
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced C(sp3)–H activation for a C–C bond forming reaction of 3,4-dihydroquinoxalin-2(1H)-one with nucleophiles using oxygen with a photoredox catalyst or under catalyst-free conditions†
Pavan Sudheer Akulaa,
Bor-Cherng Hong *a and
Gene-Hsiang Leeb
*a and
Gene-Hsiang Leeb
aDepartment of Chemistry and Biochemistry, National Chung Cheng University, Chia-Yi 621, Taiwan, Republic of China. E-mail: chebch@ccu.edu.tw; Fax: +886 5 2721040; Tel: +886 5 2729174
bInstrumentation Center, National Taiwan University, Taipei, 106, Taiwan
First published on 29th May 2018
Abstract
A convenient photocatalyzed oxidative coupling reaction of 4-alkyl-3,4-dihydroquinoxalin-2(1H)-one and its derivatives with a variety of nucleophiles was developed with a ruthenium photoredox catalyst and oxygen under a household compact fluorescent light. With a slower reaction rate, the cross coupling transformation can be achieved in the absence of an external photocatalyst with a similar isolated yield. An application to the synthesis of natural product cephalandole A was also demonstrated.
Introduction
Carbon–carbon bond-forming reactions constitute one of the foundations in organic synthesis and have attracted extensive exploration since the dawn of organic chemistry.1 For instance, the direct C–H activation of aryl compounds with transition metals has recently received much attention across the chemistry fields.2 Moreover, the emerging strategy for the visible-light photoredox-catalyzed C–C bond forming reactions further enrich the realm of this transformation,3 such as the visible-light photoredox catalyzed indole alkylation4 and the oxidative dehydrogenation of N-hetereocycles.5On the other hand, quinoxalin-2(1H)-one and benzo[b][1,4]oxazin-2-one systems constitute prevailing scaffolds frequently found in many biologically active naturally occurring and unnatural molecules. Consequently, many of them serve as key building blocks in pharmaceutical syntheses, and the derivatives display a wide spectrum of biological activities (Fig. 1).6 Owing to these merits, the synthesis of