
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of sulfamic acid supported on Cr-MIL-101 as a heterogeneous acid catalyst and efficient adsorbent for methyl orange dye
Sohier A. El-Hakama,
Salem E. Samraa,
Shady M. El-Dafrawya,
Amr A. Ibrahima,
Reda S. Salama *b and
Awad I. Ahmeda
*b and
Awad I. Ahmeda
aChemistry Department, Faculty of Science, Mansoura University, Mansoura, Egypt
bBasic Science Department, Faculty of Engineering, Delta University for Science and Technology, Gamasa, Egypt. E-mail: reda.salama@deltauniv.edu.eg; dr.reda.salama@gmail.com; Tel: +20-1061391656
First published on 5th June 2018
Abstract
Typical highly porous metal–organic framework (MOFs) materials based on chromium benzenedicarboxylates (Cr-BDC) were prepared through a one-pot hydrothermal synthesis, and were then modified by loading the appropriate ratio of sulfamic acid (SA) using a simple impregnation technique. Pure and modified MIL-101 was characterized by XRD, TEM, SEM and FT-IR measurements. TEM and SEM measurements confirmed that the MIL-101 particles preserved their regular octahedral structure after loading with different weight contents of sulfamic acid. The total number of acid sites and Brønsted to Lewis acid sites ratio (B/L) were examined using potentiometric titration and pyridine adsorption. The acid strength and surface acidity of SA/MIL-101 gradually increased after the modification of Cr-MIL-101 by sulfamic acid crystals up to 55 wt%, then decreased again. The catalytic performance of the solid catalysts was confirmed in the synthesis of 14-phenyl-14H-dibenzo [a,j] xanthene and 7-hydroxy-4-methyl coumarin. In the two reactions, the sample with 55% sulfamic acid loaded on MIL-101 displayed the highest catalytic activity and acidity. The adsorption behaviors of sulfamic acid loaded on MIL-101 materials for methyl orange (MO) as an anionic dye were studied, and were exceptionally suitable for the Langmuir adsorption isotherm. All loaded adsorbents showed high adsorption capacity for methyl orange at 25 °C. The results indicate that the adsorption capacity was modified by changing the amount of sulfamic acid loaded on MIL-101.
1. Introduction
One of the most vital features of green chemistry is the effective use of renewable resources, the removal of waste and the avoidance of polluting and toxic solvents and reagents in the production and application of chemical products. Nowadays, heterogeneous catalysts are attracting much more attention than homogeneous catalysts in synthetic organic chemistry because of the numerous advantages of these materials compared to homogeneous catalysts; e.g., the facility of recovery, good reproducibility and reusability.1 The substitution of liquid acid catalysts, which are corrosive, toxic and difficult to recover from the reaction mixture, with efficient solid materials could support the goal of catalysis in the removal of risky substances.Metal–organic frameworks (MOFs) are a class of highly crystalline porous hybrid materials that are prepared from the coordination of metal clusters or ions as nodes with polyfunctional organic ligands as linkers to form infinite 1, 2 or 3-dimensional frameworks;2–4 they are also known as porous coordination polymers (PCPs). MOFs have attracted much attention in the last few years because of their incredible properties, which include high structure porosity exhibiting large specific surface area (up to several thousand m2 g−1), high pore volume (up to 90% of the crystal volume) and low framework density.5–10 These properties allow MOFs to have potential applications in separation processes and gas storage,11 sensor technology,12,13 catalysis,14 ion exchange,15 drug delivery,16 and luminescence.17
One of the various MOFs is chromium(III) terephthalate, also known as Cr-MIL-101 (MIL for Material of Institute Lavoisier), which has suitable resistance to high temperatures up to 300 °C and solubility in water and common solvents.18–20 Cr-MIL-101 is fabricated from chromium trimeric octahedral clusters and carboxylate moieties (terephthalic acid) that offer a unique high surface area, and it has a rigid zeotype crystal structure with two types of quasi-spherical cages with diameter of 2.9 and 3.4 nm accessible through window sizes at about 1.2 and 1.6 nm, respectively. (The smaller cages have pentagonal windows, while the larger cages are related to both hexagonal and pentagonal windows).21 Additionally, coordinatively unsaturated sites (CUS) with relatively mild Lewis acid properties exist due to the presence of chromium clusters that are not attached to the organic linkers.22 The acidic CUS sites and two types of large cavities permit the post-synthesis modification of MIL-101 with active species such as nano-particles (NPS), organic molecules, polyoxometalates (POM) and superacids. These could potentially improve the Brønsted acid, Lewis acid, and redox sites for a range of acid-catalyzed and redox reactions including hydrogenation,23 esterification,24,25 as well as epoxidation reactions.26
The absence of functional groups and selective sites in most of the stable metal–organic frameworks has limited the use of MOFs in applications such as catalysis. In order to functionalize MOFs with various active sites including heteropoly acids, mineral acids or metal nanoparticles have been loaded into MIL-101 using different methods.27–31 One of the best low-cost, green and effective acids is sulfamic acid (NH2SO3H), which is commercially available and popularly utilized as an effective catalyst in organic synthesis.32,33
Xanthenes, especially 14-phenyl-14H-dibenzo [a,j] xanthene, have received much attention from the pharmaceutical manufacturing industry. This is because they have a great range of pharmacological and biological features like anti-inflammatory,34 antiviral,35 antibacterial and antifungal36 activities, in addition to their use in photodynamic therapy,37 dyes,38 visualization of biomolecules using pH-sensitive fluorescent materials39 and utility in laser technologies40 due to their interesting spectroscopic properties. Xanthenes are synthesized by the condensation reaction of aromatic aldehydes with β-naphthol in the presence of acid catalysts like heteropoly acid supported MCM-41,41 boric acid,42 silica bonded N-propyl sulfamic acid43 and succinimide-N-sulfonic acid.44
Coumarin and its derivatives attract major interest due to their significance in synthetic organic chemistry. Numerous products that contain the coumarin nucleus act as the bases for several biological activities such as antispasmodic, diuretic, anticoagulant, antibacterial and anticarcinogenic.45,46 These compounds were prepared by the Pechmann condensation in the presence of acid catalysts like sulfated titania supported on MCM-41,47 nanostructured sulfated tin oxide48 and tungsto-phosphoric acid supported mesoporous zirconia.49
Pigments and dyes attract great interest in the manufacturing industry.50 The release of these dyes into water has caused global concern because of their overall environmental risks.51 Additionally, numerous dyes are very poisonous to many organisms and cause direct damage to aquatic communities. Among the reasons dye decomposition is difficult is the high stability of dyes to oxidation and light.52 In order to remove dye materials from polluted water, numerous chemical, physical and biological methods have been examined. Among the proposed methods, the adsorption technique is considered to be the most popular method for dye removal due to its being remove several coloring materials simultaneously and it does not require high temperatures.53
One of the most popular anionic/acidic dyes is methyl orange (MO), which we used to examine the adsorption properties of our modified and unmodified MIL-101. Methyl orange is used in printing, dyeing textiles, dyestuff and so on53 and it is also used in the acid–base titration of mineral acids and strong bases as an indicator. Methyl orange is considered as an azo dye as it contains the azo group (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) in its structure. These types of azo dyes are the most frequently used dyes; they are not highly toxic but may cause allergies and hypersensitivity.54
N) in its structure. These types of azo dyes are the most frequently used dyes; they are not highly toxic but may cause allergies and hypersensitivity.54
In recent work, MIL-101 was synthesized with different contents of sulfamic acid using the impregnation technique. The physiochemical properties of the resulting catalysts were identified by several techniques such as XRD, SEM, TEM, Fourier transform-IR, non-aqueous titration with n-butylamine and pyridine adsorption. The catalytic activities of the prepared solid acid samples were examined by using them as catalysts for the synthesis of 7-hydroxy-4-methyl coumarin and the synthesis of 14-phenyl-14H-dibenzo [a,j] xanthene. However, so far, there have been a few reports on the usage of MIL-101 in the adsorption of methyl orange. Due to the high nitrogen content, the adsorption behaviors of the modified MIL-101 materials were determined for removing methyl orange from water.
2. Experimental
2.1 Materials
All inorganic and organic chemicals used in the synthesis of catalysts are commercially available (Merck) and were used without further purification: N,N-dimethylformamide ((CH3)2NCHO), ethanol (C2H5OH), terephthalic acid (C6H4(COOH)2), sulfamic acid (NH2SO3H) and chromium trinitrate (Cr(NO3)3·9H2 O).2.2 Preparation of MIL-101 (Cr)
Cr-MIL-101 was prepared as reported previously, with slight modification.55 A mixture of Cr(NO3)3·9H2O (2000 mg, 2.0 mmol), 1,4-benzene dicarboxylic acid (TPA) (830 mg, 2.0 mmol) and distilled water (24 mL) was sonicated until a dark blue suspension was formed, the suspension was transferred into a Teflon-lined autoclave bomb and placed in an oven for 8 h at 220 °C. After cooling, the suspension was centrifuged to isolate the green powder of MIL-101 and the resulting product was soaked in N,N-dimethylformamide for one day to remove unreacted terephthalic acid encapsulated inside the pores, then the solid product was washed with DMF and hot ethanol, filtered off and finally dried in an oven at 150 °C for 5 h.56Sulfamic acid loaded on MIL-101 was prepared by the direct impregnation method, in which 1 g of the as synthesized MIL-101 was suspended in 20 mL of aqueous solution with different weight percentages of sulfamic acid (15%, 25%, 40%, 55%, 70%, 85% SA/MIL-101) under vigorous stirring for 4 h and impregnated for one day. The product was filtered, washed, and dried at 100 °C overnight. Then, 55 wt% of SA/MIL-101 was calcined in a muffle furnace in air at calcination temperatures of 150, 250 and 350 °C for 4 hours at a rate of 5 °C per minute.
2.3 Catalyst characterization
2.3.5.1 Non-aqueous titration. Measurements of acid strength and the total acidity of solid materials were reported using potentiometric titration.47 The activated catalysts (0.1 g) were suspended in 10 mL acetonitrile for 2 h. Then, the suspension was titrated with 0.05 N n-butylamine in acetonitrile. The Orion 420 digital model was used to measure the electrode potential variation using a double junction electrode.
2.3.5.2 FTIR spectra of chemisorbed pyridine. Brønsted and Lewis acid sites existing on the material surfaces were detected by FT-IR spectra of chemisorbed pyridine. About 0.1 g of the samples was degassed at 200 °C for 3 h under high vacuum and then the samples were cooled to 30 °C. The dry pyridine was then flashed inside the vacuum system and the samples were maintained under these conditions for one month. The excess pyridine was removed by evaporation at 70 °C (ref. 59) then the samples were analyzed using a Perkin-Elmer system 2000 FT-IR spectrophotometer at 1400–1700 cm−1 after mixing 0.005 g of the sample with 0.1 g KBr to make 30 mm diameter self-supporting discs.
2.3.6.1 The synthesis of 14-phenyl-14H-dibenzo [a,j] xanthene. In a typical experiment, a mixture of β-naphthol (2 mmol), benzaldehyde (1 mmol), and catalyst (50 mg) were added in a 25 mL round bottom flask and heated at 120 °C in an oil bath. After the reaction was completed (monitored by TLC), the product was dissolved in chloroform and filtered off by a simple filtration process to recover the catalyst and the crude solid product was purified by recrystallization in ethanol to produce pure 14-phenyl-14H-dibenzo [a,j] xanthene, which was identified by FT-IR and melting point.
2.3.6.2 The synthesis of 7-hydroxy-4-methyl coumarin. A mixture of resorcinol (1.1 g, 10 mmol) and ethyl acetoacetate (2.5 mL, 20 mmol) was placed in a 25 mL round bottom flask, followed by catalyst powder and was activated at 120 °C for 2 h. The flask was placed in an oil bath and refluxed at 120 °C for 2 h. The resultant mixture was filtered and poured into a 50 mL beaker containing crushed ice. The product was characterized by FT-IR spectroscopy and melting point.
The concentration of MO adsorbed at equilibrium (qe in mg g−1), was calculated from the equation
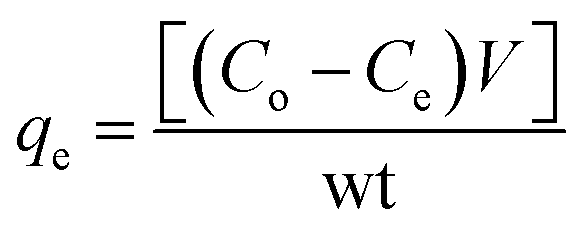
3. Results and discussion
3.1 X-ray diffraction pattern (XRD)
The XRD patterns of the as synthesized MIL-101 and modified MIL-101 with various sulfamic acid contents are shown in Fig. 1 and 2. All samples displayed good crystallinity through the intensity of the peaks and agreed with the previously published XRD patterns for MIL-101.60 The main diffraction peaks of pure MIL-101 were at 2θ of 3.14°, 5.17°, 8.40°, 9.25° and 16.49°, which were found to be similar to the standard data for MIL-101.61 Low angle XRD patterns of the various materials illustrated in Fig. 1(X), demonstrated that there were no notable changes in the intensity of the peaks of the original MIL-101 material and the modified samples of MIL-101 by sulfamic acid. This approach is in agreement with the results of Ferey et al.21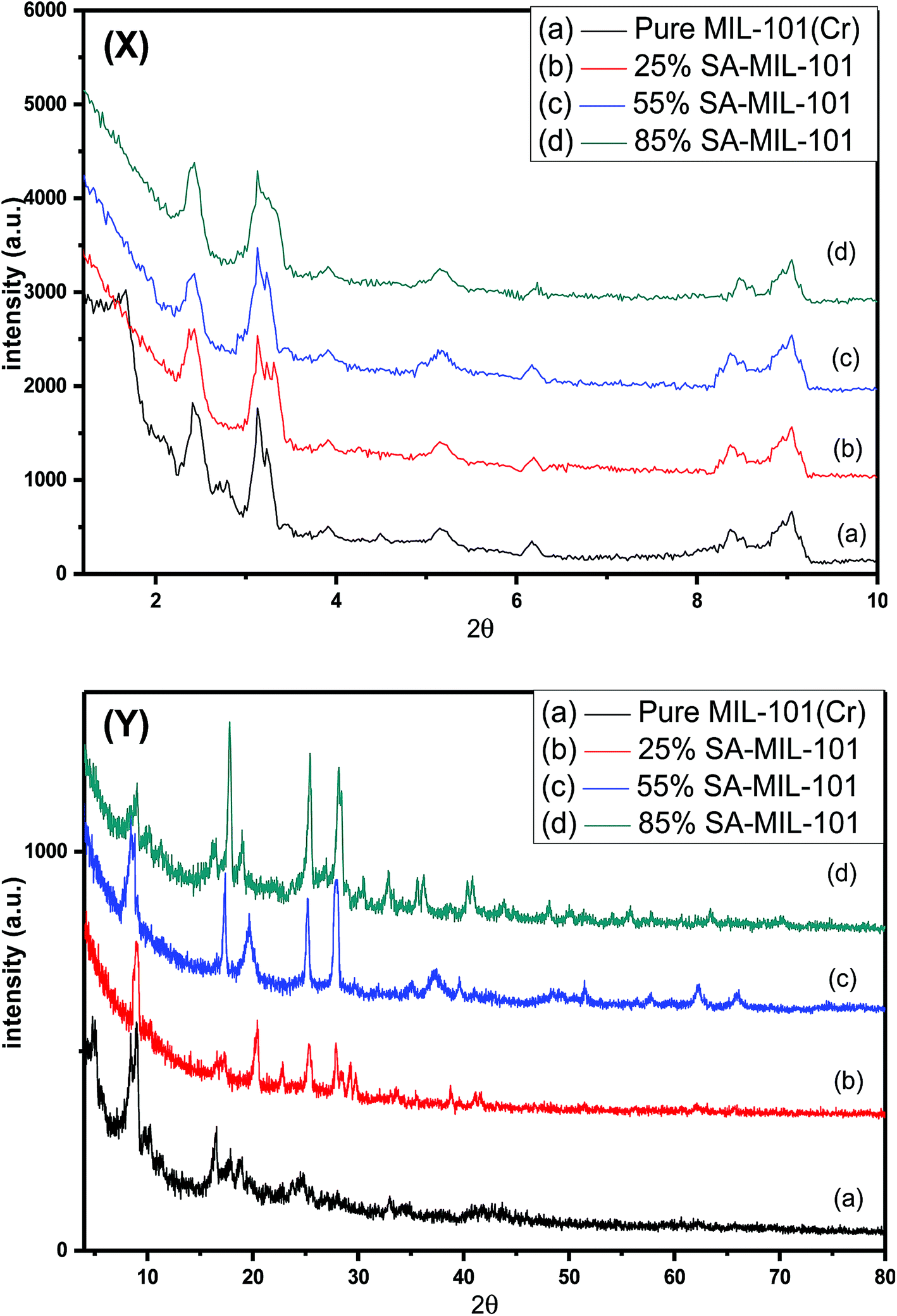 | ||
| Fig. 1 (X) Low angle and (Y) high angle X-ray diffraction patterns of (a) Cr-MIL-101, (b) 25% SA@MIL-101, (c) 55% SA@MIL-101 and (d) 85% SA@MIL-101. | ||
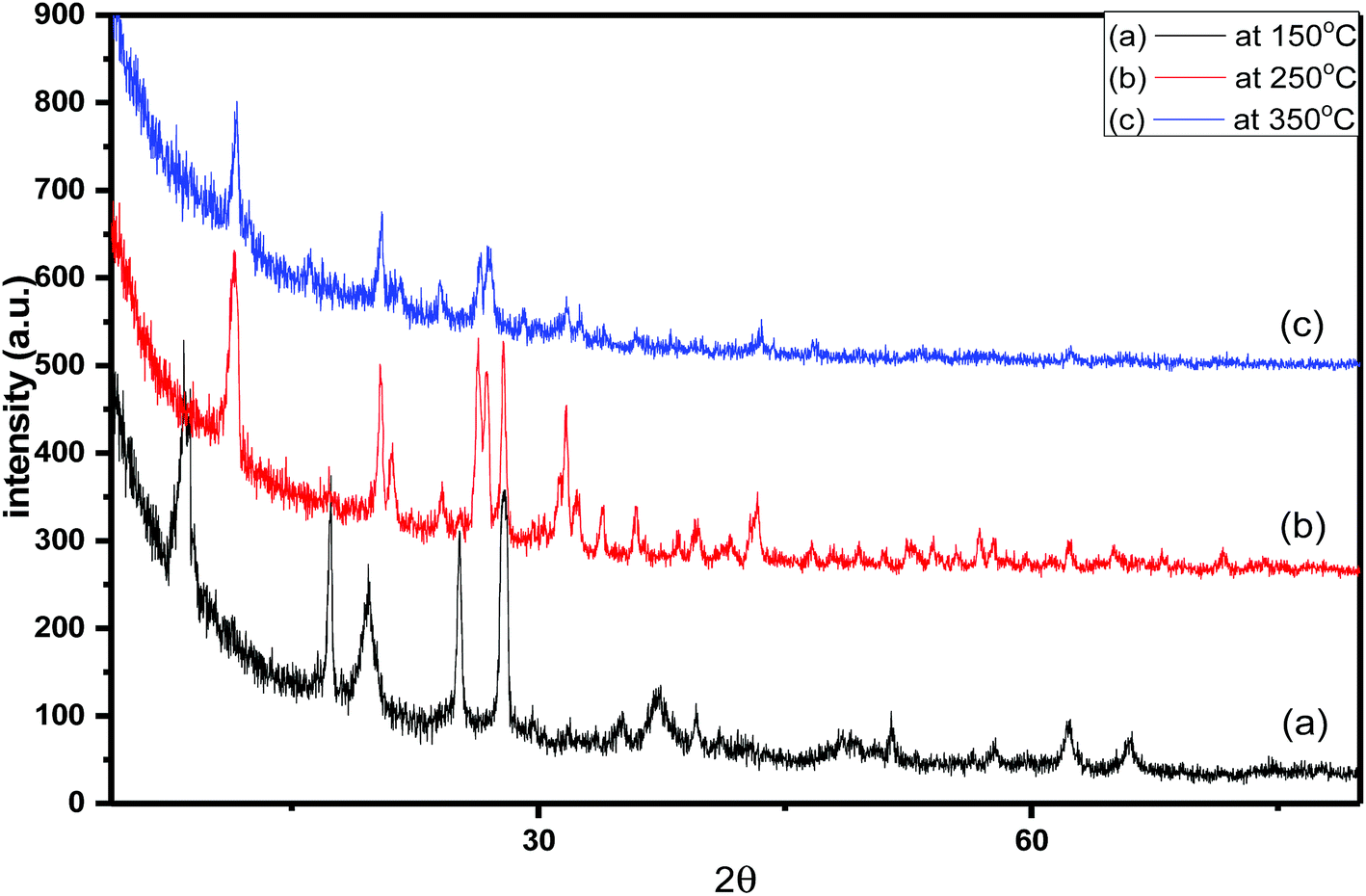 | ||
| Fig. 2 X-ray diffraction pattern of 55% sulfamic acid loaded on MIL-101 at (a) 150 °C, (b) 250 °C and (c) 350 °C. | ||
The wide-range XRD patterns were obtained by scanning the samples from 5 to 80 degrees as presented in Fig. 1(Y), which displays a number of strong and sharp peaks at 2θ = 20.3°, 25.3°, 28.2°, 37.5°, 40.5°, 43.2°, 51.4° and 63.7°, reflecting well-crystallized sulfamic acid crystals.62,63 The intensities of these peaks increased with increasing the sulfamic acid content; the peaks at 2θ of 25.3° and 28.2° also became more obvious at 85% SA/MIL-101. These results show that the sulfamic acid was well dispersed on Cr-MIL-101.
The diffraction patterns corresponding to samples of 55 wt% SA/MIL-101 calcined at 150°, 250°, 350 °C are shown in Fig. 2. As the calcination temperature increased, the intensities of the main peaks decreased and there was some shifting in the peak positions, which may be related to the decomposition of sulfamic acid at higher temperature and the evolution of SO3 from sulfamic acid bound to the Cr-MIL-101 surface.47
3.2 FT-IR spectra
FT-IR is an efficient analytical technique for confirming the presence of functional groups. Fig. 3 shows the FTIR spectra of pure and modified MIL-101 of the spectrum of Cr-MIL-101 in Fig. 3a indicates that there are sharp peaks in the range of 1400–1600 cm−1 that are related to the stretching mode vibration of (CC) bound to the aromatic rings. Weak and narrow bands at 1017 and 749 cm−1 are related to the δ(C–H) and γ(C–H) vibrations of aromatic rings, respectively; the existence of aromatic rings shows that the organic ligand (linker) is present in the final product. The weak peaks in the spectral range of 400–700 cm−1 are most likely because of the out plane and in-plane bending modes of carboxylic (COO−1) groups.61 The strong peak at 1626 cm−1 is due to the vibration of the (CO) group, while the broad peaks in the spectral range of 2400–3400 may be due to crystalline water or acidic OH of carboxylic groups. The peak with moderate strength at 598 cm−1 is attributed to the (Cr–O) vibration, which proves that Cr-MIL-101 was actually synthesized. The difference in spectra was clearly observed after modification with sulfamic acid as shown in Fig. 3b–d. Two weak bands were observed at 3137 and 3439 cm−1, which are related to the symmetrical and asymmetrical stretching vibrations of amine groups.64 On the other hand, new bands appeared at 1185 and 1297 cm−1, which are related to the (OSO) symmetric and asymmetric stretching modes, respectively; the increasingly weak band due to increasing sulfamic acid content appeared at 1053 cm−1 and was assigned to (S–O) stretching vibration.65 The existence of a new vibrational peak at 665 cm−1 was related to NH2 and N–H wagging for sulfamic acid.62 The FTIR spectra of modified MIL-101 (Cr) confirmed the presence of sulfamic acid loaded on Cr-MIL-101.
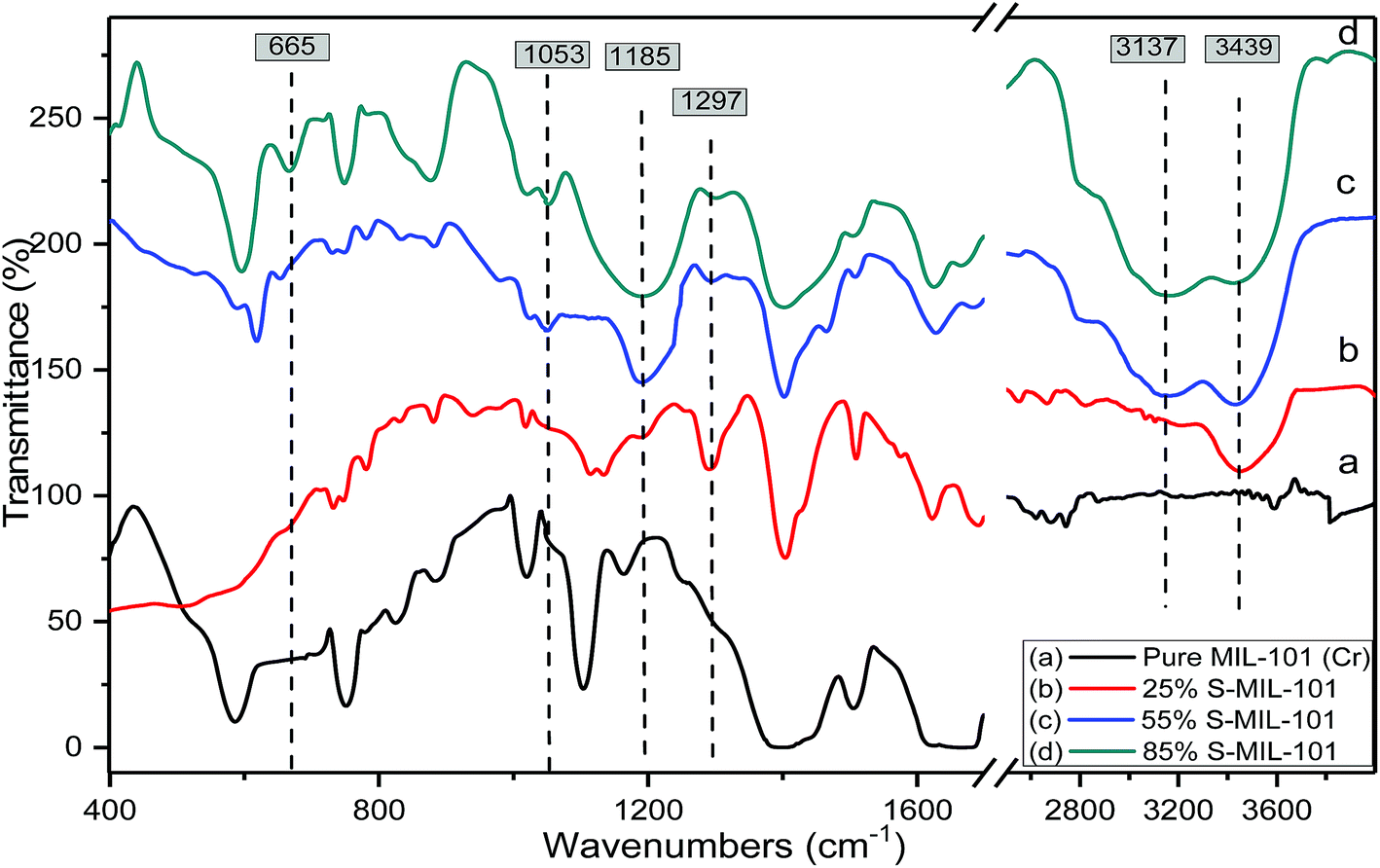 | ||
| Fig. 3 FTIR spectra of (a) Cr-MIL-101; (b) 25% SA@MIL-101; (c) 55% SA@MIL-101 and (d) 85% SA@MIL-101. | ||
Fig. 4 shows the effect of the calcination temperature of 55% sulfamic acid on the IR spectra of MIL-101 (Cr). When the calcination temperature increased from 150 °C to 350 °C, the intensity of the 1185 cm−1 band decreased sharply and the other three peaks at 665, 1053 and 1297 cm−1 nearly disappeared at 350 °C due to the decomposition of sulfamic acid.
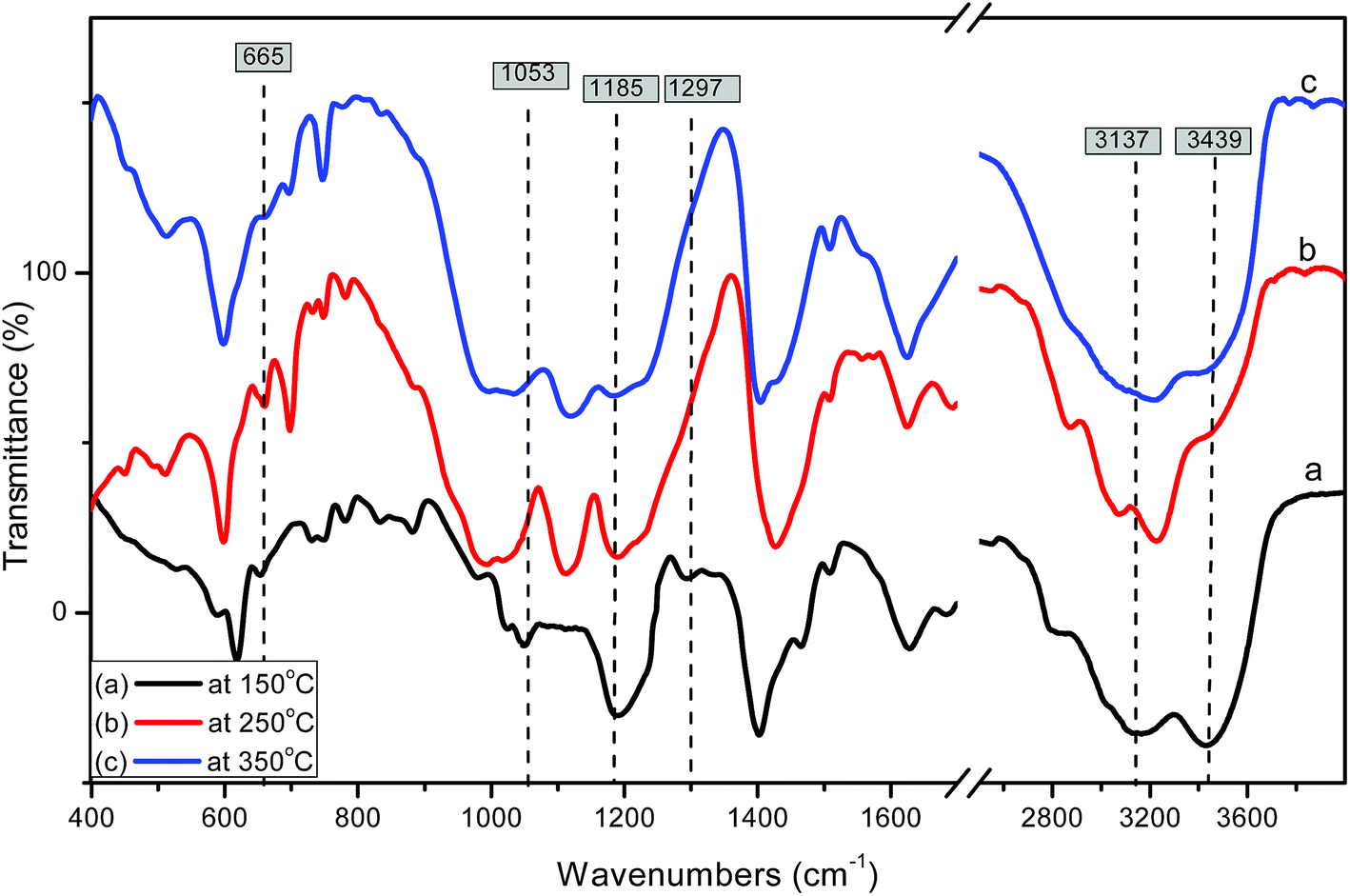 | ||
| Fig. 4 FTIR spectra of 55% sulfamic acid loaded on MIL-101 at (a) 150 °C; (b) 250 °C and (c) 350 °C. | ||
3.3 TEM and SEM images
The particle size and surface morphology of pure and modified MIL-101 samples were characterized by using SEM and TEM and the results are shown in Fig. 5 and 6. The typical TEM and SEM images of pure and modified MIL-101 samples showed that the crystals had a regular octahedral structure with smooth surface and average diameters of 300–500 nm.66 As also depicted in Fig. 6, it is clear that there was no aggregation of sulfamic acid crystals in TEM images up to 55 wt% of sulfamic acid. However, some aggregation was observed on the MIL-101 surface in the case of 85 wt% SA/MIL-101, which could be due to formation of aggregates of sulfamic acid crystals. | ||
| Fig. 5 SEM images of (A) pure MIL-101, (B) 25 wt%, (C) 55 wt%, (D) 85 wt% sulfamic acid/MIL-101. | ||
 | ||
| Fig. 6 TEM images of (A) pure MIL-101, (B) 25 wt%, (C) 55 wt%, (D) 85 wt% sulfamic acid/MIL-101. | ||
3.4 BET measurements
The resulting pore modifications were observable in the N2 adsorption isotherms of pristine MIL-101 (Cr) and sulfamic acid loaded on MIL-101 (Cr). The N2 adsorption isotherms and pore-size distribution curves are shown in Fig. 7. The BET surface area and pore volume of the pristine MIL-101 (Cr) are 2989 m2 g−1 and 1.645 cm3 g−1, respectively, which are in agreement with earlier reports.67 After modification with sulfamic acid, the SA/MIL-101 samples exhibited a significant decrease in the N2 adsorption amounts and the corresponding BET surface areas were decreased to 944 m2 g−1 with the increase in the sulfamic acid content. On the other hand, the pore size and the pore volume gradually decreased with the increase in the sulfamic acid contents as shown in Table 1. This could be due to the pore blockage produced by the deposition of sulfamic acid crystals inside the pores. As can be seen from the pore size distribution curves (Fig. 7Y), the incorporation of sulfamic acid resulted in a decrease in the pore volume and there was a slight shift to a smaller pore diameter compared to Cr-MIL-101.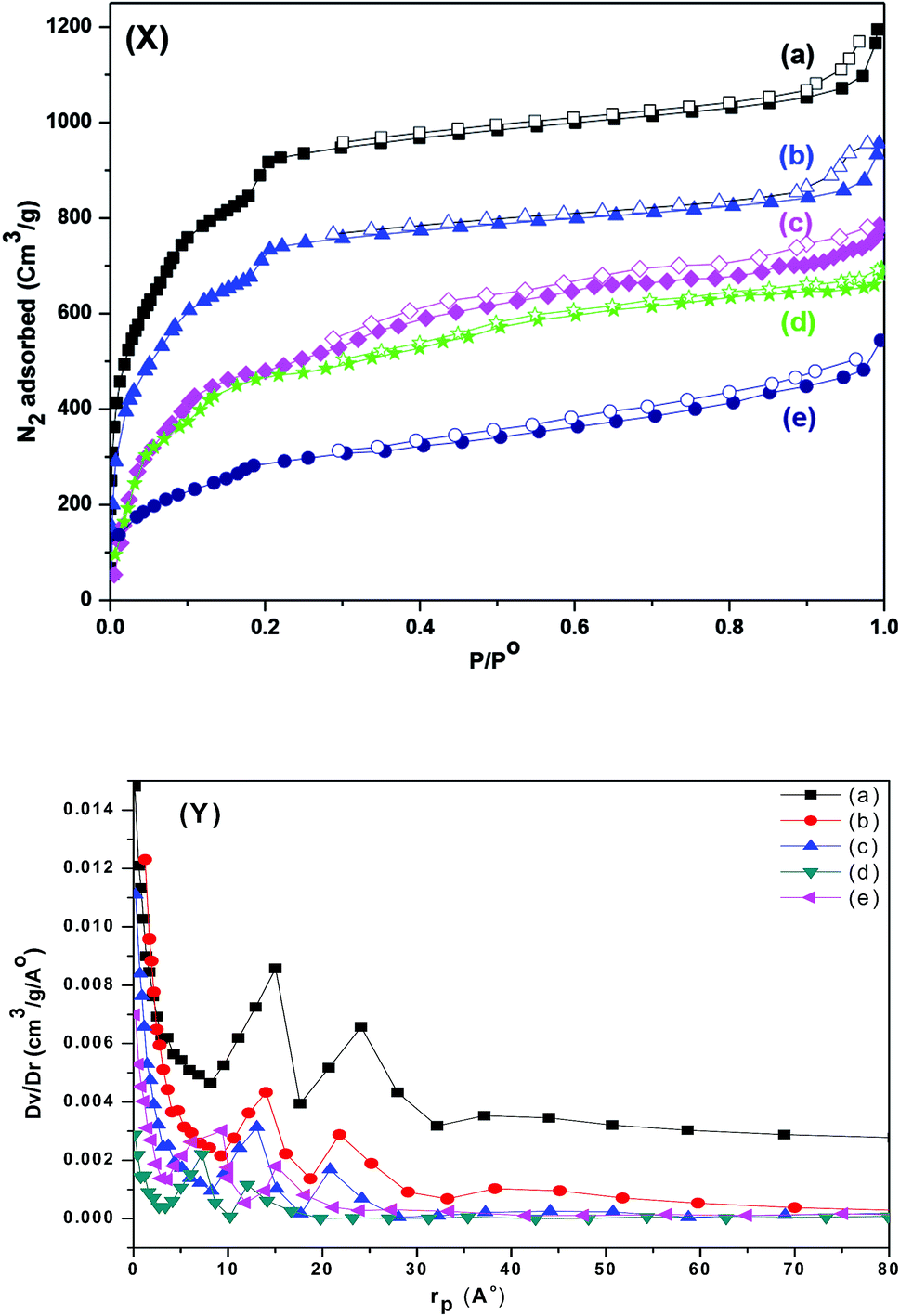 | ||
| Fig. 7 (X) N2 adsorption–desorption isotherm at 77 K; (Y) the corresponding pore size distribution of (a) Cr-MIL-101; (b) 15 wt% SA/MIL-101; (c) 25 wt% SA/MIL-101, (d) 55 wt% SA/MIL-101 and (e) 85 wt% SA/MIL-101. | ||
| Sample name | SBET (m2 g−1) | Vpore (cm3 g−1) |
|---|---|---|
| Pure Cr-MIL-101 at 150 °C | 2989 | 1.645 |
| 15% SA-MIL-101 at 150 °C | 2391 | 1.276 |
| 25% SA-MIL-101 at 150 °C | 1730 | 1.012 |
| 55% SA-MIL-101 at 150 °C | 1387 | 0.877 |
| 85% SA-MIL-101 at 150 °C | 944 | 0.639 |
3.5 Surface acidity studies
where NA is Avogadro's number.
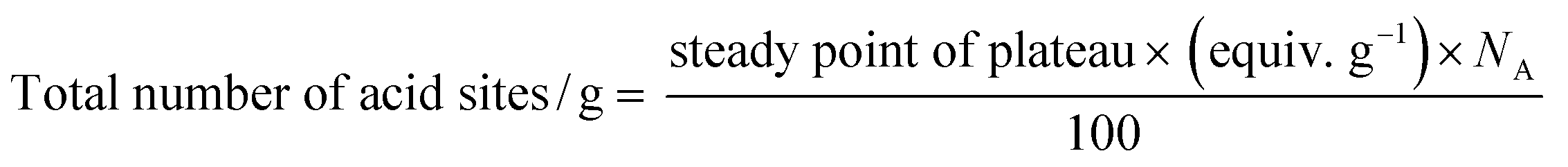
On the other hand, the acid strength of the catalyst sites may be categorized according to the following scale: Ei > 100 mv (very strong sites), 0 < Ei < 100 mv (strong sites), −100 < Ei < 0 mv (weak sites) and Ei < −100 mv (very weak sites).69 Fig. 8 shows that the addition of sulfamic acid increases the acidity strength of Cr-MIL-101 and generated strong acid sites on the surfaces. Pure Cr-MIL-101 showed a maximum acidity of Ei = 148 mv; however, the incorporation of sulfamic acid in MIL-101 was accompanied by a progressive increase in acidic strength up to 55% SA/MIL-101, where Ei = 533 mv as shown in Table 2, then a decrease. Also, the total number of acid sites/gram were varied in the same trend as the total surface acidity as shown in Fig. 9. These results indicate that the distribution of sulfamic acid loaded on the Cr-MIL-101 surface was the maximum and was responsible for higher acidity. The super acidic property is related to the double bond nature of (SO) in the complex formed by interaction of MIL-101 with sulfamic acid. In other words, the acid strength of the SA/MIL-101 catalysts became stronger with the inductive effect of (SO).70
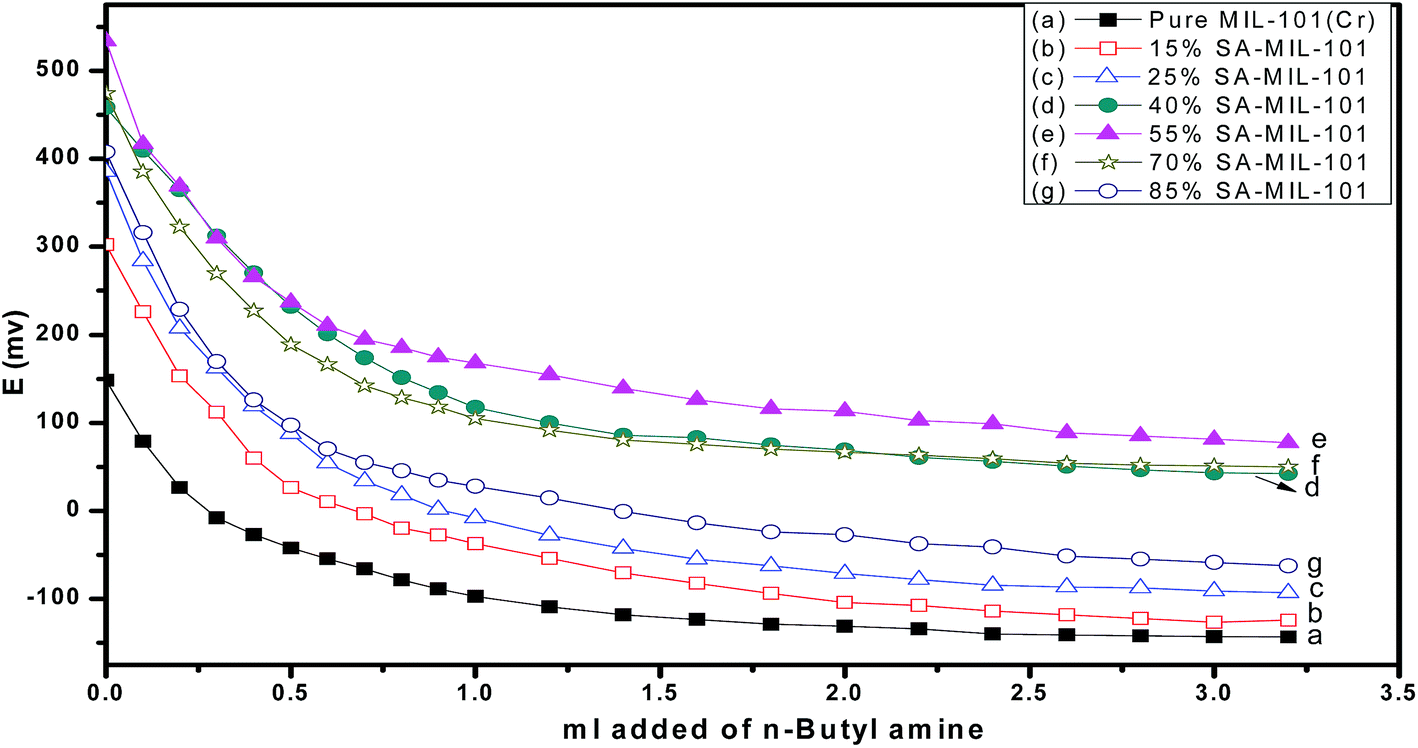 | ||
| Fig. 8 Potentiometric titration curves of catalysts calcined at 150 °C. | ||
| Sample name | Initial potential (Ei) | No. of acid sites in mequiv. g−1 (×1019) | B/L ratio | 14-Phenyl-14H-dibenzo [a,j] xanthene | 7-Hydroxy-4-methyl coumarin |
|---|---|---|---|---|---|
| Pure Cr-MIL-101 at 150 °C | +148.2 | 2.031 | — | 38.2 | 0 |
| 15% SA-MIL-101 at 150 °C | +302.4 | 3.437 | — | 55.9 | 10.5 |
| 25% SA-MIL-101 at 150 °C | +385.4 | 3.612 | 2.43 | 67.1 | 25.4 |
| 40% SA-MIL-101 at 150 °C | +458.4 | 3.987 | 2.93 | 79.8 | 39.5 |
| 55% SA-MIL-101 at 150 °C | +533.8 | 4.425 | 4.86 | 93.7 | 72.8 |
| 70% SA-MIL-101 at 150 °C | +474.3 | 4.244 | — | 83.3 | 44.9 |
| 85% SA-MIL-101 at 150 °C | +407.5 | 3.847 | 2.53 | 69.2 | 28.1 |
| 55% SA-MIL-101 at 250 °C | +364.0 | 3.533 | 2.31 | 61.1 | 51.1 |
| 55% SA-MIL-101 at 350 °C | +308.0 | 3.441 | 2.11 | 47.5 | 38.2 |
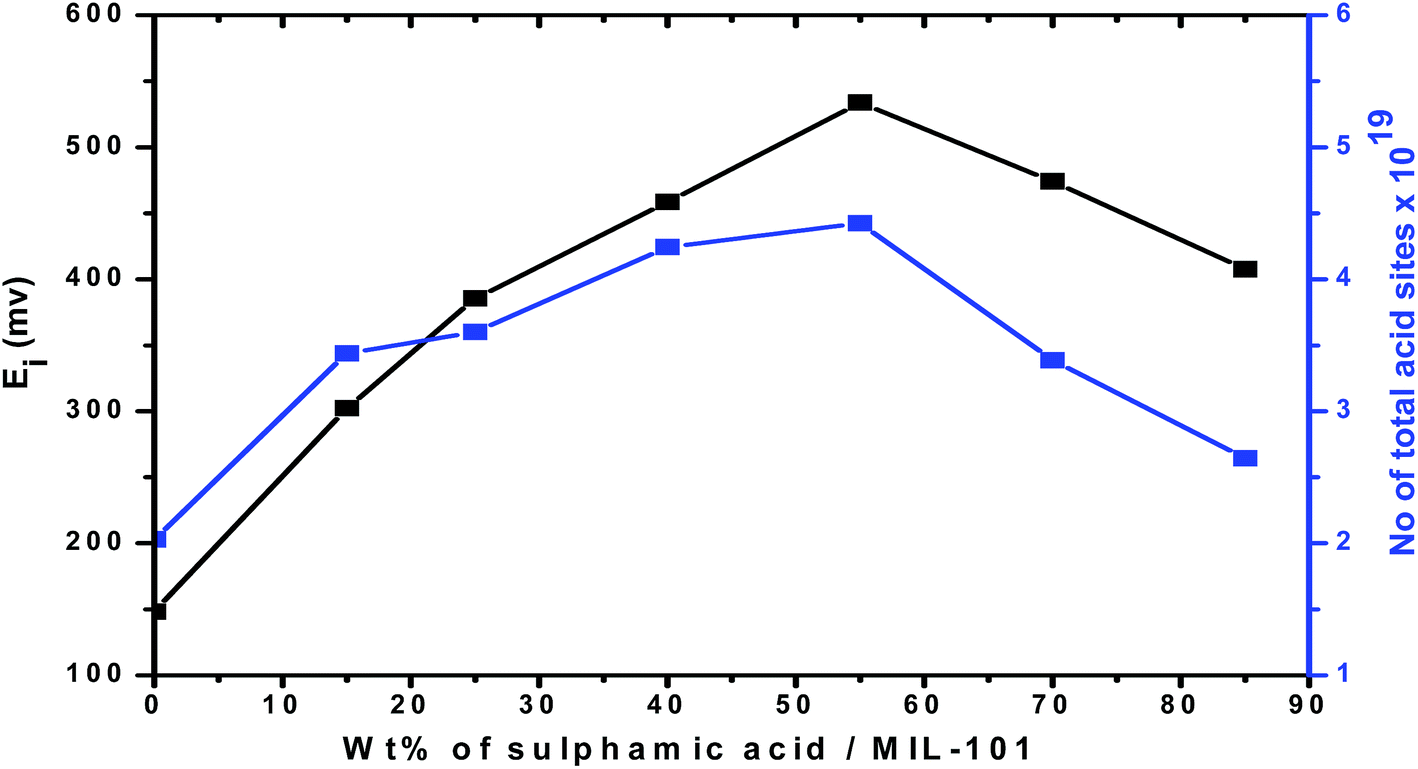 | ||
| Fig. 9 Total number of acid sites and initial potential vs. weight percent of sulfamic acid over MIL-101. | ||
Fig. 10 and 11 show the gradual loss of surface acidity and total number of acid sites at higher calcination temperatures for 55% SA/MIL-101, which may be attributed to the partial evolution of SO3 that decomposed from sulfamic acid bound to the Cr-MIL-101 surface. This was further confirmed by FTIR spectra and XRD.
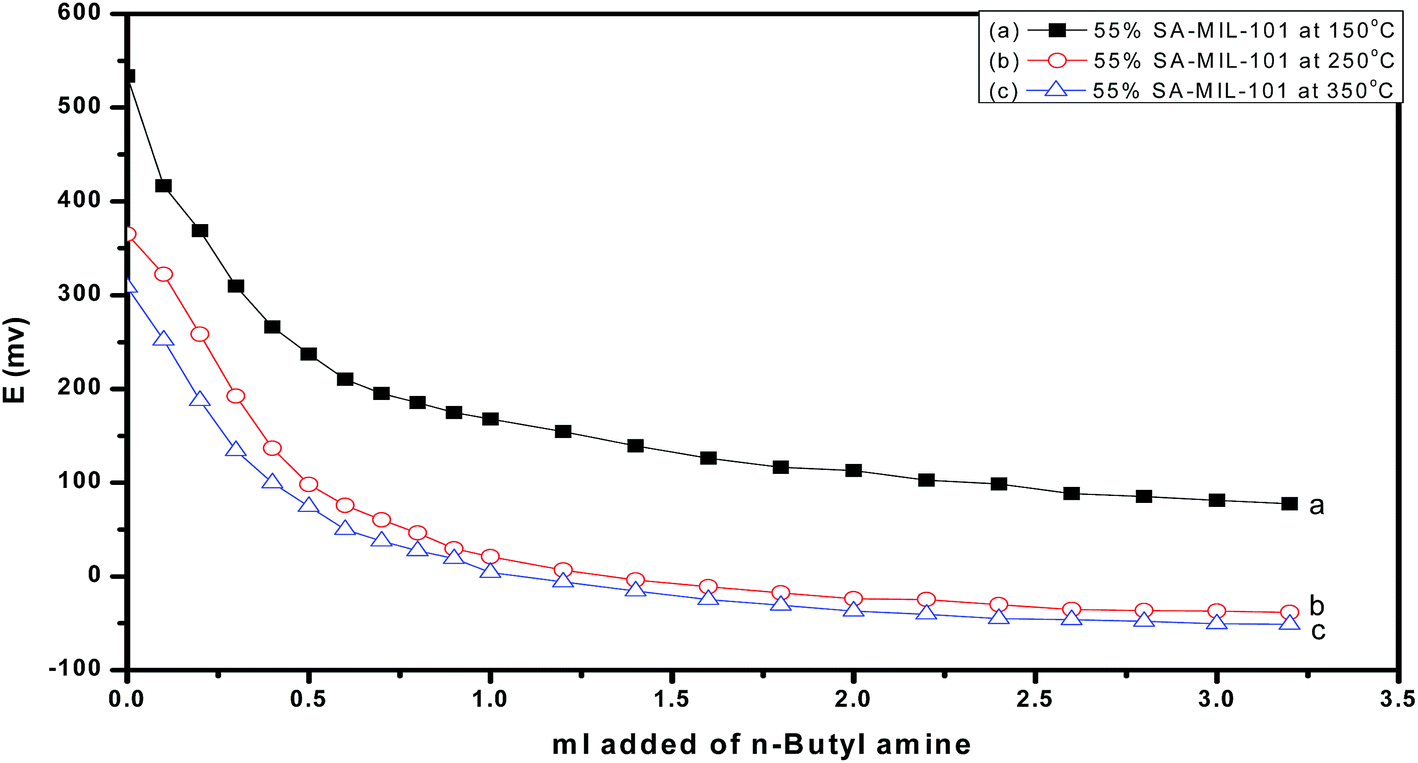 | ||
| Fig. 10 Potentiometric titration curves of 55% SA-MIL-101 calcined at different temperatures. | ||
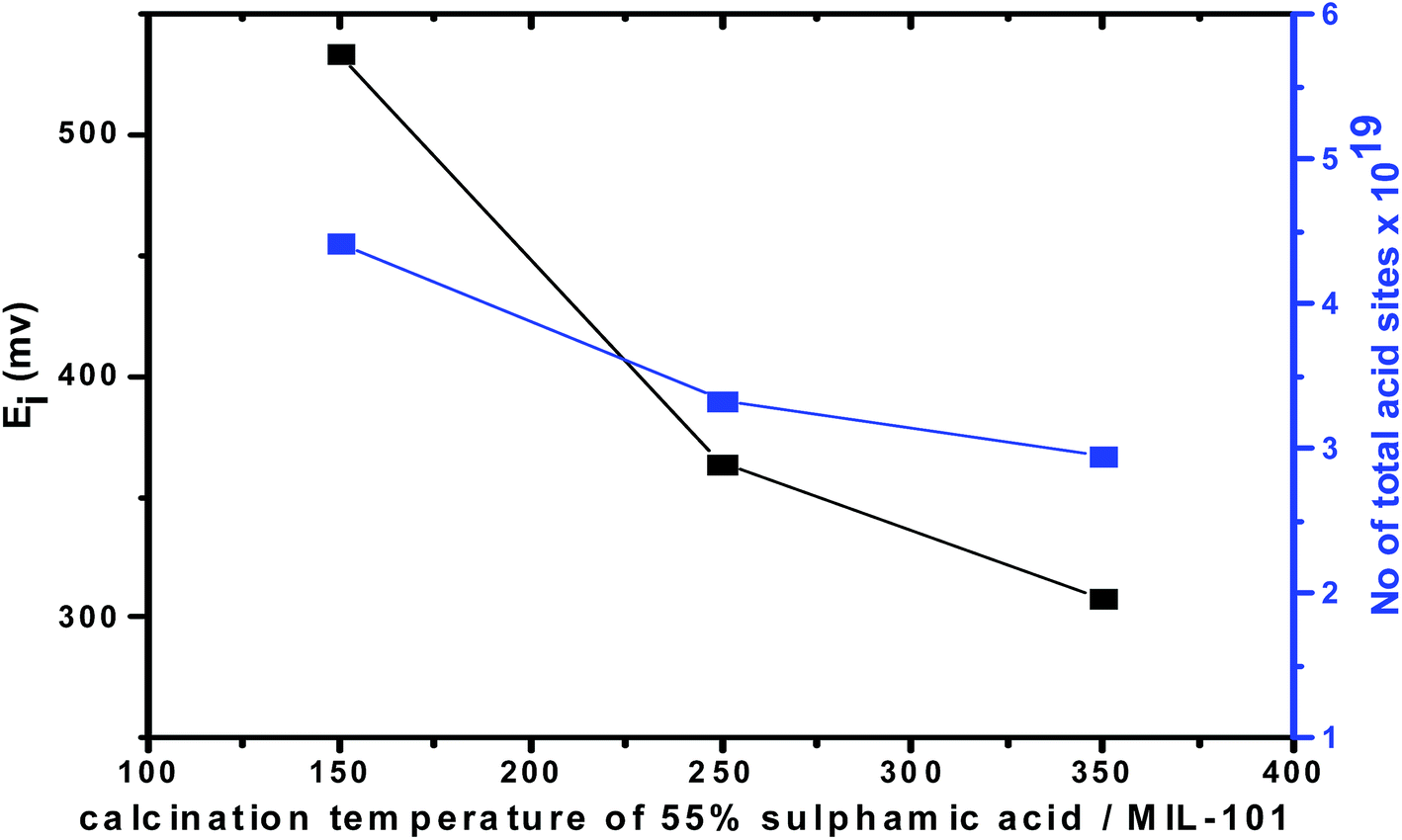 | ||
| Fig. 11 Total number of acid sites and initial potential vs. calcination temperature of 55% SA/MIL-101. | ||
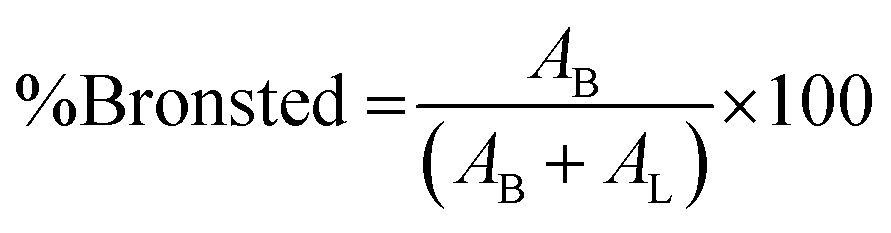
where AB is the integrated area of the peak at 1488 cm−1, attributed to the pyridinium cation adsorbed on the Bronsted acid site, and AL is the intensity at 1438 cm−1 of the pyridine molecule coordinated to the Lewis acid site.
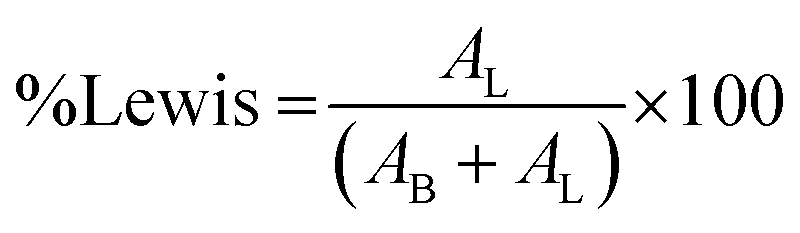
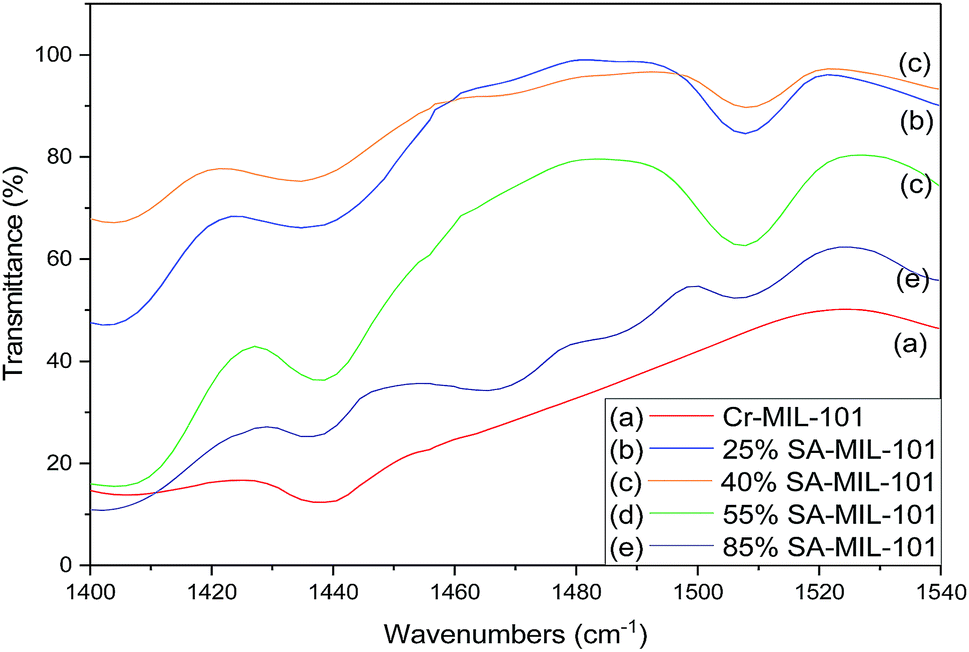 | ||
| Fig. 12 FT-IR spectra of chemisorbed pyridine (showing both Lewis (L) and Brønsted (B) acid sites) on (a) Cr-MIL-101, (b) 25% SA-MIL-101, (c) 40% SA-MIL-101, (d) 55% SA-MIL-101 and (e) 85% SA-MIL-101 calcined at 150 °C. | ||
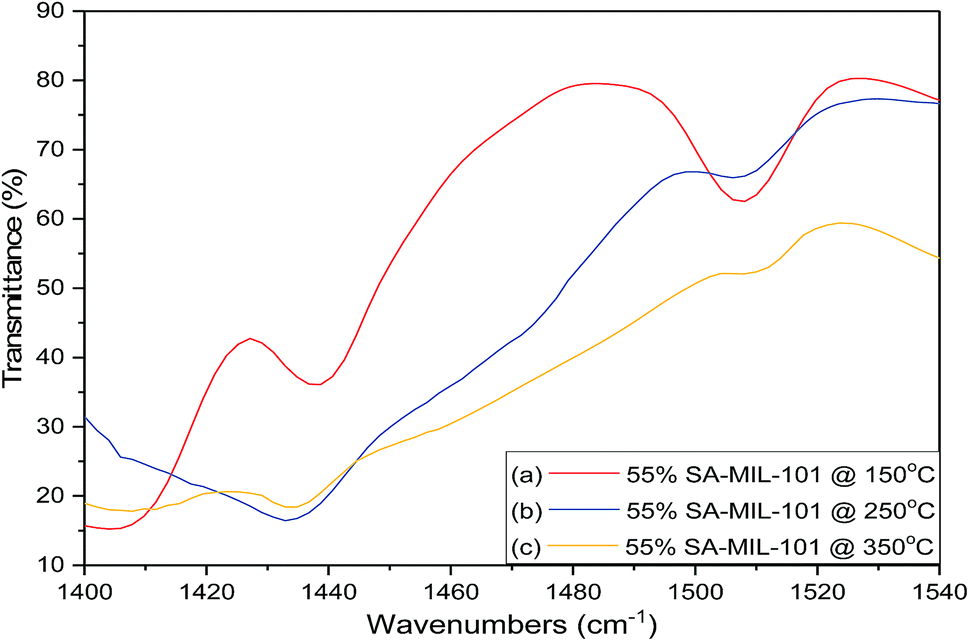 | ||
| Fig. 13 FT-IR spectra of chemisorbed pyridine (showing both Lewis (L) and Brønsted (B) acid sites) on 55% SA-MIL-101 calcined at (a) 150 °C; (b) 250 °C and (C) 350 °C. | ||
From these data, pure Cr-MIL-101 contained only Lewis acid sites, observed at 1438 cm−1, while both Lewis and Brønsted acid sites were observed in all the samples with various ratios depending on the sulfamic acid content. The ratios of Brønsted/Lewis (B/L) sites that are shown in Table 2 were calculated from the ratio of the peak area at 1488 cm−1 and 1438 cm−1, respectively. Brønsted/Lewis acid (B/L) ratios increased with the increase in sulfamic acid content up to 55% SA/MIL-101, then decreased again. The B/L ratio decreased with an increase in calcination temperature and this expected decrease may be due to the evolution of SO3 from sulfamic acid bound to Cr-MIL-101 surface.47
3.6 Catalytic studies
3.6.1.1 The effects of reaction molar ratio and reaction time. The molar ratio of the reaction during the synthesis of the desired xanthenes was studied by using 55% SA-MIL-101 as shown in Fig. 14. After completion of the reaction (monitored by TLC), it was observed that the percentage yield of the desired xanthene increased from 48.2% to 93.7% with the increase of the molar ratio of benzaldehyde
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β-naphthol from 1:1 to 1:2. A further increase to 1:3 and 1:4 was accompanied by a significant loss in the product yield of the desired xanthenes to 77.4% and 56.8%, respectively. The reduction in catalytic activity could be elucidated based on the known fact that the increase in the concentration of β-naphthol inhibits the reaction by blocking the active sites on the catalyst surface.72
:β-naphthol from 1:1 to 1:2. A further increase to 1:3 and 1:4 was accompanied by a significant loss in the product yield of the desired xanthenes to 77.4% and 56.8%, respectively. The reduction in catalytic activity could be elucidated based on the known fact that the increase in the concentration of β-naphthol inhibits the reaction by blocking the active sites on the catalyst surface.72
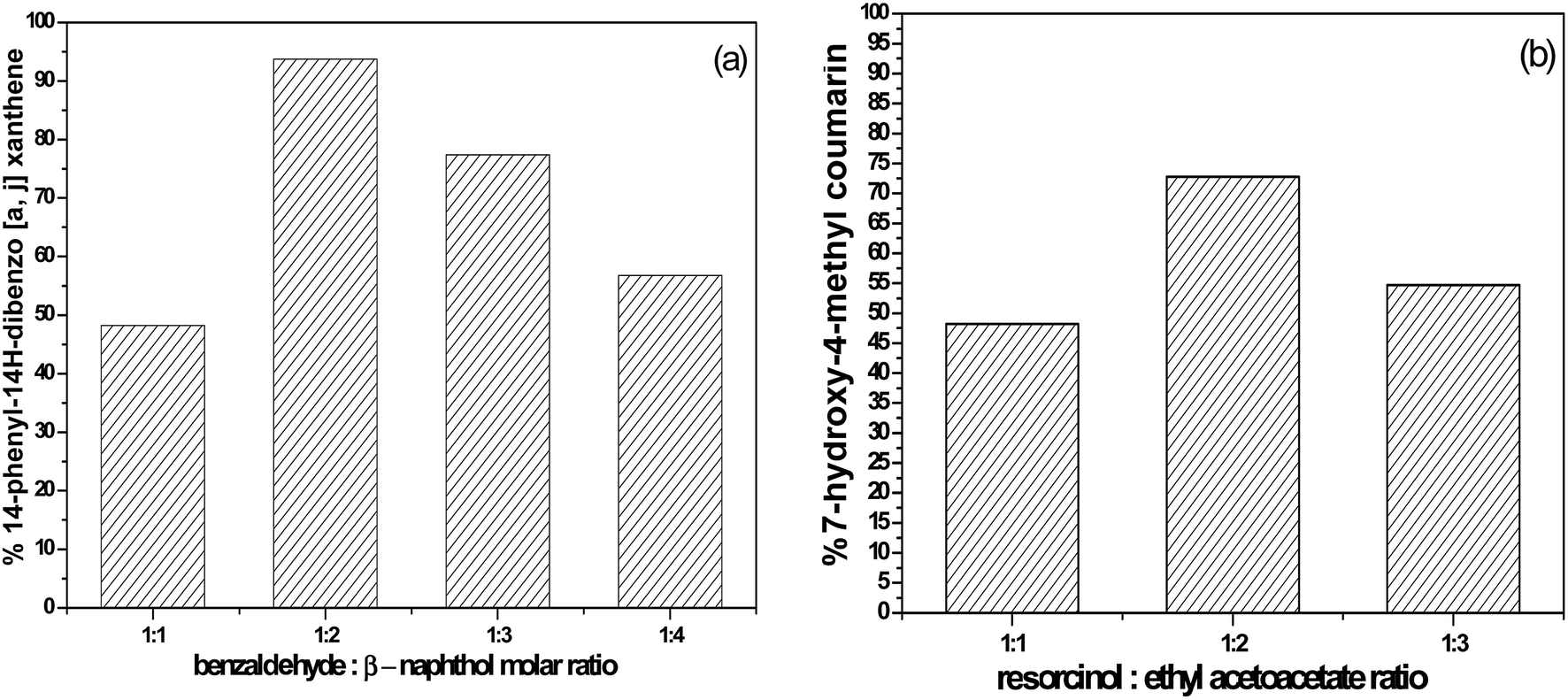 | ||
| Fig. 14 The effect of the molar ratio of (a) benzaldehyde:β-naphthol and (b) resorcinol:ethyl acetoacetate on the catalytic activity over 55% SA-MIL-101. | ||
The reaction time for the synthesis of xanthene over 55% SA-MIL-101 was monitored by TLC, which indicated that all the reactants disappeared after 90 minutes and just one product was formed. The reaction was studied using 0.05 g of supported catalysts under the following reaction conditions: temperature = 125 °C; time = 1.5 h and molar ratio = 1:2 (benzaldehyde:β-naphthol).
3.6.1.2 Effects of sulfamic acid content and calcination temperature. The catalytic activities of unmodified and sulfamic acid-modified Cr-MIL-101, calcined at 150 °C for 2 h in the synthesis of the desired xanthene were studied at 125 °C using the reactant molar ratio of 1
:2 (benzaldehyde:β-naphthol) for 90 min. Pure Cr-MIL-101 showed a slightly lower activity because the active sites of MIL-101 have a very weak acidic nature. However, MIL-101 modified with various wt% of sulfamic acid showed that the percentage yield of the desired xanthene increased with an increase in the sulfamic acid content up to 55 wt% SA-MIL-101, which showed the highest activity with 94% of desired xanthene. There was a subsequent decrease with the further increase in sulfamic acid content, which may be due to the decrease in the surface acidity of the supported catalysts. It was also noticed that the reaction yield increased with the increase in Brønsted to Lewis acid sites ratio as shown in Table 2. These results indicate that the formation of the desired xanthenes depends on the B/L ratio.The effect of the calcination temperatures (150, 250 and 350 °C) on the yield of desired xanthene over the 55% SA-MIL-101 catalyst is also shown in Table 2. Examination of the table shows that with the increase in the calcination temperature, the percent yield decreases because of the reduction in the catalyst acidity and Brønsted/Lewis acid sites ratio as a result of the decomposition of sulfamic acid and evolution of SOx moieties.
The results obtained in this work using sulfamic acid loaded on MIL-101 were compared with other reported results in literature; i.e., with nano-ZnO,73 sulfamic acid,74 tungstate sulfuric acid75 and L-proline, DCE76 in the synthesis of the desired xanthene. From these results, sulfamic acid supported on MIL-101 acted as efficacious catalyst with regard to the obtained product, yield and the reaction times.
3.6.1.3 Reusability of the catalyst. The reusability of SA-MIL-101 catalysts was examined; a typical recycling study on the sample with 55 wt% SA-MIL-101 was performed by separating the catalyst from the reaction, washing with ethanol and drying at 120 °C. After that, the stability of the catalyst was investigated via studying the stability of the characteristic IR bands of the sulfate groups before and after using the catalyst 4 times. There were small changes in the peak intensity, which were related to the SO3H group as shown in Fig. 16. The catalyst showed a small but significant loss in activity after four cycles as shown in Fig. 15 for the preparation of the desired xanthene; this is because xanthene synthesis depends on both Bronsted acid sites (SO3H) and Lewis acid sites.
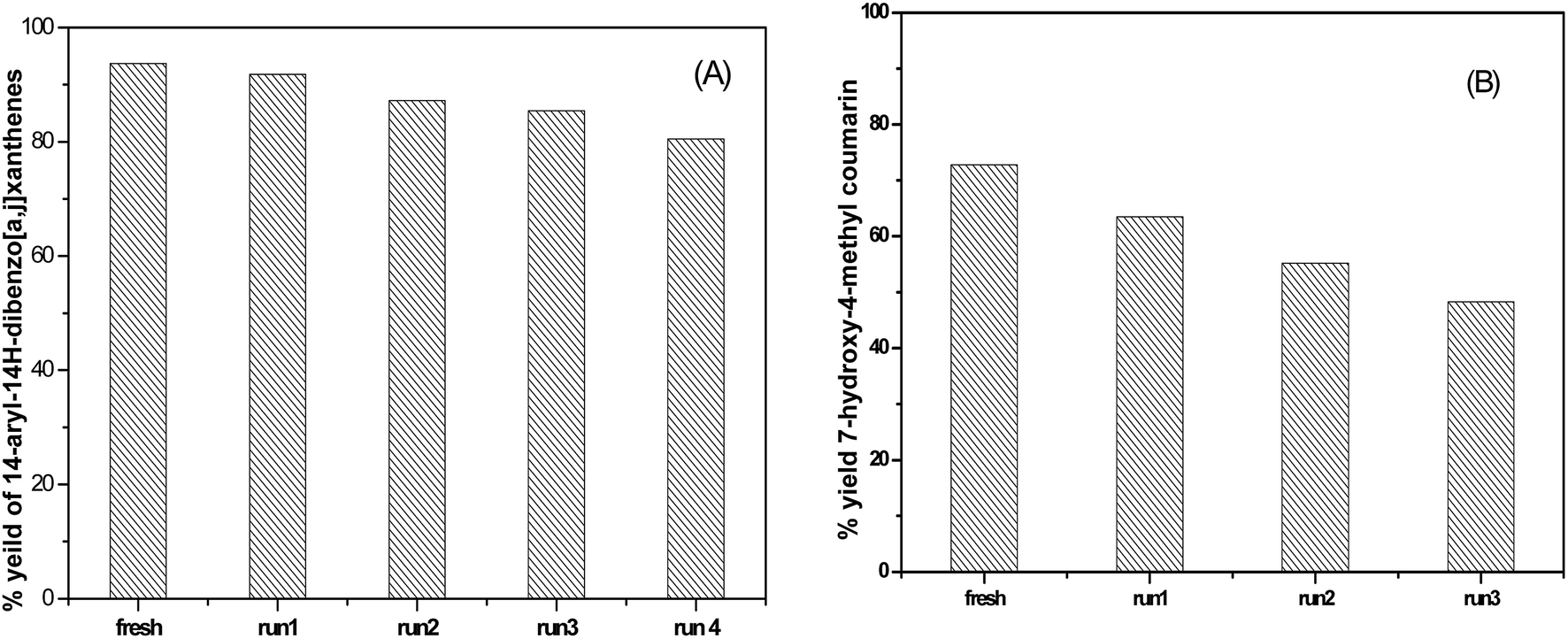 | ||
| Fig. 15 The reusability of 55% SA-MIL-101 in the synthesis of (A) 14-phenyl-14H-dibenzo [a,j] xanthene and (B) 7-hydroxy-4-methylcoumarin. | ||
3.6.2.1 The effect of the molar ratio. The effect of the molar ratio on the formation of the desired coumarin was investigated using 100 mg of 55% SA-MIL-101, as shown in Fig. 14. The yield of the desired coumarin increased from 48.2 to 72.8% with the increase in the molar ratio of resorcinol
:ethyl acetoacetate from 1:1 to 1:2. A further increase to 1:3 was accompanied by a significant decrease in the percentage yield of the desired coumarin to 54.7%. The decrease in the catalytic activity with the further increase in the ethyl acetoacetate molar ratio was due to the saturation of the catalyst surface with the ethyl acetoacetate, which blocked the acid adsorption centers. The efficiency of the catalyst was therefore reduced due to the competitive adsorption of the ethyl acetoacetate on the acid sites with resorcinol.59
3.6.2.2 The effect of sulfamic acid content and calcination temperature. The synthetic reaction was conducted over pure and modified MIL-101 using sulfamic acid at 120 °C for two hours, with the molar ratio of the reactants being 1
:2 (resorcinol:ethyl acetoacetate). Pure MIL-101 did not have considerable catalytic activity in the reaction, which may be attributed to the weakly acidic active sites on the MIL-101 framework. On the other hand, MIL-101 modified with various amounts of sulfamic acid showed higher activity. Table 2 shows that the percentage yield of the desired coumarin gradually increased with increasing the amount of sulfamic acid loaded on Cr-MIL-101 until the maximum yield was reached (72.8%) at 55 wt% SA-MIL-101, and then decreased. The decrease in the catalyst activity may be due to the aggregation of sulfamic acid crystals, as shown in the TEM images, which resulted in the decrease in the surface acidity of the samples and the B/L ratio. Table 2 shows that the sharp decrease in both the acidity and catalytic activity with further increase in the calcination temperature may be attributed to the decomposition of sulfamic acid and the evolution of SOx moieties.A comparison was made of the results obtained in this work using sulfamic acid loaded on MIL-101 with those achieved using polyaniline-sulfate,77 aniline-sulfosalicylate,77 amberlyst-15 (ref. 78) and heteropoly acid/MIL-101 catalysts.68 The results indicate that SA/MIL-101 acts as an effective catalyst in the synthesis of the desired coumarin with respect to yields and products.
3.6.2.3 The reusability of the catalyst. Upon completion of the reaction, the reusability of the 55% SA-MIL-101 sample was investigated and the catalyst was found to be re-usable for at least three times as shown in Fig. 15, where it can be seen that the catalytic activity decreased from 72.9% to 48.4% after three uses. This decrease may be due to the removal of some Brønsted acid sites (SO3H) as evidenced in the leaching test shown in Fig. 16, since the synthesis of coumarin basically depends on Brønsted acid sites.79
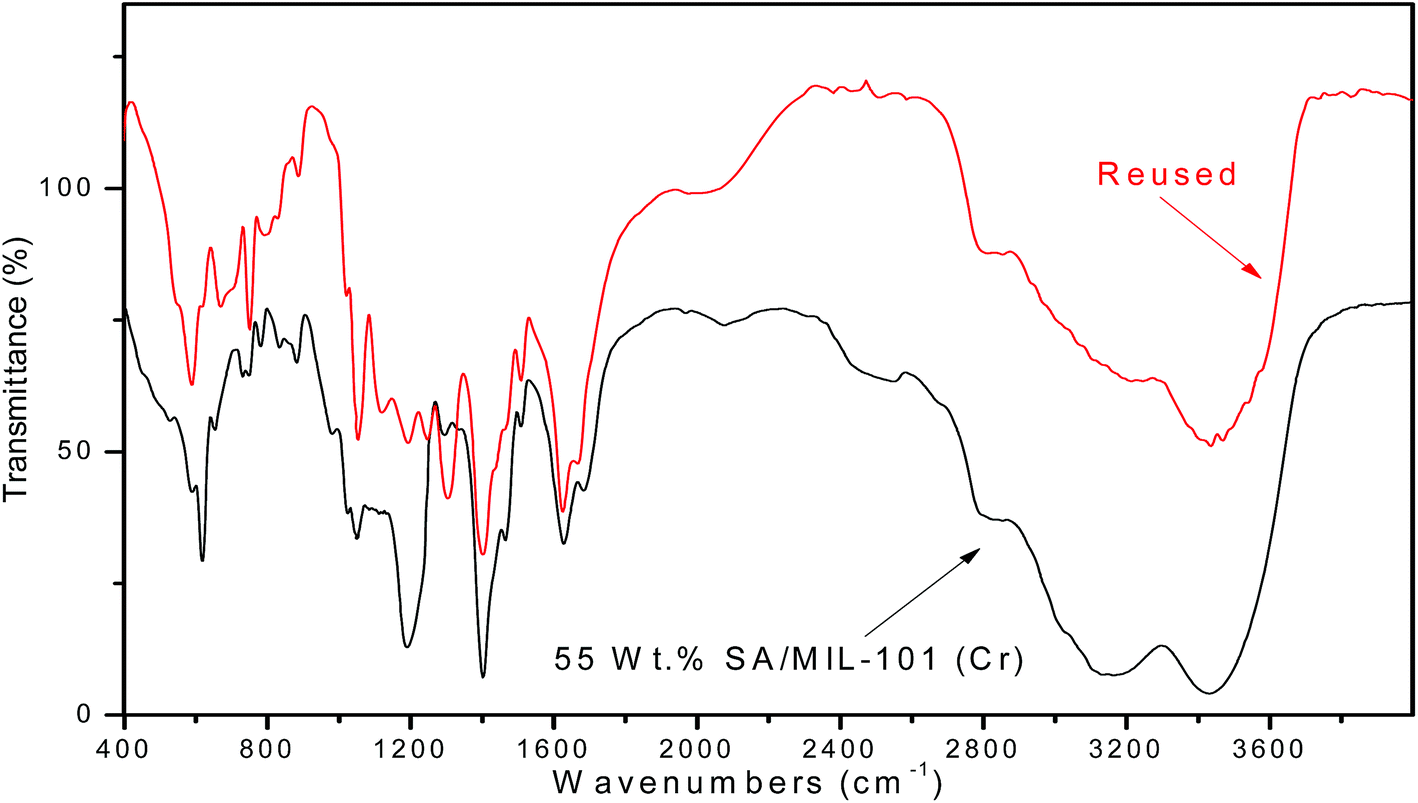 | ||
| Fig. 16 FT-IR spectra of fresh and recovered 55% sulfamic acid/MIL-101 (Cr). | ||
3.7 Adsorption of methyl orange
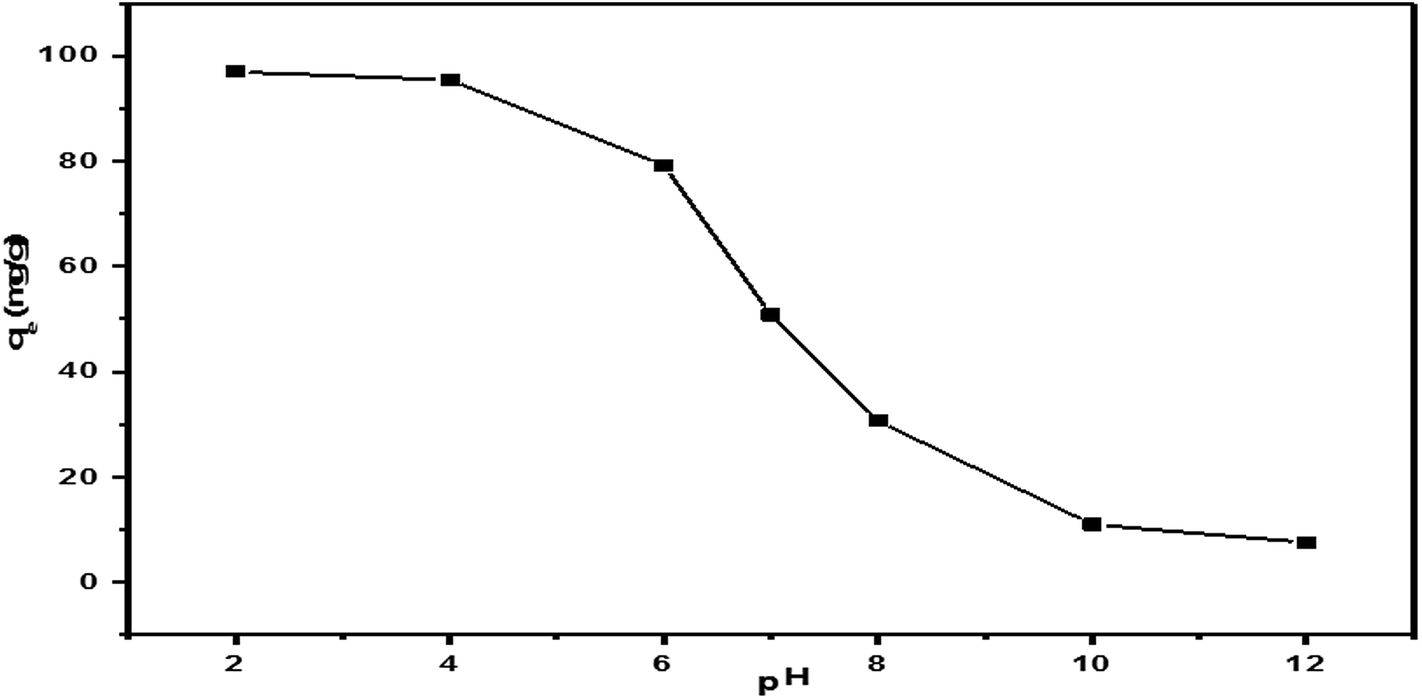 | ||
| Fig. 17 The effect of the initial pH on the adsorption of MO on the 55% SA-MIL-101 sample (adsorbent dosage = 50 mg, initial MO concentration = 100 ppm, solution volume = 50 mL, temperature = 25 °C, contact time = 90 min). | ||
The adsorption mechanism may be as follows:
Firstly, the methyl orange dye (R–SO3Na) decomposes into anions when dissolved in an aqueous solution.
| R–SO3Na + H2O → R–SO3− + Na+ |
Secondly, the adsorption process is achieved due to electrostatic attractions between the MO anions and the adsorbent surface.83
| RNH3+ + RSO3− → RNH3+O3SR |
Thus, pH close to 2 was selected as optimal for the following adsorption experiments.
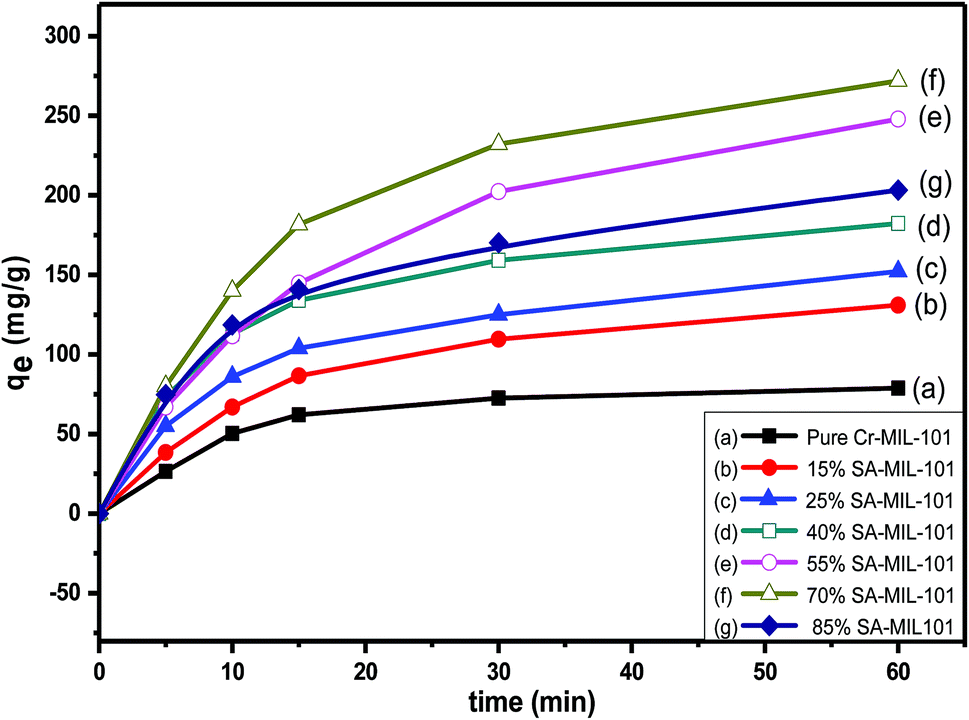 | ||
| Fig. 18 The effect of contact time on MO removal using initial dye concentration (Co = 200 ppm) over different modified MIL-101 adsorbents. | ||
Numerous steps were used to observe the adsorption kinetics controlling sorption processes like diffusion control, chemical reaction and mass transfer. Pseudo 1st order and pseudo 2nd order models were used for MO adsorption over modified and unmodified Cr-MIL-101. The similarity between the predicted values and experimental data was expressed by the correlation coefficients R2.84 The formula for the pseudo 1st order kinetic model is given by eqn (1):
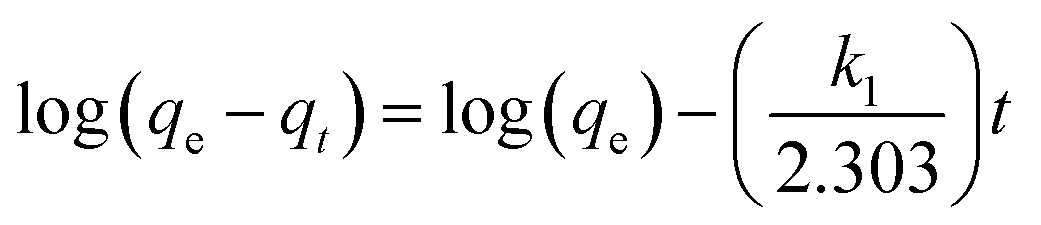 | (1) |
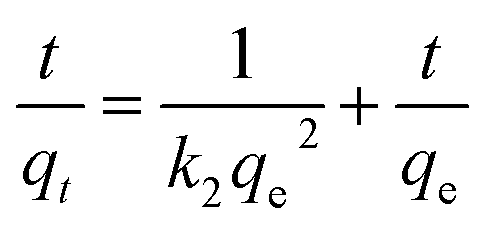 | (2) |
| Sample name | Experimental qm (mg g−1) | Pseudo first order kinetics | Pseudo second order kinetics | ||||
|---|---|---|---|---|---|---|---|
| R2 | K1 (min−1) | qe (mg g−1) | R2 | K2 (g (mg min)−1) | qe (mg g−1) | ||
| Pure Cr-MIL-101 at 150 °C | 94.6 | 0.9151 | 0.0105 | 44.1 | 0.9991 | 0.00064 | 95.7 |
| 15% SA-MIL-101 at 150 °C | 143.5 | 0.9069 | 0.0137 | 67.7 | 0.9997 | 0.00059 | 147.9 |
| 25% SA-MIL-101 at 150 °C | 179.9 | 0.8909 | 0.0117 | 81.9 | 0.9999 | 0.00048 | 183.5 |
| 40% SA-MIL-101 at 150 °C | 200.8 | 0.8272 | 0.0104 | 70.1 | 0.9999 | 0.00070 | 201.6 |
| 55% SA-MIL-101 at 150 °C | 272.5 | 0.8522 | 0.0138 | 111.4 | 0.9993 | 0.00031 | 280.9 |
| 70% SA-MIL-101 at 150 °C | 291.5 | 0.8832 | 0.0157 | 110.4 | 0.9991 | 0.00035 | 300.3 |
| 85% SA-MIL-101 at 150 °C | 230.4 | 0.77325 | 0.0100 | 89.6 | 0.9997 | 0.00047 | 232.1 |
| 55% SA-MIL-101 at 250 °C | 188.7 | 0.9022 | 0.0139 | 91.9 | 0.9991 | 0.00037 | 196.1 |
| 55% SA-MIL-101 at 350 °C | 143.1 | 0.9192 | 0.0224 | 74.8 | 0.9996 | 0.00054 | 147.9 |
Previously, it was stated that the insertion of nitrogen- or oxygen-containing functional groups into carbon materials might be helpful to enhance the adsorption capacity of materials for dyes.86,87 The introduced nitrogen- or oxygen-containing groups may coat the metal organic framework material with more basic sites and create more positively charged surfaces that could improve the interaction between anionic dye (MO) and MOFs though interactions like covalent bonding, electrostatic interaction, H-bonding, dipole–dipole interaction, and so on.
In order to detect the role of functional groups containing nitrogen in the adsorption capacity of pure and modified MIL-101, the percentage removal of methyl orange at different initial concentrations was also investigated as shown in Fig. 19. The percentage removal of methyl orange was reduced progressively when the concentration of methyl orange increased from 25 ppm to 500 ppm. At a moderately low concentration (25 ppm), all adsorbents could totally remove methyl orange from aqueous solution. At a higher concentration of dye (250 ppm), 55% and 70% SA-MIL-101 could still cause the complete removal of dye. Evidently, both 55% and 70% SA-MIL-101 material showed higher adsorption ability for MO removal compared to other adsorbents. This may be due to the high percentage of amine groups on the surface of the modified Cr-MIL-101.
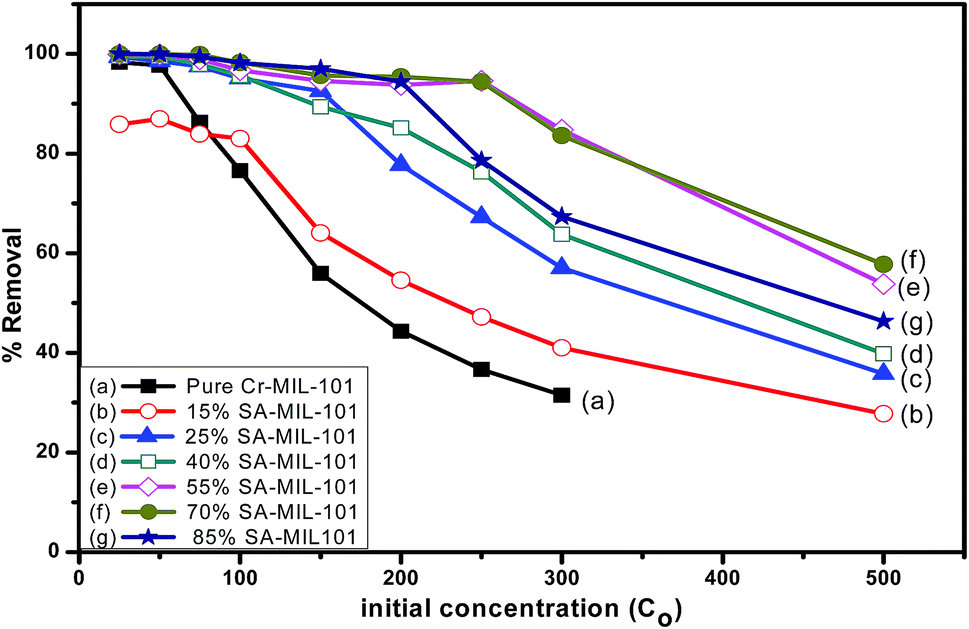 | ||
| Fig. 19 The effect of methyl orange concentration on the adsorption performance over different adsorbents. | ||
The adsorption behavior of methyl orange on pure and modified MIL-101 was further studied, and two adsorption isotherm equations, the Langmuir and Freundlich isotherm models, were applied based on the adsorption equilibrium data.
Firstly, the Langmuir isotherm is given by eqn (3):
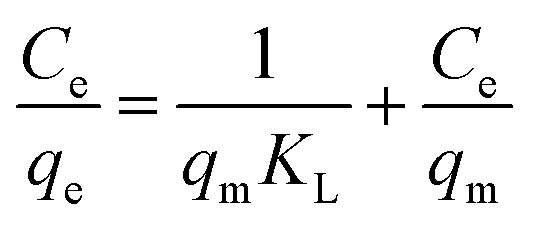 | (3) |
The Langmuir model assumes the formation of a monolayer over the surface of the adsorbent without attraction between adsorbed molecules. Maximum adsorption capacity qm and Langmuir constants KL values are listed in Table 4, which were calculated from the isotherm shown in Fig. 20. The main features of the Langmuir adsorption isotherm can be expressed in terms of the equilibrium parameter or separation factor (RL), which is defined by eqn (4):
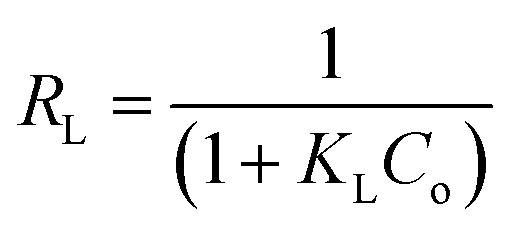 | (4) |
| Sample name | Langmuir isotherm | Freundlich isotherm | |||||
|---|---|---|---|---|---|---|---|
| R2 | RL | KL (L mg−1) | qm (mg g−1) | R2 | 1/n | KF | |
| Pure Cr-MIL-101 at 150 °C | 0.9982 | 0.0139 | 0.2362 | 94.6 | 0.8708 | 0.1867 | 37.948 |
| 15% SA-MIL-101 at 150 °C | 0.9954 | 0.0080 | 0.2478 | 143.5 | 0.8342 | 0.3488 | 21.704 |
| 25% SA-MIL-101 at 150 °C | 0.9995 | 0.0076 | 0.2596 | 179.9 | 0.8975 | 0.2; 547 | 54.347 |
| 40% SA-MIL-101 at 150 °C | 0.9994 | 0.0074 | 0.2665 | 200.8 | 0.9278 | 0.2584 | 60.234 |
| 55% SA-MIL-101 at 150 °C | 0.9987 | 0.0067 | 0.2957 | 272.5 | 0.9234 | 0.2708 | 82.719 |
| 70% SA-MIL-101 at 150 °C | 0.9980 | 0.0064 | 0.3115 | 291.5 | 0.9283 | 0.2402 | 98.356 |
| 85% SA-MIL-101 at 150 °C | 0.9964 | 0.0066 | 0.3014 | 230.4 | 0.9346 | 0.1677 | 99.289 |
| 55% SA-MIL-101 at 250 °C | 0.9992 | 0.0076 | 0.2619 | 188.7 | 0.9129 | 0.2330 | 62.127 |
| 55% SA-MIL-101 at 350 °C | 0.9968 | 0.0173 | 0.1132 | 143.1 | 0.9337 | 0.2283 | 42.729 |
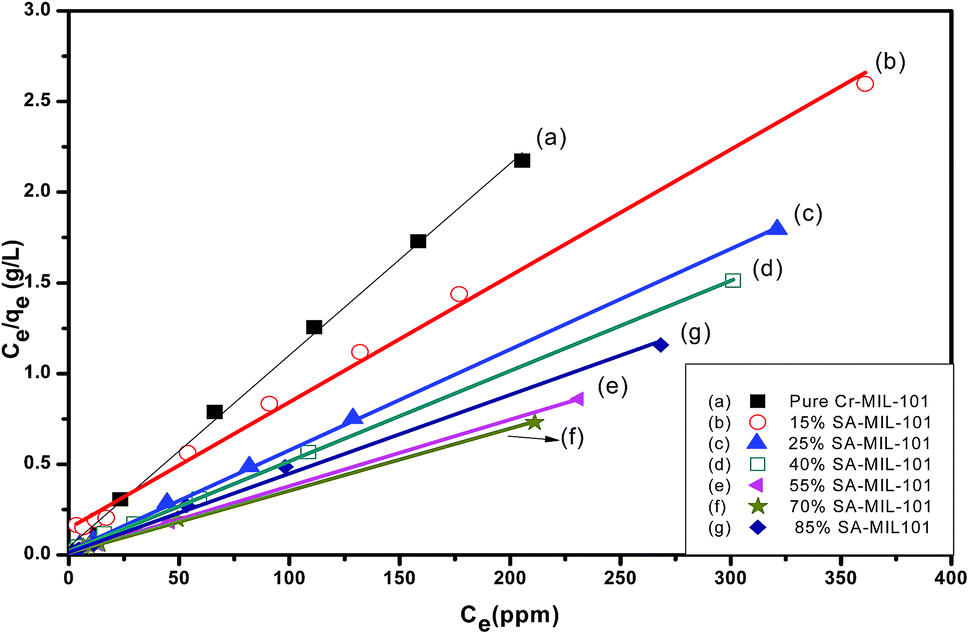 | ||
| Fig. 20 Langmuir isotherms for the adsorption of MO onto modified and unmodified Cr-MIL-101. | ||
Secondly, the surface heterogeneity was examined by the Freundlich model, which is given by eqn (5):
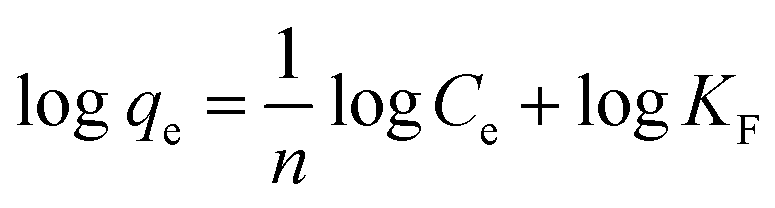 | (5) |
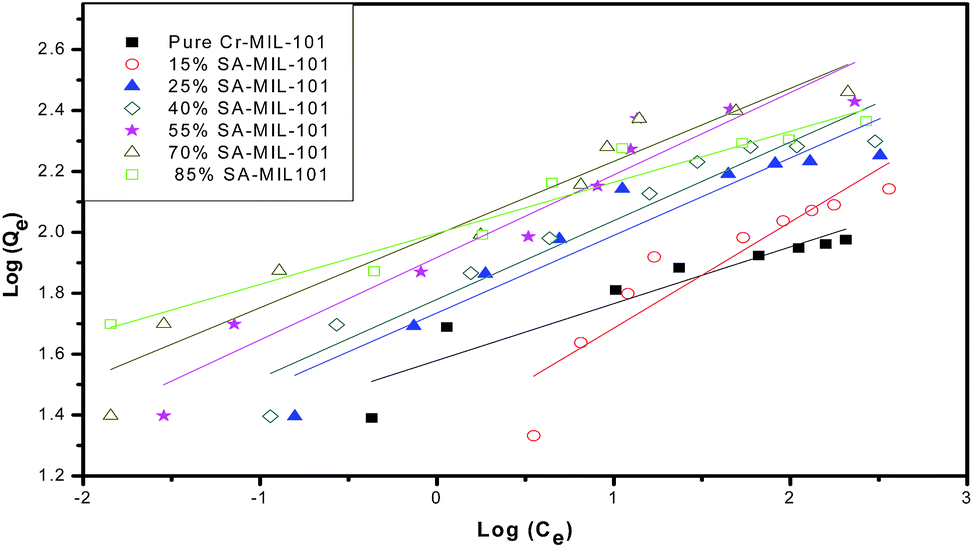 | ||
| Fig. 21 Freundlich isotherms for the adsorption of MO onto modified and unmodified Cr-MIL-101. | ||
From Table 4, the maximum adsorption capacity of methyl orange over the 70% SA-MIL-101 sample calcined at 150 °C (qm) is 291.5 mg g−1. A comparison of the maximum adsorption capacity qm values for methyl orange on 70% SA-MIL-101 with previously reported N-containing adsorbents is summarized in Table 5, which indicates that modified MIL-101 has great potential for methyl orange removal from water. It is obvious that the modified MIL-101 samples used in this work are exceptional adsorbent materials for the removal of dyes from wastewater.
4. Conclusions
In this study, a series of sulfamic acid-functionalized Cr-MIL-101 catalysts was prepared for the first time via a facile impregnation method and characterized by various techniques. We demonstrated that Cr-MIL-101 has particle size of about 300–500 nm and a crystalline structure. The modification of Cr-MIL-101 by sulfamic acid crystals progressively increased the surface acidity and Brønsted/Lewis acid ratio up to 55 wt% SA loading. The catalytic performance of SA/MIL-101 catalysts was examined in the synthesis xanthenes and coumarin derivatives. The catalytic performance was maximum at 55 wt% sulfamic acid loading. The decrease in the catalytic activity at higher loading (>55 wt%) may be due to the decrease in acid strength and B/L ratio. The existence of small amounts of reachable basic sites, like N-containing functional groups on the surface of SA/MIL-101 materials, plays a great role in influencing the adsorption activity of methyl orange. The adsorption isotherms were well fitted to the Langmuir equation and the adsorption capacity of SA/MIL-101 reached 291 mg g−1 for the 70% SA/MIL-101 sample.Conflicts of interest
There are no conflicts to declare.References
- D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef PubMed.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef PubMed.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef PubMed.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- A. Corma, H. García and F. X. L. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef PubMed.
- F. Gandara, H. Furukawa, S. Lee and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 5271–5274 CrossRef PubMed.
- G. Blaniţa, M. Streza, M. D. Lazar and D. Lupu, Int. J. Hydrogen Energy, 2017, 42, 3064–3077 CrossRef.
- Z. Dou, J. Yu, Y. Cui, Y. Yang, Z. Wang, D. Yang and G. Qian, J. Am. Chem. Soc., 2014, 136, 5527–5530 CrossRef PubMed.
- M. G. Campbell and M. Dinca, Sensors, 2017, 17, 1108 CrossRef PubMed.
- L. T. L. Nguyen, K. K. A. Le, H. X. Truong and N. T. S. Phan, Catal. Sci. Technol., 2012, 2, 521–528 Search PubMed.
- C. K. Brozek, L. Bellarosa, T. Soejima, T. V. Clark, N. Lopez and M. Dinca, Chem.–Eur. J., 2014, 20, 6871–6874 CrossRef PubMed.
- D. Cunha, M. Ben Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaım, G. Maurink, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767–2776 CrossRef.
- D. Tian, Y. Li, R. Y. Chen, Z. Chang, G. Y. Wang and X. H. Bu, J. Mater. Chem. A, 2014, 2, 1465–1470 Search PubMed.
- J. Kim, Y. R. Lee and W. S. Ahn, Chem. Commun., 2013, 49, 7647–7649 RSC.
- R. B. Ferreira, P. M. Scheetz and A. L. B. Formiga, RSC Adv., 2013, 3, 10181–10184 RSC.
- Y. Fang, G. Zhao, W. Dai, L. Ma and N. Ma, Microporous Mesoporous Mater., 2017, 251, 51–57 CrossRef.
- A. Demessence, P. Horcajada, C. Serre, C. Boissiere, D. Grosso, C. Sanchez and G. Ferey, Chem. Commun., 2009, 46, 7149–7151 RSC.
- D. Ma, B. Li, K. Liu, X. Zhang, W. Zou, Y. Yang, G. Li, Z. Shi and S. Feng, J. Mater. Chem. A, 2015, 3, 23136–23142 Search PubMed.
- I. E. Ertas, M. Gulcan, A. Bulut, M. Yurderi and M. Zahmakiran, Microporous Mesoporous Mater., 2016, 226, 94–103 CrossRef.
- G. Akiyama, R. Matsuda, H. Sato, M. Takata and S. Kitagawa, Adv. Mater., 2011, 23, 3294–3297 CrossRef PubMed.
- Y. D. Zang, J. Shi, F. M. Zhang, Y. J. Zhong and W. D. Zhu, Catal. Sci. Technol., 2013, 3, 2044–2049 Search PubMed.
- L. Bromberg, Y. Diao, H. Wu, S. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664–1675 CrossRef.
- W. Du, G. Chen, R. Nie, Y. Li and Z. Hou, Catal. Commun., 2013, 41, 56–59 CrossRef.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef PubMed.
- M. G. Goesten, J. J. Alcaniz, E. V. R. Fernandez, K. Gupta, E. Stavitski, H. V. Bekkum, J. Gascon and F. Kapteijn, J. Catal., 2011, 281, 177–187 CrossRef.
- Y. D. Zang, J. Shi, X. M. Zhao, L. C. Kong, F. M. Zhang and Y. J. Zhong, React. Kinet., Mech. Catal., 2013, 109, 77–89 CrossRef.
- D. Jiang, L. L. Keenan, A. D. Burrows and K. J. Edler, Chem. Commun., 2012, 48, 12053–12055 RSC.
- A. Rostami and A. Yari, J. Iran. Chem. Soc., 2012, 9, 489–493 CrossRef.
- A. Rostami and J. Akradi, Chin. Chem. Lett., 2011, 22, 1317–1320 CrossRef.
- A. A. Napoleon and F. R. Khan, Med. Chem. Res., 2014, 23, 4749–4760 CrossRef.
- K. M. Naidu, B. S. Krishna, M. A. Kumar, P. Arulselvan, S. I. Khalivulla and O. Lasekan, Molecules, 2012, 17, 7543–7555 CrossRef PubMed.
- R. M. Kalla, M. Akkarapall and H. Osman, J. Drug Metab. Toxicol., 2014, 5, 1–7 Search PubMed.
- A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817–840 CrossRef PubMed.
- D. S. Pellosi, B. M. Estevao, J. Semensatoa, D. Severinob, M. S. Baptistab, M. J. Politi, N. Hiokaa and W. Caetanoa, J. Photochem. Photobiol., A, 2012, 247, 8–15 CrossRef.
- S. Q. Zhang and W. B. Euler, Sens. Actuators, B, 2016, 225, 553–562 CrossRef.
- G. Harichandran, S. D. Amalraj and P. Shanmugam, J. Mol. Catal. A: Chem., 2014, 392, 31–38 CrossRef.
- J. Mondal, M. Nandi, A. Modak and A. Bhaumik, J. Mol. Catal. A: Chem., 2012, 363–364, 254–264 CrossRef.
- Z. K. Jaberi and M. Keshavarzi, Chin. Chem. Lett., 2010, 21, 547–549 CrossRef.
- F. Rashedian, D. Saberib and K. Niknam, J. Chin. Chem. Soc., 2010, 57, 998–1006 CrossRef.
- F. Shirini and N. G. Khaligh, Dyes Pigm., 2012, 95, 789–794 CrossRef.
- M. B. Halli, R. B. Sumathi and M. Kinni, Spectrochim. Acta, Part A, 2012, 99, 46–56 CrossRef PubMed.
- S. Emami and S. Dadashpour, Eur. J. Med. Chem., 2015, 102, 611–630 CrossRef PubMed.
- S. A. El-Hakam, S. E. Samra, S. M. El-Dafrawy, A. A. Ibrahim and R. S. Salama, Int. J. Mod. Chem., 2013, 5(1), 55–70 Search PubMed.
- A. I. Ahmed, S. A. El-Hakam, A. S. Khder and W. S. Abo El-Yazeed, J. Mol. Catal. A: Chem., 2013, 366, 99–108 CrossRef.
- A. I. Ahmed, A. A. El-Khouly and Y. S. Samy, Int. J. Mod. Chem., 2013, 5(2), 87–100 Search PubMed.
- M. M. Ayad and A. Abu El-Nasr, J. Phys. Chem. C, 2010, 114(34), 14377–14383 Search PubMed.
- L. Zhou, C. Gao and W. Xu, ACS Appl. Mater. Interfaces, 2010, 2(5), 1483–1491 Search PubMed.
- S. Chen, J. Zhang, C. Zhang, Q. Yue, Y. Li and C. Li, Desalination, 2010, 252, 149–156 CrossRef.
- E. Haque, J. E. Lee, I. T. Jang, Y. K. Hwang, J. Chang, J. Jegal and S. H. Jhung, J. Hazard. Mater., 2010, 181, 535–542 CrossRef PubMed.
- L. Obeid, A. Bée, D. Talbot, S. B. Jaafar, V. Dupuis, S. Abramson, V. Cabuil and M. Welschbillig, J. Colloid Interface Sci., 2013, 410, 52–58 CrossRef PubMed.
- X. Li, Y. Mao, K. Leng, G. Ye, Y. Sun and W. Xu, Microporous Mesoporous Mater., 2017, 254, 114–120 CrossRef.
- X. Hu, Y. Lu, F. Dai, C. Liu and Y. Liu, Microporous Mesoporous Mater., 2013, 170, 36–44 CrossRef.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef.
- A. I. Ahmed, S. A. El-Hakam, M. A. Abd Elghany and W. S. Abo El-Yazeed, Appl. Catal., A, 2011, 407, 40–48 CrossRef.
- Y. Jin, J. Shi, F. Zhang, Y. Zhong and W. Zhu, J. Mol. Catal. A: Chem., 2014, 383–384, 167–171 CrossRef.
- R. Fazaeli, H. Aliyan, M. Moghadam and M. Masoudinia, J. Mol. Catal. A: Chem., 2013, 374–375, 46–52 CrossRef.
- B. Kannan, P. R. Seshadri, P. Murugakoothan and K. Ilangovan, Indian J. Sci. Technol., 2013, 6, 4357–4361 Search PubMed.
- R. A. Gharde and D. T. Chunarkar, International Journal of Advanced Research in Computer Engineering & Technology, 2012, 1, 230–233 Search PubMed.
- M. Anbia and V. Hoseini, J. Nat. Gas Chem., 2012, 21, 339–343 CrossRef.
- X. Liu, H. Li, H. Pan, H. Zhang, S. Huang, K. Yang, W. Xue and S. Yang, J. Energy Chem., 2016, 25, 523–530 CrossRef.
- L. Qin, Z. Li, Z. Xu, X. Guo and G. Zhang, Appl. Catal., B, 2015, 179, 500–508 CrossRef.
- G. Chen, S. Wu, H. Liu, H. Jiang and Y. W. Li, Green Chem., 2013, 15, 230–235 RSC.
- E. Rafiee, M. Joshaghani, S. Eavania and S. Rashidzadeh, Green Chem., 2008, 10, 982–989 RSC.
- A. S. Khder, H. M. A. Hassan and M. S. El-Shall, Appl. Catal., A, 2014, 487, 110–118 CrossRef.
- A. I. Ahmed, S. A. El-Hakam, S. E. Samra, A. A. EL-Khouly and A. S. Khder, Colloids Surf., A, 2008, 317, 62–70 CrossRef.
- H. M. Altass and A. S. Khder, J. Mol. Catal. A: Chem., 2016, 411, 138–145 CrossRef.
- C. L. Hill and C. M. P. McCarthy, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef.
- J. Safaei-Ghomi and M. A. Ghasemzadeh, Chin. Chem. Lett., 2012, 23, 1225–1229 CrossRef.
- B. Rajitha, B. S. Kumar, Y. T. Reddy, P. N. Reddy and N. Sreenivasulu, Tetrahedron Lett., 2005, 46, 8691–8693 CrossRef.
- B. Karami, K. H. Eskandari, S. Gholipour and M. Jamshidi, Org. Prep. Proced. Int., 2013, 45, 220–226 CrossRef.
- B. Das, J. Kashanna, R. A. Kumar and P. Jangili, Synth. Commun., 2012, 42, 2876–2884 CrossRef.
- S. Palaniappan and R. C. Shekhar, J. Mol. Catal. A: Chem., 2004, 209, 117–124 CrossRef.
- A. J. Hoefnagel, E. A. Gunnewegh, R. S. Downing and H. v. Bekkum, J. Chem. Soc., Chem. Commun., 1995, 225–226 RSC.
- S. Kim, D. Kang, C. H. Lee and P. H. Lee, J. Org. Chem., 2012, 77, 6530–6537 CrossRef PubMed.
- W. Song, B. Gao, X. Xu, L. Xing, S. Han, P. Duan, W. Song and R. Jia, Bioresour. Technol., 2016, 210, 123–130 CrossRef PubMed.
- M. V. Subbaiah and D. S. Kim, Ecotoxicol. Environ. Saf., 2016, 128, 109–117 CrossRef PubMed.
- K. Fouad and B. Omar el Farouk, Desalin. Water Treat., 2015, 53, 3711–3723 CrossRef.
- D. H. K. Reddy and S. M. Lee, Adv. Colloid Interface Sci., 2013, 201–202, 68–93 CrossRef PubMed.
- R. Lafi and A. Hafiane, J. Taiwan Inst. Chem. Eng., 2016, 58, 424–433 CrossRef.
- J. R. Deka, C. L. Liu, T. H. Wang, W. C. Chang and H. M. Kao, J. Hazard. Mater., 2014, 278, 539–550 CrossRef PubMed.
- Y. Liu, G. Cui, C. Luo, L. Zhang, Y. Guo and S. Yan, RSC Adv., 2014, 4, 55162–55172 RSC.
- J. Ma, F. Yu, L. Zhou, L. Jin, M. Yang, J. Luan, Y. Tang, H. Fan, Z. Yuan and J. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 5749–5760 Search PubMed.
- H. Li, N. An, G. Liu, J. Li, N. Liu, M. Jia, W. Zhang and X. Yuan, J. Colloid Interface Sci., 2016, 466, 343–351 CrossRef PubMed.
- K. Y. Foo and B. H. Hameed, Chem. Eng. J., 2010, 156, 2–10 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |