DOI:
10.1039/C8RA03230K
(Review Article)
RSC Adv., 2018,
8, 19456-19464
Combining transition metals and transient directing groups for C–H functionalizations
Received
15th April 2018
, Accepted 14th May 2018
First published on 29th May 2018
Abstract
In the domain of synthetic chemistry, C–H bond activation has always remained in the spotlight for researchers over the last few decades. Although different strategies have been employed to chemically trigger unactivated C–H bonds, transition metal catalyzed directing group (DG) aided C–H bond activation is the most explored pathway of all because of its ability to perform diverse site selective functional metamorphosis. Despite its popularity, tedious synthetic methodology requiring additional steps for the installation and removal of DGs from the target substrate diminishes its efficacy. However, replacement of directing groups by transient directing groups (tDGs) reduces the hurdle to a greater extent without compromising the product yield and selectivity. In this report we have depicted the intense journey of transient directing groups with three (Rh, Ru, and Pd) prevalent second row transition metals.
 Trisha Bhattacharya | Trisha Bhattacharya was born in 1993 in the state of West Bengal, India. She obtained her B.Sc. degree in chemistry from Asutosh College, University of Calcutta in 2015. Then she completed her M.Sc in chemistry at the Indian Institute of Technology, Hyderabad, in 2017 following which she joined Prof. Debabrata Maiti’s group at the Indian Institute of Technology, Bombay. Presently, she is pursuing her PhD on the topic of transition metal catalyzed distal C–H activation. |
 Sandeep Pimparkar | Sandeep was born and raised in Nasik, Maharashtra. After his M.Sc in Organic Chemistry he joined Prof. Maiti’s group in Jan 2015 as a collaborative PhD student between IIT Bombay and Monash University, Australia. His research interest is in Catalytic Chemistry for the development of sustainable and eco-friendly methodologies for the synthesis of important classes of bio-active motifs. |
 Debabrata Maiti | Debabrata Maiti received his PhD from John Hopkins University (USA) in 2008 under the supervision of Prof. Kenneth D. Karlin. After postdoctoral studies at the Massachusetts Institute of Technology (MIT) with Prof. Stephen L. Buchwald (2008–2010), he joined the Department of Chemistry at IIT Bombay in 2011, where he is currently an Associate Professor. His research interests are focused on the development of new and sustainable synthetic and catalytic methods. |
1 Introduction
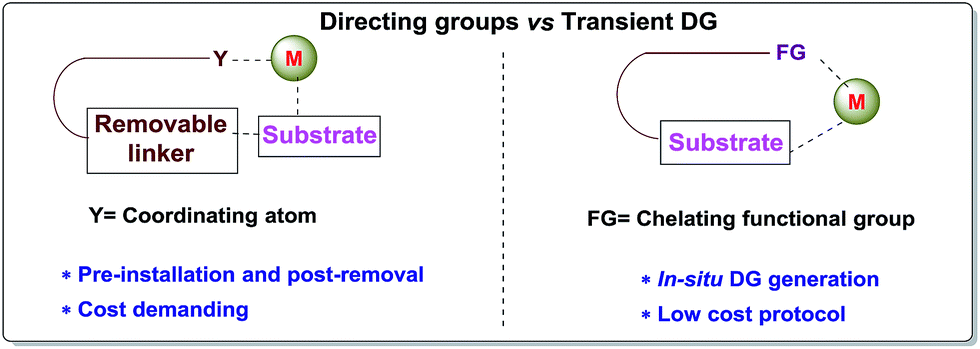
Carbon–hydrogen bonds are nature’s most ubiquitous functional unit. Regardless of their easy accessibility, C–H bonds are quite inert due to a very low polarity difference between the two constituent atoms and a high C–H bond dissociation energy. Therefore direct conversion of C–H bonds into C–C or C–heteroatom bonds poses a great challenge to the synthetic community. Therefore, transition metal catalyzed C–H activation has become one of the most exciting topics in the last couple of decades in the domain of organometallic chemistry.1–3 Even with its indisputable evolution, the major complication lies in achieving position-selective C–H functionalization.4 Irrespective of different chemical environments, all C–H bond energy values more or less coincide in the same range, hence it becomes difficult to perform a regioselective C–H bond transformation. Although a series of different strategies have been employed to address this concern, the use of directing groups (DGs) is still an irreplaceable solution.5 Directing groups are none other than a variety of σ-coordinating groups that can easily bind to the metal center forming a stable metallacycle which then selectively reaches the proximal C–H bond through its tuned orientation. Employing appropriate directing groups, versatile functionalizations have been achieved at ortho-,6,7meta-8–11 and recently at para-12–15 positions of different aromatic rings. This strategy is also useful for selective functionalizations at various positions (α, β, γ and δ) of different aliphatic systems.16–22 Although enormous progress has been observed in DG assisted C–H bond activation, the major drawback of this approach is the installation of the DG in the substrate and also the removal at the end, and these often demand tedious synthetic procedures. To resolve this problem, two alternative solutions have been applied: (i) use of a transient mediator23 in a stoichiometric ratio and (ii) implementation of a traceless directing group.24,25 As well as these two approaches, an alternative way is to develop a functionally capable directing group which can reversibly connect with the substrate as well as to a metal. The comparison between the concepts of covalently linked directing groups and transient directing groups is shown in Fig. 1.
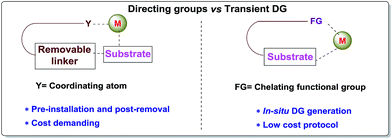 |
| | Fig. 1 Directing groups vs. transient directing groups. | |
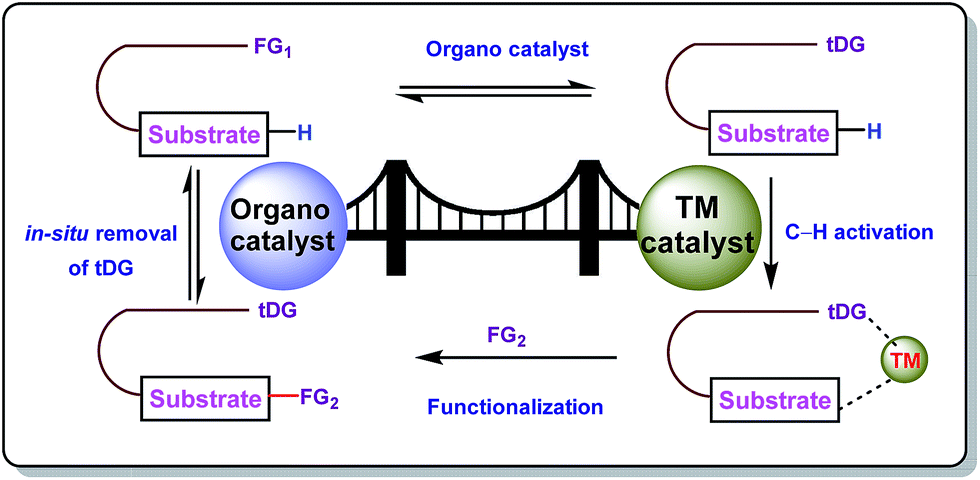
The concept of transient directing groups relies on the reversible binding of an organocatalyst with a substrate containing a particular functional group. The newly generated entity effectively coordinates with the metal center in a monodentate or bidentate fashion to generate the metallacycle which upon reaction with the coupling partner, furnishes the site selective functionalization (Fig. 2). Previously, detailed reviews on transient DG mediated C–H activation have been reported by Besset26 and Ackermann27 independently from two different perspectives. This work reviews the momentous evolution of transition metal catalyzed C–H activation mediated by transient modifiers. In this mini review we have categorized the entire theme into three major parts which are (a) rhodium catalyzed, (b) ruthenium catalyzed, and (c) palladium catalyzed transient DG assisted C–H activation, with detailed mechanisms and substrate scopes as well as their limitations.