these frameworks has drawn considerable investigations. For example, Huo and his co-workers reported an FeCl2-catalyzed oxidative C–C bond formation of 3,4-dihydro-1,4-benzoxazin-2-ones with malonic ester, ketone or indoles (Scheme 1).7 Xiao and co-workers developed visible light photocatalytic cross-coupling reactions of amines and carbonyls for the alkylation of 3,4-dihydroquinoxalin-2 (1H)-ones.8 Zhang et al. revealed a direct C–H arylation of quinoxalin-2(1H)-ones with Ph2IBF4.9 Alternatively, a copper-catalyzed direct coupling of benzoxazin-2-ones with indoles was reported by Lee and coworkers.10 Nevertheless, despite these advances, an efficient, green, and convenient protocol for the synthesis of these systems remains a topic of high interest in chemical research.11
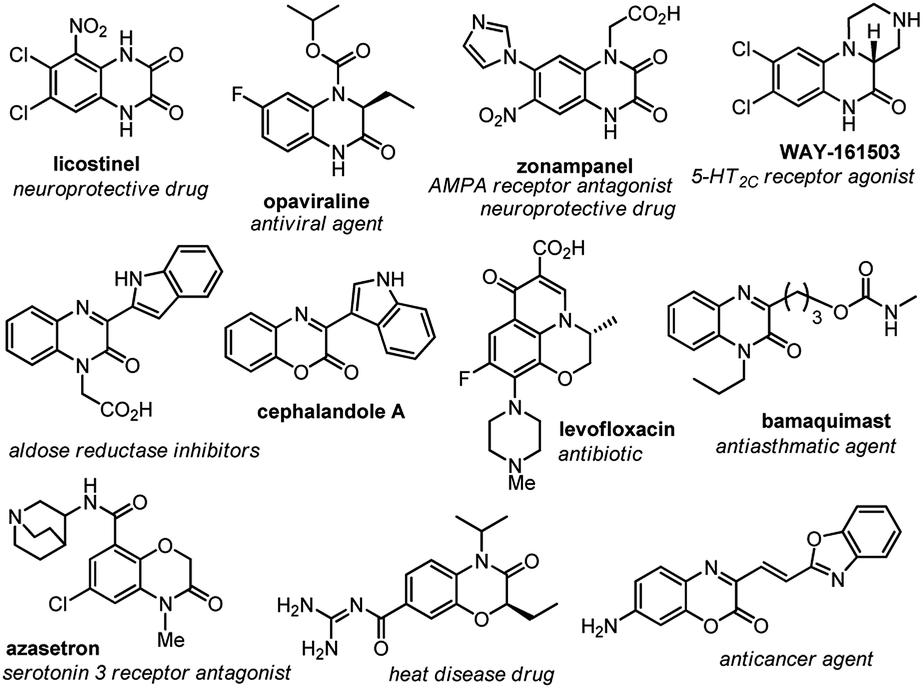 | ||
| Fig. 1 Selected examples of naturally occurring and pharmaceutically active quinoxalin-2(1H)-one or benzo[b][1,4]oxazin-2-ones. | ||
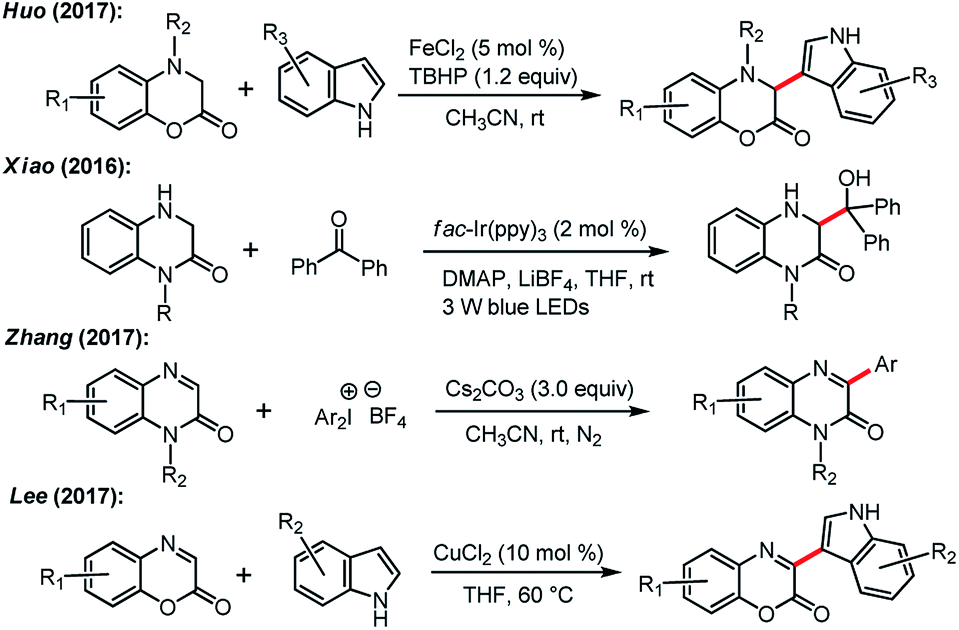 | ||
| Scheme 1 Direct synthesis of quinoxalin-2(1H)-one or benzo[b][1,4]oxazin-2-one derivatives via oxidative coupling. | ||
Informed by this background and as a continuation of our efforts in exploring the visible-light photoredox C–C bond-forming reactions,12 we envisaged that a direct C(sp3)–C(sp2) or C(sp3)–C(sp3) oxidative coupling of the quinoxalin-2(1H)-one (or benzo[b][1,4]oxazin-2-ones) and alkyl groups could be achieved for the efficient synthesis of these scaffolds. We herein present the outcome of this exploration.
Results and discussion
Initially, a solution of 1c, indole (2a, 2 equiv.), and Ru(bpy)3Cl2·6H2O (2 mol%) in CH3CN (0.05 M) under a balloon of oxygen was irradiated with a household 24 W compact fluorescent plug-in light (CFL) bulb with a distance of around 10 cm at room temperature for 4 h and gave 54% yield of the expected adduct 3ca after chromatographic purification (Table 1, entry 1). Increasing the reaction concentration to 0.1 M, or reducing the indole 2a to 1 equivalent under the same reaction conditions gave lower yields (40% and 15% yields, respectively, Table 1, entries 2–3).
