
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of novel two-stage structure MnO micrometer particles as lithium-ion battery anode materials
Xian-Yinan Peiab,
Dong-Chuan Mo ab,
Shu-Shen Lyuab,
Jian-Hui Zhangc and
Yuan-Xiang Fu*ab
ab,
Shu-Shen Lyuab,
Jian-Hui Zhangc and
Yuan-Xiang Fu*ab
aSchool of Chemical Engineering and Technology, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: fuyx8@mail.sysu.edu.cn; Tel: +86-20-84111073
bGuangdong Engineering Technology Research Centre for Advanced Thermal Control Material and System Integration (TCMSI), Guangzhou 510275, P. R. China
cSchool of Chemistry, Sun Yat-sen University, Guangzhou 510275, P. R. China
First published on 10th August 2018
Abstract
MnO micrometer particles with a two-stage structure (composed of mass nanoparticles) were produced via a one-step hydrothermal method using histidine and potassium permanganate (KMnO4) as reagents, with subsequent calcination in a nitrogen (N2) atmosphere. When the MnO micrometer particles were utilized in lithium-ion batteries (LIBs) as anode materials, the electrode showed a high reversible specific capacity of 747 mA h g−1 at 100 mA g−1 after 100 cycles, meanwhile, the electrode presented excellent rate capability at various current densities from 100 to 2000 mA g−1 (∼203 mA h g−1 at 2000 mA g−1). This study developed a new approach to prepare two-stage structure micrometer MnO particles and the sample can be a promising anode material for lithium-ion batteries.
Introduction
Transition-metal oxides including MxOy1–6 (where M is Fe, Co, Ni, Mn, Zn, Cu, etc.), binary7–9 and ternary oxides10,11 have attracted growing interest as promising anode material candidates for lithium-ion batteries (LIBs) due to their advantages in electrochemistry. As a typical representative of MxOy, MnO and its composites have been widely studied as anode materials for LIBs due to the high theoretical capacity of MnO (756 mA h g−1), low cost and natural abundance of Mn on earth.12–14 Nevertheless, just like the other pure MxOy anodes, MnO also suffers from inherent low electric conductivity and dramatic volume changes during lithium cycling, which constitutes the major obstacle for its practical applications in LIBs.Extensive research and testing has since demonstrated that some effective strategies to overcome the pure MnO anode material disadvantages15–18 depend on fabrication of MnO/carbonaceous composites doped with other types of materials, and construction of different morphologies and crystal structures of pure MnO. Firstly, fabrication of carbonaceous MnO composites, such as MnO/porous carbon, MnO/carbon fibers,19 MnO/carbon nanotubes20 and MnO/graphene21 have been widely reported, and the carbon materials can improve electron transport in the electrodes owing to their intrinsic high electrical conductivity. In addition, carbon materials can be effectively buffer the volume change of MnO during the lithium cycling. Secondly, MnO with doped other materials, such as various metals22 or metal oxides,18,23 can improve the structural stability of the MnO hybrid materials during the charge and discharge process. Last but not the least, to structure different morphologies of pure MnO, it is particularly more beneficial to improve the Li+ transport in the electrodes with microstructure or nanostructure, due to the short lithium diffusion length. As a consequence, many scholars have always been fabricated microparticles, microspheres,24 nanoflakes,13 nanospheres25,26 and three-dimensional (3D) structures27,28 of MnO, and lots of MnO anode materials display outstanding electrochemical performances have been reported.13,24,29–31 He et al.24 synthesized porous MnO microspheres, which delivered a high reversible capacity up to 1234.2 mA h g−1 after 300 cycles at 0.2 C. Li's group29 prepared porous micrometer-sized MnO cubes, which displayed the reversible lithium storage capacity of 615.9 mA h g−1 at 200 mA g−1. Therefore, it is might be a fascinating thing to design and fabricate novel structure of pure MnO as high performance anode material.
In this paper, we reported a simple hydrothermal method and a subsequent annealing scheme to obtain two-stage structure MnO micrometer particles, in which composed of mass MnO nanoparticles. The whole preparation process has merits of mild conditions, and a certain amount of nitrogen (N) can be achieved in the MnO micrometer particles. When we employed the MnO micrometer particles as anodes for LIBs, the MnO electrode exhibits high reversible capacity of 747 mA h g−1 at 100 mA g−1 after 100 cycles. Meanwhile, the MnO electrode presents excellent rate capability at different current densities range from 100 to 2000 mA g−1 (the reversible capacity can retain 203 mA h g−1 at 2000 mA g−1).