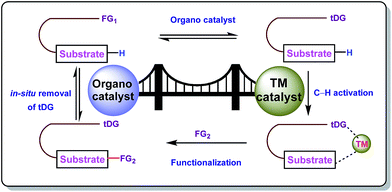 |
| | Fig. 2 Transient directing group mediated C–H activation. | |
2 Transient DG assisted C–H activation with second row transition metals
2.1 Transient DG assisted C–H activation with rhodium
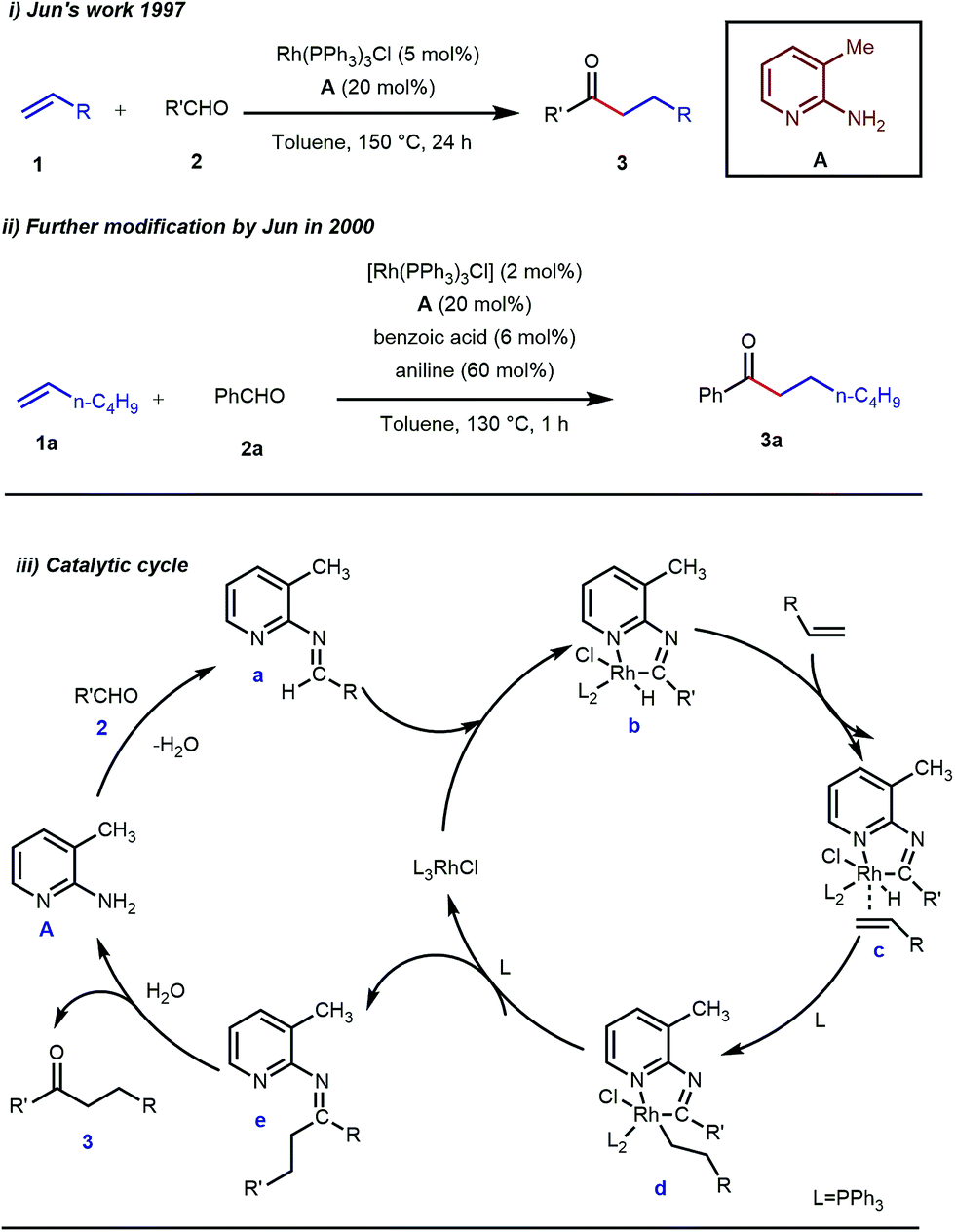
Rhodium is the most exploited transition metal in terms of transient directing group assisted C–H activation. A series of functionalizations have been reported so far which use rhodium based catalysts. The journey started back in 1979 when Suggs reported a detailed study on the hydroacylation of aromatic aldehydes in three steps where Wilkinson’s catalyst [(PPh3)3RhCl] (5 mol%) was used to activate the aldehyde C–H bond.28 Of these three steps, one key step was the formation of aldimines from the corresponding aldehydes and amines via aminal formation. Starting from 2-amino-3-picoline A three different aldimines were prepared, which after reaction with different olefins resulted in the desired hydroacylated products mediated by (iminoacyl)rhodium(III) hydrides. However, the protocol was restricted to very limited variants of aromatic aldehydes with no α-hydrogens. Later, in 1997, Jun and co-workers developed a one-step hydroacylation of aldehydes 2 with different terminal olefins 1 through the in situ generation of 3-methyl-2-aminopyridyl aldimines (Fig. 3i).29 The method was not only step-reducing, but a variety of aliphatic aldehydes were also incorporated along with aromatic aldehydes.
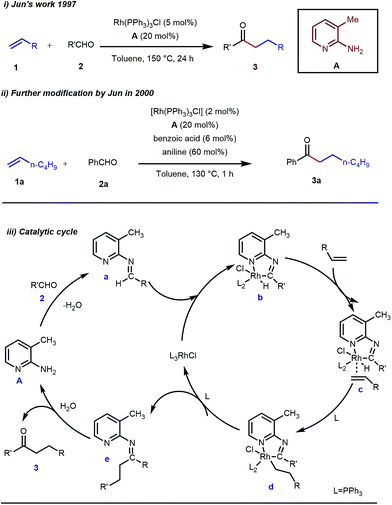 |
| | Fig. 3 Intermolecular hydroacylation (i) by Jun and co-workers in 1997 (ii) further modification of Jun's work in 2000 (iii) proposed mechanistic outline for intermolecular hydroacylation. | |
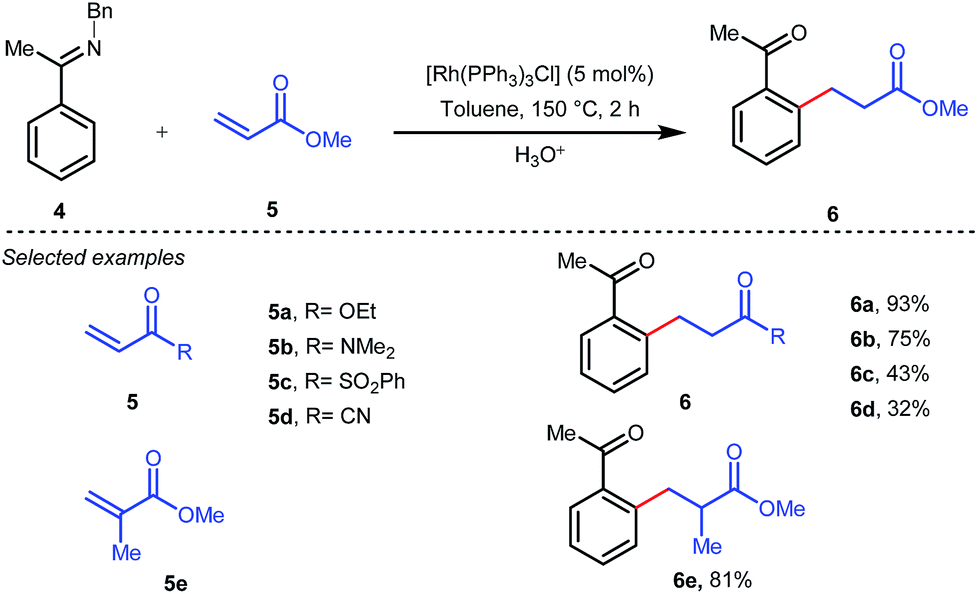
The outline of the reaction mechanism was proposed as follows: (1) generation of aldimine a from the corresponding aldehyde 2 and 2-amino-3-picoline A which acts as a co-catalyst, (2) formation of (iminoacyl)rhodium(III) hydride b by C–H activation, (3) olefin coordination at the Rh center forming intermediate c, and (4) β-migratory insertion to generate an (iminoacyl)rhodium(III) alkyl complex d followed by (5) reductive elimination to form the linear ketone 3 with regeneration of the catalyst again (Fig. 3iii). The reaction was further modified through the use of a catalytic amount of benzoic acid along with aniline as an additive where they both triggered the aldimine formation in the initial step (Fig. 3ii).30 Later, in 2002 the same analogy was applied by Jun et al. to obtain the ortho-alkylated aromatic ketones starting from a masked ketone i.e. ketimine 4 with a variety of unactivated olefins.31 The protocol covered a reasonable variety of ketones apart from acetone. The same functionalization was further carried out with activated olefins 5 in 2004 by the same group (Table 1).32 The reaction was amenable for a wide range of activated olefins covering linear 5a-b as well as branched acrylates 5e as efficient alkylating precursors. Phenyl vinyl sulfone 5c and acrylonitrile 5e also provided the ortho-alkylated acetophenones but the yields were not satisfactory. However, in this case, the ketone partner was mostly limited to acetophenone.