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Conditionb | Solvent | Time (h) | Yieldc (%) |
| a Unless otherwise noted, the reactions were performed on a 0.05 M concentration of 1c (1.0 equiv.), 2a (2.0 equiv.) with 2 mol% of catalyst in solvent under O2 atmosphere at ambient temperature (∼25–30 °C).b Light source: a 24 W household CFL light bulb (cool daylight).c Isolated yields of 3ca.d 1c (1.0 equiv.), 2a (2.0 equiv.), and the reaction was performed on a 0.1 M concentration.e 1c (1.0 equiv.), 2a (1.0 equiv.), and the reaction was performed on a 0.1 M concentration.f Trace amount of 3ca with complicated mixtures.g Under a balloon of air.h In the presence of 24 blue LEDs.i Using Ir(ppy)3 as photocatalyst. | |||||
| 1 | 2 | O2, light | CH3CN | 4 | 54 |
| 2d | 2 | O2, light | CH3CN | 4 | 40 |
| 3e | 2 | O2, light | CH3CN | 4 | 15 |
| 4 | 2 | O2, light | MeOH | 4 | 76 |
| 5 | 2 | O2, light | CH2Cl2 | 4 | 63 |
| 6 | 2 | O2, light | CHCl3 | 4 | 51 |
| 7 | 2 | O2, light | Acetone | 4 | 43 |
| 8 | 2 | O2, light | DMF | 4 | ∼0f |
| 9g | 2 | Air, light | MeOH | 7 | 71 |
| 10 | 2 | O2, dark | MeOH | 72 | Trace |
| 11 | — | O2, dark | MeOH | 72 | ∼0 |
| 12 | — | O2, light | MeOH | 14 | 69 |
| 13 | — | Air, light | MeOH | 20 | 63 |
| 14h | 2 | O2, light | MeOH | 4 | 74 |
| 15i | 2 | O2, light | MeOH | 4 | 75 |
A series of solvents were evaluated, and methanol was found to be the best choice of the solvent in the reaction to give 76% yield of 3ca, while CH2Cl2, CHCl3, acetone, and DMF afforded lesser or poor yields (entries 4–8). The reaction under air afforded similar yields, but proceeded slightly slower as the oxygen concentration decreased (Table 1, entry 9). Importantly, the reaction in the dark under oxygen atmosphere in the presence of a ruthenium catalyst or in the absence of the catalyst gave only trace amounts or no observation of the product 3ca, respectively, after 72 h (Table 1, entries 10–11). Apparently, visible light plays an essential role in the reaction. Notably, the reaction with the CFL irradiation in the absence of catalyst under the oxygen or air atmosphere still provided the adduct 3ca in yields of 69% and 63%, respectively, albeit these conditions required significantly longer reaction time, compared to the catalyst conditions (Table 1, entries 12 and 13 vs. entries 4 and 9). Briefly, entry 4 in Table 1 was identified as the best condition with respect to both the yields and the reaction time.
With the best condition in hand (Table 1, entry 4), a series of 1 and nucleophile 2 were reacted, and the most promising outcomes are summarized in Scheme 2. The reactions of 1c and indole (2a–2p) with Ru(bpy)3Cl2·6H2O (2 mol%) in MeOH under a balloon of oxygen were irradiated with a household 24 W compact fluorescent (CFL) plug-in light bulb at room temperature and provided the corresponding adduct 3ca–3cp in good yields, ranging from 60% to 82%. The reactions with the indoles (2b, 2c, 2h, and 2i), which bore electron-withdrawing substituents (e.g., F and Cl), gave lesser yields of the corresponding 3cb, 3cc, 3ch, and 3ci, probably due to the lesser nucleophilicity of these indoles. The photoredox reactions with 2-alkyl indoles (2l, 2m, and 2n) provided high yields of adducts (3cl, 3cm, and 3cn). Reactions with the protected indoles (2o and 2p) did not increase the yields (3co and 3cp). The reaction with other aromatic compounds, such as 7-azaindole (2q), naphthalen-2-ol (2r), 1H-benzo[g]indole (2s), pyrrole (2t) and phenol (2u) were feasible, although a low yield was obtained in the case of 7-azaindole (2q), and two regioisomeric products 3cu-p and 3cu-o were observed in the reaction with phenol (2u). The reactions of silyl enol ethers (2v and 2w) and 1c smoothly provided the corresponding adducts in good yields (83% and 79% yield, respectively), albeit with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stereoisomeric ratio of 3cv-1 and 3cv-2. Exposure of TMSCN 2x to 1c under the reaction conditions provided 74% yield of 3cx. On the other hand, the reactions of chloro- and fluoro-dihydroquinoxalin-2(1H)-one (1g and 1i) with indole 2a afforded the corresponding products (3ga and 3ia) in 72% and 64% yields, respectively. It is noteworthy that the reaction with propyl-substituted dihydroquinoxalinone (1j) with indole 2a gave a lesser yield of adduct 3ja (49% yield), as compared to the reactions with benzyl-substituted dihydroquinoxalinones (1c, 1d and 1e), where 3ca, 3da and 3ea were obtained in 76%, 71%, and 75% yields, respectively. Notably, the same reaction condition with benzoyl substituted dihydroquinoxalinones (1b) gave no observable product, with recovery of most of the starting compounds. Alternatively, the reaction of 3,4-dihydroquinoxalin-2(1H)-one (1a) and indole (2a) with Ru(bpy)3Cl2·6H2O (2 mol%) in MeOH was slow, 1a was consumed in 2 days, and converted to quinoxalin-2(1H)-one (8), monitored by crude 1H NMR, followed by the transformation to afford the product 4aa in 65% yield, after 5 days irradiation.13 In another batch of the reaction, where the irradiation was continued for only 2 days, and 8 was isolated in 71% yield. Apparently, the initial SET photo-oxidation and the subsequent oxidation afforded 8. The nucleophilic addition of indole towards 8, activated by acid (generated in the photo reaction condition), followed by oxidation gave the adduct 4aa.14 Later, the protocol was applied in the concise synthesis of cephalandole A (4ka).15 Irradiation of 1k and indole (2a) in the presence of Ru(bpy)3Cl2·6H2O (2 mol%) in CH3CN for 48 h provided adduct 3ka in 44% yield, along with 21% yield of the product 9.16 Subsequently, debenzylation of 3ka (H2, 10% Pd/C, THF, 16 h, rt), followed by the dilution with THF and the addition of DDQ with stirring under nitrogen atmosphere for 1 h gave 61% overall yield of cephalandole A (4ka) in a one-pot, two-step operation.
