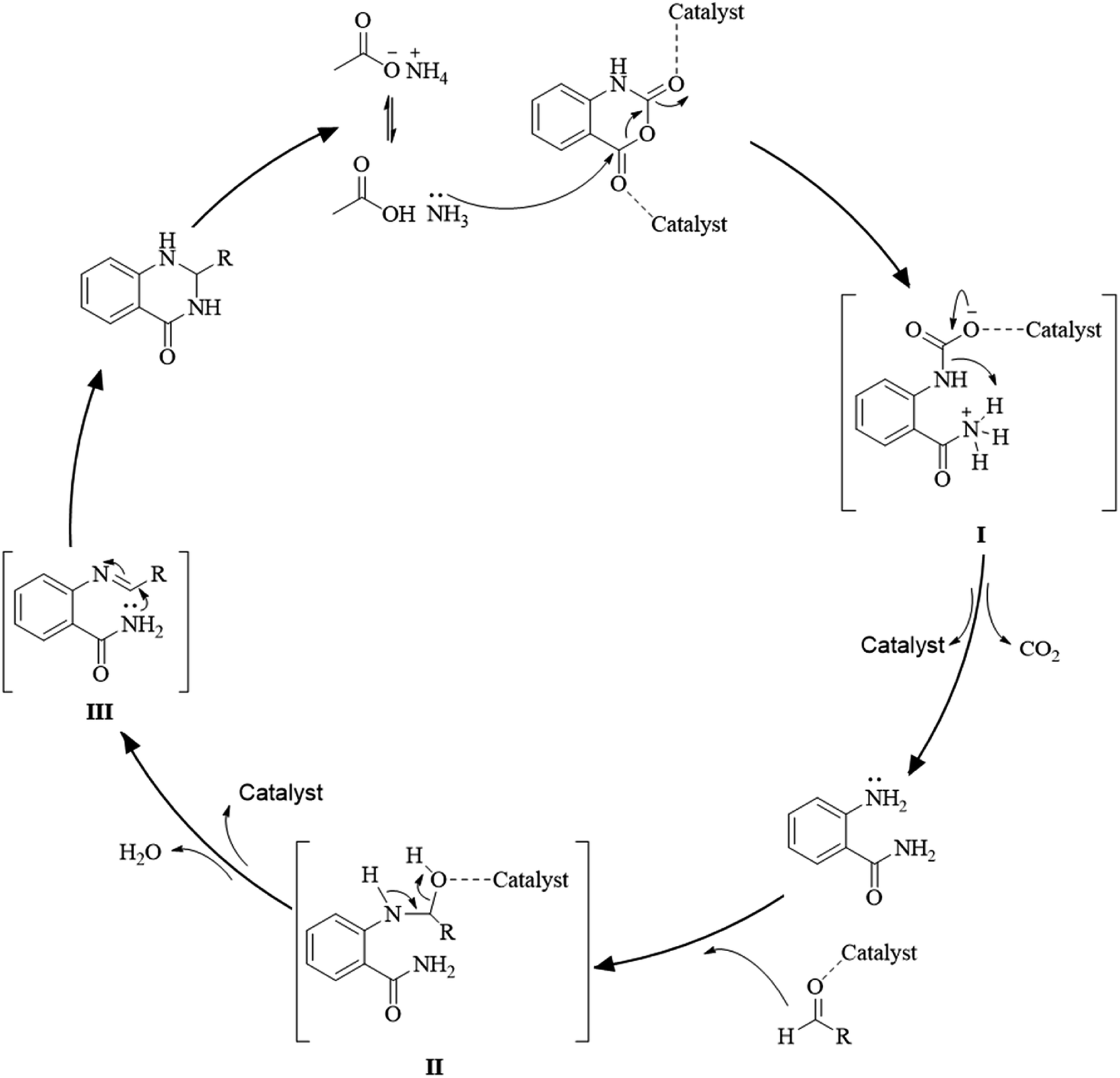
Multi-component reactions are characterized by (i) atom economy, incorporating all materials used in the process into the final product; (ii) high levels of diversity achieved simply by varying the reaction components; (iii) time-efficiency, since products are formed in a single step, allowing a fast probe of a chemical hypothesis; and (iv) simple experimental procedures, ideally there is no need to isolate the intermediates. Obviously, the adoption of such a strategy minimizes both waste production and cost.58 The plausible mechanism of the one-pot three-component synthesis of DHQs is shown in Scheme 5. Initially, the catalyst facilitates the nucleophilic attack of NH4OAc on the carbonyl carbon of the isatoic anhydride. Nucleophilic addition of ammonium leads to intermediate I while the following decarboxylation produces anthranilamide (II). Then, the reaction proceeds similarly to the cyclocondensation. The catalyst promotes the nucleophilic attack of the amino group of II on the carbonyl carbon of the aldehyde, resulting in the formation of the Schiff base (III) by removing a water molecule. Finally, the imine intramolecularly cyclizes by nucleophilic attack of the nitrogen of the amide group on the imine carbon, to furnish the corresponding DHQ derivative. Examples of various catalysts and organic solvents used in the reaction of isatoic anhydride, NH4OAc and an aldehyde are listed in Table 3.
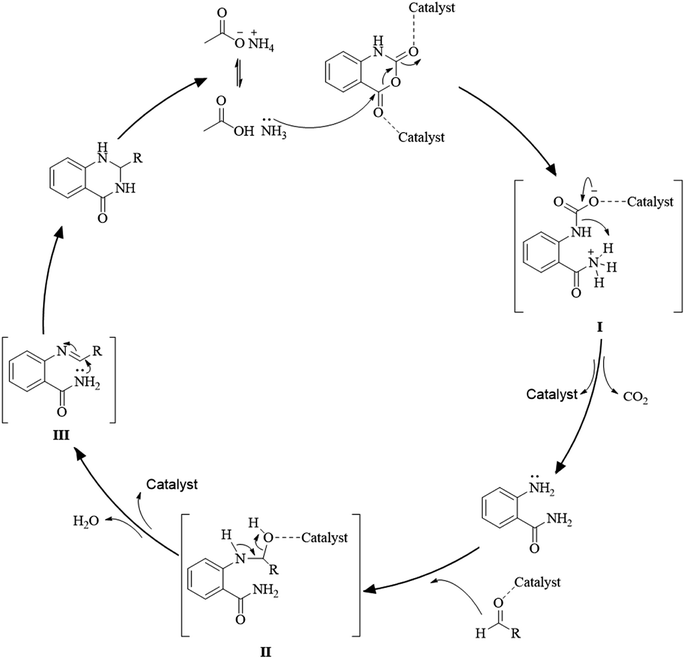 |
| | Scheme 5 Plausible mechanism of the one-pot three-component synthesis of DHQ. | |
Table 3 Reaction conditions for the one-pot three-component synthesis of DHQs
| Nature Catalyst |
Catalyst |
Conditions |
Time |
% Yield |
Ref. |
| BrØnsted acid |
EDDA |
H2O, reflux |
7–10 h |
86–93 |
59 |
| PFPAT |
Toluene, reflux |
3 h |
88–95 |
60 |
| Sulfonic acid functionality |
PTSA |
EtOH or H2O, reflux |
3–12 h |
50–71 |
61 |
| DBSA |
H2O, USI |
1–2 h |
80–91 |
62 |
| Co(m-NBS)2 |
EtOH/H2O, reflux |
0.5–4 h |
82–98 |
65 |
| TBBDA or PBBS |
EtOH/H2O, reflux |
1–3.6 h |
60–95 |
67 |
| Sulfonic acid functionality/recyclable |
Ce(SO4)2·4H2O |
Solvent-free, 120 °C |
30–50 min |
85–97 |
63 |
| Al(MS)3·4H2O |
EtOH/H2O, reflux |
0.5–6.5 h |
60–96 |
64 |
| Cu[C6H5SO3]2·6H2O |
EtOH/H2O, reflux |
0.5–6 h |
71–95 |
66 |
| I2 |
EtOH or H2O, reflux |
0.5–10 h |
56–95 |
68 |
| Solvent-free, 115 °C |
4–25 min |
94–98 |
69 |
| Lewis acid |
SrCl2·6H2O |
EtOH/H2O, reflux |
0.5–6 h |
42–94 |
70 |
| Cu(OTf)2 |
Toluene, reflux |
12–18 h |
50–85 |
72 |
| Yb(OTf)3 |
DMSO, 90 °C |
15 h |
36–41 |
73 |
| Lewis acid/recyclable |
Ga(OTf)3 |
EtOH, 70 °C |
35–60 min |
71–91 |
71 |
| Recyclable |
β-CD |
H2O, 60 °C/reflux |
1.5–5 h |
78–92 |
76–78 |
| β-CD-SO3H |
H2O, rt → 50 °C |
25 min |
80–97 |
79 |
| Starch solution |
EtOH, 70 °C |
4–8 h |
73–94 |
80 |
| Starch sulfate |
Solvent-free, 100 °C |
5–55 min |
75–96 |
81 |
| KAl(SO4)2·12H2O |
EtOH, reflux |
4–6 h |
70–83 |
82 |
| Citric acid |
H2O, 80 °C |
1–7 h |
50–94 |
83 |
| VB1 |
EtOH, reflux |
2–6 h |
75–94 |
84 |
| Amberlyst-15 |
Solvent-free, MWI |
3–7 min |
69–87 |
85 |
| Heterogeneous/recyclable |
Fe3O4 NPs |
H2O, reflux |
1.5–6 h |
51–88 |
88 |
| Al/Al2O3 NPs |
Solvent-free, 115 °C |
8–30 min |
65–98 |
89 |
| CuO NPs |
EtOH/H2O, reflux or USI |
10–30 min |
73–95 |
90 |
| In2O3 NPs |
H2O, 80 °C |
4 h |
78–88 |
91 |
| AIN NPs |
Drop of H2O, 130 °C |
3–7 h |
62–73 |
92 |
| HAP NPs |
H2O, 110 °C |
0.7–2 h |
80–90 |
93 |
| SPNP |
H2O, reflux |
1–6 h |
79–97 |
95 and 96 |
| SiO2–FeCl3 |
Solvent-free, 80 °C |
9 min to 2 h |
45–91 |
97 |
| Fe3O4-SBA-15 |
EtOH, reflux |
1.5–4 h |
65–78 |
98 |
| Titanium-SiO2 |
H2O, 100 °C |
2–8 h |
86–95 |
99 |
| Solid acidic Catalyst |
SSA |
EtOH, reflux |
3–7 h |
73–92 |
100 |
| H2O, 80 °C or solvent-free |
3–6 h |
70–86 |
101 |
| SBSSA |
EtOH, 80 °C |
0.5–4 h |
75–90 |
102 |
| SBNPSA |
EtOH, reflux |
2.5–3 h |
81–88 |
103 |
| LPCAHS-SiO2 |
H2O, 80 °C |
1.5–4 h |
72–95 |
104 |
| MCM-41-SO3H |
Solvent-free, 115 °C |
4–20 min |
75–98 |
105 |
| SPC |
Solvent-free, 70 °C |
2.5–3.5 h |
78–86 |
106 |
| H3BO3-MCM-41 |
Solvent-free, 80 °C |
0.3–1 h |
76–94 |
107 |
| Cellulose-H3BO3 |
Solvent-free, rt |
3–40 min |
79–92 |
108 |
| Al(H2PO4)3 |
Solvent-free, 100 °C |
9–17 min |
80–93 |
109 |
| H3PO4–Al2O3 |
Solvent-free, 100 °C |
4min to 3 h |
70–90 |
110 |
| PTA-DEAEC |
EtOH, reflux |
5–7 h |
60–91 |
111 |
| Copolymer-PTSA |
EtOH, reflux |
5–7 h |
70–94 |
112 |
| Montmorillonite K-10 |
EtOH, reflux |
0.5–7 h |
70–95 |
113,114 |
| Heterogeneous/recyclable |
Cu-CNTs |
Solvent-free, MWI |
5–23 min |
87–99 |
116 |
| Co-CNTs |
Solvent-free |
5–20 min |
85–96 |
117 |
| Co-MWCNTs |
EtOH, USI |
6–20 min |
76–97 |
118 |
| Pt-MWCNTs |
EtOH, USI |
8–20 min |
88–96 |
119 |
| 732-resin |
EtOH/H2O, 90 °C |
0.5–5 h |
85–95 |
122 |
| La3+/4 Å |
AcCN, reflux |
24 h |
45–95 |
123 |
| Acid-surfactant-Combined |
Zn(PFO)2 |
EtOH/H2O, reflux |
6–7 h |
77–86 |
120 |
| TA-SDS |
EtOH/H2O, grinding, rt |
1 min to 24 h |
87–93 |
121 |
Conventional reaction conditions
In general, the one-pot three-component syntheses of DHQs were performed under acid-catalyzed conditions (Table 3). BrØnsted acid catalysts, such as ethylenediamine diacetate59 and pentafluorophenylammonium triflate60 were used under reflux conditions, giving DHQ derivatives with 86–95% yield in about 10 h or less. Catalysts bearing sulfonic acid functionalities were also widely used in the multi-component reactions.61–66 They significantly reduced the reaction time, generating DHQs with 50–98% yield. Recently, the use of N-halo sulfonamides as catalysts, broadly reported in organic synthesis, has been attempted in the one-pot three-component reaction.67 Metal-catalyzed multi-component synthesis of DHQ derivatives was also reported.68–73 Generally, the use of metals or Lewis acids as catalysts did not lead to great yields (<90%), except when I2 is used under solvent-free conditions at 115 °C69 (Table 3).
Recyclable catalysts
To improve the one-pot three-component reaction using green chemistry, the use of recyclable catalysts was introduced to further minimize waste production (Table 3). Some Lewis acid catalysts had the advantage of being easily recovered after the reaction and recycled several times without considerable loss of reactivity.63,64,66,71
β-Cyclodextrin (β-CD) is a cyclic oligosaccharide, composed of seven glucose units connected “head-to-tail” by 1,4-links. This cyclic heptamer has a truncated cone shape, with a hydrophobic cavity in the center. Thus, β-CD is used in numerous applications in drug formulation. It is well known that hydrophobic and van der Waals interactions are involved in the inclusion of complex formation between guest molecules and β-CD.74 Due to its hydrophobic cavity, β-CD is widely used as a catalyst for a variety of organic reactions, providing a microenvironment whereby it catalyzes reactions through the formation of non-covalent interactions.75 β-CD is a suitable catalyst in the one-pot three-component synthesis of various DHQs producing 78–92% yield in aqueous media.76–78 DHQs were also obtained in less than half an hour in the presence of an inexpensive, safe and recyclable sulfonic acid-functionalized β-CD (β-CD-SO3H) as an efficient catalyst in green media.79
Starch is a renewable, biodegradable, and relatively inexpensive material. A starch aqueous solution in EtOH was employed as safe, non-toxic and reusable catalyst for the preparation of DHQ derivatives with 73–94% yields.80 Starch sulfate also reduced the reaction time to less than 1 hour.81
Furthermore, alum (KAl(SO4)2·12H2O),82 citric acid,83 and thiamine hydrochloride (VB1),84 catalyzed the one-pot three-component reaction in aqueous media. The protocols were longer (up to 7 h) but reported to be environmentally safe.
Amberlyst-15 is a cationic exchange resin. These types of resins, especially the macroporous ones, are recyclable catalysts for various organic syntheses, including the preparation of DHQs. Amberlyst-15 presents several advantages over conventional catalyst with respect to corrosion, product recovery, and selectivity.85
Heterogeneous and reusable catalysts
Recently, the development of heterogeneous organic reactions has been gaining popularity. They are characterized by ease of handling, separation, recycling, and environmentally safe disposal.86 In the last decade, the field of nanoscience and nanotechnology has had tremendous growth. Nanoparticles (NPs) are defined as materials having 1–50 nm diameter, a size range where metals can show size-dependent properties. Because of their interesting structures and high catalytic activities, due to the wide surface/volume ratio that provides many active sites per unit area, NPs and particularly magnetic particles have emerged as useful heterogeneous catalysts in terms of selectivity, reactivity, and improved yields of products. In addition, the magnetic properties of NPs make complete recovery of the catalyst possible by means of an external magnetic field.87 As reported in Table 3, some metal and metal oxide NPs, exhibiting high surface/volume ratio, quantum size and quantum tunnel effects, efficiently catalyzed the one-pot three-component synthesis of DHQs, in 0.5–6 h.88–91 Nano-sized aluminum nitride, a non-toxic, low-cost and highly pure powder, was used as a solid source of ammonia in the multi-component synthesis of DHQs.92 Hydroxyapatite (HAP) NPs show ion-exchange ability, adsorption capacity, and acid–base properties. Because of their higher surface areas and lower particle size, HAP NPs provide greater catalytic activity in the synthesis of DHQs.93 Although NPs have advantages such as simplified isolation of the product, easy recyclability and recovery of the catalyst, the naked NPs could aggregate into large clusters, limiting their use. This problem could be solved by immobilizing the NPs on mesoporous substrates characterized by large surface area, high chemical and thermal stability and good compatibility, such as silica, polymers and carbon.94 The integration of mesoporous silica with magnetic NPs is certainly of great interest for practical applications. As shown in Table 3, different silica-supported NPs were used as catalysts for the one-pot three-component reaction, leading to 45–97% yield in 1–8 h.95–99 The use of solid acids, as non-toxic, low-cost and reusable catalysts, has also emerged in the synthesis of DHQs (Table 3). Among them, several acids bearing sulfonic moiety supported on silica catalyzed the multi-component synthesis under different conditions, providing DHQs with >70% yield in 0.5–7 h.100–104 MCM-41 is another type of ordered mesoporous silica material. Various acid-functionalized MCM-41,105–107 cellulose,108 and alumina,109,110 were used as solid recyclable acidic catalysts for the one-pot three-component synthesis of DHQs, under solvent-free conditions, affording 70–98% yield in less than 1 hour. On the other hand, solid acids used in refluxing EtOH lengthened the reaction time to 5–7 hours.111,112 Montmorillonite K-10 is one of the most important smectites, a phyllosilicate mineral species, used as a catalyst in organic synthesis. It is a clay with both BrØnsted and Lewis acid sites, with a high cation-exchange capacity. Montmorillonite K-10 is considered a solid acid that acts as heterogeneous catalyst for diverse syntheses, including the one-pot three-component cyclocondensation, and can be easily removed from the reaction mixture.113,114 Other interesting solid supports used in heterogeneous catalysis are carbon nanotubes (CNTs), due to the porosity, inertness, and low interactions between catalyst and support and good mechanical strength.115 The deposition of metal NPs on the external surface of CNTs and multi-walled CNTs (MWCNTs) are attractive for catalysis, since they increase the reactive surface area. As reported in Table 3, CNTs supporting transition metals efficiently catalyzed the one-pot three-component synthesis of DHQs, obtained with 76–99% yield in less than half an hour.116–119 About the same yield was obtained using acid-surfactant-combined catalysts.120,121 A strong acidic cation-exchange resin (732-resin),122 and a 4 Å molecular sieve modified with lanthanum(III) (La3+/4 Å),123 were employed as heterogeneous and recyclable catalysts for the multi-component synthesis of DHQs.
Greener and convenient approaches to obtain DHQ derivatives
In the new century, there has been an increasing demand for the development of sustainable chemistry. In 1998, Anastas and Warner published the “Twelve Principles of Green Chemistry”,124 whose main purpose was the pollution prevention. In this regard, green chemistry was committed to (i) decrease pollution-generating chemicals; (ii) limit the use of dangerous chemicals and exhaustible feedstock materials and scarce resources; and (iii) reduce the harmful effects of final products.125–127 The development of cleaner and safer chemical processes started with the use of heterogeneous and recyclable catalysts or alternative solvents, which are not volatile, flammable or toxic. Then, it moved to performing reactions under catalyst- and/or solvent-free conditions. Finally, it incorporated the replacement of conventional thermal equipment by non-conventional sources, such as microwave or ultrasound irradiations.