Table 1
Ortho-alkylation of masked ketones with activated olefins
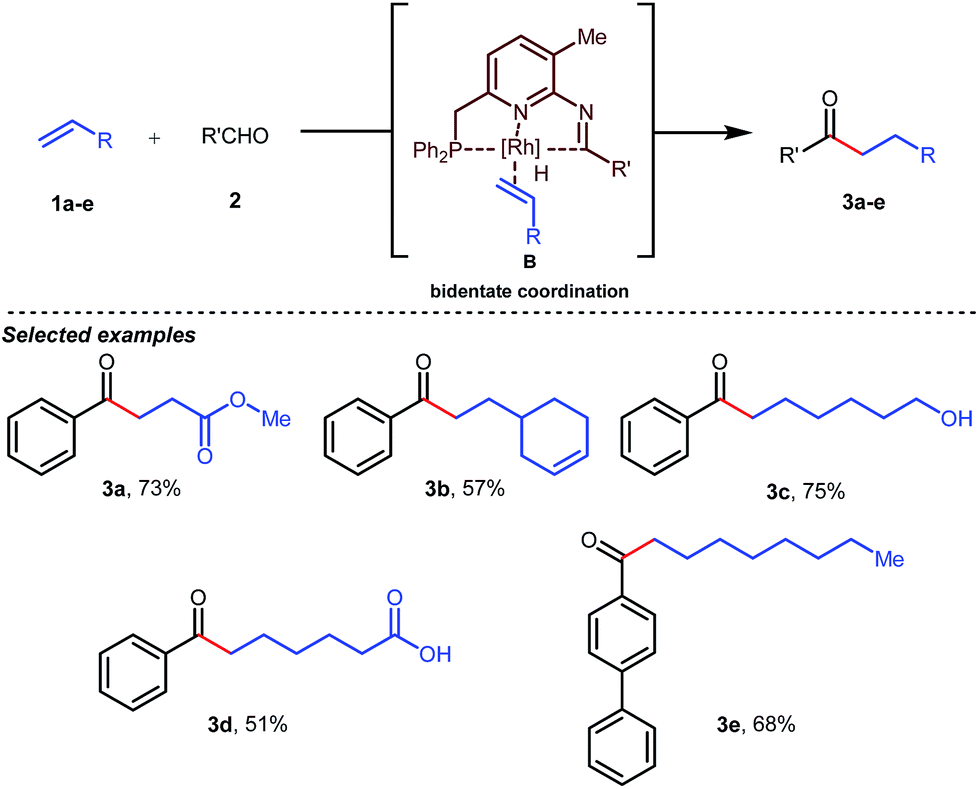
Jun’s catalytic system was further revised with an additional phosphine tether which overall offered bidentate coordination to the metal center and subsequently promoted the C–H activation step to provide intermolecular hydroacylated products (Table 2).33
Table 2 Intermolecular hydroacylation with an in situ generated bidentate tDG
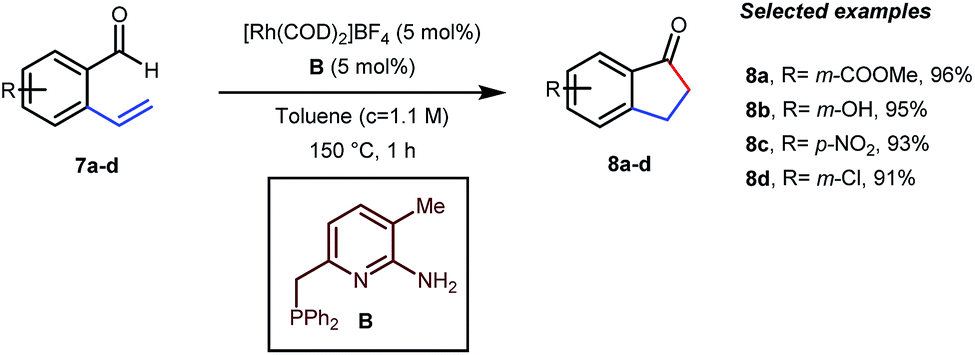
In the acylated products, different functional groups like ester 3a, internal alkene 3b, alcohol 3c, and acid 3d were well-tolerated. After the successful intermolecular hydroacylation, the authors intended to extend the scope of this work further to achieve intramolecular hydroacylation, for which they chose o-vinylbenzaldehydes 7a–d in the presence of 5 mol% of [Rh(COD)2]BF4 catalyst and 5 mol% of ligand B (Table 3). Different functional groups including carboxylic ester 8a, free phenol 8b, nitro group 8c, and chloride 8d were well-suited to the reaction conditions, earning excellent yields.
Table 3 Intramolecular hydroacylation with an in situ generated bidentate transient DG
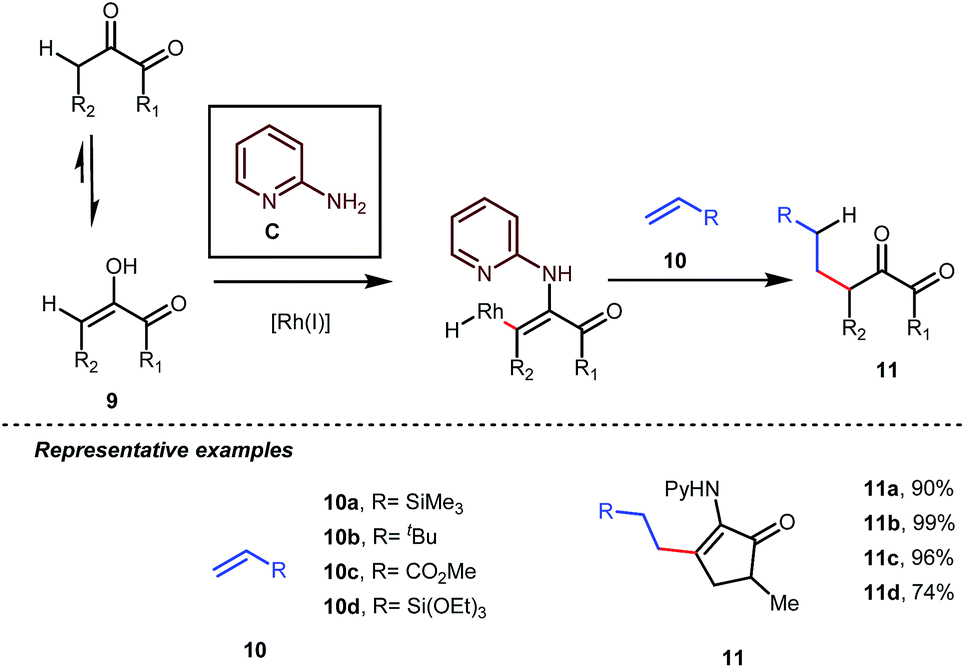
Influenced by all of these developments, Dong and co-workers developed an α-alkylation of 1,2-diketones with different types of aromatic as well as aliphatic olefins in the presence of stoichiometric amounts of 2-aminopyridine C (Table 4).34
Table 4 α-Alkylation of diketones using 2-aminopyridine as a transient modifier
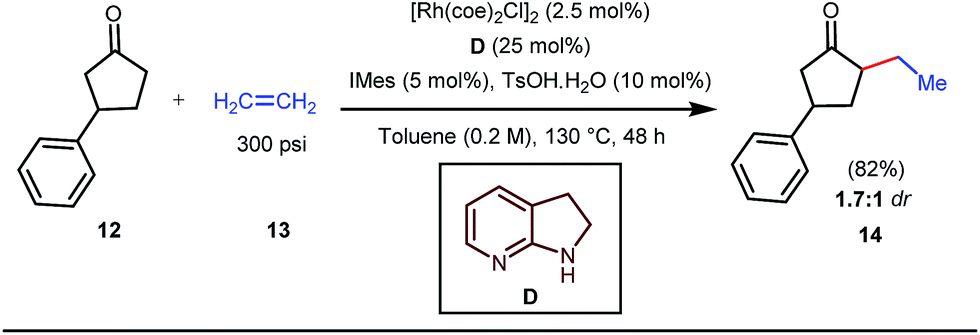
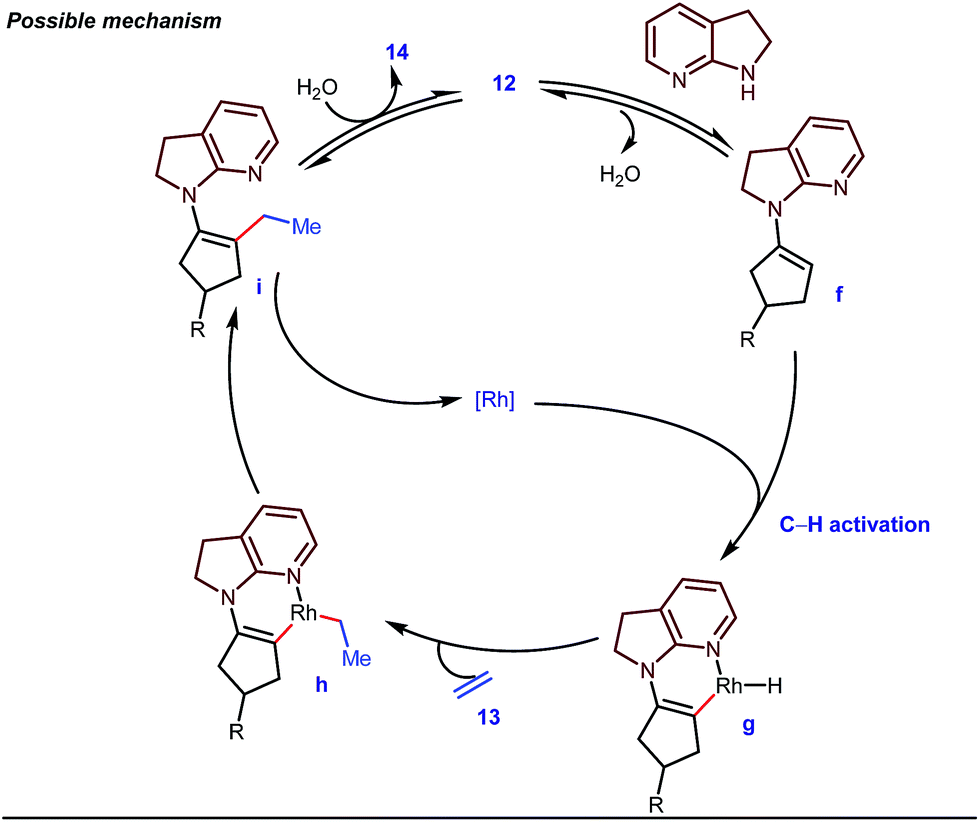
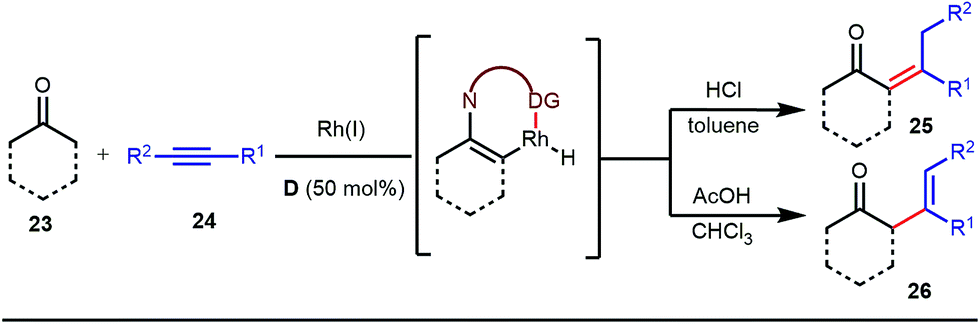
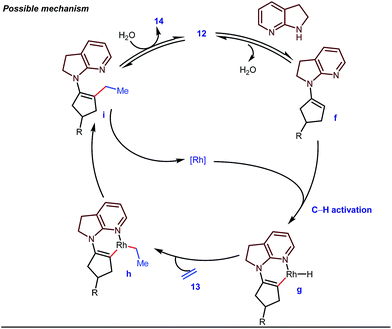
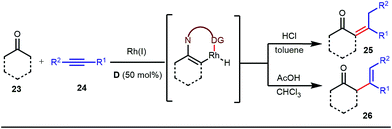
The reaction continued through an enamine mediated C–H activation. A range of electron rich as well as electron deficient olefin coupling sources were equally fruitful. Regardless of several difficulties with the ketimine formation, in 2014, Mo and Dong reported α-alkylation of cyclic ketone derivatives with a modified rhodium(I) complex [Rh(coe)Cl]2 using simple ethylene gas where 7-azaindoline acted as an effective transient directing group (Fig. 4).35 The transformation was highly regioselective for the less crowded 5-position of the cyclic ketone, overriding the electronic bias of the substituents present on the ketones. In particular, this work excluded the possibility of forming other side products. The use of the Rh(I) dimer complex and ligand D was found to be essential in this case. The enamine formation in the initial step was promoted by 10 mol% TsOH·H2O which again supported the proof-of-concept. To accelerate the oxidative addition of enamine C–H bonds, a sterically hindered electron rich NHC ligand (IMes) was employed (5 mol%). A plausible mechanistic outline for this reaction is shown in Fig. 5. At the very beginning, an in situ enamine f was formed, followed by oxidative addition of the Rh complex, generating g. In the next step, olefin 13 coordinated with the metal to form complex h, which after reductive elimination generated i. Hydrolysis of this intermediate accomplished the desired ketones 14.