:1 stereoisomeric ratio of 3cv-1 and 3cv-2. Exposure of TMSCN 2x to 1c under the reaction conditions provided 74% yield of 3cx. On the other hand, the reactions of chloro- and fluoro-dihydroquinoxalin-2(1H)-one (1g and 1i) with indole 2a afforded the corresponding products (3ga and 3ia) in 72% and 64% yields, respectively. It is noteworthy that the reaction with propyl-substituted dihydroquinoxalinone (1j) with indole 2a gave a lesser yield of adduct 3ja (49% yield), as compared to the reactions with benzyl-substituted dihydroquinoxalinones (1c, 1d and 1e), where 3ca, 3da and 3ea were obtained in 76%, 71%, and 75% yields, respectively. Notably, the same reaction condition with benzoyl substituted dihydroquinoxalinones (1b) gave no observable product, with recovery of most of the starting compounds. Alternatively, the reaction of 3,4-dihydroquinoxalin-2(1H)-one (1a) and indole (2a) with Ru(bpy)3Cl2·6H2O (2 mol%) in MeOH was slow, 1a was consumed in 2 days, and converted to quinoxalin-2(1H)-one (8), monitored by crude 1H NMR, followed by the transformation to afford the product 4aa in 65% yield, after 5 days irradiation.13 In another batch of the reaction, where the irradiation was continued for only 2 days, and 8 was isolated in 71% yield. Apparently, the initial SET photo-oxidation and the subsequent oxidation afforded 8. The nucleophilic addition of indole towards 8, activated by acid (generated in the photo reaction condition), followed by oxidation gave the adduct 4aa.14 Later, the protocol was applied in the concise synthesis of cephalandole A (4ka).15 Irradiation of 1k and indole (2a) in the presence of Ru(bpy)3Cl2·6H2O (2 mol%) in CH3CN for 48 h provided adduct 3ka in 44% yield, along with 21% yield of the product 9.16 Subsequently, debenzylation of 3ka (H2, 10% Pd/C, THF, 16 h, rt), followed by the dilution with THF and the addition of DDQ with stirring under nitrogen atmosphere for 1 h gave 61% overall yield of cephalandole A (4ka) in a one-pot, two-step operation.
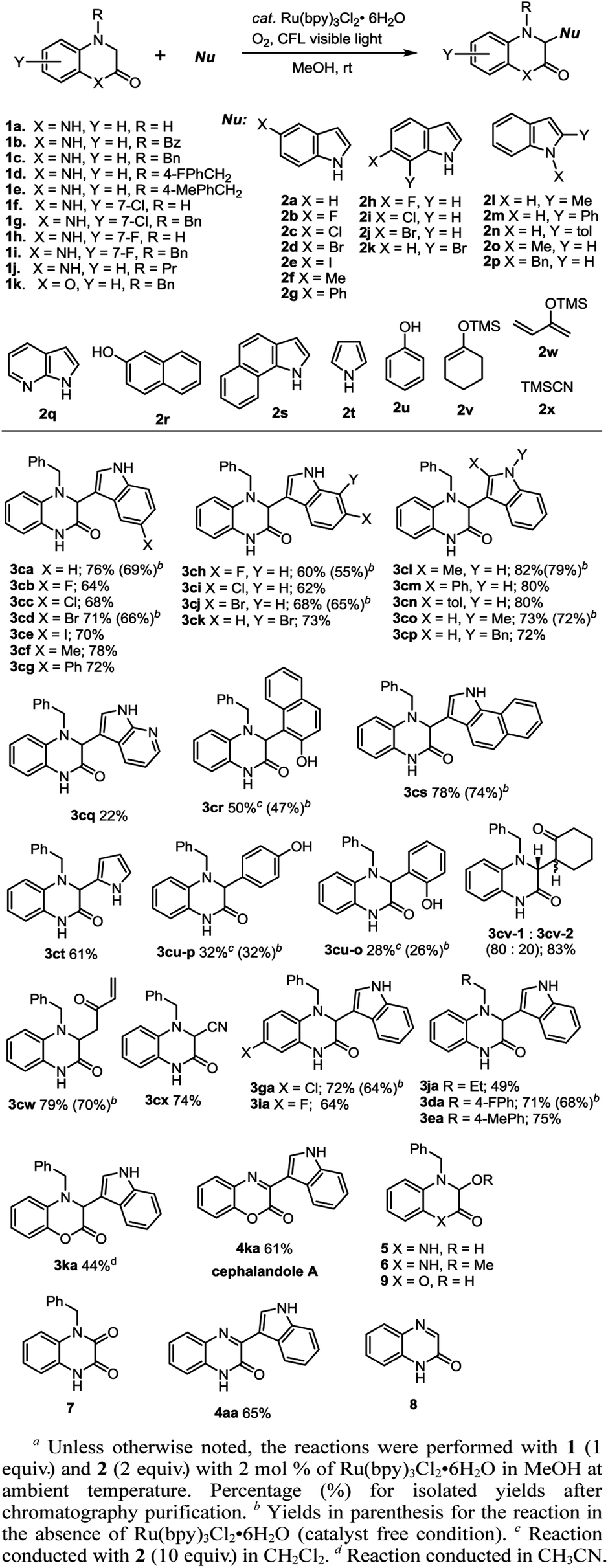 | ||
| Scheme 2 Examples of the photoredox cross-coupling reaction.a | ||
On the other hand, as previously observed in Table 1, some reactions were studied in the absence of photocatalyst (catalyst-free condition) with CFL light and successfully provided the corresponding adducts, albeit in slightly lower yields (as shown in the parenthesis in Scheme 2 and ESI, Scheme S1†), but required longer reaction time for the completion. The feasibility of this catalyst-free and visible-light photo reaction is of particular interest