Experimental
Materials synthesis
The MnO sample was fabricated by annealing of MnCO3 precursor under N2 atmosphere, which was prepared by a simple hydrothermal method. First, to fabricate MnCO3 precursor by hydrothermal process, 0.01 mol KMnO4 was dissolved in 30 ml deionized water in a beaker, 0.025 mol histidine was dissolved in 30 ml deionized water in another beaker. Then, the glycine aqueous solution was slowly dropwise added into the KMnO4 aqueous solution, stirring rapidly for 30 min. The as-prepared mixture was sealed in a Teflon-lined autoclave in an oven at 180 °C for 15 h, and the MnCO3 particles were obtained after being washed several times by deionized water and being dried in a vacuum oven at 80 °C for 12 h. Subsequently, the MnO particles were acquired after the heat treatment of MnCO3 particles in a N2 atmosphere. The schematic of the method as shown in Fig. 1.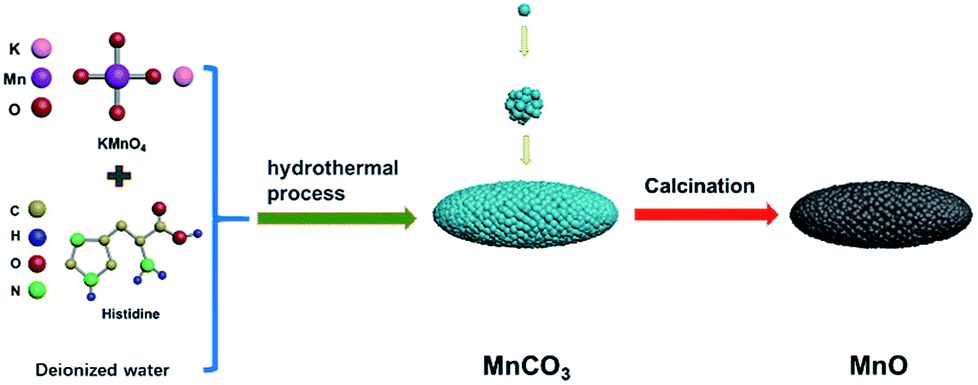 | ||
| Fig. 1 Schematic representation of preparation process. | ||
Characterization
The crystal structures of the samples were conducted by X-ray powder diffraction (XRD, Rigaku, D/max 2200VPC) equipped with Cu Kα radiating from 5° to 80°. Scanning electron microscopy (SEM) images were analyzed by a Field Emission Scanning electron microscopy (FEI, Quanta 400F). Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy images (HRTEM) of the samples were characterized by JEM-2010HR (JEOL). The thermogravimetric analysis (TGA) was carried out on a TG 209 Tarsus F3 analyzer (Netzsch) from room temperature to 850 °C at a heating rate of 10 °C min−1. The BET specific surface area of the sample was characterized by nitrogen sorption studies using the Micromeritics ASAP 2460 at −77 °C. The X-ray photoelectron spectroscopy (XPS) was affirmed by ESCA Lab250 X-ray photoelectron spectrometer (Thermo Scientific), and the mapping of the MnO particles were tested by the Gemini500 Field-Emission-SEM.Electrochemical measurement
The electrochemical performance of the as-prepared MnO samples were performed using a 2032-type coin half-cell. The working electrodes were prepared by mixing the MnO particles, conductive carbon black and polyvinylidene fluoride with a mass ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 into a certain amount N-methyl pyrrolidone (NMP). The as-prepared slurry was coated on a pure Cu foil and dried at 120 °C for 10 h in the vacuum oven. After being cut into Φ 14 mm wafers, the CR 2032-type coin cells were assembled in a highly pure argon-filled glovebox using the above-mentioned working electrodes. The lithium metal foils were used as the counter electrode, LiPF6 in ethylene carbonate/ethyl methyl carbonate/dimethyl carbonate (vol = 1:1:1) was used as the electrolyte and Celgard 2400 was used as the separator. The galvanostatic charge/discharge performances between 0.1 and 3.0 V (vs. Li/Li+) were conducted on the batteries testing system (LANHE CT2001A) at 25 °C. The cyclic voltammetry (CV) tests (from 0.1 V to 3 V with scan rate: 0.1 mV s−1) were performed on the electrochemical workstation (CHI660E).
:10:10 into a certain amount N-methyl pyrrolidone (NMP). The as-prepared slurry was coated on a pure Cu foil and dried at 120 °C for 10 h in the vacuum oven. After being cut into Φ 14 mm wafers, the CR 2032-type coin cells were assembled in a highly pure argon-filled glovebox using the above-mentioned working electrodes. The lithium metal foils were used as the counter electrode, LiPF6 in ethylene carbonate/ethyl methyl carbonate/dimethyl carbonate (vol = 1:1:1) was used as the electrolyte and Celgard 2400 was used as the separator. The galvanostatic charge/discharge performances between 0.1 and 3.0 V (vs. Li/Li+) were conducted on the batteries testing system (LANHE CT2001A) at 25 °C. The cyclic voltammetry (CV) tests (from 0.1 V to 3 V with scan rate: 0.1 mV s−1) were performed on the electrochemical workstation (CHI660E).
Results and discussion