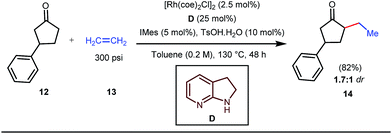 |
| | Fig. 4 α-Alkylation of cyclic ketones with the 7-azaindoline tDG precursor. | |
 |
| | Fig. 5 Proposed mechanism for α-alkylation of cyclic ketones using 7-azaindoline as a tDG. | |
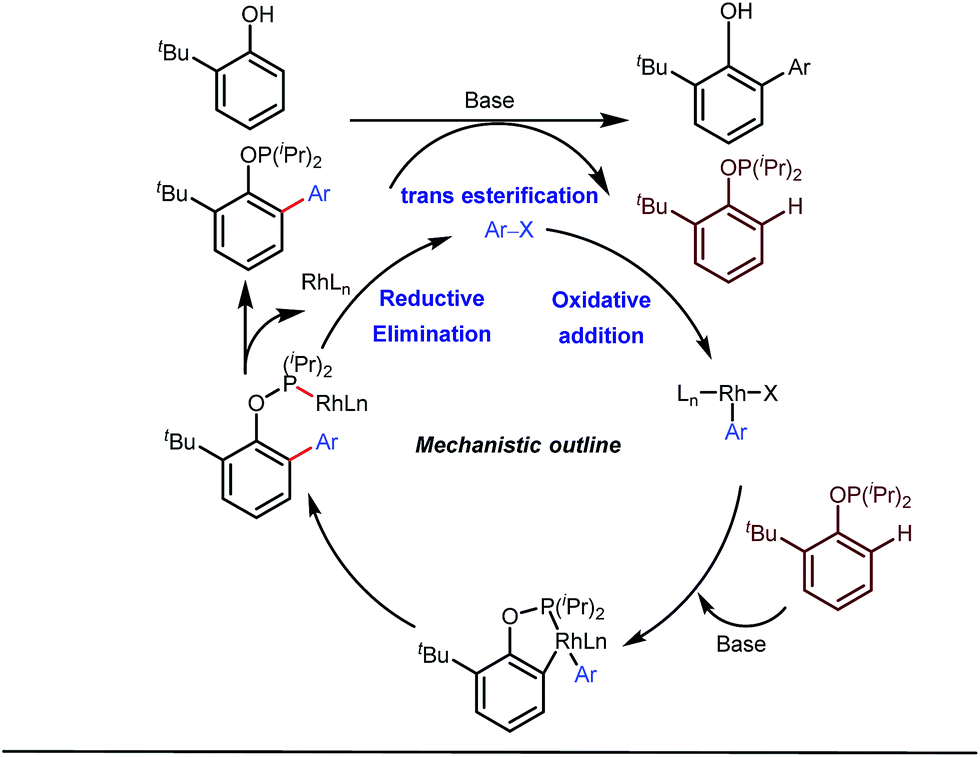
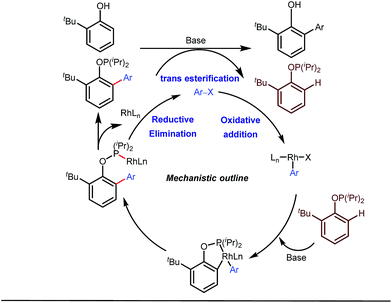
Apart from imine based transient directing groups, Bedford reported Rh(I) catalyzed ortho-arylation of phenols via a phosphinite based transient directing group (Fig. 6).36 Previous methods for the preparation of ortho-arylated phenols were limited to Suzuki or Stille coupling reactions which demand stoichiometric quantities of coupling partners like boronic acid derivatives or organotin compounds. On the other hand, the incorporation of aryloxide groups into phosphite or phosphinite systems assured the formation of thermodynamically and kinetically stable low-strain five membered metallacycles for the ortho-C–H activation of phenols. The authors envisaged two intertwined catalytic pathways involving directed ortho-C–H arylation of phenol assimilated with a PR2(OAr) organocatalyst, followed by transesterification of the phosphinite ligand, to release substituted phenol (Fig. 6). Bedford’s elegant work was inspired by Lewis’s work in 1985 where they elegantly demonstrated ruthenium catalyzed ortho-ethylation and deuteration of phenols involving in situ reversible transesterification of triarylphosphite ligands with phenol. In 2008, Breit et al. used the same phosphinites as temporary directing groups to execute hydroformylation of homoallylic alcohols 17 to furnish γ-lactones and γ-lactols with highly branched regioisomers .37
 |
| | Fig. 6 Rh-catalyzed ortho-arylation of phenols using a phosphite tDG. | |
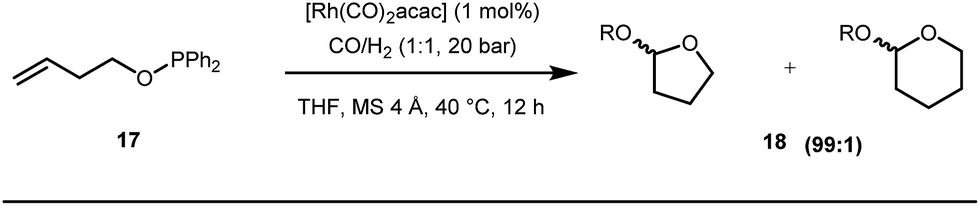
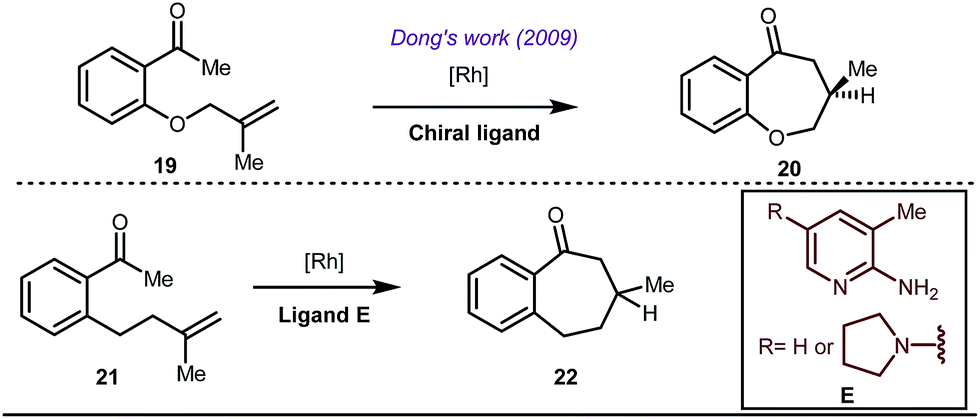
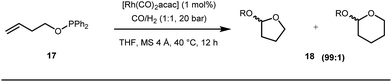
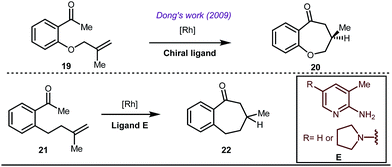
Early methodologies were selective for the linear hydroformylation of alkenes whereas the use of phosphine or phosphite ligands revised the regiochemistry in favour of branched isomers (Fig. 7). Indeed, both E and Z alkenes provided the same selective branched lactols 18 under the optimized reaction conditions . Interestingly, Tan and co-workers also achieved selective branched hydroformylation of homoallylic alcohols through an amino phosphine based ligand.38 In 2012, Douglas excellently exploited a similar approach for the design of seven membered cyclic benzoketones via intramolecular hydroacylation.39 The report was influenced by Dong’s work in 2009, where the presence of a heteroatom in the ketone ring was essential (Fig. 8).40
 |
| | Fig. 7 Rh-catalyzed selective branched lactone synthesis by Breit and co-workers. | |