due to its green and cost-effective strategy.17 The UV-vis absorption spectrum of 1c in MeOH showed no obvious absorption at low concentrations in the visible region. However, upon increasing the concentration (to 0.05 M, the concentration used in the experiments), a small absorption band appeared in the visible region (380–480 nm), which implied that, due to aggregation, 1c could absorb visible light and undergo photo-activation.18,19 Consequently, the UV-vis absorption spectrum of the 1:2 mixture of 1c and indole (2a) in methanol, after 30 min, showed a significant broad absorption in the UV-vis around 380–520 nm. Indeed, the mixture was colorless in the dark for hours, but turned to pale yellow after 30 min CFL irradiation.19 The structure of adducts 3ca, 3cd, 3ga, 5, and 7 was unequivocally confirmed by their single-crystal X-ray crystallographic diffraction analyses (Fig. 2).19
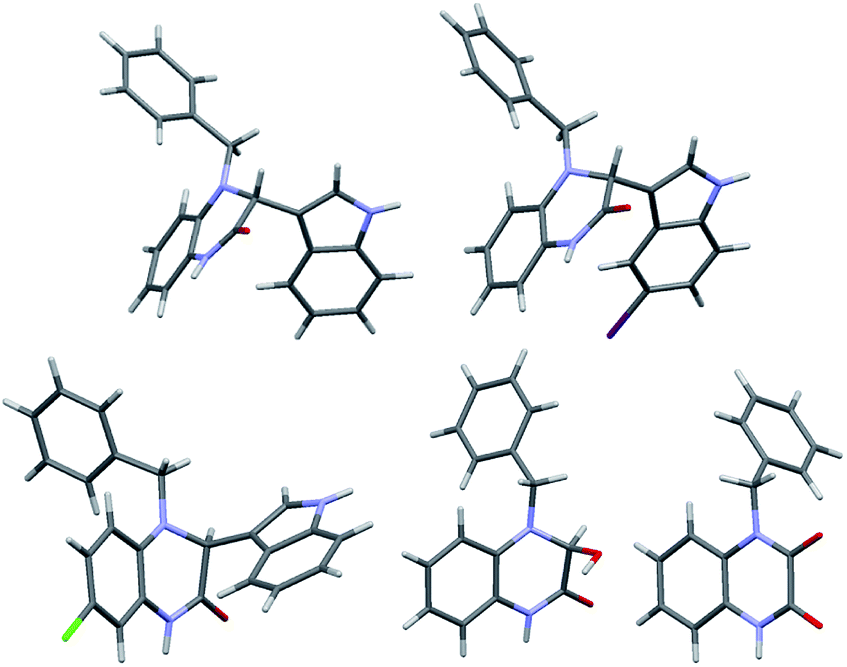 | ||
| Fig. 2 Stereo plots of the X-ray crystal structures of 3ca, 3cd, 3ga, 5 and 7: C, gray; O, red; N, blue; Br, purple; Cl, green. | ||
To shed some light on the reaction mechanism, the light/dark experiment was conducted by on–off switching of the light at different intervals for the reactions of 1c to 3ca under standard conditions (Fig. S2†).19 The reaction ceased during the periods in the dark and restarted when the light source was switched on once again. The observations excluded a light-initiated chain process and clearly revealed the essential role of visible-light photocatalysis through the reaction process.20 In addition, a radical trapping experiment was carried out to probe the radical nature of this reaction. A methanolic solution of 1c and indole with 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), an efficient radical scavenger and a radical mechanism indicator, was irradiated with 24 W CFL light under oxygen atmosphere. The reaction was inhibited, and even after 48 h irradiation less than 8% of the product 3ca was observed (estimated by crude 1H NMR), and most of 1c was recovered after chromatographic purification. In addition, MS analysis of the reaction mixture showed the presence of the 394.2 molecular ion which reflected the presence of [1c–TEMPO + H]+. This observation further reveals the involvement of a radical process in the reaction mechanism.21