As shown in Fig. 1, fusiform MnO micrometer particles in this study were prepared by hydrothermal method, followed by a calcination treatment. Firstly, glycine aqueous solution was slowly dropwise added into the KMnO4 aqueous solution, then MnCO3 micrometer particles would be produced after the hydrothermal process. Followed by annealing to the MnCO3 sample in N2, MnO micrometer particles were finally obtained.Fig. 2a shows the MnCO3 micrometer particles with fusiform shape, and the X-ray diffraction pattern of MnCO3 indexed as a rhombohedral structure according to the JCPDS card no. 99-0089 in Fig. 2b, meanwhile, and a broad apparent diffraction peak corresponding to carbonaceous material32 at 2θ = 20–30° was observed in the patterns of MnCO3. The as-prepared MnO micrometer particles were acquired by calcination of the MnCO3 particles base on TGA result of the MnCO3 particles precursor. Fig. 2c reveals the TGA curve of the MnCO3 precursor in a nitrogen (N2) atmosphere from room temperature to 850 °C at a heating rate of 10 °C min−1. There appears a weight loss keeps slowly decreasing before 350 °C and a large weight loss from 350 °C to 420 °C then the weight also slowly reduces until the temperature rises to 850 °C. The total weight loss of the sample about 57.5% in the whole process, which combines with the weight loss (38.2%) calculated from eqn (1), carbonaceous material decomposition and something adsorbent.29
| MnCO3 → MnO + CO2 | (1) |
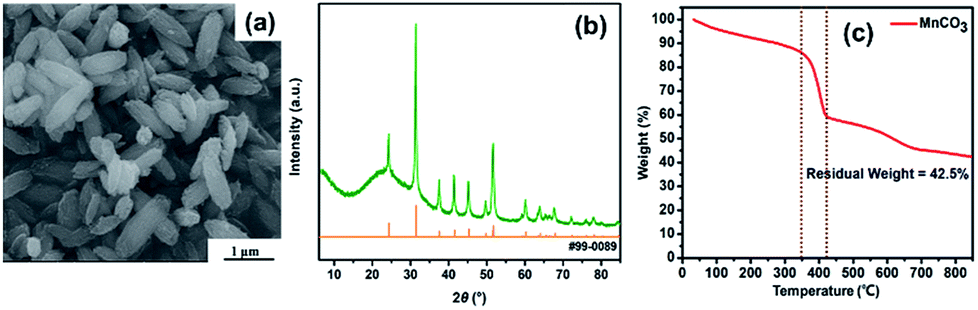 | ||
| Fig. 2 SEM image (a), XRD pattern (b) and the TGA curve of the MnCO3 particles (c). | ||
In consideration of the TGA result change curve of MnCO3 particles, we tested the XRD pattern and morphology of sample at four different annealing temperatures (500 °C, 600 °C, 700 °C and 800 °C). Fig. 3a displays the X-ray diffraction patterns of the as-prepared MnO samples (in short: MnO-500, MnO-600, MnO-700 and MnO-800). The major diffraction peaks of all the samples can be indexed to MnO with a cubic structure according to the JCPDS card no. 75-0626.13 However, it can be seen that the diffraction peaks are not sharp in the MnO-500 and MnO-600 samples. When the annealing temperature was increased to 700 °C and above, the two samples have obviously sharp diffraction peaks, especially the MnO-800 sample. We noticed that all the four samples have a small board peak (2θ = 20–30°) that might indicate to a certain amount of carbon.16 Meanwhile, we noticed that the SEM images of the MnO-500, MnO-600 and MnO-700 samples maintain typical fusiform shape just as the MnCO3 precursor in Fig. 3b–d. However, as shown in Fig. 3e, some little powder particles exist in the MnO-800 sample except for the original fusiform shape MnO particles. Thus, it may imply that the 800 °C is too high to prepare fusiform shape MnO sample. Then, the we chose the MnO-700 sample as a representative for the subsequent characterizations.
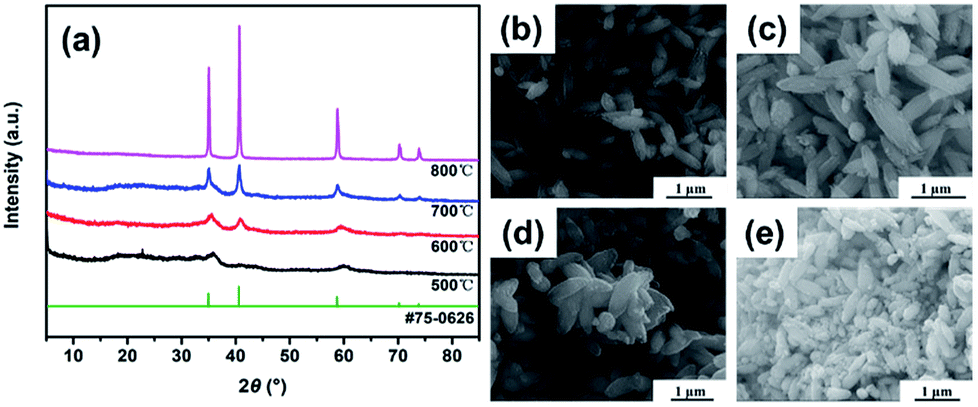 | ||
| Fig. 3 XRD results (a) and SEM images of MnO samples with different calcination temperatures, (b) is 500 °C (MnO-500), (c) is 600 °C (MnO-600), (d) is 700 °C (MnO-700) and (e) is 800 °C (MnO-800). | ||
The TEM images further reveal the morphology and structure of the as-prepared MnO sample. As shown in Fig. 4a, the MnO-700 particles displayed typical fusiform shape in agreement with SEM image. It could be seen that the MnO micrometer particle composed of many nano MnO particles in Fig. 4b. The nano MnO particles have good crystal structure for the clear lattice lines and the lattice fringe has interplanar spacing about 0.26 nm for the (111) plane of cubic MnO phase consistent with previous reports16,33 from the HRTEM image (Fig. 4c), and the lattice structure of the particles was proved by the electron diffraction pattern at the top right corner of in Fig. 4c. The two-stage structure of MnO micrometer particles can be directly demonstrated through the TEM image after ultrathin section technique. Fig. 4d displays several MnO micrometer particles in the TEM image, it could be clearly observed that many nano particles are make up the whole MnO micrometer particle, indicate to the novel two-stage structure of the sample. Fig. 4e shows the nitrogen adsorption–desorption isotherm curve of the MnO-700 sample, and the specific surface area was calculated to be 75.5 m2 g−1. The pore size distribution plot calculated from the desorption isotherm using