 |
| | Fig. 8 Rh-catalyzed intramolecular hydroacylation leading to seven membered cyclic ketones. | |
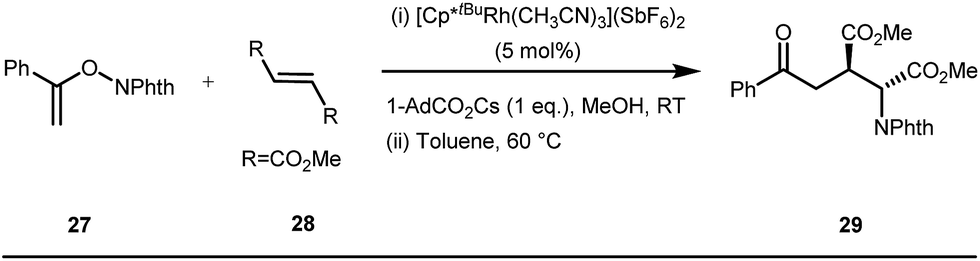
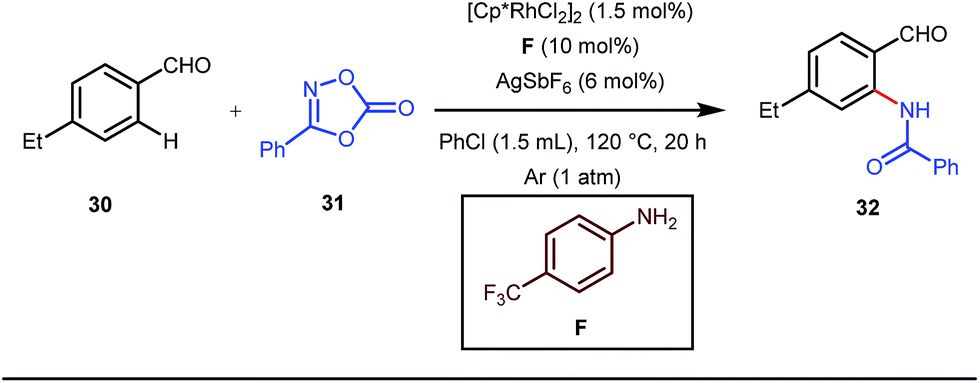
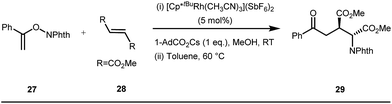
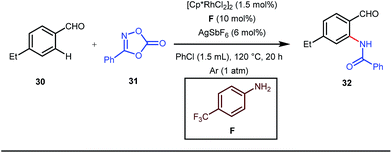
The use of a catalytic amount of ligand E accelerated the rate of hydroacylation by reducing decarbonylation of the ketones. The same Rh(I) catalyst was then implemented by Dong and co-workers to furnish α-alkylated ketones, where 2-aminopyridine acted as the key component for the intermediate imine formation. A catalytic quantity of 5-methyl-2-benzoic acid played a crucial role in improving enamine formation.41 In addition, α-alkenylation of cyclic ketones 23 was achieved with internal alkynes 24 using the same 7-azaindoline D as for the transient modifier. The methodology enabled the production of both the α,β- as well as the β,γ-enones in moderate to excellent yields (Fig. 9).42 Thereafter, in 2015, Rovis and Piou successfully implemented the syn-carboamination of doubly substituted alkenes in the presence of a Rh(III) catalyst with enoxypthalimides as the amination source (Fig. 10).43 Very recently, Jiao and co-workers demonstrated an imine based transient DG assisted amidation of benzaldehyde derivatives (Fig. 11).44 They used 3-phenyl-1,4,2-dioxazol-5-one as the potential aminating source and 4-trifluoromethyl aniline F proved its efficacy as a catalytic modifier.
 |
| | Fig. 9 Rh-catalyzed α-alkenylation of cyclic ketones using 7-azaindoline as a transient modifier. | |
 |
| | Fig. 10 Rh-catalyzed syn-carboamination using enoxypthalimides as the amine source. | |
 |
| | Fig. 11 Rh-catalyzed ortho-amination of benzaldehydes. | |
2.2 Transient DG assisted C–H activation with ruthenium
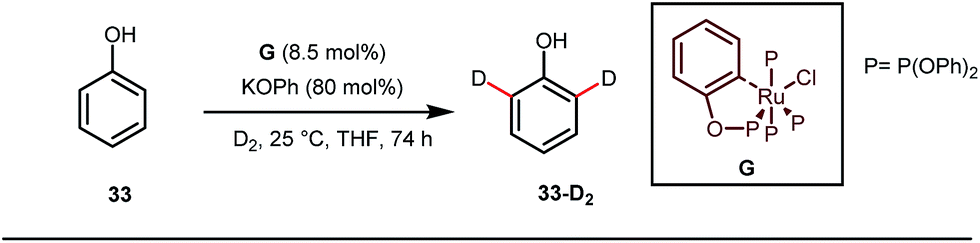
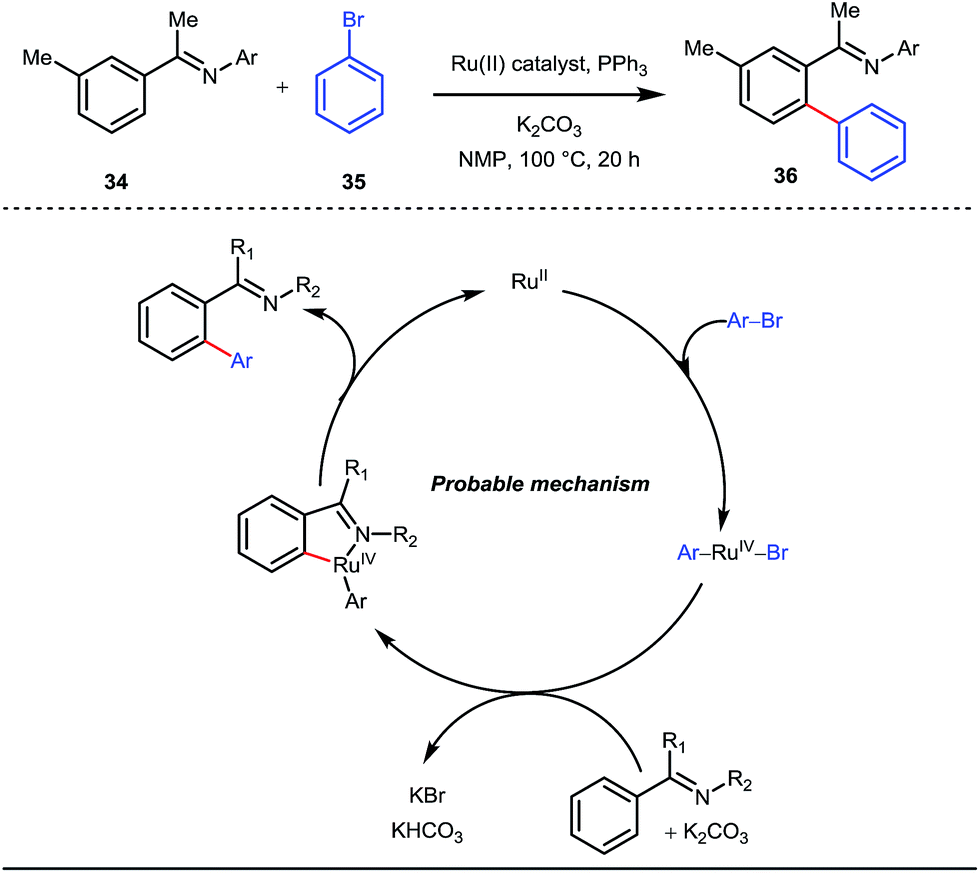
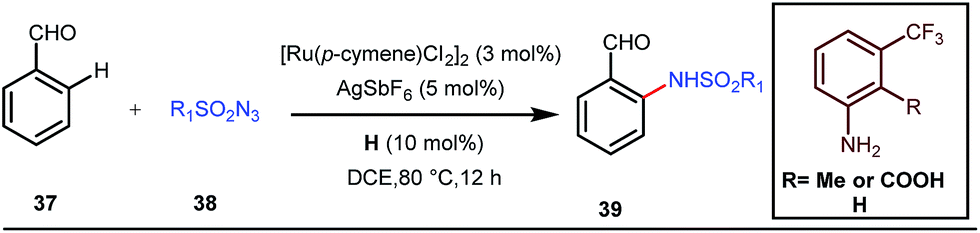
A notable advancement in this domain was achieved by Lewis and co-workers. They reported Ru-catalysed ortho-deuteration of phenol using a phosphite transient DG under a D2 atmosphere. The authors discovered that the presence of a catalytic amount of KOPh can speed up the transesterification process between phenol and triphenylphosphites via an active metallacycle G (Fig. 12).45 Ruthenium(II) catalyzed ortho-arylation of benzaldehydes between imines (masked benzaldehydes) 34 and aryl bromides 35 was also reported by Inoue et al. (Fig. 13).46 Recently, Rasheed and Zhang synthesized ortho-(sulfonyl-amino)benzaldehydes in the presence of 3 mol% [Ru(p-cymene)Cl2]2 as the catalyst using organic azides as the source of N-functionalities in conjunction with a catalytic quantity of aniline H (Fig. 14).47