Heterogeneous/recyclable catalysts
As shown in Table 4, recyclable catalysts, such as β-CD,77 amberlyst-15,128 and cerous methanesulfonate [Ce(MS)3],129 have been used for the cyclocondensation of anthranilamide and aldehydes. In general, the use of heterogeneous and reusable catalysts afforded high yields, up to 99%, in very short times. A number of solid acid catalysts, on various supports, such as silica,130–138 and clay,139,140 as well as different catalysts bearing sulfonic acid groups and anchored on cellulose,141 Wang resin,142 NPs,143,144 and other supports,145,146 were employed. Reusable acid-surfactant-combined catalysts,147,148 and heteropolyacids,149–152 have also emerged as good catalysts, due to their unique properties like high thermal stability, low cost, ease of preparation and recovery. They efficiently catalyze the synthesis of DHQs in aqueous media in 5–90 min.
Table 4 Catalysts for green chemistry approaches to synthesize various DHQs
| Nature catalyst |
Catalyst |
Conditions |
Time |
% Yield |
Ref. |
| Recyclable |
β-CD |
H2O, 60 °C |
1.5 h |
78–92 |
77 |
| Amberlyst-15 |
CH3CN, 80 °C |
10–30 min |
96–98 |
128 |
| Ce(MS)3 |
H2O, grinding technique |
0.2–3 h |
84–94 |
129 |
| Solid acid |
Sulfamic acid |
H2O, 60 °C or MeOH, rt |
15–180 min |
57–95 |
130 |
| H2SO4–SiO2 |
rt |
0.2–5 h |
93–97 |
131 |
| PPA-SiO2 |
Solvent-free, rt |
1.5–4 h |
89–93 |
132 and 133 |
| Amberlyst-15 and SiO2–HClO4 |
CH3CN, 80 °C |
45–120 min |
55–90 |
134 |
| SiO2–ZnCl2 |
Solvent-free, 100 °C |
6–80 min |
51–95 |
135 |
| CAN·SiO2 |
CH3CN, rt |
10–50 min |
78–94 |
136 |
| Boehmite-SSA |
EtOH, 80 °C |
35–130 min |
85–96 |
137 |
| Boehmite-Si-DSA |
EtOH, reflux |
30–190 min |
92–98 |
138 |
| Montmorillonite KSF |
CH3CN, rt |
20–50 min |
90–99 |
139 |
| HCNC-4 |
CH3CN, rt |
15–30 min |
82–99 |
140 |
| Cellulose-SO3H |
CH3CN, rt |
40–60 min |
77–92 |
141 |
| Wang-OSO3H |
H2O, 100 °C |
0.4–1.1 h |
78–88 |
142 |
| Fe3O4-SA-PPCA |
EtOH, 80 °C |
30–80 min |
91–95 |
143 |
| MNPs-PSA |
H2O, 70 °C |
25–170 min |
71–97 |
144 |
| SuSA |
H2O, 70 °C |
48–60 min |
86–95 |
145 |
| SO42−/ZrO2 |
EtOH, reflux |
7–160 min |
84–96 |
146 |
| Zr(DS)4 |
H2O, rt |
8–45 min |
83–97 |
147 |
| p-SAC |
H2O, rt |
18–90 min |
64–94 |
148 |
| H3PW12O40 |
H2O, rt |
8–10 min |
79–97 |
149 |
| H3PW12O40 |
EtOH/H2O, 80 °C |
12–18 h |
71–94 |
140 |
| SiO2–H3PW12O40 |
EtOH, reflux |
5–40 min |
88–98 |
151 |
| Poly(VPyPS)-PW |
EtOH, USI |
6–16 min |
74–96 |
152 |
| Organocatalyst heterogeneous/recyclable |
α-Chymotrypsin |
EtOH, 60 °C |
30–60 min |
90–98 |
153 |
| Fe3O4–GO |
EtOH, reflux |
2.5–5 h |
70–80 |
154 |
| GO nanosheets |
H2O, rt |
10–30 min |
85–97 |
155 |
| Co-CNTs |
Solvent-free, MWI |
10–35 min |
75–98 |
156 |
| Ag-CNTs |
EtOH, USI |
5–21 min |
86–97 |
157 |
| CuCl2/Fe3O4-TEDETA |
EtOH, 80 °C |
25–100 min |
94–98 |
158 |
| MCM-41-dtz-Ni |
PEG-400, rt |
10–35 min |
90–98 |
159 |
| Fe3O4/TiCl2/cellulose |
EtOH, rt |
6–15 min |
79–96 |
160 |
| Sc(OTf)3 |
PEG-400, 80 °C |
2 h |
78–90 |
161 |
Enzymes have received great attention as sustainable and biodegradable catalysts for the synthesis of biologically active compounds. Among them, α-chymotrypsin rapidly catalyzes the cyclocondensation of anthranilamide and aldehydes with 90–98% yield.153 Transition metal-based heterogeneous systems were efficiently used as recyclable catalysts for the cyclocondensation of anthranilamide and aldehydes, yielding > 70% DHQs.154–161
Alternative solvents
The choice of the solvent for a desired chemical process can have profound economic and environmental consequences. For this reason, there has been significant interest in using alternative “clean” solvents, mostly aqueous media and ionic liquids. Water is readily available, cheap, non-toxic, non-flammable and is very attractive from both an economical and environmental point of view.162 The use of ionic liquids as reaction media and catalysts also gives a solution to solvent emission and catalyst-recycling problems. Ionic liquids present many important features, such as negligible vapor pressure, non-inflammability, immiscibility with non-polar solvents, reasonable thermal and chemical stability and recyclability.163
Various catalysts were used in aqueous media to synthesize DHQ derivatives, both from the cyclocondensation of anthranilamide and aldehydes, and the one-pot three-component reaction. A hydrotropic solution164 and a deep eutectic solvent165 were used under catalyst-free conditions (Table 5). Ionic liquids were also widely employed for the synthesis of DHQ derivatives, in different reaction conditions (Table 6). Imidazolium-based ionic liquids30,31,46,166–171 afforded DHQ derivatives with 70–99% yield, in 0.5–10 h unless used under microwave irradiation (MWI).172 The triazolium-based reactions take less than half an hour to complete the one-pot three-component reaction.173 Ionic liquids bearing sulfonic acid functionality were used under solvent-free conditions and rapidly catalyzed the synthesis of DHQs.174,175 Although basic ionic liquids171–178 and a glycerol based ionic liquid with a boron core179 were also used under solvent-free conditions, they catalyzed the reaction in a longer time (10–90 min).
Table 5 Alternative solvents: aqueous media for greener approaches to synthesize of DHQs
| Nature solvent |
Solvent |
Conditions |
Time |
% Yield |
Ref. |
| Aqueous medium |
Aq. EtOH (50%) |
MES, MWI |
5–20 min |
83–96 |
22 |
| Sulfanilic acid, 70 °C |
|
|
23 |
| Malonic acid, rt |
5–37 min |
81–98 |
27 |
| EtOH/H2O |
Al(MS)3·4H2O, reflux |
0.5–6.5 h |
60–96 |
64 |
| Co(m-NBS)2, reflux |
0.5–4 h |
82–98 |
65 |
| Cu[C6H5SO3]2·6H2O, reflux |
0.5–6 h |
71–95 |
66 |
| TBBDA or PBBS, reflux |
1–3.6 h |
60–95 |
67 |
| SrCl2·6H2O, reflux |
0.5–6 h |
42–94 |
70 |
| CuO NPs, reflux or USI |
10–30 min |
73–95 |
90 |
| Zn(PFO)2, reflux |
6–7 h |
77–86 |
120 |
| TA-SDS, grinding, rt |
1 min-24 h |
87–93 |
121 |
| 732-resin, 90 °C |
0.5–5 h |
85–95 |
122 |
| H3PW12O40, 80 °C |
12–18 h |
71–94 |
150 |
| EtOH or H2O |
PTSA, rflux |
3–12 h |
50–71 |
61 |
| I2, reflux |
0.5–10 h |
56–95 |
68 |
| H2O |
NaHSO4, grinding |
0.5–7 h |
54–97 |
25 |
| I2/KI, rt |
2–12 h |
47–95 |
33 |
| CAN, rt → 60 °C |
1–8 h |
62–97 |
50 |
| EDDA, reflux |
7–10 h |
86–93 |
59 |
| DBSA, USI |
1–2 h |
80–91 |
62 |
| β-CD, 60 °C/reflux |
1.5–5 h |
78–92 |
76–78 |
| β-CD-SO3H, rt → 50 °C |
25 min |
80–97 |
79 |
| CA, 80 °C |
1–7 h |
50–94 |
83 |
| Magnetic Fe3O4 NPs, reflux |
1.5–6 h |
51–88 |
88 |
| In2O3 NPs, 80 °C |
4 h |
78–88 |
91 |
| HAP NPs, 110 °C |
0.7–2 h |
80–90 |
93 |
| SPNP, reflux |
1–6 h |
79–97 |
95 and 96 |
| Titanium-SiO2, 100 °C |
2–8 h |
86–95 |
99 |
| SSA, 80 °C |
3–6 h |
70–86 |
101 |
| LPCAHS- SiO2, 80 °C |
1.5–4 h |
72–95 |
104 |
| Ce(MS)3, grinding |
0.2–3 h |
84–94 |
129 |
| Sulfamic acid, 60 °C or MeOH, rt |
15–180 min |
57–95 |
130 |
| Wang-OSO3H, 100 °C |
0.4–1.1 h |
78–88 |
142 |
| MNPs-PSA, 70 °C |
25–170 min |
71–97 |
144 |
| SuSA, 70 °C |
48–60 min |
86–95 |
145 |
| Zr(DS)4, rt |
8–45 min |
83–97 |
147 |
| p-SAC, rt |
18–90 min |
64–94 |
148 |
| H3PW12O40, rt |
8–10 min |
79–97 |
149 |
| GO nanosheets, rt |
10–30 min |
85–97 |
155 |
| Hydrotropic solution |
NaPTS (50%) |
Catalyst-free, 60 °C |
50–95 min |
78–95 |
164 |
| Deep eutectic solvent |
L-(+)-TA-DMU |
Catalyst-free, 90 °C |
4 h |
79 |
165 |
Table 6 Alternative solvents: ionic liquids for green chemistry approaches to synthesize various DHQs
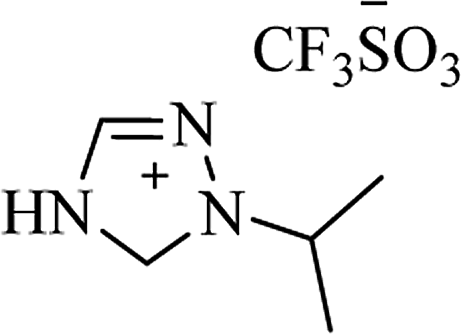
| Ionic liquid |
Structure |
Conditions |
Time |
% Yield |
Ref. |
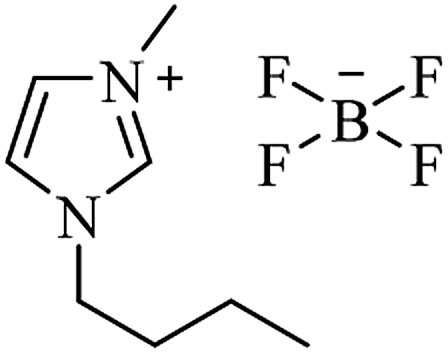
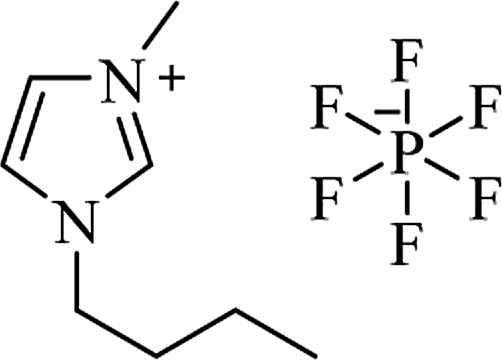
| [bmim+][BF4−] |
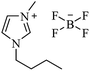 |
I2, 50 °C |
4–10 h |
76–98 |
30 |
| I2, 80 °C |
0.5–1 h |
90–99 |
31 |
| Catalyst-free, 70 °C |
1.5–2.5 h |
78–94 |
166 |
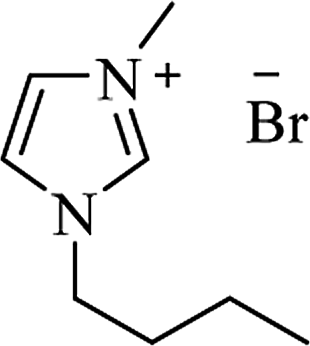
| [bmim+][Br−] |
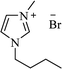 |
I2, 80 °C |
7–10 h |
82–91 |
167 |
| Yb(OTf)3, rt |
6–8 h |
85–96 |
46 |
| Catalyst-free, 80 °C |
|
|
168 |
| [bmim+][PF6−] |
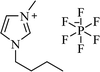 |
Solvent-free, 75 °C |
35–75 min |
77–94 |
169 |
| [bmim+][HSO4−] |
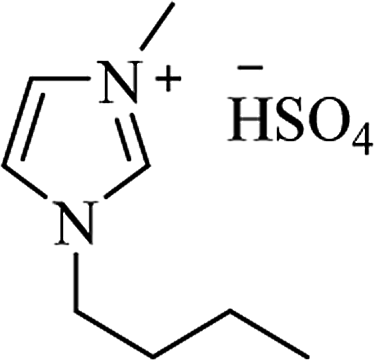
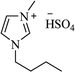 |
H2O, reflux |
3–4 h |
70–85 |
170 |
| [msim+][HSO4−] |
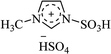 |
EtOH/H2O, reflux |
25–45 min |
80–95 |
171 |
| [bdbim+][Br−] |
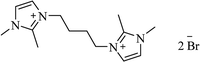 |
MWI, 100 °C |
3–7 min |
89–97 |
172 |
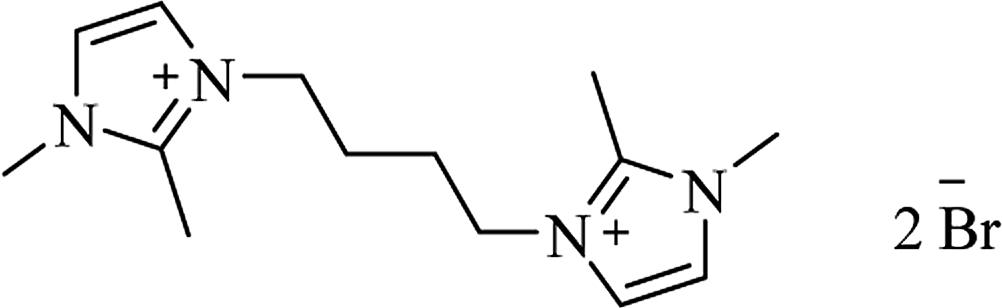
| IPTT |
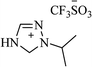 |
EtOH, 30 °C |
1–24 min |
10–96 |
173 |
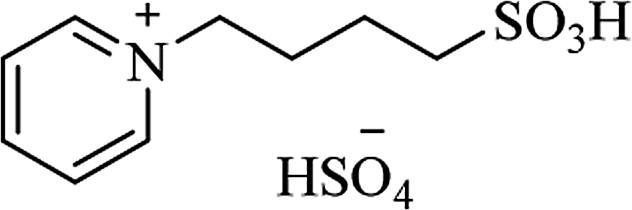
| [PY(CH2)4SO3H][HSO4] |
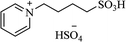 |
A300SiO2 solvent-free, 110 °C |
10–14 min |
81–90 |
174 |
| [tpps+][TS−] |
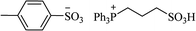 |
Solvent-free, 80 °C |
4–30 min |
85–96 |
175 |
| TBAB |
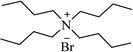 |
Solvent-free, 100 °C |
45–90 min |
72–84 |
176 |
| Solvent-free, 105 °C or H2O, 70 °C |
25–75 min |
67–91 |
177 |
| Basic ionic liquid |
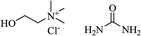 |
rt |
3–5 h |
80–90 |
178 |
| H[Gly2B] |
 |
Solvent-free, 60 °C |
10–55 min |
83–92 |
179 |
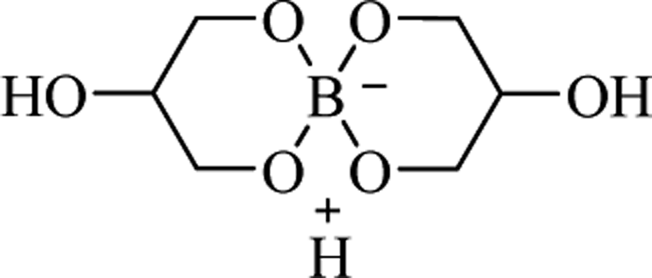
Catalyst- and solvent-free reactions
The challenge for a sustainable environment requires the development of greener and cleaner chemical processes that can avoid the use of harmful solvents and catalysts. In this sense, new strategies have been developed to synthesize DHQ derivatives in solvent- and/or catalyst-free conditions (Table 7). The cyclocondensation of anthranilamide and aldehydes was performed in the presence of sodium dihydrogen phosphate (NaH2PO4),180 acidic catalysts,181–183 and various heterogeneous systems, under solvent-free conditions, leading to various DHQs with 50–98% yield in a few minutes. On the contrary, a simple and environmentally benign procedure, without any catalyst, lengthened the reaction time to hours, although producing similar yield.184–192 The one-pot three-component reaction was also efficiently performed under solvent-free conditions in the presence of heterogeneous catalysts using conventional heating or MWI. The absence of catalyst again afforded 74–97% yield but in longer reaction times (1–6 h).193–197 Urea or thiourea was used in the multi-component synthesis of DHQs as the ammonia surrogate under catalyst-free conditions.198 A very efficient and fast synthesis of DHQs was attempted under both catalyst- and solvent-free conditions (>90% yield, in 3–10 min).199,200
Table 7 Catalyst- and/or solvent-free synthesis of DHQs
| Reaction |
Catalyst |
Conditions |
Time |
% Yield |
Ref. |
| Cyclocondensation |
H2SO4 |
Solvent-free, MWI |
Few min |
68–78 |
17 |
| H3BO3 |
Solvent-free, 120 °C |
5 min |
82–90 |
28 |
| H3BO3-MCM-41 |
Solvent-free, 80 °C |
20–65 min |
76–94 |
107 |
| Cellulose-H3BO3 |
Solvent-free, rt |
3–40 min |
79–92 |
108 |
| Co-CNTs |
Solvent-free, rt |
5–20 min |
85–96 |
117 |
| PPA-SiO2 |
Solvent-free, rt |
1.5–4 h |
89–93 |
132 |
| Co-CNTs |
Solvent-free, MWI |
10–35 min |
75–98 |
156 |
| H3BO3 or NaH2PO4 |
Solvent-free, 120 °C |
3–15 min |
50–92 |
180 |
| Citric acid |
Solvent-free, grinding |
10–20 min |
65–98 |
181 |
| Lemon juice |
Solvent-free, rt |
7–10 min |
82–85 |
182 |
| Lactic acid |
Solvent-free, 70 °C |
0.3–6 h |
80–92 |
183 |
| Catalyst-free |
L-(+)-TA-DMU, 90 °C |
4 h |
79 |
165 |
| AcOH, rt |
4 h |
35–49 |
184 |
| Dry MeOH, reflux |
5 h |
55–73 |
185 |
| TFE, reflux |
0.4–56 h |
40–98 |
186 |
| DCM, reflux |
2–3 d |
98 |
187 |
| PEG-400, 100–110 °C |
4–10 h |
78–92 |
188 |
| Glycerol, 80 °C |
1–4 min |
87–95 |
189 |
| H2O, 90 °C |
1–6 h |
67–94 |
190 and 191 |
| H2O, reflux |
0.5–27 h |
73–99 |
192 |
| One-pot Three-component |
Ce(SO4)2·4H2O |
Solvent-free, 120 °C |
30–50 min |
85–97 |
63 |
| I2 |
Solvent-free, 115 °C |
4–25 min |
94–98 |
69 |
| Starch sulfate |
Solvent-free, 100 °C |
5–55 min |
75–96 |
81 |
| Amberlyst-15 |
Solvent-free, MWI |
3–7 min |
69–87 |
85 |
| Al/Al2O3 NPs |
Solvent-free, 115 °C |
8–30 min |
65–98 |
89 |
| SiO2–FeCl3 |
Solvent-free, 80 °C |
9–120 min |
45–91 |
97 |
| SSA |
Solvent-free, rt |
3–6 h |
70–86 |
101 |
| MCM-41-SO3H |
Solvent-free, 115 °C |
4–20 min |
75–98 |
105 |
| SPC |
Solvent-free, 70 °C |
2.5–3.5 h |
78–86 |
106 |
| Cellulose-H3BO3 |
Solvent-free, rt |
3–40 min |
79–92 |
108 |
| Al(H2PO4)3 |
Solvent-free, 100 °C |
9–17 min |
80–93 |
109 |
| H3PO4–Al2O3 |
Solvent-free, 100 °C |
4–180 min |
70–90 |
110 |
| Cu-CNTs |
Solvent-free, MWI |
5–23 min |
87–99 |
116 |
| SiO2–ZnCl2 |
Solvent-free, 100 °C |
6–80 min |
51–95 |
135 |
| Catalyst-free |
NaPTS (50%), 60 °C |
3–5 h |
80–90 |
178 |
| AcOH, reflux |
1–2.5 h |
79–97 |
193 |
| TFE, reflux |
3 h |
80–97 |
194 |
| [bmim+][PF6−], 75 °C |
15–55 min |
74–93 |
195 |
| PEG-400, 120–125 °C |
1–6 h |
80–97 |
196 |
| Glycerol, 80 °C |
2 h |
88–90 |
197 |
| EtOH, reflux |
6 h |
80–92 |
198 |
| Solvent-free, 70 °C |
10 min |
87–96 |
199 |
| Solvent-free, 120 °C or MWI |
3 min |
90–97 |
200 |
New energy sources: microwave and ultrasound irradiations
MWI in organic synthesis is commonly used because it facilitates heat transfer better in chemical reactions. The efficiency of MWI heating results in a dramatic reduction in reaction times to minutes as compared to conventional heating methods taking several hours. From an economic and environmental viewpoint, the use of MWI provides unique chemical processes, characterized by enhanced reaction rates, sometimes higher yields, greater selectivity, and ease of manipulation.201,202 Previously, efforts were made to synthesize DHQs under MWI (Table 8). Among them, the cyclocondensation of anthranilamide, under acidic catalysis,203 as well as the one-pot three-component reaction, in the presence of L-proline in water.204 The ultrasound irradiation (USI)-assisted reactions have also become increasingly popular in organic synthesis. Due to faster reactions, MWI and USI allow the elimination or minimization of side products formation. They are frequently used in the pharmaceutical industry and may pave the way towards a greener and more sustainable approach to chemical synthesis.205 A variety of organic reactions were carried out within short times under USI, including the synthesis of DHQs, in the presence of amberlyst-15 (ref. 206) and in other different conditions (Table 8).