A conceivable mechanism is proposed in Scheme 3. Initially, irradiation of Ru(bpy)32+ by CFL visible light provides the active Ru(bpy)32+*, which oxidizes amine 1c via a single electron transfer (SET) process to give the radical cation A and Ru(bpy)3+. The species Ru(bpy)3+ can be oxidized by O2 and regenerated to start the new catalytic cycle.22 Deprotonation of the intermediate A affords the radical B, and the subsequent conversion provides the imine intermediate C. Subsequently, reaction of indole with imine C provide the adduct 3ca. Side reaction of the intermediate B could occur by reaction with oxygen or H2O2 to give 5 and 7. The side reaction becomes dominant when the aforementioned nucleophilic substitution of imine C occurs with weak nucleophiles (e.g., 2u, 2q, 2r). For the reaction with such a weak nucleophile, a competition of the MeOH and weak nucleophile with iminium C can occur and leads to the formation of side product 6 as well.23 Alternatively, in the absence of external ruthenium photocatalyst, substrate 1c, which acts as a photosensitizer, was slowly excited by CFL light irradiation.18a The activated 1c* underwent an energy transfer process with the ground state 3O2 to render the singlet 1O2, and regenerate the ground state 1c. The produced singlet 1O2 can undergo single-electron-transfer oxidation of 1c to give a radical cation A and O2˙−, which could further react with 1c to yield O22− and additional radical cation A followed by the subsequent reaction process.24 Alternatively, the photoreaction with the catalyst-free condition might be allowed due to the electron donor–acceptor complex.25 The results in the presence of ruthenium photocatalyst (e.g., Table 1, entry 4) indicate that the addition of an external photosensitizer significantly increases the reactivity, in consonance with the occurrence of an additional photoinduced electron transfer pathway.
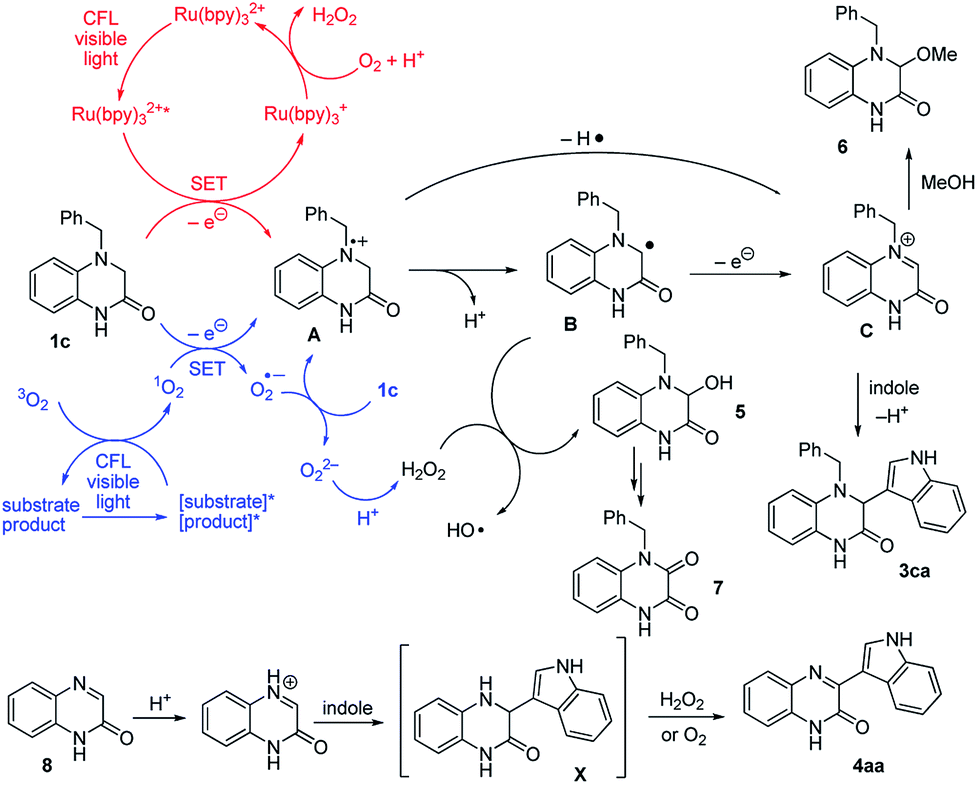 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In summary, we have established a visible-light induced C–C-bond formation reaction of 4-alkyl-3,4-dihydroquinoxalin-2(1H)-one and indoles as well as a variety of other nucleophiles. This reaction protocol not only contributed a convenient process with a household compact fluorescence lamp for the synthesis of related heterocycles, but also manifested an illustration of a catalyst-free photoreaction. The structures of several products were established by X-ray crystallographic analyses.26 Given the extensive interest of heterocycles in synthetic and medicinal chemistry and the growing interest in photocatalysis as well as the imperative need of acquiring green chemistry processes, this study could provide a valuable contribution to chemical synthesis. Further expansions of these reactions and demonstration of the process in elaborated systems are now in progress.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge financial support for this study from the Ministry of Science and Technology (MOST, Taiwan) and thank the instrument center of MOST for analyses of compounds. We thank Prof. Tzyy-Schiuan Yang (NCCU) and Mr Ching-Hsien Wang (NCCU) for the help in acquiring UV-visible absorption spectra.Notes and references
- (a) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef PubMed; (b) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040 RSC.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086 CrossRef PubMed.
- C. M. R. Volla, L. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef PubMed.
- Z.-Q. Wang, M. Hu, X.-C. Huang, L.-B. Gong, Y.-X. Xie and J.-H. Li, J. Org. Chem., 2012, 77, 8705 CrossRef PubMed.
- (a) K.-H. He, F.-F. Tan, C.-Z. Zhou, G.-J. Zhou, X.-L. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 3080 CrossRef PubMed; (b) M. K. Sahoo, G. Jaiswal, J. Rana and E. Balaraman, Chem.–Eur. J., 2017, 23, 14167 CrossRef PubMed.
- (a) R. Liu, Z. Huang, M. G. Murray, X. Guo and G. Liu, J. Med. Chem., 2011, 54, 5747 CrossRef PubMed; (b) A.-G. A. El-Helby, R. R. A. Ayyad, K. El-Adl and A. Elwan, Med. Chem. Res., 2017, 26, 2967 CrossRef; (c) X. Qin, X. Hao, H. Han, S. Zhu, Y. Yang, B. Wu, S. Hussain, S. Parveen, C. Jing, B. Ma and C. Zhu, J. Med. Chem., 2015, 58, 1254 CrossRef PubMed.
- (a) C. Huo, J. Dong, Y. Su, J. Tang and F. Chen, Chem. Commun., 2016, 52, 13341 RSC; (b) J. Dong, W. Min, H. Li, Z. Quan, C. Yang and C. Huo, Adv. Synth. Catal., 2017, 359, 3940 CrossRef.
- W. Ding, L.-Q. Lu, J. Liu, D. Liu, H.-T. Song and W.-J. Xiao, J. Org. Chem., 2016, 81, 7237 CrossRef PubMed.
- K. Yin and R. Zhang, Org. Lett., 2017, 19, 1530 CrossRef PubMed.
- R. P. Pandit, J.-J. Shim, S. H. Kim and Y. R. Lee, RSC Adv., 2017, 7, 55288 RSC.
- (a) S. Gräßle, S. Vanderheiden, P. Hodapp, B. Bulat, M. Nieger, N. Jung and S. Bräse, Org. Lett., 2016, 18, 3598 CrossRef PubMed; (b) D. Li, H. Ma and W. Yu, Adv. Synth. Catal., 2015, 357, 3696 CrossRef; (c) A. Carrër, J.-D. Brion, S. Messaoudi and M. Alami, Org. Lett., 2013, 15, 5606 CrossRef PubMed; (d) A. Carrër, J.-D. Brion, M. Alami and S. Messaoudia, Adv. Synth. Catal., 2014, 356, 3821 CrossRef; (e) V. Mamedov, N. A. Zhukova, A. I. Zamaletdinova, T. N. Beschastnova, M. S. Kadyrova, I. K. Rizvanov, V. V. Syakaev and S. K. Latypov, J. Org. Chem., 2014, 79, 9161 CrossRef PubMed; (f) F. Wang, B.-L. Hu, L. Liu, H.-Y. Tu and X.-G. Zhang, J. Org. Chem., 2017, 82, 11247 CrossRef PubMed.