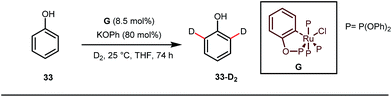 |
| | Fig. 12
Ortho-deuteration of phenol using a phosphite tDG. | |
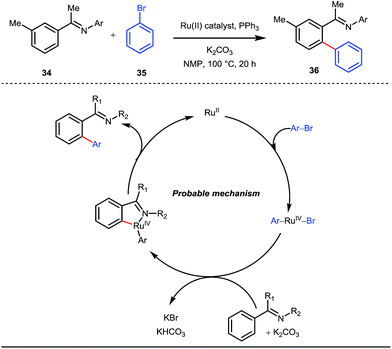 |
| | Fig. 13 Mechanistic cycle of Ru-catalyzed ortho-arylation of benzaldehydes using aryl bromides as coupling partners. | |
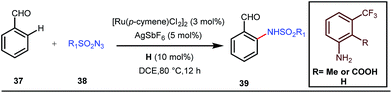 |
| | Fig. 14 Ru-catalyzed ortho-amination of benzaldehydes using tosyl azide. | |
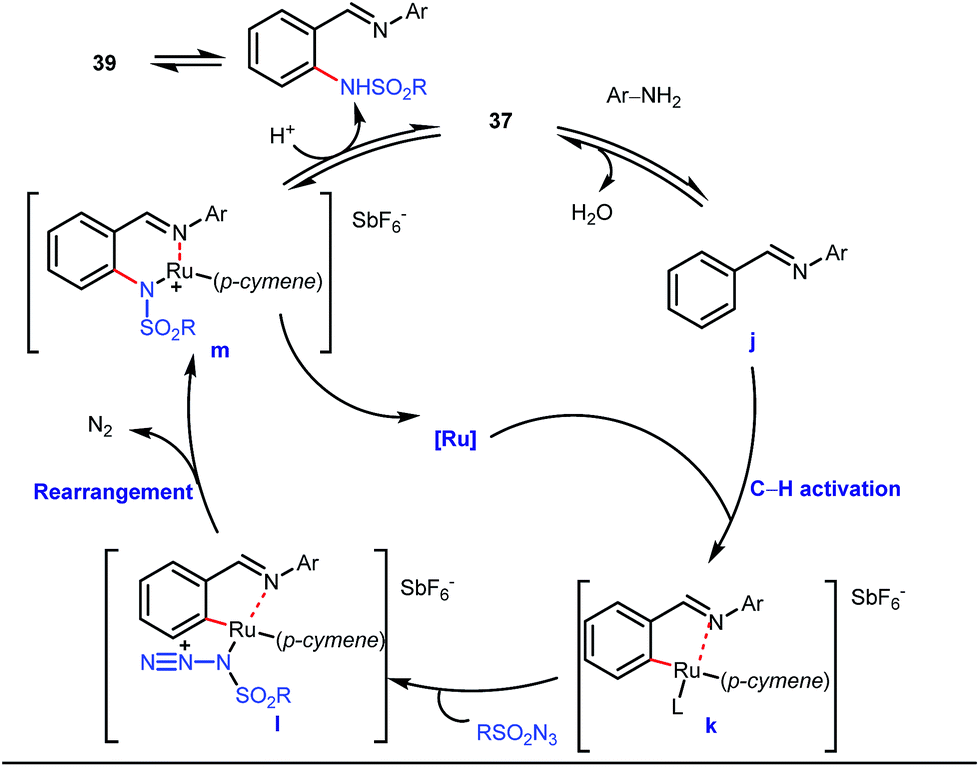
5 mol% AgSbF6 was found to be the best chloride ion scavenger in this case. With the optimized reaction conditions in hand, the authors explored the generality of this reaction. Both electron donating groups and electron withdrawing groups were well-tolerated. The catalytic cycle for this reaction included the generation of a cationic ruthenium species after ion exchange with AgSbF6 (Fig. 15). The active catalyst [Ru(p-cymene)](SbF6)2 was thus generated. Eventually, the ruthenium catalyst was bound to the in situ formed imine j which then underwent C–H activation to form complex k. Furthermore, the N center of tosyl azide was ligated to the metal center in an oxidatively additive manner to produce l. The exclusion of nitrogen and the concomitant rearrangement led to the complex m which upon treatment with acid afforded the ortho-aminated product 39.
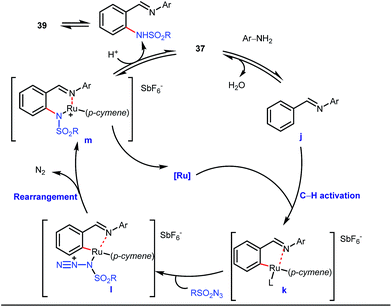 |
| | Fig. 15 Ru-catalyzed ortho-amination of benzaldehydes. | |
2.3 Transient DG assisted C–H activation with palladium
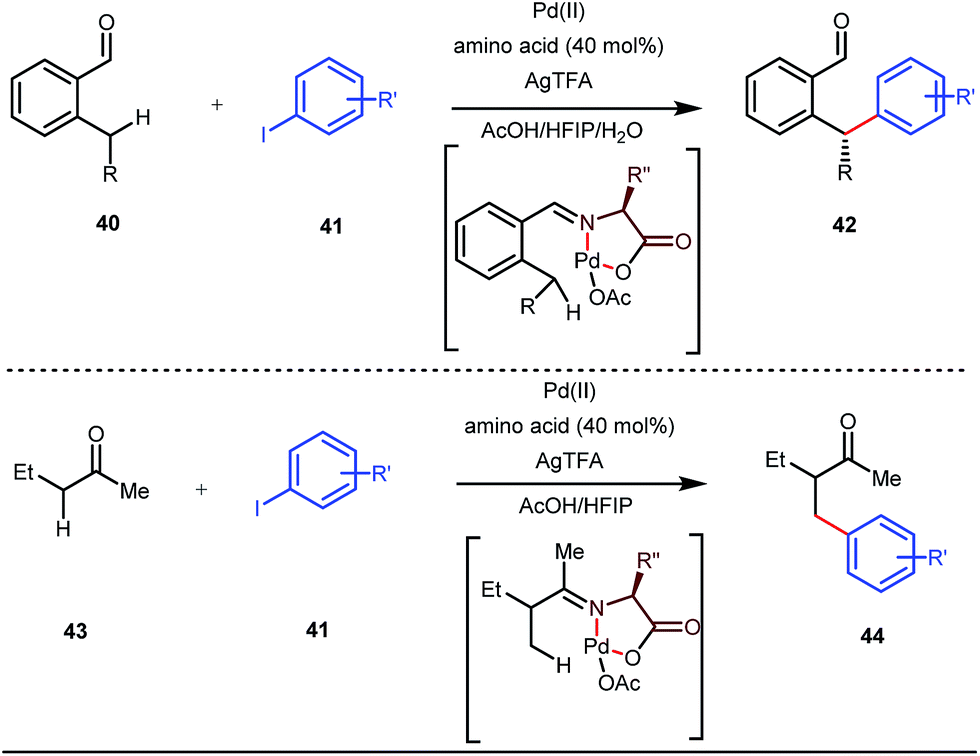
Although Rh-catalyzed transient directing group assisted C–H functionalizations have been well-explored, no such reports with palladium metal catalysts were found in the literature until the work of Yu and co-workers. They disclosed the arylation of C(sp3)–H bonds of ortho-alkyl benzaldehydes in 2016 (Fig. 16).48 However, the traditionally-used iminopyridine or iminooxazoline directing groups failed to show adequate directing abilities in this case due to their strong coordination with the Pd metal center, thus disfavouring the targeted C–H bond cleavage. However, a catalytic amount of amino acids solved the problem in the presence of a mixture of protic solvents. A proportionate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and acetic acid (AcOH) improved the product formation in the case of the aryl system while a combination of AcOH and water in a ratio of 9:1 enhanced the scope for β-arylation of aliphatic ketones.
:1) mixture of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and acetic acid (AcOH) improved the product formation in the case of the aryl system while a combination of AcOH and water in a ratio of 9:1 enhanced the scope for β-arylation of aliphatic ketones.