the Barrett–Joyner–Halenda (BJH) method (Fig. 4f) showed that MnO-700 has pores of 3 nm which is in consistent with TEM result. We estimated that the novel two-stage structure MnO sample was acquired is ascribed to the histidine with a special five-membered ring chemical structure. First, the histidine offers numerous –NH2 and –COOH for KMnO4 to produce nano MnCO3 particle, then the five-membered ring of the histidine provides carbon skeleton to interact with the nano MnCO3 particles, and gradually grow larger, afterwards the nano MnCO3 particles become to the micrometer-sized MnCO3 fusiform particles. Finally, the novel two-stage structure micrometer fusiform MnO particles were produced by annealing treatment of the MnCO3 precursor (the preparation process of schematic diagram as shown in Fig. 1). Of course, the prediction derived from our previous research result, due to we found that the glycine (with straight-chain structure) cannot prepare this structure of MnO under the same experimental conditions.34
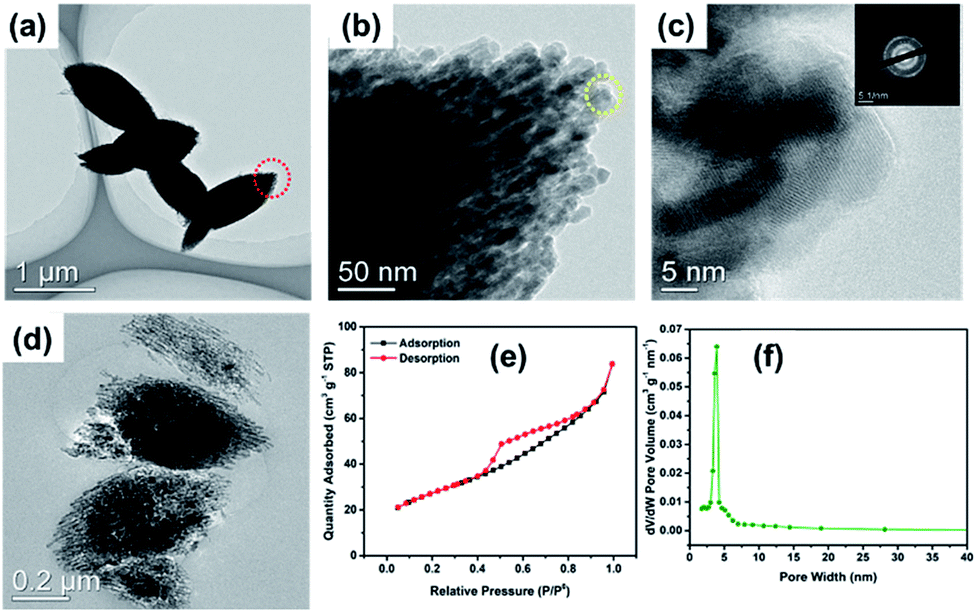 | ||
| Fig. 4 TEM (a), HRTEM (b and c) images and the ultrathin section image (d) of MnO-700; the adsorption/desorption isotherms (e) and pore size distribution (f) of the MnO-700 sample. | ||
Fig. 5a discloses the energy dispersive X-ray spectrum (EDS) of MnO-700 sample. The presence of Mn, O, C and N elements in the MnO particles was confirmed. Al element was observed due to the sample was fixed on Al foil for EDS testing. The element mapping images further reveal the evenly distribution of Mn, O, C and N elements in the MnO-700 sample, providing the evidences of the existence of N and C in the MnO sample (down of Fig. 5b). Fig. 5c shows the XPS spectrum of MnO-700 sample, whose chemical binding energies located at 284.8, 398.8, 530.1, 641.5 are ascribed to the characteristic peaks of C 1s, N 1s, O 1s, Mn 2p respectively. As shown in Fig. 5d, the two peaks located at 641.9 and 653.3 eV can be clearly distinguished as the symbols of Mn 2p3/2 and Mn 2p1/2 in the high-resolution XPS spectrum of Mn 2p, indicating the Mn2+ oxidation state, which agrees well the earlier MnO materials studies.35 The high-resolution O 1s spectrum of exhibits three peaks at 530.3, 531.4 and 532.7 eV (in Fig. 5e), the peak at 530.3 eV belongs to the bonding of Mn–O–Mn, and the peak at 531.4 eV is assigned to C–O, and the small peak at 532.7 eV maybe the existence of the hydrated surface of MnO.36 The C 1s of MnO is shifted to peaks related to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, which is ascribed to the amorphous carbon from the histidine carbon skeleton after calcination in Fig. 5f. The high resolution of N 1s peak (in Fig. 5g) could be fitted into three peaks at 398.6, 400.6 and 403.2 eV from the pyrolysis of histidine (with N atoms), which may be ascribed to pyridinic N, pyrrolic N and graphitic N bonds, respectively.33 The TGA analysis of MnO-700 was measured in air, from room temperature to 850 °C at a heating rate of 10 °C min−1. An obvious weight variation ranges from 250 °C to 600 °C in TGA curve, which was attributed to co-oxidation of carbonaceous material and MnO in the air. Certainly, the residual weight only Mn2O3 exists after 800 °C according to the early reports,33 meanwhile, the content of carbon in the MnO-700 sample is about 6.62 wt% based on the TGA result.
C, which is ascribed to the amorphous carbon from the histidine carbon skeleton after calcination in Fig. 5f. The high resolution of N 1s peak (in Fig. 5g) could be fitted into three peaks at 398.6, 400.6 and 403.2 eV from the pyrolysis of histidine (with N atoms), which may be ascribed to pyridinic N, pyrrolic N and graphitic N bonds, respectively.33 The TGA analysis of MnO-700 was measured in air, from room temperature to 850 °C at a heating rate of 10 °C min−1. An obvious weight variation ranges from 250 °C to 600 °C in TGA curve, which was attributed to co-oxidation of carbonaceous material and MnO in the air. Certainly, the residual weight only Mn2O3 exists after 800 °C according to the early reports,33 meanwhile, the content of carbon in the MnO-700 sample is about 6.62 wt% based on the TGA result.
 | ||
| Fig. 5 EDS analysis (a) and mapping image (b) of the MnO-700, (c) XPS full spectrum of the MnO-700, high resolution XPS of Mn 2p (d), O 1s (e), C 1s (f) and N 1s (g), and TGA curve of MnO-700 (h). | ||
The electrochemical measurements are carried out to evaluate the MnO-700 sample as anode material for LIBs. The charge–discharge cycling performances of the four MnO samples (MnO-500, MnO-600, MnO-700 and MnO-800) electrodes under a current density of 100 mA g−1 after 100 cycles is compared in Fig. 6a. The MnO-700 sample remains a reversible capacity of 747 mA h g−1 which is closed to the theoretical value (756 mA h g−1) of MnO,14,37 while the MnO-500 and MnO-600 sample only show low capacity of 116 and 157 mA h g−1, respectively. It is worthy to note, the MnO-800 sample exhibits slightly higher capacity than the MnO-700 sample in the first 30 cycles, subsequently, the capacity is gradually reduced, and its reversible capacity was obtained 590 mA h g−1 at the 100th cycle. Meanwhile, the MnO-700 sample exhibits the reversible capacity of 518 mA h g−1 after 200 cycles at 500 mA g−1, while the MnO-800 sample displays a capacity of 460 mA h g−1. In contrast, the MnO-600 and the MnO-500 sample only exhibit 140 and 5 mA h g−1 at the same test condition. We predicted that the MnO-700 sample exhibits the excellent cycling performances may be attribute to the good crystal and the morphology structure integrity base on the XRD and the SEM image results. Moreover, the precise mechanism of the in curve of capacity retention is not clear, but the main opinion is the oxidation of Mn2+ to a higher oxidation state due to defects or Mn aggregation during the charge and discharge cycles,34,38,39 and the increasing appearance also exhibited in the others type compounds in early reports.40,41 Then the MnO-500 and the MnO-600 samples with low capacities can be traced back to the imperfect crystal under the slightly low heat treatment temperature from the XRD result. The capacity of the MnO-700 sample higher than the MnO-800 sample may be ascribed to the MnO-800 particles easy to pulverization after repeatedly lithium cycling for the higher annealing temperature. The image of MnO-700 and MnO-800 electrode before cycle measurement are shown in Fig. 6c and d, respectively. It can be observed that the MnO-700 particles are larger than the MnO-800. Meanwhile, the same results also appeared in the after 200 cycles measurement image, in which it can be seen that the MnO-800 electrode (Fig. 6e) are more pulverized than the MnO-700 sample electrode (Fig. 6f). In addition, the TEM images of the MnO-700 (Fig. 6g) and the MnO-800 (Fig. 6h) can be more clearly confirmed the prediction.
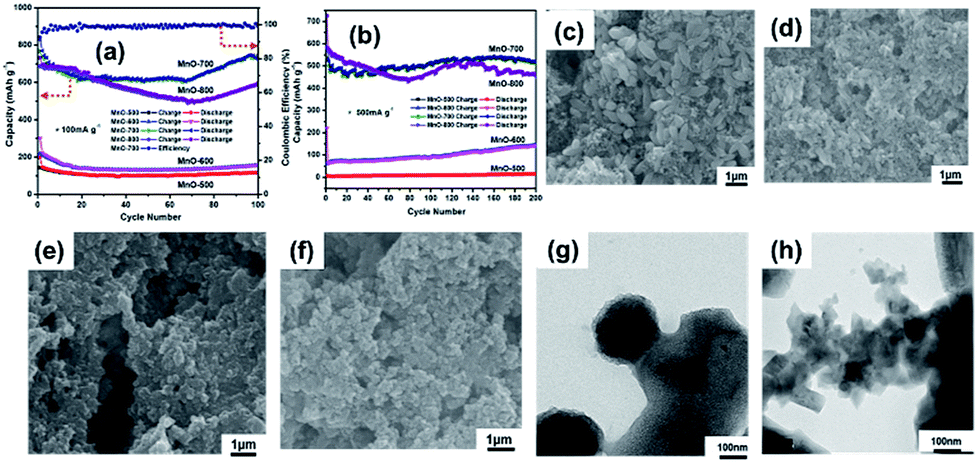 | ||
| Fig. 6 The cycling performances of MnO-500, MnO-600, MnO-700 and MnO-800 samples, (a) 100 mA g−1, (b) 500 mA g−1, SEM images of MnO-700 and MnO-800 electrodes, before (c and d) and after (e and f) 200 discharge/charge cycles at 500 mA g−1, TEM images of MnO-700 sample (g) and MnO-800 sample (h) after 200 discharge/charge cycles at 500 mA g−1. | ||
Fig. 7a shows the first five cyclic voltammogram (CV) curves the MnO-700 electrode at a scan rate of 0.1 mV s−1 at room temperature. A clear peak located around 0.3 V in the first catholic scan can be attributed to the decomposition of MnO into Mn and the formation of amorphous Li2O and the solid electrolyte interphase (SEI), and the broad peak at about 1.25 V is observed in the initial anodic scan curve, corresponding to reversible formation of MnO. The second loop of CV result is similar with the first scan, only the peaks location has slightly changed. However, the peak disappears in the following three cycles, indicating that the cell stability during further cycling.8,42 The discharge/charge curves of MnO-700 sample are shown in Fig. 7b. In the first discharge curve shows a plateau at 0.3 V, attributed to the reduction of Mn2+ to Mn0. The initial efficiency is 72.9%, which can be ascribed to effect of the novel two-stage structure of the MnO particles with C and N elements. Subsequently, the MnO electrode shows good capacity retention from the fifth cycle.16 Fig. 7c displays the rate performance of the MnO-700 sample, it can be found that the MnO-700 electrode exhibits the initial reversible capacity of 729 mA h g−1 at 100 mA g−1. Then, the reversible capacity of the sample decreases gradually with the current density increasing. When the current density at 2000 mA g−1, the reversible capacity can maintain 203 mA h g−1. In addition, the reversible capacity can revert to 702 mA h g−1 when the current density back to 100 mA g−1.
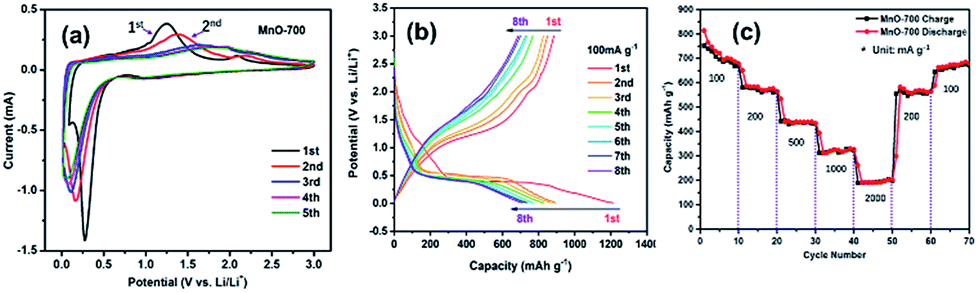 | ||
| Fig. 7 The CV curves of MnO-700 at 0.1 mV s−1 (a), the charge and discharge cycle curves of the MnO-700 electrode at 100 mA g−1 (b) and the rate performance of the MnO-700 sample (c). | ||
The excellent electrochemical performances of the pure MnO electrodes can be attributed to the combined effect of the novel two-stage structure and the existence of carbon and N element. Firstly, the novel two-stage structure can be suffered the volume expansion of MnO particles during lithium cycling, and the sub-micropores of the sample can enhance Li+ transport rate in the electrodes, owing to the short lithium diffusion length. After that, the carbon can improve the electronic conductivity of the active materials and prevents the aggregation of MnO particles during the charge and discharge process.43 Finally, the N element can further facilitate the transfer rate of Li+ diffusion and the electron transport between the interface of electrolyte and electrode.44 Meanwhile, comparison with the mentioned samples in Table 1, the MnO-700 electrode exhibits more excellent cycling performances than most of the pure MnO samples and some MnO composites. Moreover, for comparison the others pure MxOy, binary and ternary oxide anode materials in LIBs, the MnO-700 sample also delivers competitive reversible capacity. The above results indicated the novel two-stage structure fusiform MnO particles can be a promising anode material in LIBs through the easy strategy.
| Sample name | Current density | Capacity retention (mA h g−1) | Cycle number (times) | Reference |
|---|---|---|---|---|
| Fusiform MnO particles | 100 mA g−1 | 747 | 100 | This work |
| Nanocrystalline MnO film | 0.125 C | 472 | 25 | 31 |
| MnO particles | 0.125 C | 650 | 150 | 2 |
| Porous MnO nanoflakes | 246 mA g−1 | 648.3 | 100 | 13 |
| MnO thin-film | 0.05 C | ∼700 | 100 | 45 |
| Porous MnO spheres | 100 mA g−1 | 960 | 100 | 46 |
| Porous MnO/C microsphere | 50 mA g−1 | 690 | 50 | 30 |
| MnO/NC composites | 100 mA g−1 | 775.4 | 200 | 47 |
| MnO/C microspheres | 100 mA g−1 | 625 | 60 | 16 |
| MnO/carbon nanofiber | 200 mA g−1 | 398 | 200 | 48 |
| MnO/nitrogen-doped carbon | 100 mA g−1 | 578 | 60 | 14 |
| 3D/carbon-matrix/MnO/C | 100 mA g−1 | 1159 | 50 | 49 |
| C/MnO/Mn3N2 | 100 mA g−1 | 626 | 60 | 50 |
| MnO/carbon nanotube | 100 mA g−1 | 650 | 30 | 20 |
| MnO/ZnO/RGO | 100 mA g−1 | 660 | 100 | 17 |
| MnO/RGOS hybrid | 100 mA g−1 | 665.5 | 50 | 21 |
| TNP-Fe2O3 | 100 mA g−1 | 712 | 100 | 51 |
| Fe2O3 nanoparticles | 100 mA g−1 | 303.2 | 50 | 52 |
| Mesoporous Fe2O3 particles | 100 mA g−1 | 1009 | 230 | 53 |
| ZnO–NiO microspheres | 100 mA g−1 | 733 | 50 | 54 |
| ZnO–MnO composite | 0.1 C | 625 | 50 | 18 |
| TiO2@ZnO nanosheet | 200 mA g−1 | 340.2 | 100 | 55 |
| NiO–Ni nanocomposite | 100 mA g−1 | 690 | 50 | 56 |
| Hollow NiO microspheres | 100 mA g−1 | 560 | 45 | 57 |
| Porous NiO film | 0.1 C | 570 | 50 | 58 |
| Crinkly NiO layers | 100 mA g−1 | 744 | 50 | 59 |
| CuO nanotube array | 0.1 C | 581 | 50 | 60 |
| Cu2O–CuO–TiO2 nanocages | 50 mA g−1 | 700 | 85 | 61 |
Conclusions
In summary, the novel two-stage structure fusiform MnO micrometer particles comprising mass nano MnO particles with a certain of C and N elements were successfully prepared via a facile hydrothermal and a subsequent annealing scheme. The MnO-700 sample exhibited excellent electrochemical performances in terms of reversible capacity (high specific capacity of 747 mA h g−1 at 100 mA g−1 after 100 cycles), cycling stability and rate capability (203 mA h g−1 at 2000 mA g−1) when used as the anode material for LIBs. The results show that the novel two-stage structure MnO sample can be a promising anode material for LIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Natural Science Foundation of China (Grant No. 51676212) Fundamental Research Funds for the Central Universities (Grant No. 17lgpy68) and Pearl River S&T Nova Program of Guangzhou (Grant No. 201710010043).Notes and references
- J. C. Guo, Q. Liu, C. S. Wang and M. R. Zachariah, Adv. Funct. Mater., 2012, 22, 803–811 CrossRef.
- K. F. Zhong, X. Xia, B. Zhang, H. Li, Z. X. Wang and L. Q. Chen, J. Power Sources, 2010, 195, 3300–3308 CrossRef.
- Y. Z. Fan, R. M. Liu, W. Du, Q. Y. Lu, H. Pang and F. Gao, J. Mater. Chem., 2012, 22, 12609–12617 RSC.
- S. K. Liu, Z. X. Chen, K. Xie, Y. J. Li, J. Xu and C. M. Zheng, J. Mater. Chem. A, 2014, 2, 13942–13948 RSC.
- Y. Mao, Q. Y. Kong, B. K. Guo, L. Shen, Z. X. Wang and L. Q. Chen, Electrochim. Acta, 2013, 105, 162–169 CrossRef.
- S. Li, Y. Xiao, X. Wang and M. H. Cao, Phys. Chem. Chem. Phys., 2014, 16, 25846–25853 RSC.
- M. V. Reddy, C. Yu, F. Jiahuan, K. P. Loh and B. V. R. Chowdari, RSC Adv., 2012, 2, 9619–9625 RSC.
- M. V. Reddy, C. T. Cherian, K. Ramanathan, K. C. W. Jie, T. Y. W. Daryl, T. Y. Hao, S. Adams, K. P. Loh and B. V. R. Chowdari, Electrochim. Acta, 2014, 118, 75–80 CrossRef.
- D. Darbar, M. V. Reddy, S. Sundarrajan, R. Pattabiraman, S. Ramakrishna and B. V. R. Chowdari, Mater. Res. Bull., 2016, 73, 369–376 CrossRef.
- A. S. Hameed, H. Bahiraei, M. V. Reddy, M. Z. Shoushtari, J. J. Vittal, C. K. Ong and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2014, 6, 10744–10753 CrossRef PubMed.
- M. V. Reddy, C. Y. Quan, K. W. Teo, L. J. Ho and B. V. R. Chowdari, J. Phys. Chem. C, 2015, 119, 4709–4718 CrossRef.
- S. J. Ma, D. M. Chen and W. L. Wang, Phys. Chem. Chem. Phys., 2016, 18, 19130–19136 RSC.
- X. W. Li, D. Li, L. Qiao, X. H. Wang, X. L. Sun, P. Wang and D. Y. He, J. Mater. Chem., 2012, 22, 9189–9194 RSC.
- H. Liu, Z. H. Li, Y. R. Liang, R. W. Fu and D. C. Wu, Carbon, 2015, 84, 419–425 CrossRef.
- Z. Y. Cai, L. Xu, M. Y. Yan, C. H. Han, L. He, K. M. Hercule, C. J. Niu, Z. F. Yuan, W. W. Xu, L. B. Qu, K. N. Zhao and L. Q. Mai, Nano Lett., 2015, 15, 738–744 CrossRef PubMed.
- X. Z. Wang, S. Qiu, G. X. Lu, C. Z. He, J. R. Liu, L. Q. Luan and W. Liu, CrystEngComm, 2014, 16, 1802–1809 RSC.
- F. Jiang, L. W. Yang, Y. Tian, P. Yang, S. W. Hu, K. Huang, X. L. Wei and J. X. Zhong, Ceram. Int., 2014, 40, 4297–4304 CrossRef.
- M. S. Song, S. Nahm, W. I. Cho and C. Lee, Phys. Chem. Chem. Phys., 2015, 17, 23496–23502 RSC.
- T. Wang, H. G. Li, S. J. Shi, T. Liu, G. Yang, Y. M. Chao and F. Yin, Small, 2017, 13, 1604182 CrossRef PubMed.
- S. D. Xu, Y. B. Zhu, Q. C. Zhuang and C. Wu, Mater. Res. Bull., 2013, 48, 3479–3484 CrossRef.
- Y. J. Mai, D. Zhang, Y. Q. Qiao, C. D. Gu, X. L. Wang and J. P. Tu, J. Power Sources, 2012, 216, 201–207 CrossRef.
- J. Lee, H. Z. Zhu, W. Deng and Y. Wu, Nano Res., 2017, 10, 1033–1043 CrossRef.
- N. N. Wang, Y. J. Zhai, X. J. Ma and Y. T. Qian, RSC Adv., 2015, 5, 61148–61154 RSC.
- X. W. Li, X. N. Shang, D. Li, H. W. Yue, S. Y. Wang, L. Qiao and D. Y. He, Part. Part. Syst. Charact., 2014, 31, 1001–1007 CrossRef.
- L. L. Zhang, D. H. Ge, G. L. Qu, J. W. Zheng, X. Q. Cao and H. W. Gu, Nanoscale, 2017, 9, 5451–5457 RSC.
- J. Yue, X. Gu, L. Chen, N. N. Wang, X. L. Jiang, H. Y. Xu, J. Yang and Y. T. Qian, J. Mater. Chem. A, 2014, 2, 17421–17426 RSC.
- X. H. Ma, Q. Y. Wan, X. Huang, C. X. Ding, Y. Jin, Y. B. Guan and C. H. Chen, Electrochim. Acta, 2014, 121, 15–20 CrossRef.
- W. Zhang, J. Z. Sheng, J. Zhang, T. He, L. Hu, R. Wang, L. Q. Mai and S. C. Mu, J. Mater. Chem. A, 2016, 4, 16936–16945 RSC.
- X. Y. Fan, S. H. Li and L. Lu, Electrochim. Acta, 2016, 200, 152–160 CrossRef.
- K. F. Zhong, B. Zhang, S. H. Luo, W. Wen, H. Li, X. J. Huang and L. Q. Chen, J. Power Sources, 2011, 196, 6802–6808 CrossRef.
- X. Q. Yu, Y. He, J. P. Sun, K. Tang, H. Li, L. Q. Chen and X. J. Huang, Electrochem. Commun., 2009, 11, 791–794 CrossRef.