- (a) B.-C. Hong, C.-W. Lin, W.-K. Liao and G.-H. Lee, Org. Lett., 2013, 15, 6258 CrossRef PubMed; (b) C.-W. Lin, B.-C. Hong, W.-C. Chang and G.-H. Lee, Org. Lett., 2015, 17, 2314 CrossRef PubMed; (c) C.-C. Chen, B.-C. Hong, W.-S. Li, T.-T. Chang and G.-H. Lee, Asian J. Org. Chem., 2017, 6, 426 CrossRef.
- Reaction does not proceed in the dark..
- For related synthesis of 8 derivatives from 4aa and their analogs, see (a) X. Zeng, C. Liu, X. Wang, J. Zhang, X. Wang and Y. Hu, Org. Biomol. Chem., 2017, 15, 8929 RSC; (b) S. Paul, J. H. Ha, G. E. Park and Y. R. Lee, Adv. Synth. Catal., 2017, 359, 1515 CrossRef; (c) Ref. 9.; (d) Ref. 11c..
- (a) C. Huo, J. Dong, Y. Su, J. Tang and F. Chen, Chem. Commun., 2016, 52, 13341 RSC; (b) P. K. Jaiswal, V. Sharma, J. Prikhodko, I. V. Mashevskaya and S. Chaudhary, Tetrahedron Lett., 2017, 58, 2077 CrossRef.
- The reaction in MeOH gave only 15% yield of the product, and the reaction does not proceed in the dark. In the absence of photocatalyst, the reaction was not proceeded..
- (a) W. Liu and C.-J. Li, Synlett, 2017, 28, 2714 CrossRef; (b) M. L. Deb, C. D. Pegu, P. J. Borpatra, P. J. Saikia and P. K. Baruah, Green Chem., 2017, 19, 4036 RSC; (c) J. F. Franz, W. B. Kraus and K. Zeitler, Chem. Commun., 2015, 51, 8280 RSC; (d) S. Borra, D. Chandrasekhar, S. Khound and R. A. Maurya, Org. Lett., 2017, 19, 5364 CrossRef PubMed.
- For related examples of the high concentration induced visible-light photocatalysis, see (a) S. Naskar and I. Das, Adv. Synth. Catal., 2017, 359, 875 CrossRef; (b) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef PubMed.
- See the ESI† for details..
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- E. G. Bagryanskaya and S. R. A. Marque, Chem. Rev., 2014, 114, 5011 CrossRef PubMed.
- After the reaction, the solution mixture was analyzed by calorimetric H2O2 Mquant™ test strip to be around 20 mg L−1, and the solution was found to be around pH 4.0..
- Pure 6 was isolated and characterized, but gradually decomposed into 5, via the reaction of intermediate C and moisture..
- For related mechanism, see (a) W. Ji, P. Li, S. Yang and L. Wang, Chem. Commun., 2017, 53, 8482 RSC; (b) S. Yang, P. Li, Z. Wang and L. Wang, Org. Lett., 2017, 19, 3386 CrossRef PubMed; (c) S. Li, B. Zhu, R. Lee, B. Qiao and Z. Jiang, Org. Chem. Front., 2018, 5, 380 RSC.
- For other peculiar catalyst-free photoredox reactions, see (a) C.-W. Hsu and H. Sundén, Org. Lett., 2018, 20, 2051 CrossRef PubMed; (b) L. Marzo, S. Wang and B. König, Org. Lett., 2017, 19, 5976 CrossRef PubMed; (c) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef PubMed; (d) M. L. Deb, C. D. Pegu, P. J. Borpatra, P. J. Saikia and P. K. Baruah, Green Chem., 2017, 19, 4036 RSC; (e) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019 CrossRef PubMed; (f) S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adán and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485 CrossRef PubMed; (g) P. Liu, W. Liu and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 14315 CrossRef PubMed; (h) A. U. Meyer, A. Wimmer and B. König, Angew. Chem., Int. Ed., 2017, 56, 409 CrossRef PubMed; (i) L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809 CrossRef PubMed; (j) W.-T. Xu, B. Huang, J.-J. Dai, J. Xu and H.-J. Xu, Org. Lett., 2016, 18, 3114 CrossRef PubMed.
- During the preparation of this manuscript, a related photocatalysis of 3,4-dihydro-2H-benzo[b][1,4]oxazin-2-one was published, G.-Y. Zhang, K.-K. Yu, C. Zhang, Z. Guan and Y.-H. He, Eur. J. Org. Chem., 2018, 525 CrossRef . Nevertheless, the current studies provide more examples, including the 3,4-dihydroquinoxalin-2(1H)-one, extensive study and the catalyst free condition, as well as the X-ray analysis of adducts.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, spectroscopic characterization. CCDC 1816891–1816895. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03259a |
| This journal is © The Royal Society of Chemistry 2018 |