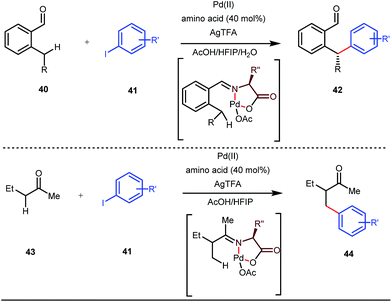 |
| | Fig. 16 Pd-catalyzed β-arylation of aromatic and aliphatic ketones. | |
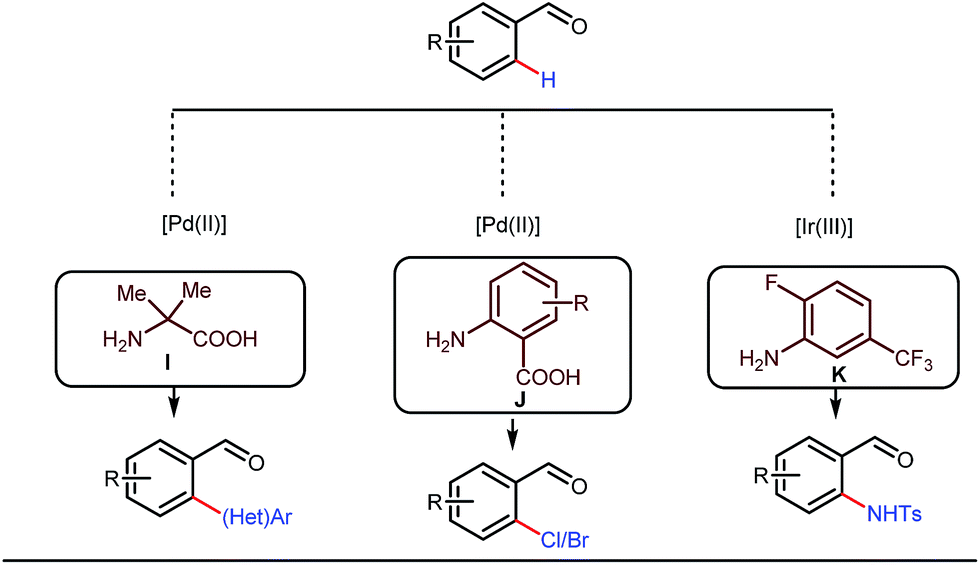
This functionalization was further extended for the synthesis of fluorenones by the Sorensen group using low-cost anthranilic acid as a transient DG.49 In addition to benzaldehydes, ortho-arylation of aromatic ketones via a glycine transient DG was also reported by Jin et al. in 2017.50 More recently, Yu et al. revealed a series of Pd-catalyzed functionalizations at the ortho-position of the benzaldehydes using a bidentate amino acid based transient DG, followed by an Ir(III) catalyzed C–H amidation with monodentate aniline K as an effective transient directing group (Fig. 17).51 A library of functionalized compounds was synthesized with a wide substrate scope and remarkable applications. In a similar way, Hu,52 Yu,53 Bull,54 and Li and Ge55 also achieved β-arylation of unactivated C(sp3)–H bonds where acetohydrazide and β-alanine turned out to be some of the most efficient active transient modifiers (Fig. 18i).
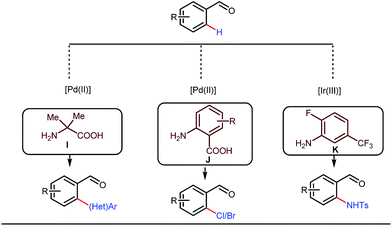 |
| | Fig. 17 Pd and Ir-catalyzed ortho-functionalizations of benzaldehydes using mono- and bidentate transient ligands. | |
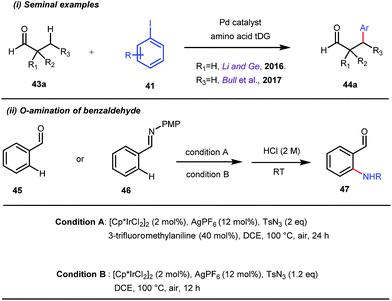 |
| | Fig. 18 (i) Seminal examples of Pd-catalyzed β-arylations of aliphatic aldehydes; (ii) ortho-amination of benzaldehydes using tosyl azide in the presence of a Pd catalyst. | |
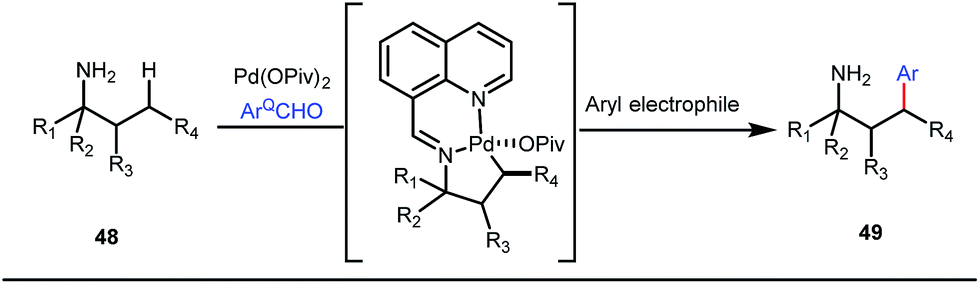
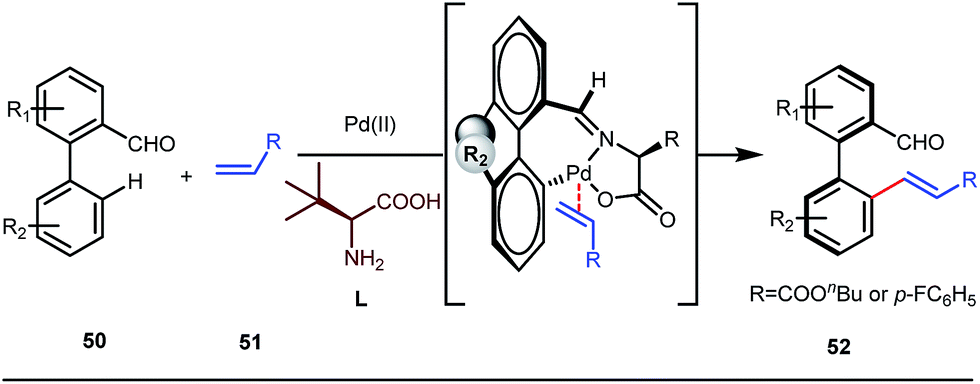
It is worth mentioning that these studies involved arylation of both methyl as well as methylene C(sp3)–H bonds of the aliphatic carbonyl system. Even though transient DG assisted C–H activation is widespread using Rh and Pd metal catalysts, in 2016, Shi et al. first described a transient auxiliary mediated C–H amidation of aryl aldehydes using tosyl azide as the aminating source.56 Initially they started with simple aldehyde 45 as their model substrate which failed to give the desired aminated product. Then, they replaced the aldehyde with imine 46 which successfully led to the product in 91% yield. Realizing the importance of the iminyl group, the authors explored the use of aldehydes directly as the substrate, with 3-trifluoromethylaniline in a catalytic amount to carry out the reaction via intermediate imine formation which furnished ortho-aminated aryl aldehydes as expected, in excellent yields (Fig. 18ii). Until 2016, all in situ generated imine based transient templates were utilized to functionalize different aromatic and aliphatic aldehydes or ketone substrates where a catalytic quantity of amine derivatives was always added to the reaction mixture to form the intermediate imine DG. In 2016, Dong and co-workers first thought to switch ligand i.e. they chose free amines as their substrates and added quinoline-8-carbaldehyde in trace amounts to obtain the imine DG.57 The exo DG enabled efficacious arylation at the γ-position of the free amines (Fig. 19). Later, Ge et al. also discovered γ-C–H arylation of free amines with different aryl iodides as aryl coupling partners.58 The use of glyoxylic acid as the temporary directing group furnished diverse arylated free amines. A similar strategy was implemented thereafter by the Yu group in 2016 with Boc protected amines to accomplish γ-C(sp3)–H arylation of free primary amines using 2-hydroxynicotinaldehyde as the transient DG.59 In 2017, Shi proposed an atropselective synthesis of biaryls with axial chirality via palladium catalyzed olefination using a chiral transient DG.60 This strategy involved commercially obtainable tert-leucine as the catalytic chiral auxiliary which enabled the formation of chiral biaryls with outstanding enantioselectivity (95–99% ee) (Fig. 20).
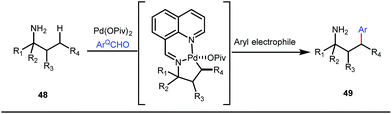 |
| | Fig. 19 Pd-catalyzed γ-arylation of free amines. | |
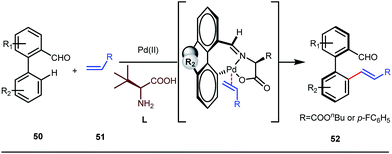 |
| | Fig. 20 Pd-catalyzed olefination of biaryls using a chiral amino acid transient DG. | |
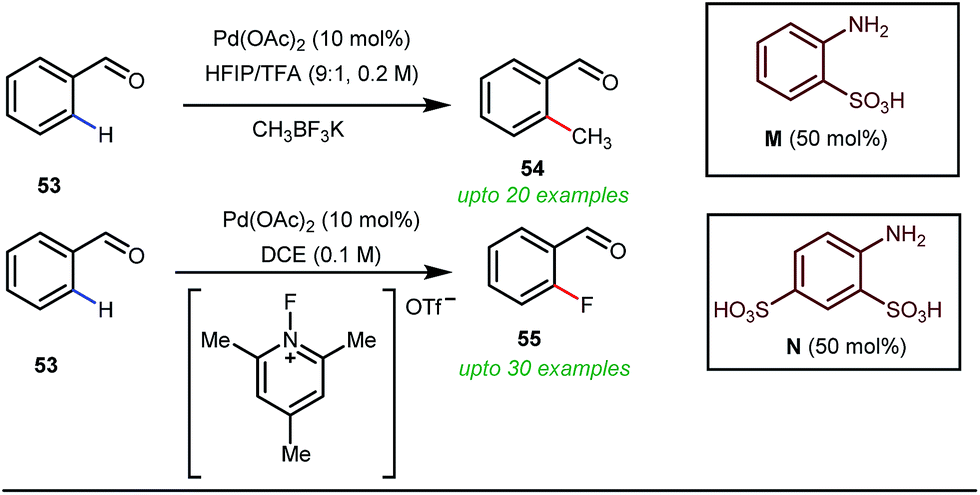
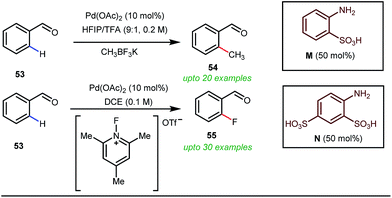
Notably, in 2018, Sorensen and co-workers developed reaction conditions for Pd-catalyzed ortho-methylation and fluorination of benzaldehydes 53 utilizing potassium methyltrifluoroborate as the methyl source or 1-fluoro-2,4,6-trimethylpyridinium triflate as the effective fluorinating agent in the presence of orthanilic acid M (45 mol%) or aniline-2,4-disulfonic acid N (50 mol%) respectively (Fig. 21).61
 |
| | Fig. 21 Pd-catalyzed ortho-methylation and fluorination of benzaldehydes. | |
For the first time, orthanilic acid has been used as a potential transient DG for C–H functionalization. Interestingly, employment of one extra sulfonic acid group in N facilitated the C–F reductive elimination step by increasing the electrophilicity of the Pd(IV) center.