Table 8 New energy sources for greener approaches to synthesize DHQs
| Reaction |
Energy source |
Catalyst |
Conditions |
Time |
% Yield |
Ref. |
| Cyclocondensation |
MWI |
H2SO4 |
Solvent-free |
Few min |
68–78 |
17 |
| MES |
Aq. EtOH (50%), 600W |
5–20 min |
83–96 |
22 |
| Cu-CNTs |
Solvent-free, 300W |
5–23 min |
87–99 |
116 |
| Catalyst-free |
Solvent-free, 300W |
3 min |
90–97 |
200 |
| PTSA |
AcOH, 300W |
5–20 min |
66–95 |
203 |
| USI |
Poly(VPyPS)-PW |
EtOH, rt |
6–16 min |
74–96 |
152 |
| Ag-CNTs |
EtOH, 75 °C |
5–21 min |
86–97 |
157 |
| Amberlyst-15 |
CH3CN, rt |
1–5 min |
95–98 |
206 |
| One-pot three-component |
MWI |
Amberlyst-15 |
Solvent-free, 360W |
3–7 min |
69–87 |
85 |
| Co-CNTs |
Solvent-free, > 500W |
10–35 min |
75–98 |
156 |
| Catalyst-free |
[bdbim+][Br−], 100 °C |
45–90 min |
72–84 |
172 |
| Catalyst-free |
Solvent-free, 300W |
1–6 h |
80–97 |
200 |
| L-Proline |
H2O, 100 °C, 250W |
7–8 min |
84–95 |
204 |
| USI |
DBSA |
H2O, 40–42 °C |
1–2 h |
80–91 |
62 |
| CuO NPs |
EtOH/H2O, reflux |
10–30 min |
73–95 |
90 |
| Co-MWCNTs |
EtOH, 35 kHz, 40 °C |
6–20 min |
76–97 |
118 |
| Pt-MWCNTs |
EtOH, 60 °C |
8–20 min |
88–96 |
119 |
Alternative synthetic strategies
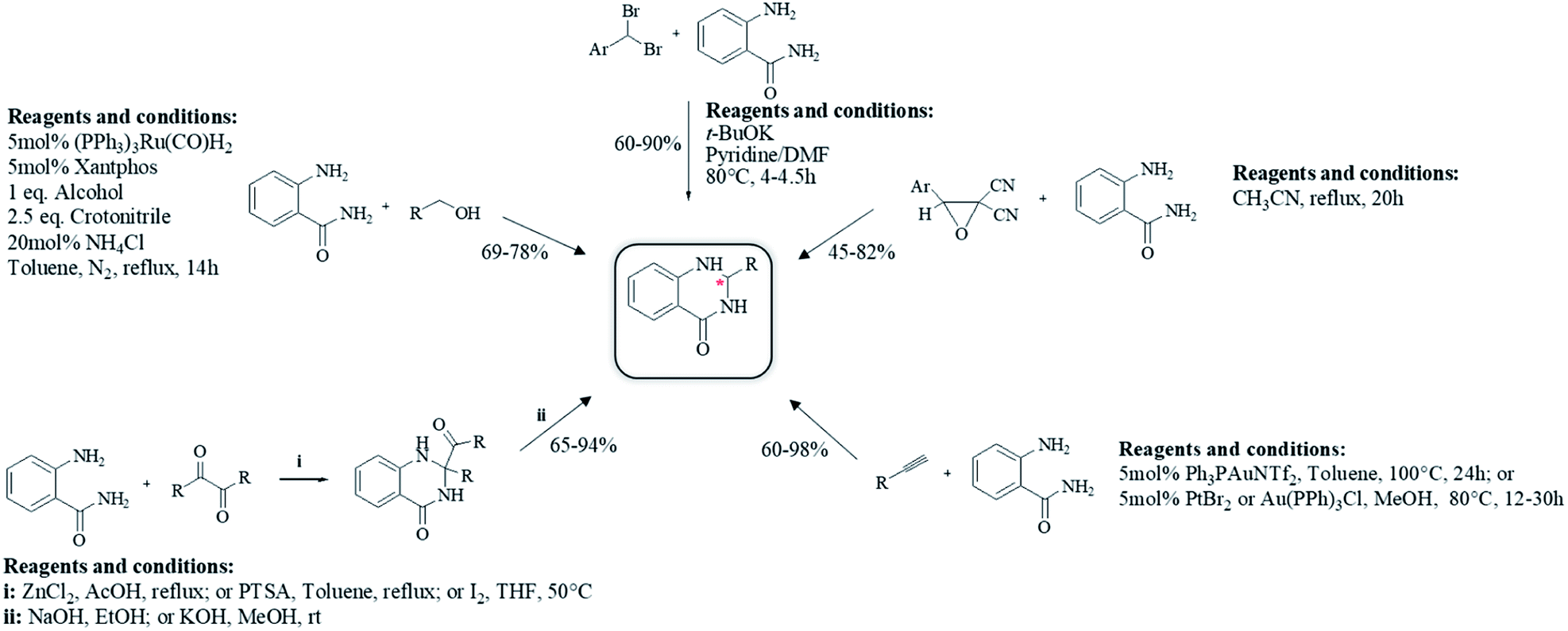
Although the cyclocondensation of anthranilamide and the one-pot three-component reaction of isatoic anhydrides, ammonium acetates and aldehydes are the main ways to synthesize DHQ derivatives, other synthetic strategies have also been developed (Table 9).
Table 9 Reaction conditions for the alternative synthetic strategies
| Starting material |
Substrate |
Conditions |
Time |
% Yield |
Ref. |
| Anthranilamide |
Benzyl |
i: ZnCl2, AcOH, reflux |
3 h |
76 |
207 |
| ii: NaOH, EtOH, rt |
1 h |
88 |
| 2-Oxo(alkyl)acetate |
i: PTSA, toluene, reflux |
4–7 h |
65–80 |
208 |
| ii: KOH, MeOH, rt |
24 h |
94 |
| 4′-Bromo acetophenone alcohol |
I2, THF, 50 °C |
6 h |
86 |
209 |
| 5mol% (PPh3)3Ru(CO)H2, 5mol% xantphos, 2.5eq. crononitrile, 20mol% NH4Cl, toluene, N2, reflux |
14 h |
69–78 |
210 |
| Gem-dibromomethylarene |
t-BuOK, Py/DMF, 80 °C |
4–4.5 h |
60–90 |
211 |
| Dicyanoepoxide |
CH3CN, reflux |
20 h |
45–82 |
212 |
| Terminal alkynes |
5mol% Ph3PauNTf2, toluene, 100 °C |
12 h |
60–97 |
213 |
| Alkynes |
5mol% PtBr2 or Au(PPh3)Cl, MeOH, 80 °C |
24 h |
70–98 |
214 |
| 2-Amino-benzonitrile |
Aldehyde |
ZnCl2, DMF, reflux |
1–24 h |
47–88 |
215–217 |
| ChOH, H2O, 80 °C |
0.5–2 h |
82–96 |
218 |
| K3PO4, H2O, 100 °C |
8 h |
28–80 |
219 |
| 1,3-Dipropylimidazole, solvent-free, rt |
1–2 h |
18–98 |
220 |
| i: 20% KOH, reflux |
7 h |
39–49 |
221 |
| ii: AcOH, rt |
4 h |
|
| Amberlyst A26 OH, EtOH/H2O, 50–60 °C |
2.5–4 h |
75–93 |
222 |
| o-Nitrobenzamide |
Aldehyde |
TiCl4/Zn, THF, reflux |
2 h |
79–91 |
223 |
| TiCl4/Sm, THF, reflux |
2 h |
71–92 |
224 |
| Fe, AcOH, 115 °C |
30 min |
73–94 |
225 |
| SnCl2·H2O, EtOH, rt |
2–4 h |
82–86 |
226 |
| SmI2, THF, rt |
3–4 h |
60–85 |
227 |
| o-Nitrobenzamide o-Azidobenzamide |
Aldehyde |
SmI2, MeOH, reflux |
20 h |
69–89 |
228 |
| o-Azidobenzamide |
Aldehyde |
SmI2, THF/MeOH, rt→reflux |
2–4 h |
69–88 |
229 |
| 2-Aminobenzoic acid |
Amine |
i: SiCl4, Py, rt |
6–24 h |
36–81 |
230 |
| ii: SMEAH, toluene, reflux |
24 h |
|
| iii: ClCO2Et, Py, rt |
48 h |
46–88 |
| Aldehyde |
i: Triphosgene, dry THF, rt |
30 min |
72 |
231 |
| ii: 28% aq. NH4OH, THF, rt |
2 h |
94 |
| iii: PTSA, MeOH, rt |
2 h |
84 |
| 2-Halobenzamide |
Aldehyde |
CuBr, L-proline, DMSO, 100 °C |
5 h |
85 |
232 |
| Aniline |
CuBr2, K2CO3, DMF, 130 °C |
4 h |
67 |
233 |
| Quinazolinone |
— |
NaBH4, diglyme, 85 °C |
1 h |
50 |
234 |
| — |
NaBH4, AcOH, 50 °C |
48 h |
50–52 |
235 |
| — |
NaBH4CN, AcOH, rt |
24 h |
33 |
236 |
| 2-Thioxo-4(3H)QZ |
— |
NiCl2, NaBH4, dry MeOH, rt |
0.5–24 h |
79–92 |
237 |
| 2-Phenyl-ethyl-anthranilate |
— |
NH4OAc, AcOH, 50 °C → 90 °C |
90–150 min |
76–81 |
238 |
| Benzylaniline |
Anthranilamide |
O2, AcOH, rt |
24 h |
62–83 |
239 |
Cyclocondensation of anthranilamide and different substrates
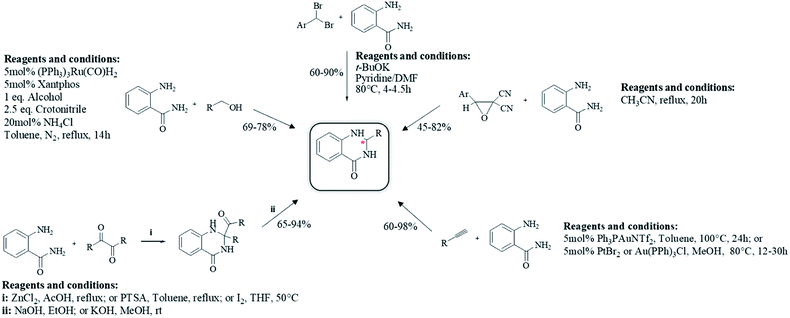
Anthranilamide was the most common starting material for the preparation of DHQs. Other than aldehydes, other substrates were used for the cyclocondensation with anthranilamide in different conditions to give DHQ derivatives (Scheme 6).
 |
| | Scheme 6 Alternative synthetic strategies of cyclocondensation of anthranilamide and different substrates. | |
First, the cyclocondensation of anthranilamide and oxo-compounds, such as benzil,207 2-oxo(alkyl)acetates,208 and 4′-bromoacetophenone,209 were attempted to give DHQs in different conditions. The obtained 2,2-disubstituted DHQ was then cleaved in ethanolic/methanolic hydroxide to give the respective derivative. Alcohols were used in a ruthenium-catalyzed cyclocondensation with anthranilamide.210 Cyclocondensations of anthranilamide and gem-dibromomethylarenes, as aldehyde equivalents, were performed in the presence of potassium tert-butoxide (t-BuOK), in anhydrous pyridine and N,N-dimethylformamide (DMF).211 Anthranilamide also reacts with dicyanoepoxide to give DHQs in refluxing CH3CN.212
The direct hydroamination/hydroarylation and double hydroamination of alkynes, followed by the cyclocondensation with anthranilamide was also exploited for the synthesis of DHQ derivatives.213,214 These alternative strategies afforded DHQs with 45–97% yield in 1–24 h (Table 9).
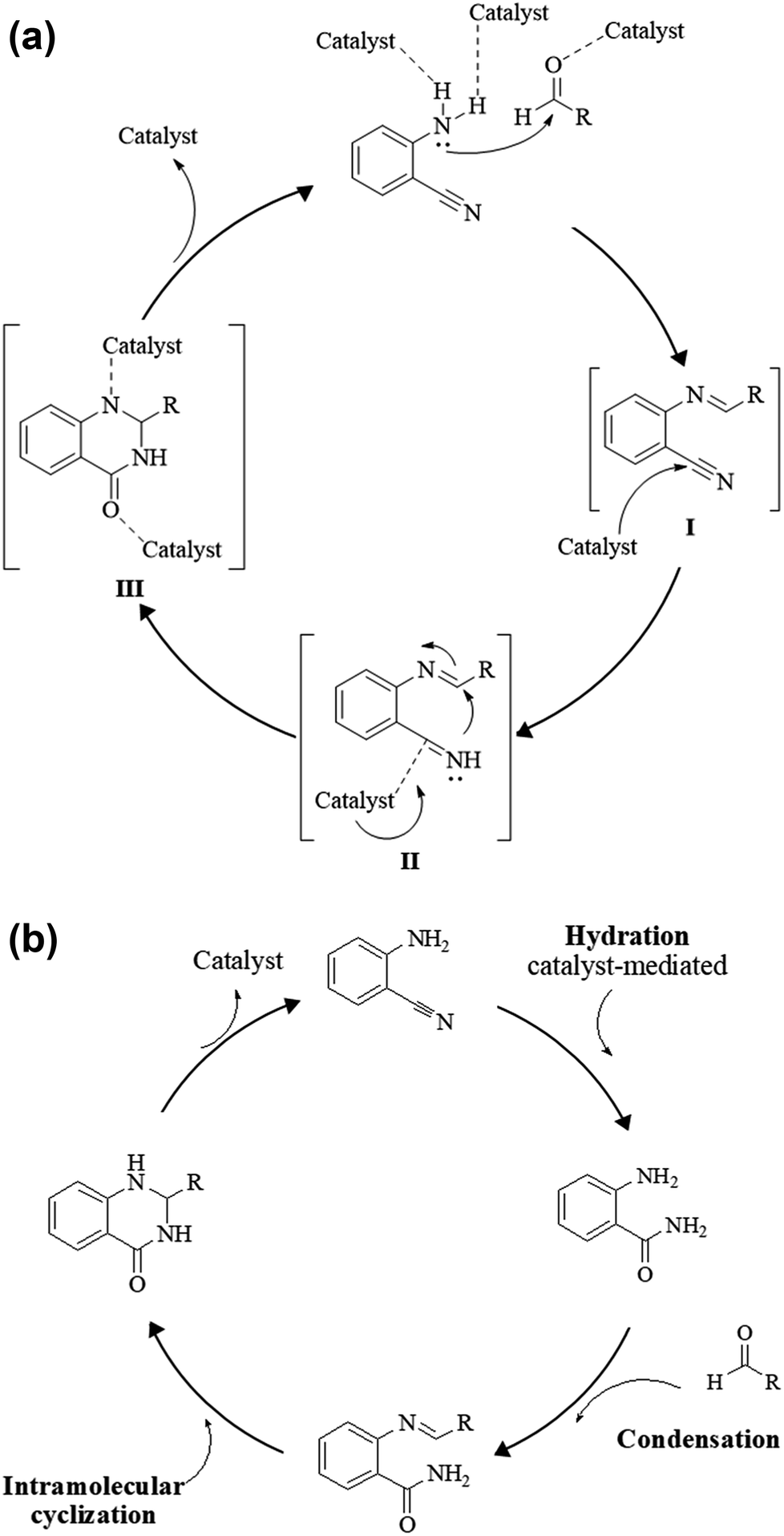
Reductive cyclocondensation of 2-aminobenzonitrile/o-nitrobenzamide/o-azidobenzamide and aldehydes
An alternative method starting from 2-aminobenzonitrile and aldehydes was developed to synthesize DHQs (Scheme 7) and two plausible mechanisms have been proposed. The first was the formation of a Schiff base and subsequent cyclocondensation in the presence of various catalysts (Scheme 8a), yielding 18–98% DHQs in 0.5–8 h (Table 9).215–220 The second proposed mechanism was the hydration of the 2-aminobenzonitrile to anthranilamide in different conditions (Scheme 8b) by means of potassium hydroxide (KOH),221 or amberlyst A26 OH.222
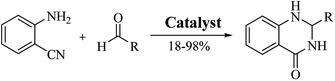 |
| | Scheme 7 Cyclocondensation of 2-aminobenzonitrile and an aldehyde for the preparation of DHQs. | |
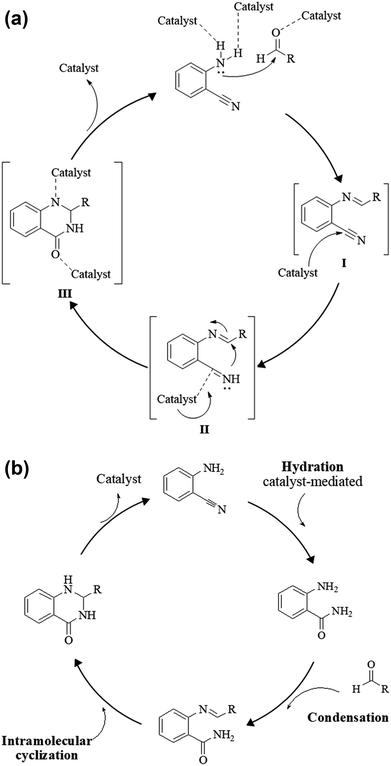 |
| | Scheme 8 (a) Plausible mechanism of the cyclocondensation of 2-aminobenzonitrile and an aldehyde through formation of Schiff base. (b) Plausible mechanism of the cyclocondensation of 2-aminobenzonitrile and an aldehyde through hydration to anthranilamide. | |
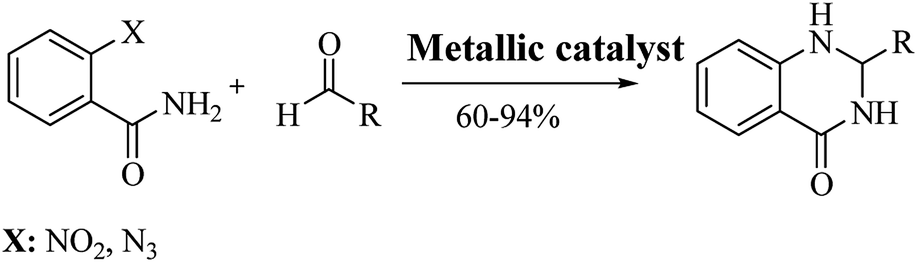
The reductive cyclization of o-nitrobenzamide223–227 and/or o-azidobenzamide228,229 and aldehydes yield DHQ derivatives in the presence of metallic catalysts with 60–94% yield in short reaction times (Scheme 9; Table 9).
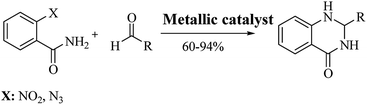 |
| | Scheme 9 Reductive cyclocondensation of o-nitrobenzamide/o-azidobenzamide and an aldehyde. | |
Other strategies
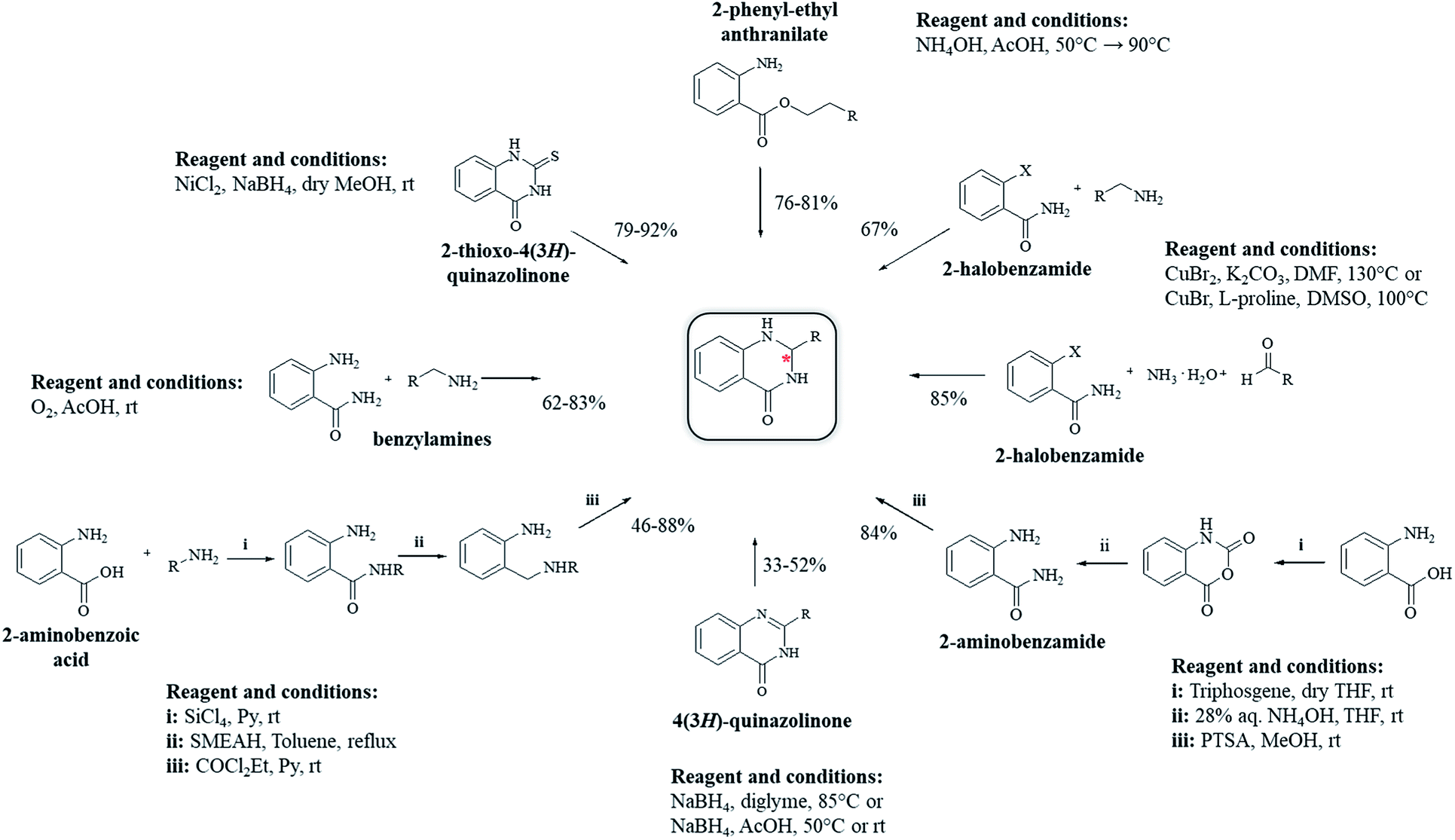
Other strategies have been developed to synthesize DHQ derivatives starting from various substrates (Scheme 10) giving 39–94% yield in 0.5–48 h (Table 9). One such method employed 2-aminobenzoic acid as a starting material to prepare DHQ derivatives. The condensation of the 2-aminobenzoic acid with amines in the presence of SiCl4 led to the corresponding substituted anthranilamides, subsequently reduced with sodium bis(2-methoxyethoxy)-aluminum hydride to o-aminobenzylamines. Then, the latter compounds were cyclized into DHQs by means of ethylchloroformate/pyridine.230 Another strategy converted the 2-aminobenzoic acid in isatoic anhydride by means of triphosgene in dry THF, then converted to anthranilamide using 28% NH4OH solution. The cyclocondensation with the aldehyde in the presence of p-toluenesulfonic acid, in refluxing MeOH, gave the respective DHQ.231 The copper-catalyzed cyclocondensation of 2-halobenzamide and aldehyde in the presence of aqueous ammonia,232 and 2-halobenzamide with anilines,233 also gave DHQs derivatives. DHQs could also be prepared by reduction from 4(3H)-quinazolinones (QZs) with NaBH4,234,235 or NaBH4CN.236 DHQs were also obtained by reductive desulfurization of 2-thioxo-4(3H)-quinazolinones with nickel boride, using nickel(II) chloride (NiCl2) and NaBH4.237 The intramolecular cyclization of 2-phenyl-ethyl anthranilate in the presence of NH4OAc led to the respective DHQs.238 The oxidation of benzylamines to the corresponding N-benzylbenzaldimines was also investigated and used for the synthesis of DHQ derivatives.239
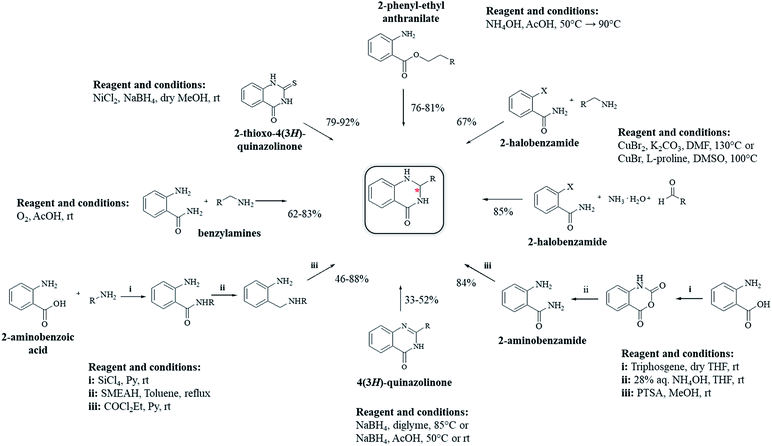 |
| | Scheme 10 Alternative synthetic strategies from diverse starting materials. | |
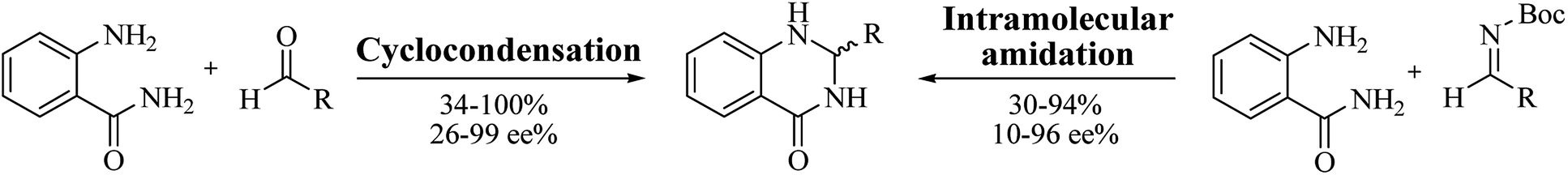
Enantioselective synthesis of DHQ derivatives
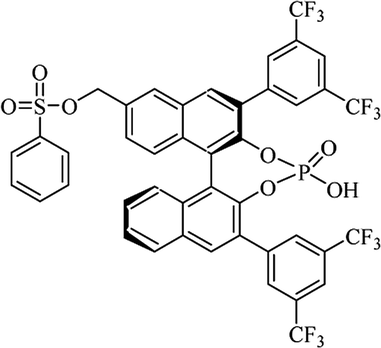
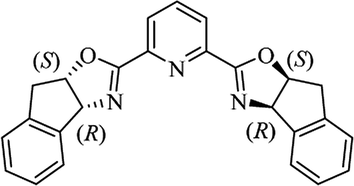
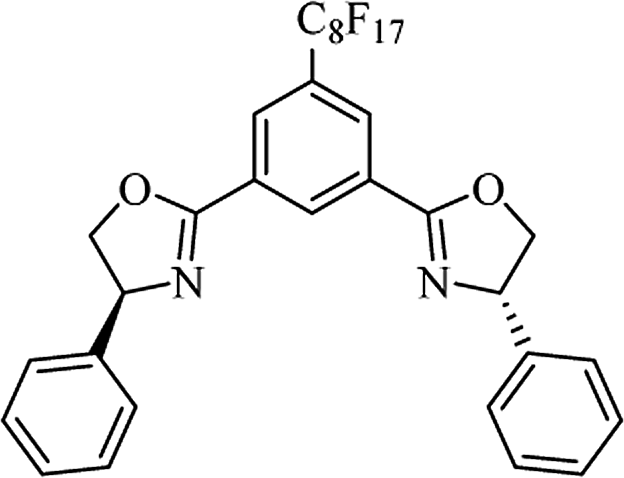
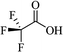
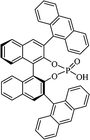
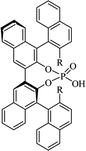
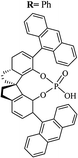
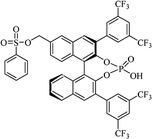
Most of the reported methods allow the synthesis of DHQ derivatives as racemic mixtures. In certain examples, the (S)-enantiomer of DHQs had better antiproliferative activity, compared to the (R)-enantiomer, even though the racemic mixture showed a similar potency to the pure S-enantiomer.3 Chinigo et al. first proved that a (S)-enantiomer of DHQ binds to tubulin, showing antiproliferative activity in different cancer cell lines. They obtained the pure (S)-enantiomer using trifluoracetic acid in CH3CN with 34–86% yield and 79–91% enantiomeric excess percentage (ee%) in 1.5 h.3 Although the enantioselective synthesis of DHQs is difficult due to an unstable aminal stereo-center that is sensitive to racemization, some asymmetric strategies have been developed to obtain pure (S)-enantiomers of DHQs (Table 10). Mainly, they consist of the cyclocondensation of anthranilamide and aldehydes in the presence of chiral catalysts that promote the formation of pure enantiomers, or the intramolecular amidation of N-Boc imines and anthranilamide (Scheme 11). The use of chiral phosphoric acidic catalysts is common in the enantioselective synthesis of DHQs, although they need a longer time (15–48 h) to complete the reaction. Various chiral phosphoric acids were employed to catalyze the cyclocondensation of anthranilamide and aldehydes,240–243 affording 67–99% yield and 26–99 ee%. An amidation of N-Boc imines and anthranilamide,244,245 gave pure (S)-enantiomer in 10–96 ee%. Scandium(III)-catalytic systems were also effectively employed for the enantioselective cyclocondensation of anthranilamide and an aldehyde (Table 10).246,247
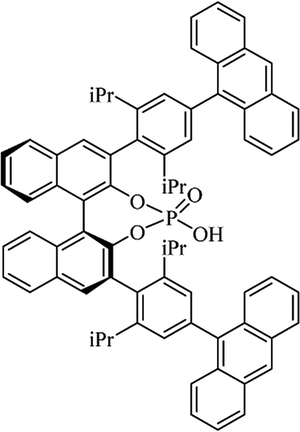
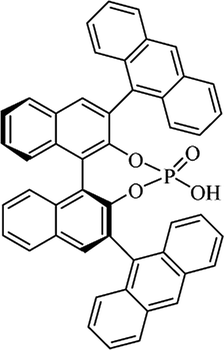
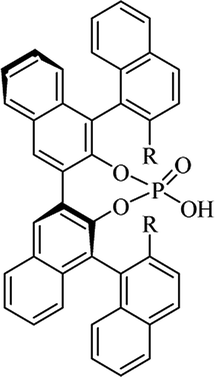
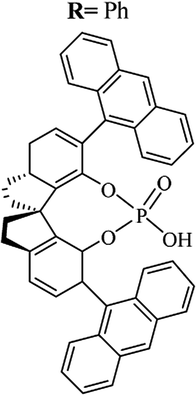
Table 10 Catalysts for the enantioselective synthesis of DHQs
| Catalyst |
Structure |
Conditions |
Time |
% Yield |
ee% |
Ref. |
| TFA |
 |
CH3CN, 45 °C |
1.5 h |
34–85 |
79–91 |
3 |
| C8-TRIP |
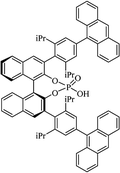 |
Toluene, 5 Å MS, −45 °C |
24 h |
67–94 |
26–98 |
240 |
| 9-Anthracenyl-TRIP |
 |
CHCl3, rt |
24–48 h |
73–99 |
90–99 |
241 |
| CHCl3, rt |
24 h |
30–94 |
10–96 |
244 |
| (R,R)-PhDAP |
 |
1,2-DFB, rt |
15 h |
99–100 |
80–86 |
242 |
| 9-Anthracenyl-SPINOL |
 |
CHCl3, 3 Å MS, rt |
24 h |
88–99 |
59–98 |
243 |
| BINOL-derived phosphoric acid |
 |
CHCl3, −15 °C |
24–36 h |
60–85 |
80–96 |
245 |
| Sc(OTf)3/Pybox |
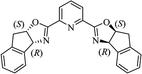 |
DCM, 4 Å MS, rt |
6–48 h |
80–94 |
86–98 |
246 |
| Sc(OTf)3/L6 |
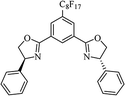 |
DCM, 4 Å MS, rt |
48 h |
81–94 |
87–98 |
247 |
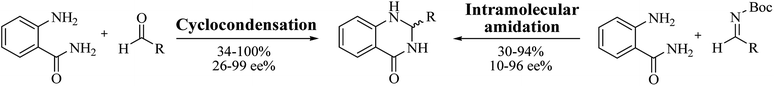 |
| | Scheme 11 Enantioselective synthesis of DHQs. | |
DHQ as an intermediate in organic chemistry
In addition to their many significant pharmacological activities, DHQs also play a central role as intermediates in organic synthesis. In particular, they can be easily oxidized to the biologically active QZs. The quinazolinone ring (Fig. 4) is frequently encountered in organic chemistry as well as in medicinal chemistry.248–254 The QZ core is present in the structure of numerous natural products, especially alkaloids,255,256 and in some drugs,257 exhibiting various pharmacological properties. QZs also represent a privileged scaffold and many protocols are reported in literature for the synthesis of this important synthon. Among them, the dehydrogenation of DHQ derivatives (Scheme 12) has emerged as an easy and fast strategy to prepare QZs under different oxidant conditions (Table 11). Initially, QZs were obtained by dehydrogenation of DHQs by means of ZnCl2 in the presence of air, with 42% yield and in 10 h.202 A catalyst-free reaction in an open flask in refluxing EtOH was completed in few hours.258 The oxidation of DHQs by the addition of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone also gave the corresponding oxidized derivatives with 83% yield.231 Various oxidizing agents were used for the dehydrogenation of DHQs to efficiently generate QZs, with 26–92% yield.259–262 Metal-catalyzed dehydrogenation was also attempted, affording the oxidized derivatives in moderate to good yields (18–85%) but in 16–24 h.263–265 Biocatalysis using laccase/N-hydroxybenzotriazole gave QZs in 62–87% yield.266
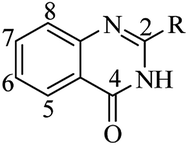 |
| | Fig. 4 Quinazolin-4(3H)-one framework. | |
 |
| | Scheme 12 Dehydrogenation of DHQs as intermediate in organic synthesis. | |
Table 11 Reaction conditions for the dehydrogenation of DHQs
| Catalyst |
Conditions |
Time |
% Yield |
Ref. |
| ZnCl2 |
AcOH, air, reflux |
10 h |
42 |
202 |
| Catalyst-free |
EtOH, air, reflux |
3 h |
65 |
258 |
| DDQ |
MeOH, reflux |
2 h |
83.5 |
231 |
| MnO2 |
DCM, rt |
2 h |
39–79 |
221 |
| MnO2 |
CHCl3, rt |
5–20 h |
26–75 |
259 |
| KMnO4 |
DMF, reflux |
2–3 h |
85–90 |
191 |
| KMnO4 |
Acetone, rt |
1 h |
|
202 |
| KMnO4 |
DMAC, MWI, 210 W |
3–5 min |
60–92 |
260 |
| O2 |
AcOH, 150 °C |
24 h |
46–72 |
239 |
| SO2 |
DMF/H2O, air or N2, 90 °C |
5 h |
65–92 |
261 |
| K2S2O8 |
CH3CN, 90 °C |
3–16 h |
55–90 |
262 |
| CuBr |
K2CO3, DMSO, air, 130 °C |
24 h |
18–85 |
263 |
| FeCl3 |
K2CO3, toluene, 120 °C |
16 h |
45–85 |
264 |
| Ph3PAuNTf2 |
Toluene, 100 °C |
24 h |
|
265 |
| Laccase/HBT |
O2, citrate buffer (pH 4.5), 45 °C |
20–30 h |
62–87 |
266 |
DHQ as a versatile fragment in drug design
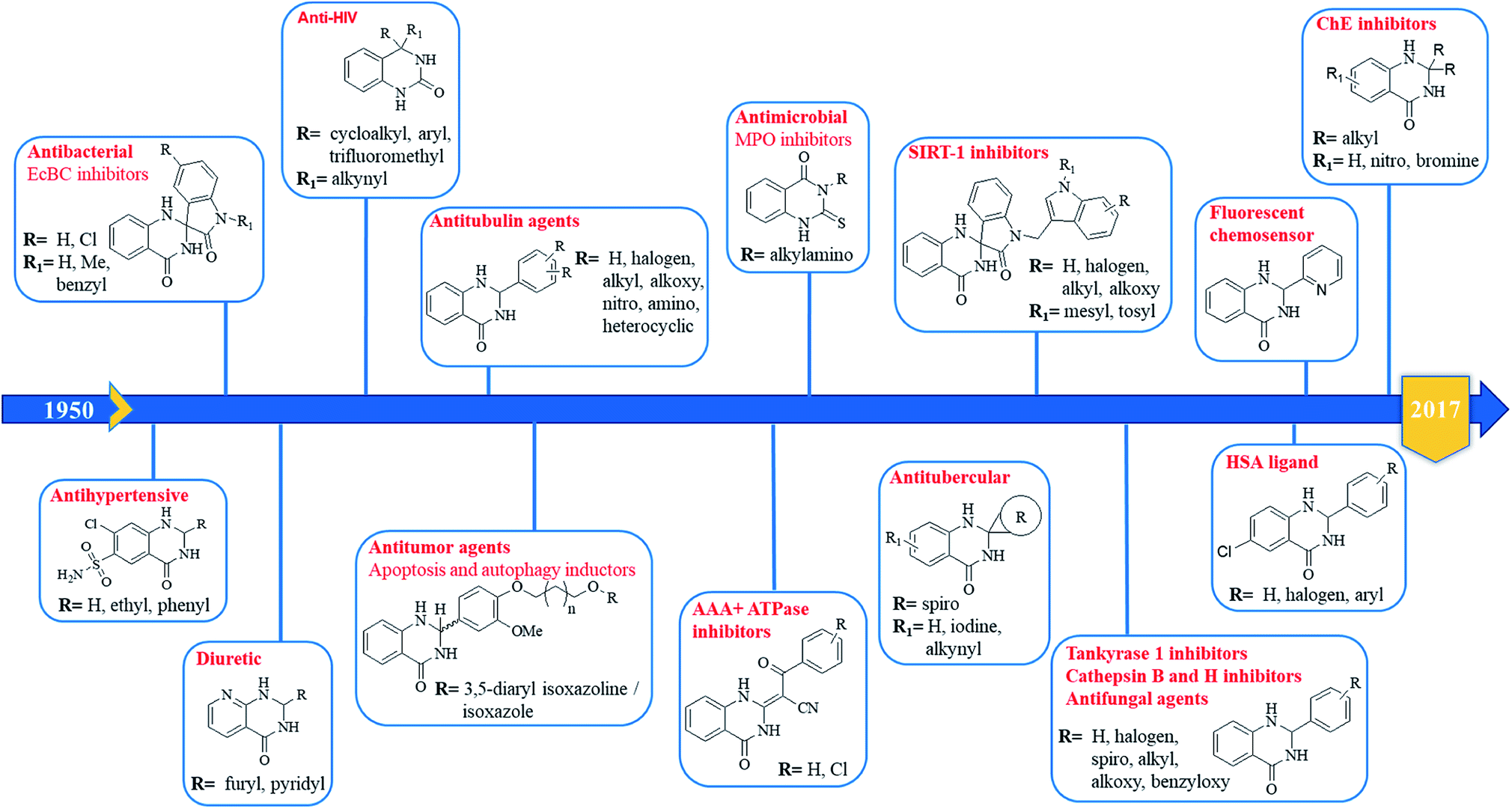
The purpose of drug discovery has always been the design and development of “magic bullets” targeting a single key biomolecule in a central pathway of a specific disease. This led to the dominant paradigm “one target, one drug”, which might be inadequate to achieve a therapeutic effect for complex diseases.267,268 For this reason, the polypharmacology research and the design of multitarget compounds is considerably emerging,269,270 contributing to overcome some of the limitations of classical approach, in term of risks and costs.271 DHQ is a versatile fragment that can be easily functionalized at different positions. The introduction of specific moieties in the DHQ nucleus leads to the ability to interact with multiple targets, ensuring diverse pharmacological properties that make it a privileged scaffold (Fig. 5).
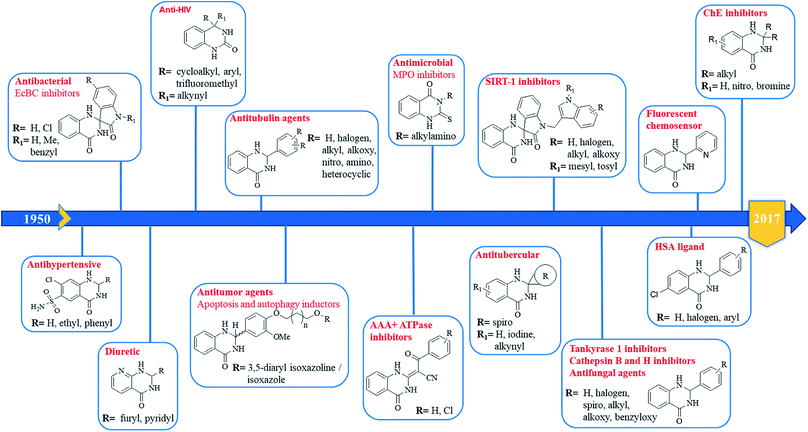 |
| | Fig. 5 Pharmacological activities of DHQ derivatives. | |
2-Aryl DHQ derivatives
The anticancer activity of DHQ derivatives was one of the first to be discovered. In 1967, Yale et al. identified DHQs as a new class of inhibitors of cell proliferation of the Earle's L cells. In particular, 2-aryl derivatives showed significant in vitro activity with low median effective dose (ED50) values of 0.1–6 μg ml−1.13 The antitumor activity of various 2-aryl DHQs against different cancer cell lines was then confirmed by several laboratories.26,36,38,109,272 Although many efforts to explain their cytotoxicity have been made, the target of the DHQ framework remains unknown. Almost three decades later, Hamel et al. used COMPARE algorithm to suggest that the antitumor effect of 2-aryl DHQs resulted from interactions with tubulin. Some derivatives inhibited the polymerization of tubulin at low micromolar concentrations and the binding of radiolabeled colchicine to tubulin at higher doses.273 Furthermore, based on the crystal structure of α,β-tubulin in complex with colchicine, through computational docking experiments and molecular dynamics, it was rationalized that (S)-DHQs may bind to tubulin better than the (R)-enantiomers, showing a better antitumor activity.3 In the same study, the accumulation of DHQs in the cytoplasm of MDA-MB-435 cells was observed through the inherent fluorescent properties of DHQs.3 In order to improve the antitumor effect of DHQs with antimitotic properties, a tumor-targeting liposomal delivery system that incorporates an anti-transferrin receptor single-chain antibody fragment was used, showing preferential targeting of tumor cells.274 2-Quinolin-5-yl DHQ induced cytochrome c mediated apoptosis and autophagy in human leukemia MOLT-4 cells as demonstrated by flow cytometry, microscopy, LC3 immunofluorescence, and western blot analysis.275 Other pathways have been suggested to clarify the antitumor activity of DHQ derivatives. DHQs bearing a phenyl substituent and piperidine/piperazine moiety on C5 and C7 respectively were identified as selective inhibitors of p38 MAP kinase. They efficiently repressed the production of TNF-α in monocyte, THP-1 cells and LPS-stimulated whole blood (IC50 values in the nanomolar range). These analogs had good clearance but low oral bioavailability in rats. However, the introduction of a bulky t-butyl substituent on the piperidine nitrogen significantly increased the oral exposure in rats.276 Other DHQ derivatives were discovered by a high-throughput screening as inhibitors of the Hedgehog pathway, involved in embryonic development and oncogenesis. The biochemical mechanism of action of these DHQs was the inhibition of the AAA+ ATPase motor cytoplasmic dynein that converts chemical energy into mechanical force and regulates many cellular processes, including ciliary trafficking, formation of mitotic spindle and organelle transport. These AAA+ ATPase inhibitors could be useful to study cellular processes that require microtubule motor.277 Different targets were proposed to cause the antitumor activity of 2-aryl DHQs. Among them are cathepsin B and H, known to facilitate invasion, angiogenesis and metastasis through degradation of extracellular matrices and are associated with cancer progression.
Some 2-aryl DHQ derivatives were identified, through molecular docking studies, as reversible inhibitors of cathepsin B and H with effective inhibitory constant (Ki) values in the nanomolar range.278 Through virtual and in vitro screening, 2-aryl DHQs were found to be potent tankyrase 2 inhibitors (IC50 ranging from low micromolar to nanomolar values). Crystal structures of tankyrase 2 with inhibitors showed that they interact with the nicotinamide-binding site of the catalytic domain of the enzyme. These compounds also inhibited Wnt/β-catenin pathway in a cell-based assay.279 Development of hybrid compounds that incorporate two pharmacophores in a single molecule with improved potency is gaining momentum. A new class of hybrids was designed and synthesized from DHQ and 3,5-diaryl isoxazoline/isoxazoles, linked through alkane spacers, and their antitumor activity was evaluated. Derivatives with 3C chain spacers showed excellent potency against lung cancer, exhibiting nanomolar IC50 values. Flow cytometric analysis revealed G2/M cell cycle arrest, usually associated with the inhibition of tubulin polymerization. Furthermore, these compounds disrupt microtubules, inhibit cyclin B1, CDK1, and induce apoptosis in cancer cells.280 N-indolylmethyl substituted spiroindoline-3,2′-DHQ were hypothesized as potential sirtuin inhibitors. Sirtuins, whose family consists of seven members (SIRT1-7), are important targets for cancer therapy, being up-regulated in several types of cancer. In particular, SIRT1 has several substrates, including p53 and NF-kB, and its inhibition leads to the re-expression of silenced tumor suppressor genes and the subsequent decrease of cancer cell growth. These new DHQ derivatives were obtained through Pd/C–Cu-mediated coupling cyclization and tested in vitro using a yeast homologue of mammalian SIRT1, Sir 2 protein, showing dose-dependent inhibition. Molecular docking analysis showed that the benzene ring of the DHQ occupied the deep hydrophobic pocket of the protein, while the NH and the sulfonyl groups form H-bonding interaction with select amino acid residues (Val412 and Gly415).281 Over half a century ago, DHQ derivatives bearing a sulfonamide moiety on C7 showed diuretic activity, causing natriuresis and chloruresis and a slight increase of potassium excretion after oral administration.282 Among them, fenquizone is a FDA-approved drug for the treatment of edema and hypertension.283–286 The substitution of C8 with N in the benzene ring of DHQ and the introduction of a pyridyl moiety on C2 led to 1,2-dihydro-2-(3-pyridyl)-3H-pyrido[2,3-d]pyrimidin-4-one derivatives. They also showed diuretic activity.287 Some DHQ derivatives also show broad-spectrum antimicrobial properties. 2-(5-Nitro-2-thienyl) DHQs showed antibacterial activity against Hemophilus vaginalis and Escherichia coli strains, responsible for bacterial vaginitis, exhibiting low minimal inhibitory concentration (MIC, ranging from 0.4 to 12.5 μg ml−1).288 More recently, some spiro-oxindole DHQs have shown significant antibacterial activity against both Gram-positive and Gram-negative bacterial strains, in particular E. coli. In order to explain the mechanism of this antibacterial activity, DHQs were docked on E. coli biotin carboxylase (EcBC) enzyme. EcBC is a known target of the fatty acid biosynthetic pathway since it catalyzes the ATP-dependent carboxylation of the vitamin biotin. Molecular docking results showed that the DHQ core fit in the hydrophobic enclosure formed by Ile437 and His236, and the different substituents form H-bonds as well as hydrophobic interactions with specific amino acid residues.289
Tuberculosis, an infectious disease caused by Mycobacterium tuberculosis, is a worldwide leading cause of death. Multiple antibiotics are needed to treat tuberculosis over a long period of time, but the development of multiple drug-resistant tuberculosis limits complete recovery. Some spiro-DHQ derivatives were tested for their in vitro inhibitory activity against Mycobacterium tuberculosis H37Rv chorismite mutase, a promising target for the identification of new antitubercular drugs. These derivatives showed a moderate inhibition at relatively high doses (30 μM).290 2-Aryl DHQs also show moderate to good antifungal activities, in particular against Candida albicans and Aspergillus niger.43 A class of 2-thioxo DHQ derivatives showed an excellent antimicrobial activity by inhibiting myeloperoxidase. This enzyme plays an important role in host defense and contributes to inflammation. They reversibly inhibited myeloperoxidase, exhibiting IC50 values in the nanomolar range.291 4-Alkynyl-3,4-dihydro-2(1H)-quinazolinones, bearing a second substituent at position 4, such as cycloalkyl, aryl and trifluoromethyl were identified as potent HIV-1 non-nucleoside reverse transcriptase inhibitors, inhibiting wild-type and various mutant forms of HIV-1. These compounds also showed a good oral bioavailability.292–294 2-Aryl DHQ derivatives, with or without substituents on N1 and N3, were found to interact with human serum albumin (HSA), using fluorescence spectroscopy. The therapeutic effects of drugs depend on their absorption, distribution, metabolism and excretion, and can be influenced by the binding affinities of drugs with HSA. In particular, strong binding can reduce free drug concentrations in plasma whereas weak binding can decrease lifetime and/or distribution of drugs. Results of site marker competitive experiments revealed that DHQs spontaneously bind to HSA on site II, subdomain IIIA, though hydrophobic forces. Various substituents in the benzene ring of DHQ could increase the interactions with HSA, forming additional van der Waals forces and H-bonds.295–297 Transition metals, such as iron, zinc and copper, play important roles in the human body. Specifically, copper is a catalytic cofactor for a variety of metalloenzymes and physiological processes. Increased levels of copper in the body could be involved in the production of reactive oxygen species (ROS), causing imbalance in cellular functions and several diseases. For this reason, the development of fluorescent chemosensors for biologically active transition metal ions are becoming attractive. In this regard, 2-aryl DHQs were efficiently used as fluorescent probes to selectively detect Cu2+ ions.298
A series of 2-disubstituted DHQ derivatives showed inhibitory activity against cholinesterases (ChE), involved in the lysis of choline-based esters that act as neurotransmitters. Acetylcholinesterase is present in chemical synapses and in red blood cell membranes, and butyrylcholinesterase is found in blood plasma. ChE inhibitors are the only effective therapeutic approach for the symptomatic treatment of Alzheimer's disease. These derivatives inhibited both enzymes (IC50 values in micromolar range), better than or comparable to the standard drug galantamine. Molecular docking studies revealed that the benzene ring of DHQs can fits into the choline-binding site while the phenyl ring at position 2 is oriented towards the peripheral anionic site. The NH group of DHQs and Asp72 form an ion-dipole interaction, while substituents in the benzene ring are involved in H-bonding interactions.16
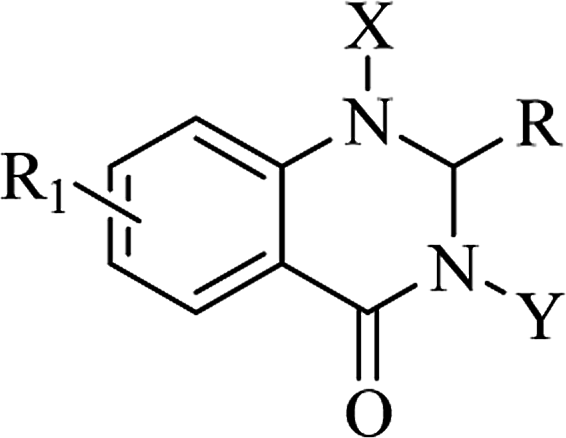
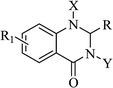
Mono- and di-substituted DHQ derivatives
As a privileged scaffold, the DHQ nucleus is amenable to modification. The most common functionalization was the single introduction of different chemical groups on heterocyclic N1 and mainly on N3, and substitutions on both positions. The resulting mono- and di-substituted derivatives showed specific activities depending on the substituents (Table 12).
Table 12 Mono- and di-substituted DHQs
|
| R |
X |
Y |
R1 |
Activity/mechanism |
Ref. |
| Alkyl, cycloalkyl |
Phenyl, benzyl |
H |
H, alkyl, alkoxy, amino, nitro |
Anti-inflammatory |
298 and 299 |
| Dimethyl |
Alkyl, cycloalkyl, aryl |
H |
Heterocycles |
PKCθ inhibitors |
301 |
| H |
Phenyl |
H |
Amide, alkylamino, sulfonamine |
p38 MAPK inhibitors |
302 |
| H, phenyl, furyl, phenylamino |
H |
Alkylamino, cycloalkylamino, hydroxy, alkoxy |
H |
Antibacterial |
311 and 312 |
| Phenyl |
H |
Alkylamino |
H |
Antimalarial |
313 |
| Thienyl, furyl, pyrrolidinyl |
H |
Phenyl |
H |
Antiviral |
314 and 315 |
| Thienyl, pyridinyl, indolyl |
H |
Phenyl |
H |
Antiprotozoal/shiga toxin |
316 |
| Phenyl |
H |
Furan-2-ylmethyl, benzyl |
H, hydroxy, methoxy |
TSHR inhibitors |
45 and 305 |
| Alkyl |
H |
Biphenyl with o-tetrazole |
H, alkoxy |
Angiotensin II receptor antagonists |
234 |
| Alky, spiro |
H |
Alkyl |
Phenyl, benzyloxy |
Antifungal/lysozyme |
317 |
| Pyridinyl-1H-pyrazolyl |
H |
Alkyl, cycloalkyl |
H, alkyl, halo |
Insecticidal/calcium channels |
318 |
| Alkyl, phenyl |
H |
Alkyl |
H, alkyl |
Anticonvulsant/Na+/Ca2+ exchanger inhibitors |
230 and 319 |
| H |
H |
Heterocycles |
H, heterocycles |
CDK5 inhibitors |
320 |
| H |
H |
Heterocycles |
H |
M1 and M4 receptors agonists |
321 |
| Keto |
H |
2-Oxoindolinyl |
H |
Antitumor |
322 |
| H |
H |
Phenyl |
Amino, phenyl |
Antitumor/p38 MAPK inhibitors |
323 |
| Keto, spiro |
H |
Alkyl, aryl |
Methoxy, oxazolyl |
IMPDH II inhibitors |
324 |
| H, methyl |
Acyl, alkyl, phenyl |
Aryl |
H |
Analgesic and anti-inflammatory |
300 and 326 |
| H |
Acyl |
Alkyl, phenyl |
H |
Choleretic and antifibrillatory |
327 |
1-Substituted DHQs can be synthesized similarly to the unsubstituted derivatives, starting from anthranilic acid284 and isatoic anhydride,299,300 bearing substituents on the amino group and the heterocyclic N respectively, or 2-fluorobenzonitrile (Scheme 13).301 Most DHQ derivatives bearing aryl substituents on N1 showed good anti-inflammatory properties.298,299 Some 1-(2-phenylethyl) DHQs were found to inhibit IL-2 production through inhibition of the protein kinase C-θ (PKCθ),301 while 1-aryl-6-substituted DHQs inhibited p38 in vitro at nanomolar concentrations.302
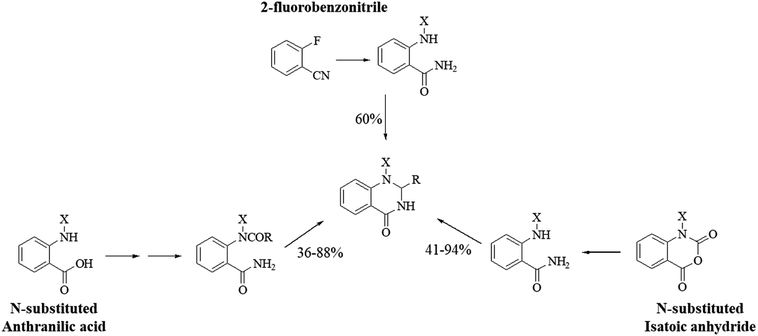 |
| | Scheme 13 Synthetic strategies for 1-substituted DHQ derivatives. | |
More commonly, 3-substituted DHQs were synthesized by cyclocondensation of 2-aminobenzamides,303–305 or 2-nitrobenzamides,306 substituted on the amide groups, with different aldehydes. 3-substituted DHQs could be also obtained through the one-pot three-component reaction of isatoic anhydride with amines and aldehydes (Scheme 14).307–310 DHQ derivatives bearing cyclic amino substituents on N3 showed antibacterial activity,311,312 while 3-alkylamino DHQs exhibited antimalarial effects.313 Different heterocycles like thienyl, furyl and pyrrolidinyl in position 2 or 3-phenyl DHQs were responsible for antiviral activity,314,315 and for the protective effect against Shiga toxin.316 3-(Furan-2-ylmethyl) derivatives were reported as thyroid-stimulating hormone receptor agonists.45,305 3-Biphenyl substituted DHQs with a tetrazole ring in the ortho position acted as angiotensin II receptor antagonists.234 3-Alkyl substituted DHQs bind to lysozyme showing antifungal activity.317 Certain 3-alkyl and 3-cycloalkyl DHQ derivatives exhibit insecticidal properties by targeting calcium channels.318 On the other hand, 3-alkyl and 3-alkylamino substituted 3,4-dihydro-2(1H)quinazolinones show anticonvulsant activity230 and inhibited the Na+/Ca2+ exchanger.319 Other 3,4-dihydro-2(1H)quinazolinones bearing heterocyclic substituents at position 3 showed neuroprotective and antipsychotic properties, inhibiting cyclin-dependent kinase 5 (CDK5),320 and targeting M1 and M4 muscarinic acetylcholine receptors,321 respectively. 3-(2-Oxoindolin-3-yl) DHQ derivatives showed antiproliferative activity against human tumor cell lines (IC50 in the micromolar range).322 Other 3-aryl and 3-alkyl DHQs also showed antitumor properties, inhibiting p38 MAP kinase,323 and inosine 5′-monophospate dehydrogenase type II (IMPDH II).324
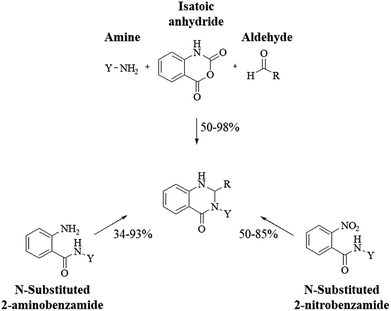 |
| | Scheme 14 Synthetic strategies for 3-substituted DHQ derivatives. | |
The less studied 1,3-disubstituted DHQs could be obtained in two different ways: through the cyclization of N-substituted isatoic anhydride and substituted arylidene anilines,299 or the reaction of 3-substituted DHQ with chloride (Scheme 15).325 1,3-Disubstituted DHQ derivatives mostly showed anti-analgesic and inflammatory activities.300,326 1-Acyl DHQs bearing alkyl and phenyl substituents on N showed choleretic and antifibrillatory activities.327
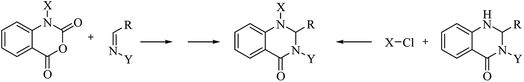 |
| | Scheme 15 Synthetic strategies for 1,3-disubstituted DHQ derivatives. | |
Conclusions
The importance of the DHQ nucleus has emerged due to its versatility as suitable substrate for functionalization and its remarkable bioactivities. Many procedures have been reported for the synthesis of DHQ derivatives as racemic mixture. Although the cyclocondensation of anthranilamide and an aldehyde and the one-pot three-component reaction of isatoic anhydride, ammonium acetate and an aldehyde represent the most common ways to obtain DHQ derivatives, several other methods have been suggested. Many approaches have been investigated, from classical to greener and more sustainable reaction conditions. Recently, enantioselective strategies have been attempted in order to obtain pure (S)-enantiomers. Furthermore, the DHQ scaffold is an important intermediate in organic chemistry and can easily be oxidized into QZ scaffold. On the other hand, various important bioactivities have been associated to the DHQ scaffold and reported in the literature. On this basis, the DHQ nucleus imposes itself as a privileged scaffold in drug design and an interesting fragment for drug discovery.
Conflicts of interest
There are no conflicts to declare.
References
- K. Imtiaz, I. Aliya, A. Waqas and S. Aamer, Eur. J. Med. Chem., 2015, 90, 124–169 CrossRef PubMed.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef PubMed.
- G. M. Chinigo, M. Paige, S. Grindrod, E. Hamel, S. Dakshanamurthy, M. Chruszcz, W. Minor and M. L. Brown, J. Med. Chem., 2008, 51, 4620–4631 CrossRef PubMed.
- B. E. Evans, K. E. Rittle, M. G. Bock, R. M. DiPardo, R. M. Freidinger, W. L. Whitter, G. F. Lundell, D. F. Veber, P. S. Anderson, R. S. L. Chang, V. J. Lotti, D. J. Cerino, T. B. Chen, P. J. Kling, K. A. Kunkel, J. P. Springer and J. Hirshfield, J. Med. Chem., 1988, 31, 2235–2246 CrossRef PubMed.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef PubMed.
- https://www.drugbank.ca.
- R. Williams, C. M. Niswender, Q. Luo, U. Le, P. J. Conn and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2009, 19, 962–966 CrossRef PubMed.
- Y. Kobayashi, Y. Nakano, K. Hoshikuma, Y. Yokoo and T. Kamiya, Planta Med., 2001, 67, 628–633 CrossRef PubMed.
- A. N. Hughes, I. Rafi, M. J. Griffin, A. H. Calvert, D. R. Newell, J. A. Calvete, A. Johnston, N. Clendeninn and A. V. Boddy, Clin. Cancer Res., 1999, 5, 111–118 Search PubMed.
- R. V. Coombs, R. P. Danna, M. Denzer, G. E. Hardtmann, B. Huegi, G. Koletar, J. Koletar, H. Ott, E. Juckniewicz, J. W. Perrine, E. I. Takesue and J. H. Trapold, J. Med. Chem., 1973, 16, 1237–1245 CrossRef PubMed.
- S. Seyedmousavi, H. Rafati, M. Ilkit, A. Tolooe, M. T. Hedayati and P. Verweij, Methods and Protocols, 2016, 1508, 107–139 Search PubMed.
- M. Shahraki, S. M. Habibi-khorassani and Y. Narouei, Am. J. Chem. Appl., 2015, 2, 83–90 Search PubMed.
- H. L. Yale and M. Kalkstein, J. Med. Chem., 1967, 10, 334–336 CrossRef PubMed.
- G. Bonola and E. Sianesi, J. Med. Chem., 1970, 13, 329–332 CrossRef PubMed.
- R. J. Ericsson and E. Reif, US pat.3651061, 1972.
- M. Sarfraz, N. Sultana, U. Rashid, M. S. Akram, A. Sadiq and M. I. Tariq, Bioorg. Chem., 2017, 70, 237–244 CrossRef PubMed.
- N. Saroja, E. Laxminarayana and K. R. S. Prasad, Indian J. Heterocycl. Chem., 2014, 24, 67–70 Search PubMed.
- S. D. Sharma and V. Kaur, Synthesis, 1989, 1989, 677–680 CrossRef.
- M. Hour, L. Huang, S. Kuo, K. Bastow, Y. Nakanishi, E. Hamel and K. Lee, J. Med. Chem., 2000, 43, 4479–4487 CrossRef PubMed.
- S. Kuo, M. Hour, L. Huang and K. Lee, US pat 6479499 B1, 2002.
- J. J. Naleway, C. M. J. Fox, D. Robinhold, E. Terpetschnig, N. A. Olson and P. Haugland, Tetrahedron Lett., 1994, 35, 8569–8572 CrossRef.
- V. B. Labade, P. V. Shinde and M. S. Shingare, Tetrahedron Lett., 2013, 54, 5778–5780 CrossRef.
- A. G. Al-Sehemi, M. Pannipara and A. Kalam, Spectrochim. Acta, Part A, 2017, 171, 97–103 CrossRef PubMed.
- X. Liu, D. Hu and H. Shen, Asian J. Chem., 2012, 24, 1365–1367 Search PubMed.
- M. Wang, J. Gao, Z. Song and L. Wang, Org. Prep. Proced. Int., 2012, 44, 159–163 CrossRef.
- R. Navudu, G. R. Mannem, T. Margani, U. M. R. Vanga and H. B. Bollikolla, Asian J. Chem., 2016, 28, 1321–1324 CrossRef.
- S. Esfandiari, M. T. Maghsoodlou, S. M. Habibi-Khorassani, S. Kiaee and J. Aboonajmi, Iranian Journal of Organic Chemistry, 2012, 4, 827–830 Search PubMed.
- M. J. Mphahlele, M. M. Maluleka and T. Khoza, Bull. Chem. Soc. Ethiop., 2014, 28, 81–90 CrossRef.
- M. Desroses, M. Scobie and T. Helleday, New J. Chem., 2013, 37, 3595–3597 RSC.
- X. Wang, K. Yang, J. Zhou and S. Tu, J. Comb. Chem., 2010, 12, 417–421 CrossRef PubMed.
- X. Wang, K. Yang, M. Zhang and C. Yao, Synth. Commun., 2010, 40, 2633–2646 CrossRef.
- Y. Nagasawa, Y. Matsusaki, T. Nobuta, N. Tada, T. Miura and A. Itoh, RSC Adv., 2015, 5, 63952–63954 RSC.
- F. Miklós, V. Hum and F. Fülöp, ARKIVOC, 2014, 2014, 25–37 CrossRef.
- D. M. Boghaei, M. M. Najafpour and V. Mckee, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, E65, o193 Search PubMed.
- M. Abdollahi-Alibeik and E. Shabani, Chin. Chem. Lett., 2011, 22, 1163–1166 Search PubMed.
- N. Ramesh, M. G. Rao, R. Valara, V. U. Rao and B. H. Babu, Med. Chem. Res., 2016, 25, 1945–1951 CrossRef.
- Y. Luo, Y. Wu, Y. Wang, H. Sun, Z. Xie, W. Zhang and Z. Gao, RSC Adv., 2016, 6, 66074–66077 RSC.
- L. Rajaka, N. R. Penumati, K. Nagaiah, Y. Poornachandra and C. G. Kumar, Synth. Commun., 2015, 45, 1893–1901 CrossRef.
- K. R. Gopinath, H. S. Shekar, K. J. Rajendraprasad, H. Nagabhushana and M. Krishnappa, WJPPS, 2016, 5, 1272–1279 Search PubMed.
- M. Sharma, S. Pandey, K. Chauhan, D. Sharma, B. Kumar and P. M. S. Chauhan, J. Org. Chem., 2012, 77, 929–937 CrossRef PubMed.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef PubMed.
- J. X. Chen, H. Y. Wu and W. K. Su, Chin. Chem. Lett., 2007, 18, 536–538 CrossRef.
- S. Vasudhevan and R. J. Karunakaran, Int. J. PharmTech Res., 2013, 5, 2844–2853 Search PubMed.
- P. Sivaguru, K. Parameswaran and A. Lalitha, Tetrahedron Lett., 2016, 57, 2549–2553 CrossRef.
- E. E. Englund, S. Neumann, E. Eliseeva, J. G. McCoy, S. Titus, W. Zheng, N. Southall, P. Shin, W. Leister, C. J. Thomas, J. Inglese, C. P. Austin, M. C. Gershengorn and W. Huang, MedChemComm, 2011, 2, 1016–1020 RSC.
- S. Wang, K. Yang and X. Wang, Chin. J. Org. Chem., 2011, 8, 1235–1239 Search PubMed.
- Y. Shang, L. Fan, X. Li and M. Liu, Chin. Chem. Lett., 2015, 26, 1355–1358 CrossRef.
- S. Li, W. Jhang, T. Liou and D. Yang, Dyes Pigm., 2015, 114, 259–266 CrossRef.
- A. Shaabani, A. Maleki and H. Mofakham, Synth. Commun., 2008, 38, 3751–3759 CrossRef.
- M. Wang, J. J. Gao, Z. G. Song and L. Wang, Chem. Heterocycl. Compd., 2011, 47, 851–855 CrossRef.
- R. Venkatesh, S. Kasaboina, H. K. Gaikwad, S. Janardhan, R. Bantu, L. Nagarapu, G. N. Sastry and S. K. Banerjee, Eur. J. Med. Chem., 2015, 96, 22–29 CrossRef PubMed.
- R. Noel, J. Martinez, D. Buisson, L. Johannes, D. Gillet, J. Barbier and J. Cintrat, J. Med. Chem., 2013, 56, 3404–3413 CrossRef PubMed.
- R. M. Christie and S. Moss, J. Chem. Soc., Perkin Trans. 1, 1985, 1, 2779–2783 RSC.
- A. Ghorbani-Choghamarani, Z. Darvishnejad and M. Norouzi, Appl. Organomet. Chem., 2015, 29, 707–711 CrossRef.
- P. Hanumanthu, S. K. V. Seshavatharam, C. V. Ratnam and N. V. S. Rao, Proc. Indian Acad. Sci., 1976, 84, 57–63 Search PubMed.
- P. R. Kumar and S. Reddy, Synth. Commun., 1992, 22, 2499–2508 CrossRef.
- R. P. Staiger, C. L. Moyer and G. R. Pitcher, J. Chem. Eng. Data, 1963, 8, 454–456 CrossRef.
- A. Dömling, Chem. Rev., 2006, 106, 17–86 CrossRef PubMed.
- M. Narasimhulu and Y. R. Lee, Tetrahedron, 2011, 67, 9627–9634 CrossRef.
- S. Khaksar and M. Gholami, Res. Chem. Intermed., 2015, 41, 3709–3718 CrossRef.
- M. Baghbanzadeh, P. Salehi, M. Dabiri and G. Kozehgary, Synthesis, 2006, 2, 344–348 Search PubMed.
- B. Chen, J. Li and G. Chen, Ultrason. Sonochem., 2015, 23, 59–65 CrossRef PubMed.
- A. Davoodnia, M. Khashi and N. Tavakoli-Hoseini, Chin. J. Catal., 2014, 35, 1054–1058 CrossRef.
- Z. Song, L. Liu, Y. Wang and X. Sun, Res. Chem. Intermed., 2012, 38, 1091–1099 CrossRef.
- Z. Song, X. Wan and S. Zhao, Indian J. Chem. Technol., 2012, 19, 118–123 Search PubMed.
- M. Wang, T. T. Zhang, Y. Liang and J. J. Gao, Monatsh. Chem., 2012, 143, 835–839 CrossRef.
- R. Ghorbani-Vaghei, A. Shahriari, Y. Maghbooli and J. Mahmoudi, Res.
Chem. Intermed., 2017, 43, 983–993 CrossRef.
- L. Zeng and C. Cai, J. Heterocycl. Chem., 2010, 47, 1035–1039 CrossRef.
- S. Rostamizadeh, A. M. Amani, R. Aryan, H. R. Ghaieni and N. Shadjou, Synth. Commun., 2008, 38, 3567–3576 CrossRef.
- M. Wang, T. T. Zhang, Y. Liang and J. J. Gao, Chin. Chem. Lett., 2011, 22, 1423–1426 CrossRef.
- J. Chen, D. Wu, F. He, M. Liu, H. Wu, J. Ding and W. Su, Tetrahedron Lett., 2008, 49, 3814–3818 CrossRef.
- X. Zhu, S. R. Kang, L. Xia, J. Lee, N. Basavegowda and Y. R. Lee, Mol. Diversity, 2015, 19, 67–75 CrossRef PubMed.
- L. Sancineto, N. Iraci, S. Massari, V. Attanasio, G. Corazza, M. L. Barreca, S. Sabatini, G. Manfroni, N. R. Avanzi, V. Cecchetti, C. Pannecouque, A. Marcello and O. Tabarrini, ChemMedChem, 2013, 1941–1953 CrossRef PubMed.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef PubMed.
- S. V. Bhosale and S. V. Bhosale, Mini-Rev. Org. Chem., 2007, 4, 231–242 CrossRef.
- K. Ramesh, K. Karnakar, G. Satish, K. H. V. Reddy and Y. V. D. Nageswar, Tetrahedron Lett., 2012, 53, 6095–6099 CrossRef.
- K. Ramesh, K. Karnakar, G. Satish, B. S. P. Anil Kumar and Y. V. D. Nageswar, Tetrahedron Lett., 2012, 53, 6936–6939 CrossRef.
- D. R. Patil, P. G. Ingole, K. Singh and D. S. Dalal, J. Inclusion Phenom. Macrocyclic Chem., 2013, 76, 327–332 CrossRef.
- J. Wu, X. Du, J. Ma, Y. Zhang, O. Shi, L. Luo, B. Song, S. Yang and D. Hu, Green Chem., 2014, 16, 3210–3217 RSC.
- M. T. Maghsoodlou, N. Khorshidi, M. R. Mousavi, N. Hazeri and S. M. Habibi-Khorassani, Res. Chem. Intermed., 2015, 41, 7497–7508 CrossRef.
- H. R. Shaterian and F. Rigi, Res. Chem. Intermed., 2015, 41, 721–738 CrossRef.
- M. Dabiri, P. Salehi, S. Otokesh, M. Baghbanzadeh, G. Kozehgary and A. A. Mohammadi, Tetrahedron Lett., 2005, 46, 6123–6126 CrossRef.
- A. Ghorbani-Choghamarani and T. Taghipour, Lett. Org. Chem., 2011, 8, 470–476 CrossRef.
- Y. Chen, W. Shan, M. Lei and L. Hu, Tetrahedron Lett., 2012, 53, 5923–5925 CrossRef.
- M. P. Surpur, P. R. Singh, S. B. Patil and S. D. Samant, Synth. Commun., 2007, 37, 1965–1970 CrossRef.
- S. Ikegami and H. Hamamoto, Chem. Rev., 2009, 109, 583–593 CrossRef PubMed.
- M. Moreno-Man and R. Pleixats, Acc. Chem. Res., 2003, 36, 638–643 CrossRef PubMed.
- Z. Zhang, H. Lü, S. Yang and J. Gao, J. Comb. Chem., 2010, 12, 643–646 CrossRef PubMed.
- M. Z. Kassaee, S. Rostamizadeh, N. Shadjou, E. Motamedi and M. Esmaeelzadeh, J. Heterocycl. Chem., 2010, 47, 1421–1424 CrossRef.
- J. Zhang, D. Ren, Y. Ma, W. Wang and H. Wu, Tetrahedron, 2014, 70, 5274–5282 CrossRef.
- S. Santra, M. Rahman, A. Roy, A. Majee and A. Hajra, Catal. Commun., 2014, 49, 52–57 CrossRef.
- M. Hajjami, A. Ghorbani-Choghamarani, Z. Yousofvand and M. Norouzi, J. Chem. Sci., 2015, 127, 1221–1228 CrossRef.
- N. Ravazi and B. Akhlaghinia, New J. Chem., 2016, 40, 447–457 RSC.
- J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815–838 CrossRef PubMed.
- A. Gharib, B. R. Hashemipour, M. Jahangir, M. Roshani and R. Safaee, Org. Chem. Int., 2013, 2013, 848237 Search PubMed.
- A. Gharib, L. Vojdanifard, N. Noroozi Pesyan, B. R. Hashemi Pou Khorasani, M. Jahangir and M. Roshani, Bulg. Chem. Commun., 2014, 46, 667–679 Search PubMed.
- G. Majid, A. Kobre, M. Hamed and S. Hamid Reza, Chin. J. Chem., 2011, 29, 1617–1623 CrossRef.
- A. Maleki, M. Rabbani and S. Shahrokh, Appl. Organomet. Chem., 2015, 29, 809–814 CrossRef.
- R. Mekala, M. V. Madhubabu, G. Dhanunjaya, S. Regati, K. B. Chandrasekhar and J. Sarva, Synth. Commun., 2017, 47, 121–130 CrossRef.
- P. Salehi, M. Dabiri, M. A. Zolfigol and M. Baghbanzedeh, Synlett, 2005, 7, 1155–1157 CrossRef.
- M. Dabiri, P. Salehi, M. Baghbanzadeh, M. A. Zolfigol, M. Agheb and S. Heydari, Catal. Commun., 2008, 9, 785–788 CrossRef.
- K. Niknam, M. Mohammadizadeh and S. Mirzaee, Chin. J. Chem., 2011, 29, 1417–1422 CrossRef.
- K. Niknam, N. Jafarpour and E. Niknam, Chin. Chem. Lett., 2011, 22, 69–72 CrossRef.
- A. Ghorbani-Choghamarani and P. Zamani, J. Iran. Chem. Soc., 2012, 9, 607–613 CrossRef.
- S. Rostamizadeh, A. M. Amani, G. Mahdavinia, H. Sepehrian and S. Ebrahimi, Synthesis, 2010, 8, 1356–1360 CrossRef.
- A. Shokrolahi, A. Zali, M. A. Zare and K. Esmaeilpour, Iran. J. Catal., 2012, 2, 91–94 Search PubMed.
- P. Sivaguru, K. Parameswaran, M. Kiruthiga, P. Vadivel and A. Lalitha, J. Iran. Chem. Soc., 2015, 12, 95–100 CrossRef.
- H. R. Shaterian and F. Rigi, Res. Chem. Intermed., 2014, 40, 2983–2999 CrossRef.
- H. R. Shaterian, A. R. Oveisi and M. Honarmand, Synth. Commun., 2010, 40, 1231–1242 CrossRef.
- H. R. Shaterian, N. Fahimi and K. Aziz, Res. Chem. Intermed., 2014, 40, 1879–1898 CrossRef.
- S. Li, Q. Zhang and Y. Peng, Monatsh. Chem., 2015, 146, 1859–1864 CrossRef.
- A. Saffar-Teluri and S. Bolouk, Monatsh. Chem., 2010, 141, 1113–1115 CrossRef.
- P. Salehi, Synth. Commun., 2006, 36, 2287–2292 CrossRef.
- S. M. Roopan, F. N. Khan, J. S. Jin and R. S. Kumar, Res. Chem. Intermed., 2011, 37, 919–927 CrossRef.
- F. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef.
- J. Safari and S. Gandomi-Ravandi, J. Mol. Catal. A: Chem., 2013, 371, 135–140 CrossRef.
- J. Safari and S. Gandomi-Ravandi, C. R. Chim., 2013, 16, 1158–1164 CrossRef.
- J. Safari and S. Gandomi-Ravandi, J. Mol. Struct., 2014, 1072, 173–178 CrossRef.
- J. Safari and S. Gandomi-Ravandi, J. Saudi Chem. Soc., 2017, 21, S415–S424 CrossRef.
- L. Wang, L. Hu, J. Shao, J. Yu and L. Zhang, J. Fluorine Chem., 2008, 129, 1139–1145 CrossRef.
- R. Sharma, A. K. Pandey and P. M. S. Chauhan, Synlett, 2012, 23, 2209–2214 CrossRef.
- M. Wang, T. T. Zhang, J. J. Gao and Y. Liang, Chem. Heterocycl. Compd., 2012, 6, 897–902 CrossRef.
- Á. Magyar and Z. Kell, Catal. Lett., 2016, 146, 1153–1162 CrossRef.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- D. L. Hjeresen, D. L. Schett and J. M. Boese, J. Chem. Educ., 2000, 77, 1543–1547 CrossRef.
- R. Mestres, Environ. Sci. Pollut. Res., 2005, 12, 128–132 CrossRef PubMed.
- J. L. Tucker, Org. Process Res. Dev., 2006, 10, 315–319 CrossRef.
- P. V. N. S. Murthy, D. Rambabu, G. Rama Krishna, C. Malla Reddy, K. R. S. Prasad, M. V. Basaveswara Rao and M. Pal, Tetrahedron Lett., 2012, 53, 863–867 CrossRef.
- M. Wang, J. Gao, Z. Song and L. Wang, J. Heterocycl. Chem., 2012, 49, 1250–1253 CrossRef.
- A. Rostami and A. Tavakoli, Chin. Chem. Lett., 2011, 22, 1317–1320 CrossRef.
- A. D. Roy, K. Jayalakshmi, S. Dasgupta, R. Roy and B. Mukhopadhyay, Magn. Reson. Chem., 2008, 46, 1119–1126 CrossRef PubMed.
- H. R. Shaterian and A. R. Oveisi, Chin. J. Chem., 2009, 27, 2418–2422 CrossRef.
- M. Singh and N. Raghav, Bioorg. Chem., 2015, 59, 12–22 CrossRef PubMed.
- S. B. Bharate, N. Muppararu, S. Manda, J. B. Bharate, R. Mudududdla, R. R. Yadav and R. A. Vishwakarma, ARKIVOC, 2012, 2012, 308–318 CrossRef.
- M. Ghashang, Orient. J. Chem., 2012, 28, 1213–1218 CrossRef.
- S. D. Dindulkar, J. Oh, V. M. Arole and Y. T. Jeong, C. R. Chimie, 2014, 17, 971–979 CrossRef.
- A. Ghorbani-Choghamarani and B. Tahmasbi, New J. Chem., 2016, 40, 1205–1212 RSC.
- M. Hajjami, A. Ghirbani-Choghamarani, R. Ghafouri-Nejad and B. Tahmasbi, New J. Chem., 2016, 40, 3066–3074 RSC.
- B. A. Dar, A. K. Sahu, P. Patidar, Y. Parial, P. Sharma, M. Sharma and B. Singh, Am. J. Chem., 2012, 2, 248–254 CrossRef.
- B. A. Dar, A. K. Sahu, P. Patidar, P. R. Sharma, N. Mupparapu, D. Vyas, S. Maity, M. Sharma and B. Singh, J. Ind. Eng. Chem., 2013, 19, 407–412 CrossRef.
- B. V. Subba Reddy, A. Venkateswarlu, C. Madan and A. Vinu, Tetrahedron Lett., 2011, 52, 1891–1894 CrossRef.
- A. V. Dhanunjaya, B. P. Vykunteswararao, T. Bhaskarkumar, N. R. Jogdand, D. Kalita, J. K. D. Lilakar, V. Siddaiah, P. D. Sanasi and A. Raghunadh, Tetrahedron Lett., 2015, 56, 4714–4717 CrossRef.
- A. Ghorbani-Choghamarani and G. Azadi, RSC Adv., 2015, 5, 9752–9758 RSC.
- A. Rostami, B. Tahmasbi, H. Gholami and H. Taymorian, Chin. Chem. Lett., 2013, 24, 211–214 CrossRef.
- M. Ghashang, S. S. Mansoor and K. Aswin, Res. Chem. Intermed., 2015, 41, 3447–3460 CrossRef.
- M. Abdollahi-Alibeik and E. Shabani, J. Iran. Chem. Soc., 2014, 11, 351–359 CrossRef.
- H. R. Safaei, M. Shekouhy, S. Khademi, V. Rahmanian and M. Safaei, J. Ind. Eng. Chem., 2014, 20, 3019–3024 CrossRef.
- M. Rahman, I. Ling, N. Abdullah, R. Hashim and A. Hajra, RSC Adv., 2015, 5, 7755–7760 RSC.
- X. Zong, Y. Zhao, W. Luo, X. H. Yu, J. K. Wang and Y. Pan, Chin. Chem. Lett., 2010, 21, 778–781 CrossRef.
- M. Tajbakhsh, R. Hosseinzadeh, P. Rezaee and M. Tajbakhsh, Chin. J. Catal., 2014, 35, 58–65 CrossRef.
- H. Alinezhadi, E. Soleymani and M. Zare, Res. Chem. Intermed., 2017, 43, 457–466 CrossRef.
- J. Wang, Y. Zong, R. Fu, Y. Niu, G. Yue, Z. Quan, X. Wang and Y. Pan, Ultrason. Sonochem., 2014, 21, 29–34 CrossRef PubMed.
- S. Zhang, Z. Xie, L. Liu, M. Liang and Z. Le, Chin. Chem. Lett., 2017, 28, 101–104 CrossRef.
- A. Maleki and S. Shahrokh, 1st International Electronic Conference on Materials, 26 May–10 June 2014 Search PubMed.
- N. Kausar, I. Roy, D. Chattopadhyay and A. R. Das, RSC Adv., 2016, 6, 22320–22330 RSC.
- J. Safari and S. Gandomi-Ravandi, J. Mol. Catal. A: Chem., 2014, 390, 1–6 CrossRef.
- J. Safari and S. Gandomi-Ravandi, RSC Adv., 2014, 4, 11654–11660 RSC.
- A. Ghorbani-Choghamarani and M. Norouzi, J. Mol. Catal. A: Chem., 2014, 395, 172–179 CrossRef.
- F. Havasi, A. Ghorbani-Choghamarani and F. Nikpour, Microporous Mesoporous Mater., 2016, 224, 26–35 CrossRef.
- B. B. F. Mirjalili, A. Bamoniri and S. Azad, J. Iran. Chem. Soc., 2017, 14, 47–55 CrossRef.
- P. Sivaguru, K. Parameswaran and A. Lalitha, Tetrahedron Lett., 2016, 57, 2549–2553 CrossRef.
- W. Wei, C. C. K. Keh, C. Li and R. S. Varma, Clean Technol. Environ. Policy, 2005, 7, 62–69 CrossRef.
- A. K. Chakraborti and S. R. Roy, J. Am. Chem. Soc., 2009, 131, 6902–6903 CrossRef PubMed.
- A. Patil, M. Barge, G. Rashinkar and R. Salunkhe, Mol. Diversity, 2015, 19, 435–445 CrossRef PubMed.
- S. K. Ghosh and R. Nagarajan, RSC Adv., 2015, 6, 27378–27387 RSC.
- M. Dabiri, P. Salehi and M. Baghbanzadeh, Monatsh. Chem., 2007, 138, 1191–1194 CrossRef.
- M. Zhang, Y. Liu and X. Wang, Chin. J. Org. Chem., 2014, 34, 1682–1686 CrossRef.
- M. Li and C. Feng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, E65, o2145 Search PubMed.
- J. Chen, W. Su, M. Liu and C. Jin, Green Chem., 2007, 9, 972–975 RSC.
- N. B. Darvatkar, S. V. Bhilare, A. R. Deorukhkar, D. G. Raut and M. M. Salunkhe, Green Chem. Lett. Rev., 2010, 3, 301–306 CrossRef.
- H. Alinezhad, M. Tajbakhsh and N. Ghobadi, Res. Rev. Mater. Sci. Chem., 2014, 3, 123–135 Search PubMed.
- M. Nikpassand, L. Zare Fekri, K. F. Sina, S. Z. Abed and O. Marvi, Russ. J. Gen. Chem., 2015, 85, 1959–1964 CrossRef.
- S. Nagarajan, T. M. Shaikh and E. Kandasamy, New J. Chem., 2015, 39, 9693–9699 RSC.
- G. Yassaghi, A. Davoodnia, S. Allameh, A. Zare-Bidaki and N. Tavakoli-Hoseini, Bull. Korean Chem. Soc., 2012, 33, 2724–2730 CrossRef.
- F. S. Toosi and M. Khakzadi, Res. Chem. Intermed., 2015, 41, 311–317 CrossRef.
- A. Davoodnia, S. Allameh, A. R. Fakhari and N. Tavakoli-Hoseini, Chin. Chem. Lett., 2010, 21, 550–553 CrossRef.
- M. A. B. Fard, A. Mobinikhaledi and M. Hamidinasab, Synth. React. Inorg. Met.-Org. Chem., 2014, 44, 567–571 CrossRef.
- O. Obaiah, N. N. Kebbahalli, R. M. Goravanahalli, P. S. Chottanahalli, R. S. Kanchugarakoppal and M. Kempegowda, Eur. J. Chem., 2014, 5, 671–675 CrossRef.
- H. R. Safaei, M. Shekouhy, V. Shafiee and M. Davoodi, J. Mol. Liq., 2013, 180, 139–144 CrossRef.
- Z. Karimi-Jaberi and L. Zarei, S. Afr. J. Chem., 2012, 65, 36–38 Search PubMed.
- Q. Ding, J. Zhang, J. Chen, M. Liu, J. Ding and H. Wu, J. Heterocycl. Chem., 2012, 49, 375–380 CrossRef.
- V. B. Ningdale, U. N. Chaudhary and K. A. Shaikh, Arch. Appl. Sci. Res., 2013, 5, 82–87 Search PubMed.
- S. Zhaleh, N. Hazeri and M. T. Maghsoodlou, Res. Chem. Intermed., 2016, 42, 6381–6390 CrossRef.
- D. Maitraie, G. Venkat Reddy, V. V. V. N. S. Rama Rao, S. Ravi Kanth, P. Shanthan Rao and B. Narsaiah, J. Fluorine Chem., 2002, 118, 73–79 CrossRef.
- O. A. Maloshitskaya, J. Sinkkonen, V. V. Alekseyev, K. N. Zelenin and K. Pihlaja, Tetrahedron, 2005, 61, 7294–7303 CrossRef.
- R. Z. Qiao, B. L. Xu and Y. W. Wang, Chin. Chem. Lett., 2007, 18, 656–658 CrossRef.
- Y. Zheng, M. Bian, X. Deng, S. Wang and Z. Quan, Arch. Pharm. Chem. Life Sci., 2013, 346, 119–126 CrossRef PubMed.
- P. Yerram, R. Chowrasia, S. Seeka and S. J. Tangenda, Eur. J. Chem., 2013, 4, 462–466 CrossRef.
- R. Ramesh, N. Nagasundaram, D. Meignasundar, P. Vadivel and A. Lalitha, Res. Chem. Intermed., 2017, 43, 1767–1782 CrossRef.
- Z. Xie, S. Zhang, G. Jiang, D. Sun and Z. Le, Green Chem. Lett. Rev., 2015, 8, 95–98 CrossRef.
- M. Tímea, F. Miklós, L. Lázár and F. Fülöp, ARKIVOC, 2016, 2016, 247–258 CrossRef.
- M. Wang, T. T. Zhang and Z. G. Song, Chin. Chem. Lett., 2011, 22, 427–430 CrossRef.
- Z. Karimi-Jaberi and R. Arjmandi, Monatsh. Chem., 2011, 142, 631–635 CrossRef.
- S. Khaksar and S. M. Talesh, C. R. Chimie, 2012, 15, 779–783 CrossRef.
- K. R. Rao, R. Mekala, A. Raghnadh, S. B. Meruva, S. P. Kumar, D. Kalita, E. Laxminarayana, B. Prasad and M. Pal, RSC Adv., 2015, 5, 61575–61579 RSC.
- M. Hajjami, F. Bakhti and E. Ghiasbeygi, Croat. Chem. Acta, 2015, 88, 197–205 CrossRef.
- A. Olyaei, F. Rahbarian and M. Sadeghpour, Chem. Heterocycl. Compd., 2015, 51, 899–902 CrossRef.
- P. P. Naidu, A. Raghunadh, K. R. Rao, R. Mekala, J. M. Babu, B. R. Rao, V. Siddaiah and M. Pal, Synth. Commun., 2014, 44, 1475–1482 CrossRef.
- L. Gao, H. Ji, L. Rong, D. Tang, Y. Zha, Y. Shi and S. Tu, J. Heterocycl. Chem., 2011, 48, 957–960 CrossRef.
- A. D. Gupta, S. Samanta and A. K. Mallik, Org. Prep. Proced. Int., 2015, 47, 356–360 CrossRef.
- V. Polshettiwar and R. S. Varma, Pure Appl. Chem., 2008, 80, 777–790 CrossRef.
- H. M. Hügel, Molecules, 2009, 14, 4936–4972 CrossRef PubMed.
- F. Li, Y. Feng, Q. Meng, W. Li, Z. Li, Q. Wang and F. Tao, ARKIVOC, 2007, 2007, 40–50 CrossRef.
- K. Kumari, D. S. Raghuvanshi and K. N. Singh, Indian J. Chem., 2012, 51B, 860–865 Search PubMed.
- R. S. Varma, Green Chem. Lett. Rev., 2007, 1, 37–45 CrossRef.
- D. Rambabu, S. K. Kumar, B. Y. Sreenivas, S. Sandra, A. Kandale, P. Misra, M. V. B. Rao and M. Pal, Tetrahedron Lett., 2013, 54, 495–501 CrossRef.
- J. A. Moore, G. J. Sutherland, R. Sowerby, E. G. Kelly, S. Palermo and W. Webster, J. Org. Chem., 1969, 34, 887–892 CrossRef.
- M. Soural, P. Funk, L. Kvapil, P. Hradil, J. Hlaváč and V. Bertolasi, ARKIVOC, 2010, 2010, 255–265 CrossRef.
- M. Zhang, K. Yang and X. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, E66, o1069 Search PubMed.
- A. J. A. Watson, A. C. Maxwell and J. M. J. William, Org. Biomol. Chem., 2012, 10, 240–243 Search PubMed.
- K. H. Narasimhamurthy, S. Chandrappa, K. S. Sharath, K. B. Harsha, H. Ananda and K. S. Rangappa, RSC Adv., 2014, 4, 34479–34486 RSC.
- A. E. O. Lalami, S. Boukhris, N. Habbadi, N. B. Larb and A. Souizi, J. Maroc. Chim. Heterocycl., 2002, 1, 37–43 Search PubMed.
- N. T. Patil, R. D. Kavthe, V. S. Raut, V. S. Shinde and B. Sridhar, J. Org. Chem., 2010, 75, 1277–1280 CrossRef PubMed.
- N. T. Patil, P. G. V. V. Lakshmi and V. Singh, Eur. J. Org. Chem., 2010, 4719–4731 CrossRef.
- J. Tang, J. Li, L. Zhang, S. Ma, D. Shi, Q. Zhang, L. Yang, X. Wang, X. Liu and C. Liu, J. Heterocycl. Chem., 2012, 49, 533–542 CrossRef.
- J. Tang, D. Shi, L. Zhang, Q. Zhang and J. Li, Synth. Commun., 2010, 40, 632–641 CrossRef.
- L. Zhang, X. Yang, D. Shi and J. Chen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, E64, o450 Search PubMed.
- P. N. Borase, P. B. Thale and G. S. Shankarling, RSC Adv., 2016, 6, 63078–63083 RSC.
- X. Wy, S. Oschatz, A. Block, A. Spannenberg and P. Langer, Org. Biomol. Chem., 2014, 12, 1865–1870 Search PubMed.
- H. Chai, J. Li, L. Yang, M. Liu, D. Yang, Q. Zhang and D. Shi, Chin. J. Chem., 2014, 32, 865–870 CrossRef.
- C. Balakumar, P. Lamba, D. P. Kishore, B. L. Narayana, K. V. Eao, K. Rajwinder, A. R. Rao, B. Shireesha and B. Narsajah, Eur. J. Med. Chem., 2010, 45, 4904–4913 CrossRef PubMed.
- F. Tamaddon and F. Pouramini, Synlett, 2014, 25, 1127–1131 CrossRef.
- D. Shi, L. Rong, J. Wang, Q. Zhuang, X. Wang and H. Hu, Tetrahedron Lett., 2003, 44, 3199–3201 CrossRef.
- D. Shi, J. Wang, L. Rong, Q. Zhuang, S. Tu and H. Hu, J. Chem. Res., 2003, S, 671–673 CrossRef.
- R. A. Bunce and B. Nammalwar, J. Heterocycl. Chem., 2011, 48, 991–997 CrossRef.
- M. Wang, G. Dou and D. Shi, J. Comb. Chem., 2010, 12, 582–586 CrossRef PubMed.
- G. Cai, X. Xu, Z. Li, W. P. Weber and P. Lu, J. Heterocycl. Chem., 2002, 39, 1271–1272 CrossRef.
- W. Su and B. Yang, Aust. J. Chem., 2002, 55, 695–697 CrossRef.
- W. Su and B. Yang, J. Chem. Res., 2002, S, 604–605 CrossRef.
- M. J. Kornet, J. Heterocycl. Chem., 1992, 29, 103–105 CrossRef.
- N. Ho, R. S. Harapanhalli, B. A. Dahman, K. Chen, K. Wang, S. J. Adelstein and A. L. Kassis, Bioconjugate Chem., 2002, 13, 357–364 CrossRef PubMed.
- H. Asakwa and M. Matano, Chem. Pharm. Bull., 1979, 27, 1287–1298 CrossRef.
- Z. Xu, Y. Zhang, H. Fu, H. Zhong, K. Hong and W. Zhu, Bioorg. Med. Chem. Lett., 2011, 21, 4005–4007 CrossRef PubMed.
- J. I. Levin, P. S. Chan, T. Bailey, A. S. Katocs and A. M. Venkatesan, The synthesis of 2,3-dihydro-4(1H)-quinazolinone angiotensin II receptor antagonists, Bioorg. Med. Chem. Lett., 1994, 4, 1141–1146 CrossRef.
- S. Guo, Y. Li, L. Tao, W. Zhang and X. Fan, RSC Adv., 2014, 4, 59289–59296 RSC.
- B. Hu, L. Wang, J. Xiang, L. Yang and Y. Tang, Chin. Chem. Lett., 2015, 26, 369–372 CrossRef.
- J. M. Khurana and G. Kukreja, J. Heterocycl. Chem., 2003, 40, 677–679 CrossRef.
- P. Hradil, L. Kvapil, J. Hlaváč, T. Weidlich, A. Lyčka and K. Lemr, J. Heterocycl. Chem., 2000, 37, 831–837 CrossRef.
- T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, Green Chem., 2013, 15, 2713–2717 RSC.
- X. Cheng, S. Vellalath, R. Goddard and B. List, J. Am. Chem. Soc., 2008, 130, 15786–15787 CrossRef PubMed.
- M. Rueping, A. P. Antonchick, E. Sugiono and K. Grenader, Angew. Chem., Int. Ed., 2009, 48, 908–910 CrossRef PubMed.
- T. Honjo, R. J. Phipps, V. Rauniyar and D. Toste, Angew. Chem., Int. Ed., 2012, 51, 9684–9688 CrossRef PubMed.
- D. Huang, X. Li, F. Xu, L. Li and X. Lin, ACS Catal., 2013, 3, 2244–2247 CrossRef.
- D. Cheng, Y. Tian and S. Tian, Adv. Synth. Catal., 2012, 354, 995–999 CrossRef.
- B. Zhang, L. Shi and R. Guo, Catal. Lett., 2015, 145, 1718–1723 CrossRef.
- M. Prakash and V. Kesavan, Org. Lett., 2012, 14, 1896–1899 CrossRef PubMed.
- T. Deng, H. Wang and C. Cai, J. Fluorine Chem., 2015, 169, 72–77 CrossRef.
- J. I. Levin, A. M. Venkatesan, P. S. Chan, J. S. Baker, G. Francisco, T. Bailey, G. Vice, A. Katocs, F. Lai and J. Coupet, Bioorg. Med. Chem. Lett., 1994, 4, 1135–1140 CrossRef.
- Y. Kurogi, Y. Inoue, K. Tsutsumi, S. Nakamura, K. Nagao, H. Yoshitsugu and Y. Tsuda, J. Med. Chem., 1996, 39, 1433–1437 CrossRef PubMed.
- V. J. Ram, Farhanullah, B. K. Tripathi and A. K. Srivastava, Bioorg. Med. Chem., 2003, 11, 2439–2444 CrossRef PubMed.
- A. R. Maarouf, E. R. El-Bendary and F. E. Goda, Arch. Pharm. Pharm. Med. Chem., 2004, 337, 527–532 CrossRef PubMed.
- S. M. Mosaad, M. K. Mohsen, M. M. K. Emad, A. Nageh, I. A. E. Sherein and F. A. Marwa, Eur. J. Med. Chem., 2010, 45, 3311–3319 CrossRef PubMed.
- M. N. Noolvi, H. M. Patel, V. Bhardwaj and A. Chauhan, Eur. J. Med. Chem., 2011, 46, 2327–2346 CrossRef PubMed.
- Z. Xu, Y. Zhang, H. Fu, H. Zhong, K. Hong and W. Zhu, Bioorg. Med. Chem. Lett., 2011, 21, 4005–4007 CrossRef PubMed.
- S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef.
- U. A. Kshirsagar, Org. Biomol. Chem., 2015, 13, 9336–9352 Search PubMed.
- T. Panneer Selvam and P. Vijayaraj Kumar, Res. Pharm., 2015, 1, 1–21 Search PubMed.
- R. Pater, J. Heterocycl. Chem., 1971, 8, 699–702 CrossRef.
- Y. Peng, Y. Zeng, G. Oiu, L. Cai and V. W. Pike, J. Heterocycl. Chem., 2010, 47, 1240–1245 CrossRef.
- M. Bakavoli, O. Sabzevari and M. Rahimizadeh, Chin. Chem. Lett., 2007, 18, 1466–1468 CrossRef.
- A. Romero, J. Salazar and S. E. López, Synthesis, 2013, 45, 2043–2050 CrossRef.
- J. K. Laha, K. S. S. Tummalapalli, A. Nair and N. Patel, J. Org. Chem., 2015, 80, 11351–11359 CrossRef PubMed.
- B. Hu, L. Wang, L. Yang, J. Xiang and Y. Tang, Eur. J. Org. Chem., 2015, 4504–4509 CrossRef.
- M. Kumar, Richa, S. Sharma, V. Bhatt and N. Kumar, Adv. Synth. Catal., 2015, 357, 2862–2868 CrossRef.
- L. Wang, B. Hu, J. Xiang, J. Cui, X. Hao, T. Liang and Y. Tang, Tetrahedron, 2014, 70, 8588–8591 CrossRef.
- M. Heidary, M. Khoobi, S. Ghasemi, Z. Habibi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 1789–1794 CrossRef.
- M. L. Bolognesi, Curr. Med. Chem., 2013, 20, 1639–1645 CrossRef PubMed.
- M. L. Bolognesi and A. Cavalli, ChemMedChem, 2016, 11, 1190–1192 CrossRef PubMed.
- R. Morphy, C. Kay and Z. Rankovic, Drug Discovery Today, 2004, 9, 641–651 CrossRef PubMed.
- J. L. Mendina-Franco, M. A. Giulianotti, G. S. Welmarker and R. A. Houghten, Drug Discovery Today, 2013, 18, 495–501 CrossRef PubMed.
- R. Morphy and Z. Rankovic, J. Med. Chem., 2005, 48, 6523–6543 CrossRef PubMed.
- N. M. Abdel Gawad, H. H. Georgey, R. M. Youssef and N. A. El-Sayed, Eur. J. Med. Chem., 2010, 45, 6058–6067 CrossRef PubMed.
- E. Hamel, C. M. Lin, J. Plowman, H. Wang, K. Lee and K. D. Paull, Biochem. Pharmacol., 1996, 51, 53–59 CrossRef PubMed.
- S. H. Hwang, A. Rait, K. F. Pirollo, Q. Zhou, V. M. Yenugonda, G. M. Chinigo, M. L. Brown and E. H. Chang, Mol. Cancer Ther., 2008, 7, 559–568 CrossRef PubMed.
- S. Kumar, S. K. Guru, A. S. Pathania, N. Mupparapu, A. Kumar, F. Malik, S. B. Bharate, Q. N. Ahmed, R. A. Vishwakarma and S. Bhushan, Toxicol. Rep., 2014, 1, 1013–1025 CrossRef PubMed.
- J. E. Stelmach, L. Liu, S. B. Patel, J. V. Pivnichny, G. Scapin, S. Singh, C. E. C. A. Hop, Z. Wang, J. R. Strauss, P. M. Cameron, E. A. Nichols, S. J. O'Keefe, E. A. O'Neil, D. M. Schmatz, C. D. Schmartz, C. M. Thompson, D. M. Zaller and J. B. Doherty, Bioorg. Med. Chem. Lett., 2003, 13, 277–280 CrossRef PubMed.
- A. J. Firestone, J. S. Weinger, M. Maldonado, K. Barlan, L. D. Langston, M. O'Donnell, V. I. Gelfand, T. M. Kapoor and J. K. Chen, Nature, 2012, 484, 125–129 CrossRef PubMed.
- M. Singh and N. Raghav, Bioorg. Chem., 2015, 59, 12–22 CrossRef PubMed.
- J. Nkizinkiko, B. V. S. S. Kumar, V. U. Jeankumar, T. Haikarainen, J. Koivunen, C. Madhuri, P. Yogeeswari, H. Venkannagari, E. Obaji, T. Pihlajaniemi, D. Sriram and L. Lehtiö, Bioorg. Med. Chem., 2015, 23, 4139–4149 CrossRef PubMed.
- A. Kamal, E. V. Bharathi, J. S. Reddy, M. J. Ramaiah, D. Dastagiri, M. K. Reddy, A. Viswanath, T. L. Reddy, T. B. Shaik, S. N. C. V. L. Pushpavalli and M. P. Bhadra, Eur. J. Med. Chem., 2011, 46, 691–703 CrossRef PubMed.
- D. Rambabu, G. Raja, B. Y. Sreenivas, G. P. K. Seerapu, K. L. Kumar, G. S. Deora, D. Haldar, M. V. B. Rao and M. Pal, Bioorg. Med. Chem. Lett., 2013, 1351–1357 CrossRef PubMed.
- E. Cohen, B. Klarberg and J. R. Vaughan, Organic Chemical Research Section, Lederle Laboratories Division, American Cyanamid Company, Pearl Rever, New York, 1959, vol. 81, pp. 5508–5509 Search PubMed.
- C. Ferrando, J. M. Foy, F. W. Pratt and I. R. Purvis, J. Pharm. Pharmacol., 1981, 33, 219–222 CrossRef PubMed.
- A. Marzo, G. Quadro and E. Treffner, J. Chromatogr. B: Biomed. Sci. Appl., 1983, 272, 95–102 CrossRef.
- A. Cupisti, F. Ciardella, E. Morelli, L. Dani, S. Lupetti, S. Luchi, M. Meola and G. Barsotti, Contrib. Nephrol., 1987, 58, 184–186 Search PubMed.
- F. V. Costa, C. Borghi, S. Boschi, A. Mussi and E. Ambrosini, J. Clin. Pharmacol., 1990, 30, 254–261 CrossRef PubMed.
- H. A. Parish and R. D. Gilliom, J. Med. Chem., 1982, 25, 98–102 CrossRef PubMed.
- R. J. Alaimo and H. E. Russell, J. Med. Chem., 1972, 15, 335–336 CrossRef PubMed.
- J. Zhang, J. Zhao, L. Wang, J. Liu, D. Ren and Y. Ma, Tetrahedron, 2016, 72, 936–943 CrossRef.
- D. Rambabu, S. K. Kumar, B. Y. Sreenivas, S. Sandra, A. Kandale, P. Misra, M. V. B. Rao and M. Pal, Tetrahedron Lett., 2013, 54, 495–501 CrossRef.
- Y. Li, T. Ganesh, B. A. Diebold, Y. Zhu, J. W. McCoy, S. M. E. Smith, A. Sun and D. Lambeth, ACS Med. Chem. Lett., 2015, 6, 1047–1052 CrossRef PubMed.
- T. J. Tucker, T. A. Lyle, C. M. Wiscount, S. F. Britcher, S. D. Young, W. M. Sanders, W. C. Lumma, M. E. Goldman, J. A. O'Brien, R. G. Ball, C. F. Homnick, W. A. Schleif, E. A. Emini, J. R. Huff and P. S. Anderson, J. Med. Chem., 1994, 37, 2437–2444 CrossRef PubMed.
- J. W. Corbett, S. S. Ko, J. D. Rodgers, S. Jeffrey, L. T. Bacheler, R. M. Klabe, S. Diamond, C. Lai, S. R. Rabel, J. A. Saye, S. P. Adams, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, Antimicrob. Agents Chemother., 1999, 43, 2893–2897 Search PubMed.
- J. W. Corbett, S. S. Ko, J. D. Rodgers, L. A. Gearhart, N. A. Magnus, L. T. Bacheler, S. Diamond, S. Jeffrey, R. M. Klabe, B. C. Cordova, S. Garber, K. Logue, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, J. Med. Chem., 2000, 43, 2019–2030 CrossRef PubMed.
- F. Liu, Y. Wang, L. Wang, J. Ou, M. Wang and S. Liu, Molecules, 2012, 17, 2000–2014 CrossRef PubMed.
- Y. Wang, M. Zhu, F. Liu, X. Wu, D. Pan, J. Liu, S. Fan, Z. Wang, J. Tang, R. Na, Q. X. Li, R. Hua and S. Liu, Molecules, 2016, 21, 1373 CrossRef PubMed.
- Y. Wang, M. Zhu, J. Liu, R. Na, F. Liu, X. Wu, S. Fan, Z. Wang, D. Pan, J. Tang, Q. X. Li, R. Hua and S. Liu, Appl. Sci., 2017, 7, 200 CrossRef.
- P. N. Borase, P. B. Thale and G. S. Shankarling, Dyes Pigm., 2016, 134, 276–284 CrossRef.
- K. Ozaki, Y. Yamada, T. Oine, T. Ishizuka and Y. Iwasawat, J. Med. Chem., 1985, 28, 568–576 CrossRef PubMed.
- Y. S. Sadanandam, K. R. M. Reddy and A. B. Rao, Eur. J. Med. Chem., 1987, 22, 169–173 CrossRef.
- O. I. El-Sabbagh, S. M. Ibrahim, M. M. Baraka and H. Kothayer, Arch. Pharm. Chem. Life Sci., 2010, 343, 274–281 Search PubMed.
- T. Katoh, T. Takai, T. Yukawa, T. Tsukamoto, E. Watanabe, H. Mototani, T. Arita, H. Hayashi, H. Nakagawa, M. G. Klein, H. Zou, H. Sang, G. Snell and Y. Nakada, Bioorg. Med. Chem., 2016, 24, 2466–2475 CrossRef PubMed.
- A. Schlapbach, R. Heng and F. Di Padova, Bioorg. Med. Chem. Lett., 2004, 14, 357–360 CrossRef PubMed.
- H. L. Yale, J. Heterocycl. Chem., 1977, 14, 1357–1359 CrossRef.
- S. Neumann, W. Huang, E. Eliseeva, S. Titus, C. J. Thomas and M. C. Gershengorn, Endocrinology, 2010, 151, 3454–3459 CrossRef PubMed.
- X. Zhu, S. R. Kang, L. Xia, J. Lee, N. Basavegowda and Y. R. Lee, Mol. Diversity, 2015, 19, 67–75 CrossRef PubMed.
- C. L. Yoo, J. C. Fettinger and M. J. Kurth, J. Org. Chem., 2005, 70, 6941–6943 CrossRef PubMed.
- L. Wang, L. Hu, J. Shao, J. Yu and L. Zhang, J. Fluorine Chem., 2008, 129, 1139–1145 CrossRef.
- J. M. Khurana and S. Kumar, Green Chem. Lett. Rev., 2011, 4, 321–325 CrossRef.
- A. Bharathi, S. M. Roopan, A. Kajbafvala, R. D. Padmaja, M. S. Darsana and G. N. Kumari, Chin. Chem. Lett., 2014, 25, 324–326 CrossRef.
- A. Takács, A. Fodor, J. Németh and Z. Hell, Synth. Commun., 2014, 44, 2269–2275 CrossRef.
- M. J. Namkung and T. L. Fletcher, J. Med. Chem., 1970, 13, 329–332 CrossRef PubMed.
- A. A. Mohammadi, H. Rohi and A. A. Soorki, J. Heterocycl. Chem., 2013, 50, 1129–1133 Search PubMed.
- E. R. Derbyshire, J. Min, W. A. Guiguemde, J. A. Clark, M. C. Connelly, A. D. Magalhães, R. K. Guy and J. Clardy, Antimicrob. Agents Chemother., 2014, 58, 1516–1522 CrossRef PubMed.
- C. D. S. Nelson, D. W. Carney, A. Derdowsky, A. Lipovsky, G. V. Gee, B. O'Hara, P. Williard, D. DiMaio, J. K. Sello and W. J. Atwood, mBio, 2013, 4, e00729 CrossRef PubMed.
- D. W. Carney, C. D. Nelson, B. D. Ferris, J. P. Stevens, A. Lipovsky, T. Kazakov, D. DiMaio, W. J. Atwood and J. K. Sello, Bioorg. Med. Chem., 2014, 22, 4836–4847 CrossRef PubMed.
- R. Noel, N. Gupta, V. Pons, A. Goudet, M. D. Garcia-Castillo, A. Michau, J. Martinez, D. Buisson, L. Johannes, D. Gillet, J. Barbier and J. Cintrat, J. Med. Chem., 2013, 56, 3404–3413 CrossRef PubMed.
- K. Hemalatha, G. Mdhumitha, L. Ravi, V. G. Khanna, N. A. Al-Dhabi and M. V. Arasu, J. Photochem. Photobiol., B, 2016, 161, 71–79 CrossRef PubMed.
- Y. Zhou, Q. Feng, F. Di, Q. Liu, D. Wang, Y. Chen, L. Xiong, H. Song, Y. Li and Z. Li, Bioorg. Med. Chem., 2013, 21, 4968–4975 CrossRef PubMed.
- H. Hasegawa, M. Muraoka, K. Matsui and A. Kojima, Bioorg. Med. Chem. Lett., 2003, 13, 3471–3475 CrossRef PubMed.
- R. M. Rzasa, M. R. Kaller, G. Liu, E. Magal, T. T. Nguyen, T. D. Osslund, D. Powers, V. J. Santora, V. N. Viswanadhan, H. Wang, X. Xiong, W. Zhong and M. H. Norman, Bioorg. Med. Chem., 2007, 15, 6574–6595 CrossRef PubMed.
- Y. Uruno, Y. Konishi, A. Suwa, K. Takai, K. Tojo, T. Nakako, M. Sakai, T. Enomoto, H. Matsuda, A. Kitamura and T. Sumiyoshi, Bioorg. Med. Chem., 2015, 25, 5357–5361 CrossRef PubMed.
- Y. Wang, X. Tang, Z. Shao, J. Ren, D. Liu, P. Proksch and W. Lin, J. Antibiot., 2014, 67, 395–399 CrossRef PubMed.
- J. A. Hunt, F. Kallashi, R. D. Ruzek, P. J. Sinclair, I. Ita, S. X. McCormicl, J. V. Pivnichny, C. E. C. A. Hop, S. Kumar, Z. Wang, S. J. O'Keefe, E. A. O'Neill, G. Porter, J. E. Thompson, A. Woods, D. M. Zaller and J. B. Doherty, Bioorg. Med. Chem. Lett., 2003, 13, 467–470 CrossRef PubMed.
- H. L. Birch, G. M. Buckely, N. Davies, H. J. Dyke, E. J. Frost, P. J. Glibert, D. R. Hannah, A. F. Haughan, M. J. Madigan, T. Morgan, W. R. Pitt, A. J. Ratcliffe, N. C. Ray, M. D. Richard, A. Sharpe, A. J. Taylor, J. M. Whitworth and S. C. Williams, Bioorg. Med. Chem. Lett., 2005, 15, 5335–5339 CrossRef PubMed.
- K. Okumura, T. Oine, Y. Yamada, G. Hayashi and M. Nakama, J. Med. Chem., 1968, 11, 348–352 CrossRef PubMed.
- G. Bonola, P. Da Re, M. J. Magistretti, E. Massarani and I. Setnikar, J. Med. Chem., 1968, 11, 1136–1139 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Francesca Aiello
ab,
Francesca Aiello