- T. Morishita, T. Tsumura, M. Toyoda, J. Przepiorski, A. W. Morawski, H. Konno and M. Inagaki, Carbon, 2010, 48, 2690–2707 CrossRef.
- T. Bai, H. C. Zhou, X. Y. Zhou, Q. C. Liao, S. M. Chen and J. Yang, J. Mater. Sci., 2017, 52, 11608–11619 CrossRef.
- X. Y. Pei, D. C. Mo, S. S. Lyu, J. H. Zhang and Y. X. Fu, J. Mater. Sci.: Mater. Electron., 2018, 1–9, DOI:10.1007/s10854-018-9300-0.
- D. H. Liu, H. Y. Lu, X. L. Wu, B. H. Hou, F. Wan, S. D. Bao, Q. Y. Yan, H. M. Xie and R. S. Wang, J. Mater. Chem. A, 2015, 3, 19738–19746 RSC.
- F. Wang, J. X. Cai, J. Yu, C. Li and Z. Y. Yang, ChemElectroChem, 2018, 5, 51–61 CrossRef.
- Y. M. Sun, X. L. Hu, W. Luo and Y. H. Huang, J. Mater. Chem., 2012, 22, 19190–19195 RSC.
- Y. H. Wang, X. Ding, F. Wang, J. Q. Li, S. Y. Song and H. J. Zhang, Chem. Sci., 2016, 7, 4284–4290 RSC.
- S. Zhang, L. X. Zhu, H. H. Song, X. H. Chen and J. S. Zhou, Nano Energy, 2014, 10, 172–180 CrossRef.
- M. V. Reddy, Y. Xu, V. Rajarajan, T. Ouyang and B. V. R. Chowdari, ACS Sustainable Chem. Eng., 2015, 3, 3035–3042 CrossRef.
- B. Das, M. V. Reddy, P. Malar, T. Osipowicz, G. V. Subba Rao and B. V. R. Chowdari, Solid State Ionics, 2009, 180, 1061–1068 CrossRef.
- Y. Z. Wu, P. B. Zhu, M. V. Reddy, B. V. R. Chowdari and S. Ramakrishna, ACS Appl. Mater. Interfaces, 2014, 6, 1951–1958 CrossRef PubMed.
- L. J. Zhang, G. L. Xia, Y. Q. Huang, C. Y. Wei, Y. W. Yu, D. L. Sun and X. B. Yu, Energy Storage Materials, 2018, 10, 160–167 CrossRef.
- X. Gu, J. Yue, L. Chen, S. Liu, H. Y. Xu, J. Yang, Y. T. Qian and X. B. Zhao, J. Mater. Chem. A, 2015, 3, 1037–1041 RSC.
- Z. H. Cui, X. X. Guo and H. Li, J. Power Sources, 2013, 244, 731–735 CrossRef.
- P. Remith and N. Kalaiselvi, Phys. Chem. Chem. Phys., 2016, 18, 15854–15860 RSC.
- Y. Ding, L. H. Chen, P. Pan, J. Du, Z. B. Fu, C. Q. Qin and F. Wang, Appl. Surf. Sci., 2017, 422, 1113–1119 CrossRef.
- J. G. Wang, Y. Yang, Z. H. Huang and F. Y. Kang, Electrochim. Acta, 2015, 170, 164–170 CrossRef.
- F. C. Zheng, Z. C. Yin, H. Y. Xia, G. L. Bai and Y. G. Zhang, Chem. Eng. J., 2017, 327, 474–480 CrossRef.
- Y. M. Wu, M. J. Liu, H. B. Feng and J. H. Li, Nanoscale, 2014, 6, 14697–14701 RSC.
- Y. Z. Wang, J. S. Han, X. X. Gu, S. Dimitrijev, Y. L. Hou and S. Q. Zhang, J. Mater. Chem. A, 2017, 5, 18737–18743 RSC.
- M. C. Sun, M. F. Sun, H. X. Yang, W. H. Song, Y. Nie and S. N. Sun, Ceram. Int., 2017, 43, 363–367 CrossRef.
- J. J. Zhang, T. Huang, Z. L. Liu and A. S. Yu, Electrochem. Commun., 2013, 29, 17–20 CrossRef.
- T. T. Cao, D. Fang, L. Liu, Z. P. Luo, Q. Wang, L. J. Dong and C. X. Xiong, J. Mater. Sci.: Mater. Electron., 2015, 26, 5287–5294 CrossRef.
- L. Gao, S. H. Li, D. K. Huang, Y. Shen and M. K. Wang, Electrochim. Acta, 2015, 182, 529–536 CrossRef.
- X. H. Huang, J. P. Tu, B. Zhang, C. Q. Zhang, Y. Li, Y. F. Yuan and H. M. Wu, J. Power Sources, 2006, 161, 541–544 CrossRef.
- X. H. Huang, J. P. Tu, C. Q. Zhang and F. Zhou, Electrochim. Acta, 2010, 55, 8981–8985 CrossRef.
- Z. Chen, A. Xiao, Y. Chen, C. Zuo, S. Zhou and L. Li, Mater. Res. Bull., 2012, 47, 1987–1990 CrossRef.
- J. F. Zhao, Y. Shao, J. C. Zha, H. Y. Wang, Y. Yang, S. D. Ruan, G. Yang and J. H. Chen, Ceram. Int., 2016, 42, 3479–3484 CrossRef.
- A. G. Xiao, S. B. Zhou, C. G. Zuo, Y. B. Zhuan and X. Ding, Mater. Res. Bull., 2015, 70, 795–798 CrossRef.
- G. X. Wang, Y. M. Sui, M. N. Zhang, M. Xu, Q. X. Zeng, C. Liu, X. M. Liu, F. Du and B. Zou, J. Mater. Chem. A, 2017, 5, 18577–18584 RSC.
| This journal is © The Royal Society of Chemistry 2018 |