This increased electrophilicity then attenuated the polarity difference of the Pd(IV)–F bond to execute a facile reductive elimination. Notably, the reaction has covered a broad substrate scope irrespective of electron rich and electron poor systems. For further mechanistic interest, the authors were able to trap the C–H activated metallacycle in crystal form.
3 Conclusions
In conclusion, transition metal catalyzed functionalization of C–H bonds heralds the commencement of a new horizon for the synthesis of potential complex molecular scaffolds. However, transient directing group mediated C–H activation is one of the recent advances in this field which can equally assure site-selective functional transformation of C–H bonds whilst overthrowing the technical drawbacks of directed C–H activation. This strategy has already been recognized as a significant tool for different functional alterations of C–H bonds which include arylation, amination, halogenation, alkylation, and many more. Despite its hassle-free synthetic pathway, the protocol is solely restricted to different aromatic and aliphatic aldehydes, ketones, amines and phenol derivatives. This strategy is not only functional group specific but it is also restricted to proximal C–H bonds of the respective functional groups. Systematic design of transient DGs can further extend the scope of distal C–H activation. On top of that, the strategy mainly relies on 4d transition metals like rhodium, palladium, ruthenium and iridium. Therefore experiments with 3d transition metals are highly anticipated due to their abundance as well as their eco-friendly nature. Although the approach is yet not generalized for all possible functional transformations, further remodelling will definitely shape the field of C–H activation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research is supported by (BRNS-37(2)/14/06/2018) India. Financial support was received from UGC-India (TB) and IITB-Monash Research Academy (SP).
References
- S. M. A. Shakoor, S. Kumari, S. Khullar, S. K. Mandal, A. Kumar and R. Sakhuja, J. Org. Chem., 2016, 81, 12340–12349 CrossRef CAS.
- B. Gopalakrishnan, S. Mohan, R. Parella and S. A. Babu, J. Org. Chem., 2016, 81, 8988–9005 CrossRef CAS.
- R. Manikandan and M. Jeganmohan, Chem. Commun., 2017, 53, 8931–8947 RSC.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS.
- O. Daugulis, H. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS.
- A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602–13605 CrossRef CAS.
- S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS.
- J. Yang, Org. Biomol. Chem., 2015, 13, 1930–1941 RSC.
- A. Modak, T. Patra, R. Chowdhury, S. Raul and D. Maiti, Organometallics, 2017, 36, 2418–2423 CrossRef CAS.
- S. Bag, R. Jayarajan, U. Dutta, R. Chowdhury, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 12538–12542 CrossRef CAS.
- U. Dutta, A. Modak, B. Bhaskararao, M. Bera, S. Bag, A. Mondal, D. W. Lupton, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 3162–3168 CrossRef CAS.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef CAS.
- A. Dey, S. Maity and D. Maiti, Chem. Commun., 2016, 52, 12398–12414 RSC.
- T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS.
- A. Maji, S. Guin, S. Feng, A. Dahiya, V. K. Singh, P. Liu and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 14903–14907 CrossRef CAS.
- S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, ACS Catal., 2018, 2664–2669 CrossRef CAS.
- Y. Mu, X. Tan, Y. Zhang, X. Jing and Z. Shi, Org. Chem. Front., 2016, 3, 380–384 RSC.
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef CAS.
- H. Y. Thu, W. Y. Yu and C. M. Che, J. Am. Chem. Soc., 2006, 128, 9048–9049 CrossRef CAS.
- A. Deb, S. Singh, K. Seth, S. Pimparkar, B. Bhaskararao, S. Guin, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 8171–8175 CrossRef CAS.
- X. C. Wang, W. Gong, L. Z. Fang, R. Y. Zhu, S. Li, K. M. Engle and J. Q. Yu, Nature, 2015, 519, 334–338 CrossRef CAS.
- P. X. Shen, X. C. Wang, P. Wang, R. Y. Zhu and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574–11577 CrossRef CAS.
- N. Della Ca, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef CAS.
- J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS.
- Q. Lu, S. Greßies, S. Cembellín, F. J. R. Klauck, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12778–12782 CrossRef CAS.
- Q. Zhao, T. Poisson, X. Pannecoucke and T. Besset, Synthesis, 2017, 49, 4808–4826 CrossRef CAS.
- P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CAS.
- J. William Suggs, J. Am. Chem. Soc., 1979, 101, 489 CrossRef.
- C. H. Jun, H. Lee and J. B. Hong, J. Org. Chem., 1997, 62, 1200–1201 CrossRef CAS.
- C. H. Jun, D. Y. Lee, H. Lee and J. B. Hong, Angew. Chem., Int. Ed., 2000, 39, 3070–3072 CrossRef CAS.
- C. H. Jun, C. W. Moon, J. B. Hong, S. G. Lim, K. Y. Chung and Y. H. Kim, Chem.–Eur. J., 2002, 8, 485–492 CrossRef CAS.
- S. G. Lim, J. A. Ahn and C. H. Jun, Org. Lett., 2004, 6, 4687–4690 CrossRef CAS.
- N. R. Vautravers, D. D. Regent and B. Breit, Chem. Commun., 2011, 47, 6635 RSC.
- Z. Wang, B. J. Reinus and G. Dong, J. Am. Chem. Soc., 2012, 134, 13954–13957 CrossRef CAS.
- F. Mo and G. Dong, Science, 2014, 345, 68–72 CrossRef CAS.
- R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112–114 CrossRef CAS.
- C. U. Grünanger and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 7346–7349 CrossRef.
- T. E. Lightburn, M. T. Dombrowski and K. L. Tan, J. Am. Chem. Soc., 2008, 130, 9210–9211 CrossRef CAS.
- E. V. Beletskiy, C. Sudheer and C. J. Douglas, J. Org. Chem., 2012, 77, 5884–5893 CrossRef CAS.
- Z. Shen, H. A. Khan and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2916–2917 CrossRef CAS.
- H. N. Lim and G. Dong, Angew. Chem., Int. Ed., 2015, 54, 15294–15298 CrossRef CAS.
- F. Mo, H. N. Lim and G. Dong, J. Am. Chem. Soc., 2015, 137, 15518–15527 CrossRef CAS PubMed.
- T. Piou and T. Rovis, Nature, 2015, 527, 86–90 CrossRef CAS.
- X. Wang, S. Song and N. Jiao, Chin. J. Chem., 2018, 36, 213–216 CrossRef CAS.
- L. N. Lewis, Inorg. Chem., 1985, 24, 4433–4435 CrossRef CAS.
- S. Oi, Y. Ogino, S. Fukita and Y. Inoue, Org. Lett., 2002, 4, 1783–1785 CrossRef CAS.
- O. K. Rasheed, Synlett, 2018, 29, 1033–1036 CrossRef CAS.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J. Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS.
- X. Y. Chen, S. Ozturk and E. J. Sorensen, Org. Lett., 2017, 19, 1140–1143 CrossRef CAS.
- J. Xu, Y. Liu, Y. Wang, Y. Li, X. Xu and Z. Jin, Org. Lett., 2017, 19, 1562–1565 CrossRef CAS PubMed.
- X. H. Liu, H. Park, J. H. Hu, Y. Hu, Q. L. Zhang, B. L. Wang, B. Sun, K. S. Yeung, F. L. Zhang and J. Q. Yu, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS.
- F. Ma, M. Lei and L. Hu, Org. Lett., 2016, 18, 2708–2711 CrossRef CAS PubMed.
- K. Hong, H. Park and J. Q. Yu, ACS Catal., 2017, 7, 6938–6941 CrossRef CAS PubMed.
- S. St John-Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840–4847 RSC.
- K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef CAS.
- Y. F. Zhang, B. Wu and Z. J. Shi, Chem.–Eur. J., 2016, 22, 17808–17812 CrossRef CAS.
- Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084–9087 CrossRef CAS.
- Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26–32 CrossRef CAS.
- Y. Wu, Y. Q. Chen, T. Liu, M. D. Eastgate and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS.
- Q. J. Yao, S. Zhang, B. B. Zhan and B. F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 CrossRef CAS.
- X.-Y. Chen and E. J. Sorensen, J. Am. Chem. Soc., 2018, 140, 2789–2792 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab