
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrated conversion of 1-butanol to 1,3-butadiene†
Jacob S. Kruger *,
Tao Dong,
Gregg T. Beckham and
Mary J. Biddy
*,
Tao Dong,
Gregg T. Beckham and
Mary J. Biddy
National Renewable Energy Laboratory, National Bioenergy Center, 15013 Denver West Parkway, Golden, CO 80401, USA. E-mail: Jacob.Kruger@nrel.gov
First published on 2nd July 2018
Abstract
Renewed interest in production of 1,3-butadiene from non-petroleum sources has motivated research into novel production routes. In this study, we investigated an integrated process comprising 1-butanol dehydration over a γ-Al2O3 catalyst to produce a mixture of linear butenes, coupled with a downstream K-doped Cr2O3/Al2O3 catalyst to convert the butenes into butadiene. Linear butene yields greater than 90% are achievable at 360 °C in the dehydration step, and single-pass 1,3-butadiene yields greater than 40% are achieved from 1-butene in a N2 atmosphere in the dehydrogenation step. In the integrated process, 1,3-butadiene yields are 10–15%. In all cases, linear C4 selectivity is greater than 90%, suggesting that 1,3-butadiene yields could be significantly improved in a recycle reactor. Doping the Cr2O3 catalyst with different metals to promote H2 consumption in a CO2 atmosphere did not have a large effect on catalyst performance compared to an undoped Cr2O3 catalyst, although doping with K in an N2-diluted atmosphere and with Ni in a CO2-enriched atmosphere showed slight improvement. In contrast, doping with K and Ca in a CO2-enriched atmosphere showed slightly decreased performance. Similarly, employing a CO2-enriched atmosphere in general did not improve 1,3-butadiene yield or selectivity compared to reactions performed in N2. Overall, this study suggests that an integrated dehydration/dehydrogenation process to convert 1-butanol into 1,3-butadiene could be feasible with further catalyst and process development.
Introduction
1,3-Butadiene is a large-market commodity chemical used in the manufacture of synthetic polymers and other materials.1–3 Historically, it has been produced as a byproduct of non-catalytic steam cracking of the naphtha fraction of petroleum for ethylene production, or by catalytic dehydrogenation (direct or oxidative) of n-butane and n-butenes. The advent of inexpensive light hydrocarbons, such as shale gas, has led to non-naphtha ethylene routes, concomitantly leading to supply shortages and price uncertainty of butadiene.2,4,5 These uncertainties, as well as environmental concerns surrounding fossil feedstocks, have resulted in a renewed interest in “on-purpose” catalytic dehydrogenation of C4s and in developing alternative renewable pathways to 1,3-butadiene.1,2,4 Several of these pathways have been examined, starting in the early 1900s. For example, 1,3-butadiene can be produced in yields up to 60% from ethanol over Co or Cr/MgO catalysts, and butanediols can be dehydrated to butadiene over several transition metal oxides, though butadiene yields have generally been ∼25% or less.1Several pathways to convert biomass to a renewable C4 stream exist commercially, incorporating syngas, methanol, and ethanol intermediates en route to 1-butanol, isobutanol, succinic acid, or butanediol.2,6 The pathways to convert these C4s into 1,3-butadiene are less technically developed, though the embodied chemistries are relatively mature. Specifically, 1-butanol could be dehydrated to 1- and 2-butenes, and these butenes could be dehydrogenated to 1,3-butadiene (Scheme 1).
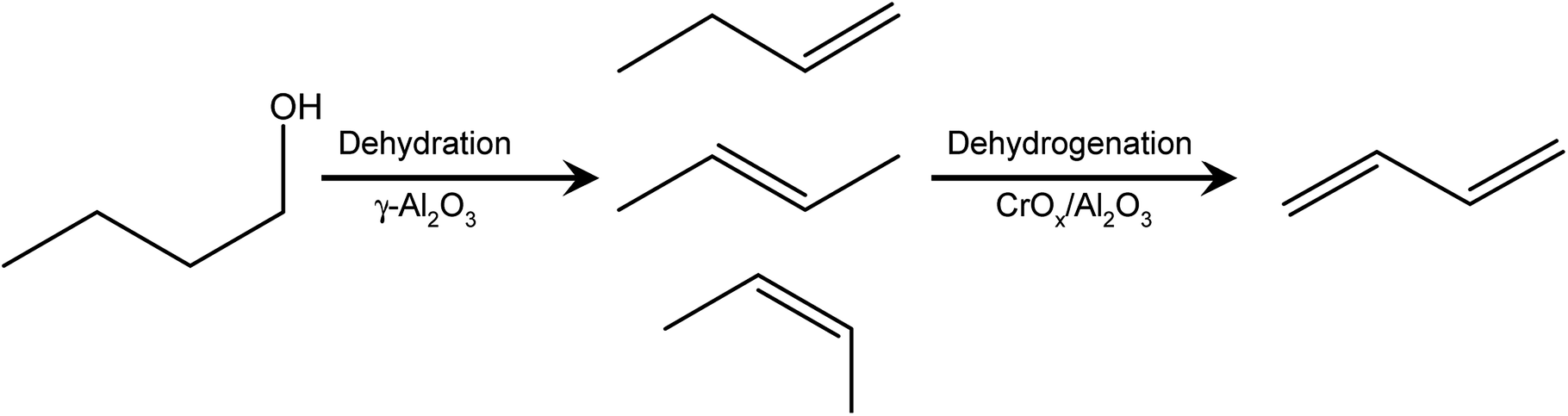 | ||
| Scheme 1 Dehydration of butanol to linear butenes, and dehydrogenation of butenes to 1,3-butadiene. | ||
A single-stage integrated process would be challenging due to the different conditions required for alcohol dehydration and olefin dehydrogenation. In particular, olefin dehydrogenation is typically carried out at temperatures greater than 550 °C and weight-hourly space velocities (WHSVs) greater than 150 h−1. High temperature is required to activate the olefins on the dehydrogenation catalyst, and the high WHSV is required to minimize unfavourable side reactions that lead to C1–C3 cracking products and coke. Additionally, dehydrogenation catalysts (Cr2O3 or Fe2O3 supported on Al2O3) are typically doped with K2CO3 or K2O, which neutralize catalyst acidity that would be necessary for alcohol dehydration. Similarly, alcohols tend to react by dehydrogenation at such temperatures to produce an undesired carbonyl, rather than by dehydration to produce a desirable olefin.7 On the other hand, 1-butanol can be dehydrated to linear butenes in greater than 95% yield over a γ-Al2O3 catalyst at temperatures of 350–410 °C and WHSVs of 1–10 h−1.8 This difference in reaction conditions may be one reason that it has been suggested,1 but never demonstrated, that an integrated dehydration–dehydrogenation process to convert 1-butanol to 1,3-butadiene could be feasible.
We hypothesized that olefin dehydrogenation could also be operated at lower temperature by decreasing the space velocity, and that the lower temperature would also be advantageous in mitigating side reactions. While cracking equilibria still strongly favour C1–C3 products at temperatures as low as 450 °C, the kinetics of these reactions are much slower.9 Thus, we were motivated to explore the integrated conversion of 1-butanol to 1,3-butadiene in a combined dehydration–dehydrogenation reactor.
Additionally, we were interested in improving yields in the butene dehydrogenation step. Linear butenes can typically be directly dehydrogenated over CrOx/Al2O3 or Fe2O3-containing catalysts in single-pass yields of 40–50% or oxidatively dehydrogenated over ZnFe2O4, Bi2MoO6, or Sn/Sb-containing catalysts in single-pass yields of 60–70%.1,9–12 The oxidative dehydrogenation shifts the dehydrogenation equilibrium toward 1,3-butadiene by converting the produced H2 to H2O. The presence of O2 and H2O concomitantly mitigate coke formation on the catalyst surface.
In dehydrogenation without O2, alternative concepts for shifting the dehydrogenation equilibrium may also be feasible. In particular, (1) coke gasification or methanation, (2) CO2 methanation, and (3) reverse water-gas shift (RWGS) are three reactions that could consume produced H2.
| C + 2H2 → CH4 | (1) |
| CO2 + 4H2 → CH4 + 2H2O | (2) |
| H2 + CO2 → H2O + CO | (3) |
Of these three, reaction (3) has proven promising in dehydrogenation of ethylbenzene to styrene and diethylbenzene to divinyl benzene13–15 and to some extent in butene dehydrogenation.16–19
To this end, certain catalysts are known to promote these desired reactions. In particular, coke gasification can be promoted by K, Ca, and Ni;20 Ni, Fe, and Mo can hydrogenate CO2 to CH4;21,22 and RWGS can be promoted by Fe2O3 and Cu/CeO2.13,23 K and Ca can also help to poison catalyst acid sites that lead to unfavourable cracking reactions, which in turn lead to coke.9
Thus, in addition to demonstrating an integrated butanol-to-butadiene process, we were motivated to explore the potential of new equilibrium-shifting catalysts for butene dehydrogenation. Herein we report process development for 1-butene dehydrogenation over a series of Cr2O3/Al2O3 catalysts doped with K, Ca, Ni, Mo, Fe, and Cu/CeO2, and integration with 1-butanol dehydration.
Experimental
Materials
1-Butene (5.4 mol% in N2) and a GC calibration standard of 1-butene, cis-2-butene, trans-2-butene, 1,3-butadiene, and n-butane (1 mol% each in N2) were purchased from Matheson Trigas. A GC calibration standard for CH4, C2H4, C2H6, C3H6, C3H8, and 1-butene was purchased from Praxair. N2, CO2, and zero-grade air were purchased from Airgas. 1-Butanol (99%) and hydrated nitrate salts of Cr, K, Ca, Ni, Fe, Cu, and Ce were purchased from Sigma-Aldrich, as was the oxalate salt of Mo. The γ-Al2O3 catalyst support was from Sasol (Puralox Nwa-155). Inert quartz chips (nominally 30–50 mesh) were from Dupré Minerals.Catalyst synthesis
Catalysts were synthesized by depositing aqueous solutions of the nitrate salts on a Sasol Puralox Nwa 155 Al2O3 support by incipient wetness. The baseline catalyst was 8.7 wt% Cr2O3. The doped catalysts had the same loading of Cr2O3 plus 1 wt% of the dopant metal, unless otherwise specified, mixed as the nitrate or oxalate salt in the same solution as the Cr. In the case of Cu/CeO2, the dopant solution was prepared separately and applied by incipient wetness to the baseline Cr2O3/Al2O3 catalyst. The metal loading for that catalyst was 0.1 wt% Cu and 0.9 wt% Ce.After depositing the salt solution, the catalysts were dried under vacuum at 40 °C overnight, then transferred to a ceramic dish and calcined by the following program: ramp to 95 °C at 25 °C min−1, hold for 1 h, ramp to 550 °C at 5 °C min−1, hold for 10 h.
Catalyst characterization
Catalysts were characterized by N2 physisorption to determine surface area, by NH3 temperature programmed desorption (TPD) to quantify acid sites, and by X-ray diffraction (XRD) to evaluate crystal structure. N2 physisorption was carried out on a Quantachrome Quantisorb SI four-station instrument. Prior to analysis, catalysts were outgassed under vacuum at 350 °C overnight. Eight adsorption points for each catalyst were recorded to establish an isotherm from which the surface area was calculated, using a 30 s equilibration time for each point.NH3 pulse chemisorption was carried out on an Altamira Instruments AMI-390 unit. Samples were treated for 2 h at 450 °C under 50 mL min−1 Ar, then cooled to 120 °C under flowing He before dosing the sample with 25 × 5 mL pulses of 10% NH3 in He. The average of the post-saturation pulses (typically the last 15–20 pulses) was used as the reference peak area. The adsorbed peak area was calculated as the sum of the difference between the observed peak area and the reference peak area for the unsaturated pulses (typically first 5–10 pulses). The NH3 areas were quantified on a TCD and converted to a quantity of adsorbed molecules assuming ideal gas behaviour of the pulse gas.
XRD was carried out on a Rigaku Ultima IV X-ray diffractometer using Cu K-α radiation. The operating voltage and current were 40 kV and 44 mA, respectively. A scan range of 5–80° 2θ, a scan speed of 5° min−1 and a point spacing of 0.05° 2θ were used.
Reactor system
The reactor system was a fixed-bed downflow stainless steel tubular reactor, 0.5′′ ID and 14′′ length. Unless otherwise specified, the catalyst bed (0.6 g) was positioned in the middle of the reactor tube, with 30–50 mesh SiO2 packed upstream and downstream to facilitate mixing and heat transfer. The particles were held in place by a plug of quartz wool on either end of the reactor, and a thermocouple was placed in the middle of the catalyst bed. In the case of the sequential dehydration–dehydrogenation, the thermocouple tip was in the middle of the dehydrogenation catalyst bed. The reactor was brought up to temperature under flowing N2, and typically allowed to equilibrate overnight before starting a reaction. To measure the temperature profile of the reactor, the top of the reactor was fitted with a thermowell that accompanied two K-type thermocouples. A control thermocouple remained in the centre of the tube while the other moved from the bottom of the tube to the top of the tube in 0.5′′ increments, and waiting 30–60 s at each point for the temperature to stabilize. When the reactor was heated to the reaction temperature using the control thermocouple in the thermowell, the wall temperature of the furnace was the same as when the control thermocouple was embedded in the catalyst bed, confirming that the temperature profile was the same during measurement and reaction.During reactor operation, a reaction-purge–regeneration-purge cycle using four mass flow controllers was implemented, with the reactor control software automatically switching between steps. The temperature, time, and flow parameters used for each cycle unless otherwise indicated are shown in Table 1.
| Parameter | Reaction | Reaction purge | Regeneration | Regeneration purge |
|---|---|---|---|---|
| Temperature | 450 °C | 450 °C | 450 °C | 450 °C |
| Duration | 72 min | 6 min | 74 min | 6 min |
| Gas feed | 100 sccm N2 or CO2 + 100 sccm 1-butene mix | 200 sccm N2 or CO2 | 100 sccm Ar + 100 sccm zero air | 200 sccm Ar |
For dehydration experiments, a 0.2 g bed of the same γ-Al2O3 material used to support the Cr2O3 catalysts was positioned 5 inches (12.7 cm) above the centre of the reactor tube. At this point, the reactor temperature was 360 °C when the axial centre of the reactor tube was at 450 °C. The butanol flow rate was 0.02 mL min−1, selected to approximate the conditions of the 5.4 mol% 1-butene mixture used in the dehydrogenation catalyst screening experiments. The N2 flow rate was 177 sccm. For integrated dehydration–dehydrogenation experiments, the configuration for both sets of experiments was combined. A 0.2 g bed of γ-Al2O3 was positioned 5 inches (12.7 cm) above the reactor centre and a 0.6 g bed of K–Cr2O3/Al2O3 was positioned at the reactor centre, the 1-butanol flow rate was 0.02 mL min−1 and the N2 flow rate was 177 sccm.
Reactor effluent passed through a condenser and then to local exhaust ventilation, with a slip stream sampled by online GCMS. The system consisted of an Agilent 6890 Plus GC equipped with a TCD, FID, and a 5973 MS, which analysed samples in parallel. The GC column was a 30 m × 0.32 mm ID GS-GASPRO column, operating in ramped flow mode with the following program: 2.3 mL min−1 for 3 min, ramp at 1 mL min−2 to 2.7 mL min−1, hold at 2.7 mL min−1. The corresponding oven program was 50 °C for 3 min, ramp at 15 °C min−1 to 75 °C, hold for 2 min, ramp at 50 °C min−1 to 250 °C, hold for 2.83 min. The inlet conditions were 250 °C, initial pressure of 10.64 psi, and a split ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The carrier gas for the system was He. Hydrocarbons were quantified on the FID, using N2 (detected on the TCD) as an internal standard, and gas mixes of authentic standards for C1–C4 compounds to develop response factors. For C5 and heavier compounds, FID response factors were calculated based on the method of Scanlon,24 though these compounds typically comprised less than 0.5% carbon yield.
:1. The carrier gas for the system was He. Hydrocarbons were quantified on the FID, using N2 (detected on the TCD) as an internal standard, and gas mixes of authentic standards for C1–C4 compounds to develop response factors. For C5 and heavier compounds, FID response factors were calculated based on the method of Scanlon,24 though these compounds typically comprised less than 0.5% carbon yield.
Yields and selectivities are reported on a carbon molar basis. Selectivities are based on the amount of butene fed rather than as a fraction of products detected.
Results
Butanol dehydration
We initially explored butanol dehydration under conditions similar to those reported by Pines and Haag.8 Using a temperature of 360 °C and WHSV of 4.86 h−1 (0.2 g γ-Al2O3 catalyst bed and 0.02 mL min−1 butanol flow rate), we were able to obtain >95% carbon balance and 91% yield to linear butenes over ∼100 min time-on-stream, as shown in Fig. 1. These were the conditions we employed in the integrated dehydration–dehydrogenation experiments.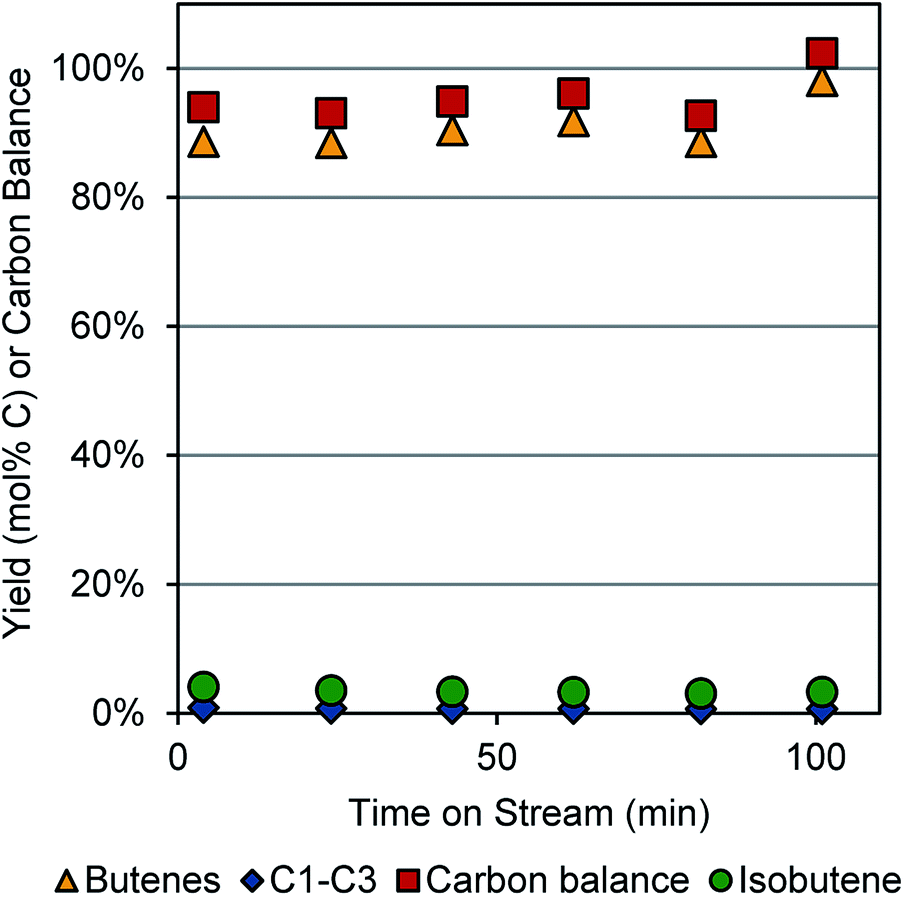 | ||
| Fig. 1 Dehydration of 1-butanol to 1- and 2-butenes over a γ-Al2O3 catalyst. Reaction conditions: 0.02 mL min−1 butanol, 177 sccm N2, 0.2 g catalyst, 350–400 °C. | ||
We also explored butanol reactivity at 450 °C, consistent with the butene dehydrogenation conditions reported below. Butanol conversion in a SiO2-packed tube (without catalyst) was 100%, but selectivity to butenes was low, and a significant amount of black solid was deposited in the reactor. Because conversion of butenes under these conditions was negligible (see below), the high conversion of butanol suggests thermal reactions through non-butene routes predominated, such as cracking, coke formation, dehydrogenation to butyraldehyde, and decarbonylation of butyraldehyde to produce propene and CO.
We attempted to mitigate these nonselective reactions by inserting a γ-Al2O3 catalyst bed upstream of the axial tube centre to convert the butanol to butenes at 450 °C. However, when the γ-Al2O3 bed was positioned within the isothermal zone of the reactor, a significant amount of isobutene was produced through skeletal isomerization. Thus, we measured the temperature profile within the reactor tube and placed the γ-Al2O3 bed 5 inches (12.7 cm) above the axial centre of the reactor, at which point the temperature was 360 °C (Fig. S1†), consistent with Pine and Haags.8 This was the location of the γ-Al2O3 bed in the integrated experiments.
Screening of butene dehydrogenation reaction conditions
To establish reaction conditions for butene dehydrogenation that would maximize butadiene selectivity, maximize single-pass yield, and minimize cracking reactions, we explored different temperatures and space velocities with the baseline Cr2O3/Al2O3 catalyst. The results of these experiments are shown in Fig. 2. Selectivity and yield are optimal at 450 °C and 0.76 h−1 weight hourly space velocity (WHSV), while both lower and higher temperatures show lower selectivity. At 400 °C, the lower selectivity is likely due to coke formation as no other compounds were observed in the effluent stream, while at 500 °C, both coke formation and cracking reactions play a role, as C1–C3 hydrocarbons were detectable, along with small amounts of C5 hydrocarbons, benzene, and toluene. These results show that high butadiene yields and selectivities can be obtained at temperatures significantly lower than those typically used for direct butene dehydrogenation. Notably, following the analysis of Tyuryaev,9 the 1-butene dehydrogenation equilibrium at a 1-butene partial pressure of 0.027 atm and temperatures of 400 °C and 450 °C would give 18–22% and 35–39% yields to butadiene, respectively. At 500 °C, 57–60% butadiene yields would be predicted. Thus, it appears that the butadiene yield at 400–450 °C is limited by equilibrium, while at 500 °C, it is limited by side reactions.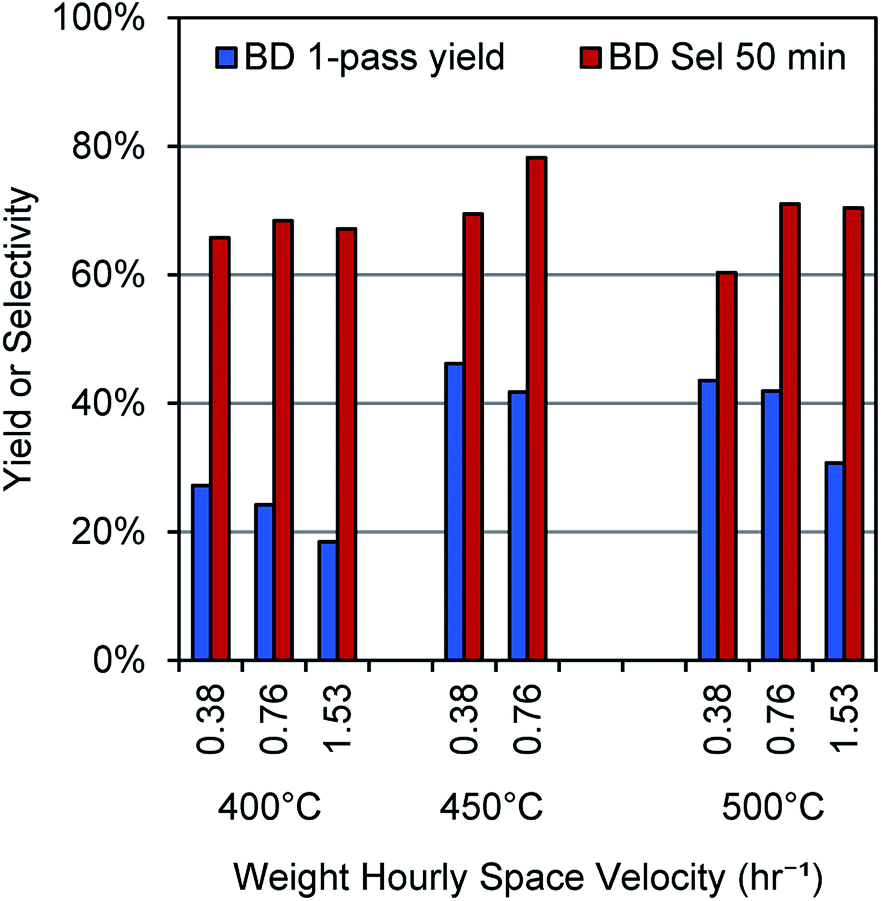 | ||
| Fig. 2 Yields and selectivity for butene dehydrogenation over a Cr2O3/Al2O3 catalyst. Results calculated at 50 min time on stream with a 2.7 mol% concentration of 1-butene in N2. | ||
Thus, we established 450 °C and a space velocity of 0.76 h−1 WHSV as the operating conditions for our remaining experiments. Notably, the butadiene selectivity and yields are comparable to common industrial catalysts reported for direct dehydrogenation.9 Under these same conditions, a control reaction (reactor tube packed with only SiO2) showed <5% conversion of 1-butene (Fig. S2†).
Effect of catalyst dopant and reaction atmosphere
In the interest of improving the butene dehydrogenation portion of the reactor by subsequently reacting produced H2, we tested a number of 1 wt% doped catalysts in N2 and CO2-rich atmospheres. The feed composition of these atmospheres were 97.3% N2 and 47.3%N2/50.0% CO2, respectively, with 2.7% 1-butene in both cases. For two reaction cycles in N2, two in CO2, and one in N2, the doped catalysts were generally comparable to the undoped Cr2O3/Al2O3 catalyst, except that the K-doped catalyst was slightly better in N2, the Ni-doped catalyst was slightly better in CO2, and the K- and Ca-doped catalysts were slightly worse in CO2. These results are shown in Fig. 3. We also tested Ni- and Mo-doped catalysts at 0.25 wt% and 5 wt% metal dopant loadings, but the results were similar to the 1 wt% loading (Fig. S3†). The corresponding selectivities are also quite high, with several catalysts giving greater than 80% selectivity at 30 min time-on-stream (TOS, Fig. S4†).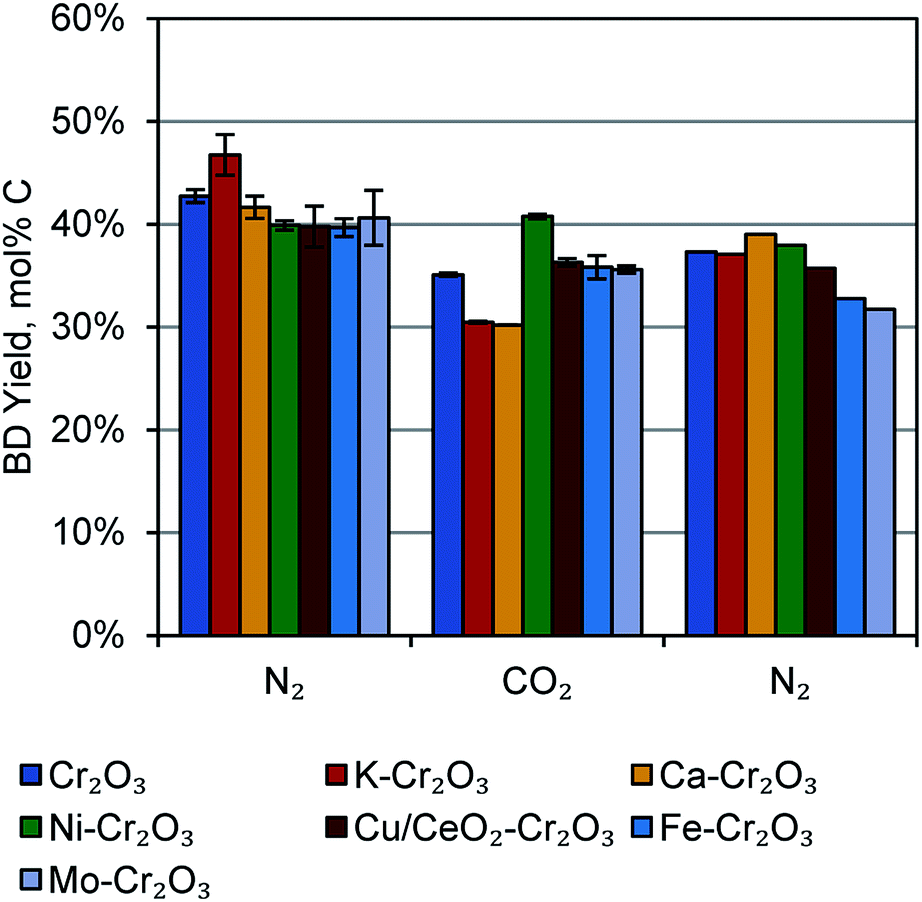 | ||
| Fig. 3 Conversion and selectivity in 1-butene dehydrogenation over 1 wt% metal-doped Cr2O3 catalysts, interpolated to 30 min time-on-stream. Left-most set: reaction cycles 1–2 in N2. Centre set: reaction cycles 3–4 in N2/CO2. Right-most set: reaction cycle 5 in N2. | ||
The CO2-enriched atmosphere did not lead to higher butadiene yields as hypothesized, despite the more favourable equilibria for H2 conversion. (For example, correlations for the water-gas shift equilibrium25 at 1 atm and 450 °C predict greater than 98% conversion of produced H2 in a 50% CO2 atmosphere). In these reactions, there was not a significantly higher amount of CH4 produced (as would be expected from methanation of C or CO2), nor was any condensate collected from the reactor knockout pot, (as may be expected from H2O production via enhanced RWGS activity). Thus, it seems that the catalysts synthesized here are either not capable of activating CO2 or not capable of catalysing the desired reactions at 450 °C. Further catalyst development will be the focus of future work.
Integration of butanol dehydration and butene dehydrogenation
With these results, we were motivated to integrate the dehydration and dehydrogenation reactions in a single reactor tube. The reactor configuration consisted of a 0.6 g bed of the 1 wt% K-doped Cr2O3/Al2O3 catalyst at the centre of the reactor tube (at 450 °C), a 0.2 g bed of γ-Al2O3 positioned 5 inches (12.7 cm) above the centre of the tube (at 360 °C), a butanol flow of 0.02 mL min−1, and an N2 flow of 177 sccm. Under these conditions, we were able to obtain ∼13% single-pass yields over 10 h TOS, as shown in Fig. 4. The gap between 200 and 300 min TOS represents a regeneration step, though the catalyst performance appeared to be decreasing only slightly with TOS. In dehydrogenation of 1-butene, catalyst performance decreased more rapidly (though maintaining higher overall yields). Thus, the integrated process with butanol feed appears to be more stable than just the dehydrogenation. The reason for this difference is not clear at this point, but it may be that the oxygen in the butanol feed is able to mitigate coke formation. Because this oxygen would likely only see the dehydrogenation catalyst as H2O, it is likely that any coke-mitigating function is due to H2O rather than 1-butanol specifically.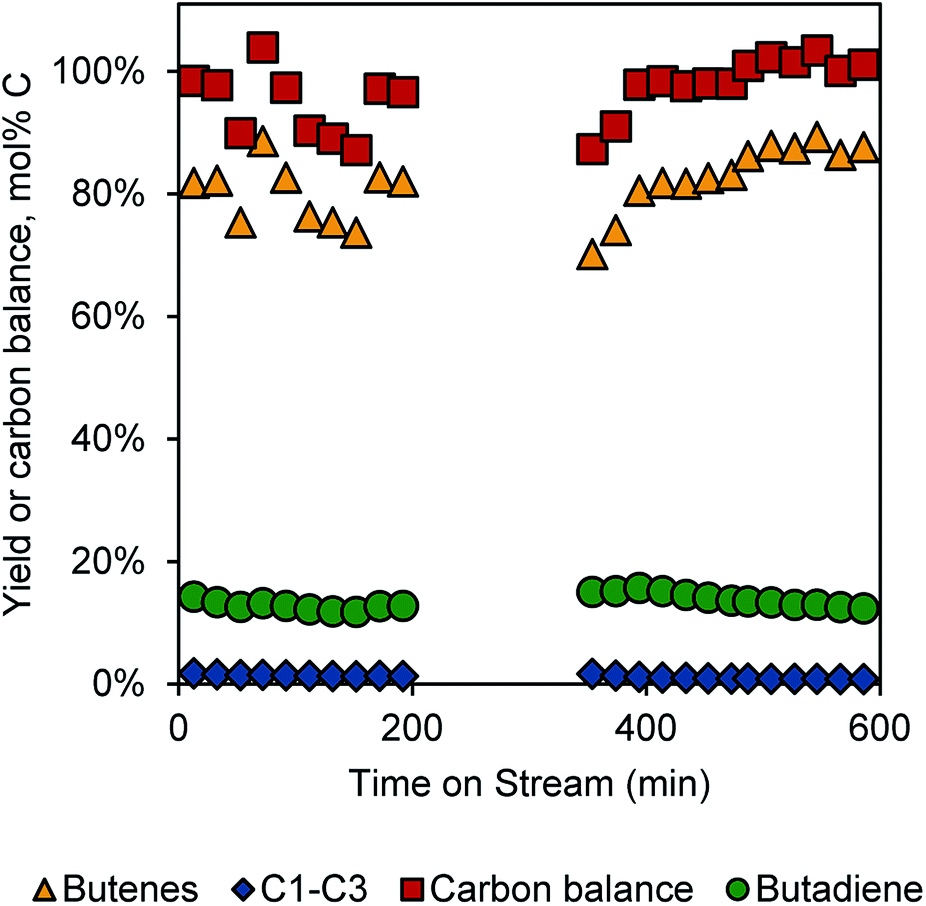 | ||
| Fig. 4 Integrated butanol dehydration and butene dehydrogenation. | ||
The yield of butadiene in the integrated process is lower than expected from the previous runs, which produced >40% yields of butadiene from 1-butene at 30 min TOS. We tentatively ascribe the lower yields to the water sensitivity of Cr2O3/Al2O3 catalysts,9 as H2O is produced in the dehydration step. Indeed, dehydrogenation activity was much lower when co-feeding 0.02 mL min−1 of H2O with the 1-butene/N2 mixture over these catalysts (Fig. S5†)
Notably, while the single-pass butadiene yield is only 13%, the average selectivity to linear C4 compounds is >95% (Fig. S6†). The remaining mass is likely due to coke formation, as some CO2 could be detected during the regeneration cycle. Thus, with a recycle reactor, the overall achievable yield of butadiene from butanol would be nearly quantitative using the present conditions.
Catalyst characterization
We characterized the fresh catalysts for surface area, acidity, and phase composition. Catalyst surface area and acidity are shown in Table 2, while phase composition is shown in Fig. S7.†| Catalyst | BET surface area (m2 g−1) | NH3 uptake (μmol g−1) |
|---|---|---|
| Al2O3 | 153 | 242 |
| Cr2O3 | 139 | 424 |
| 1K-Cr2O3 | 137 | 334 |
The γ-Al2O3 support had a surface area of 153 m2 g−1, which was reduced slightly after deposition of Cr2O3 and calcining to decompose the precursor salt. The presence of K in the Cr precursor solution does not significantly decrease the surface area further. Because high-valent Cr catalysts are known to decompose NH3 at temperatures above about 500 °C,26 we quantified acid sites by NH3 pulse chemisorption instead of NH3 TPD. The NH3 uptake on the γ-Al2O3 support suggests an acid site content of 242 μmol sites/g. Addition of Cr2O3 increases the acid site content to 424 μmol sites/g, while the presence of K in the Cr precursor solution mitigates the increase in acid sites to 334 μmol sites/g. Doping Cr oxide catalysts with K has been hypothesized to poison acid sites,9 though recent work has suggested that K also changes the surface morphology of the supported Cr oxide phase.27 Any structural changes were not apparent by XRD, as only peaks for the γ-Al2O3 support could be detected in the prepared catalysts (Fig. S7†).
Conclusions
We have integrated 1-butanol dehydration with butene dehydrogenation to produce 1,3-butadiene in a one-tube, two-stage process. Dehydration of 1-butanol gives linear butenes in >90% yield over a γ-Al2O3 catalyst at 350–410 °C. Dehydrogenation of 1-butene gives single-pass butadiene yields greater than 40% at 450 °C for a 2.7 vol% butene feed in N2, with selectivity greater than 80% at 30 min TOS. The integrated process demonstrated here gives an average single-pass butadiene yield of 13%, with overall C4 selectivity greater than 95%. With a recycle reactor configuration, overall yields of 1-butanol to butadiene would be much higher. Additionally, these results are obtained at temperatures significantly lower than those traditionally used for direct butene dehydrogenation, representing a potential route for decreasing process severity in butadiene production. Thus, the process developed here is an important step in the production of renewable butadiene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was authored by the National Renewable Energy Laboratory (NREL), operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. This work was supported by the Laboratory Directed Research and Development (LDRD) Program at NREL. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.Notes and references
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degreve, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC
.
- M. J. Biddy, C. Scarlata and C. Kinchin, Chemicals from Biomass: A Market Assessment of Bioproducts with Near-Term Potential. Technical Report NREL/TP-5100-65509, National Renewable Energy Laboratory, 2016 Search PubMed
- Z. Xi and W.-X. Zhang, Synlett, 2008, 2008, 2557–2570 CrossRef
- M. D. Jones, Chem. Cent. J., 2014, 8, 53 CrossRef PubMed
- P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef PubMed
- Nexant, Final Report: Renewable Chemicals & Materials Opportunity Assessment, Major Job Creation and Agricultural Sector Engine, Prepared for US Department of Agriculture, 2014 Search PubMed
- J. S. Kruger, R. Chakrabarti, R. J. Hermann and L. D. Schmidt, Appl. Catal., A, 2012, 411, 87–94 CrossRef
- H. Pines and W. O. Haag, J. Am. Chem. Soc., 1961, 83, 2847–2852 CrossRef
- I. Y. Tyuryaev, Russ. Chem. Rev., 1966, 35, 59 CrossRef
- J. Grub and E. Löser, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a04_431
- R. Julius, F. Robert, A. Claudia and D. Olaf, AIChE J., 2017, 63, 43–50 CrossRef
- H. Lee, J. C. Jung, H. Kim, Y.-M. Chung, T. J. Kim, S. J. Lee, S.-H. Oh, Y. S. Kim and I. K. Song, Catal. Commun., 2008, 9, 1137–1142 CrossRef
- S. Chen, A. Sun, Z. Qin and J. Wang, Catal. Commun., 2003, 4, 441–447 CrossRef
- L. Zhang, Z. Wu, N. C. Nelson, A. D. Sadow, I. I. Slowing and S. H. Overbury, ACS Catal., 2015, 5, 6426–6435 CrossRef
- Y. Yue, L. Zhang, J. Chen, D. K. Hensley, S. Dai and S. H. Overbury, RSC Adv., 2016, 6, 32989–32993 RSC
- W. Yan, Q. Y. Kouk, J. Luo, Y. Liu and A. Borgna, Catal. Commun., 2014, 46, 208–212 CrossRef
- W. Yan, J. Luo, Q.-Y. Kouk, J. E. Zheng, Z. Zhong, Y. Liu and A. Borgna, Appl. Catal., A, 2015, 508, 61–67 CrossRef
- W. Yan, Q.-Y. Kouk, S. X. Tan, J. Luo and Y. Liu, J. CO2 Util., 2016, 15, 154–159 CrossRef
- Y. Gao, B. Wang, B. Yan, J. Li, F. Alam, Z. Xiao and T. Jiang, React. Kinet., Mech. Catal., 2017, 122, 451–462 CrossRef
- A. A. Lizzio and L. R. Radovic, Ind. Eng. Chem. Res., 1991, 30, 1735–1744 CrossRef
- T. A. Le, M. S. Kim, S. H. Lee, T. W. Kim and E. D. Park, Catal. Today, 2017, 293, 89–96 CrossRef
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef
- X. Qi and M. Flytzani-Stephanopoulos, Ind. Eng. Chem. Res., 2004, 43, 3055–3062 CrossRef
- J. T. Scanlon and D. E. Willis, J. Chromatogr. Sci., 1985, 23, 333–340 Search PubMed
- C. Callaghan, PhD thesis, Worcester Polytechnic Institute, 2006
- L. Li, Z. H. Zhu, S. B. Wang, X. D. Yao and Z. F. Yan, J. Mol. Catal. A: Chem., 2009, 304, 71–76 CrossRef
- V. S. Sullivan, S. D. Jackson and P. C. Stair, J. Phys. Chem. B, 2005, 109, 352–356 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Reactor temperature profile, control reaction product distributions, additional doped catalyst results, effect of H2O-rich. See DOI: 10.1039/c8ra02977f |
| This journal is © The Royal Society of Chemistry 2018 |