
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silicon-coordinated nitrogen-doped graphene as a promising metal-free catalyst for N2O reduction by CO: a theoretical study†
Anchalee Junkaew a,
Supawadee Namuangruk*a,
Phornphimon Maitaradb and
Masahiro Ehara*c
a,
Supawadee Namuangruk*a,
Phornphimon Maitaradb and
Masahiro Ehara*c
aNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Pathum Thani 12120, Thailand. E-mail: supawadee@nanotec.or.th
bResearch Center of Nano Science and Technology, Shanghai University, Shanghai 200444, P. R. China
cInstitute for Molecular Science, Nishigo-naka 38, Myodai-ji, Okazaki, Aichi 444-8585, Japan. E-mail: ehara@ims.ac.jp
First published on 19th June 2018
Abstract
Metal-free catalysts for the transformation of N2O and CO into green products under mild conditions have long been expected. The present work proposes using silicon-coordinated nitrogen-doped graphene (SiN4G) as a catalyst for N2O reduction and CO oxidation based on periodic DFT calculations. The reaction proceeds via two steps, which are N2O reduction at the Si reaction center, producing Si–O*, which subsequently oxidizes CO to CO2. The N2O reduction occurs with an activation energy barrier of 0.34 eV, while the CO oxidation step requires an energy of 0.66 eV. The overall reaction is highly exothermic, with a reaction energy of −3.41 eV, mostly due to the N2 generation step. Compared to other metal-free catalysts, SiN4G shows the higher selectivity because it not only strongly prefers to adsorb N2O over CO, but the produced N2 and CO2 are easily desorbed, which prevents the poisoning of the active catalytic sites. These results demonstrate that SiN4G is a promising metal-free catalyst for N2O reduction and CO oxidation under mild conditions, as the reaction is both thermodynamically and kinetically favorable.
1. Introduction
Nitrous oxide (N2O) and carbon monoxide (CO) are pollutant gases which are exhausted from various combustion sources, such as vehicles and electric power plants. Because removing them requires high temperatures and large amounts of energy, catalytic materials have been extensively explored for eliminating them more efficiently. The catalytic reaction of N2O and CO, namely, the oxidation of CO by N2O or the reduction of N2O by CO, is one of the useful techniques for converting N2O and CO into less harmful products i.e., N2 and CO2. In a direct N2O (g) + CO (g) → N2 (g) + CO2 (g) reaction, the activation barrier of 2 eV and the reaction energy of −3.6 eV were determined by a density functional theory (DFT) calculation.1 Although the reduction of N2O by CO is an exothermic reaction, it is difficult to promote in the gas phase due to the large activation energy (Ea) required.2,3In the literature, various metals and metal ions such as Pt+, Ir+, Os+, Fe+, Ca+, Ge+, Sr+, Ba+, Eu+, and Yb+ have been explored for use as catalysts in the elimination of CO and N2O at low temperatures.4,5 Also, the reduction of N2O by CO have been investigated on metal cluster catalysts such as Ptn− (n = 3–4)2,6 neutral Rhn (n = 10–28),7 neutral Ptn (n = 4–12),8 Ptn+ (n = 6–8),9 Cu anions,10 Cun (n = 4–15),1 Ag7Au6 clusters,11 etc. Although metal-based catalysts perform well when eliminating CO and N2O at low temperatures, their mass-scale uses are economically limited due to their cost. Various catalytic materials, such as metals on supports, metal complexes, metal oxides and metal-free catalysts have been sought in order to reduce costs while retaining good catalytic performance.12–17
Recently, nitrogen-doped graphene (NG) became a promising metal-free catalyst; it had been proposed for various catalytic applications, such as electrochemical, oxidation and other reactions.18–21 A four pyridinic-N defect with a di-vacancy in graphene (i.e. 4N + DV) is one of the pyridinic forms that can be drawn from past experiments.22 The pyridinic-N defect provides a greater chemical reactivity than the graphitic-N and it can trap a single metal atom, such as Mg, Al, Ca, Ti, Cr, Mn, and Fe.22 This 4N + DV defective graphene (N4G) has a porphyrin-like core. Varying doping species at the porphyrin-like core can fine-tune its properties for desired applications. For instance, FeN4G, CoN4G, NiN4G, and MnN4G have been applied for the oxygen reduction reaction (ORR) applications.23–26 Later, the possible use of FeN4G as a catalyst for NO reduction via the (NO)2 adsorption mechanism was suggested by the periodic DFT method.27 Very recently, water dissociation on MN4G (M = Mg, Ba, Ti) was theoretically studied by Liu et al.28 They showed that Mg, Ba, Ti are energetically stable on the surface and TiN4G displayed promising catalytic properties for H2O dissociation.
For applications involving N2O and CO removal, p-block element doped into carbon and other support materials have been used as catalysts.13,15–17,29 Since Si is the second most abundant element on Earth,30 it is attractive to corporate with other elements for applying in various applications. From previous experimental work, Si coordinated with four nitrogen atoms has been proposed as the active site in Si-porphyrin and Si-corroles.31,32 Si-porphyrin was successfully synthesized32 and it has been further applied in a photochemical oxygenation of alkenes,30 water oxidation to hydrogen peroxide,33 and dye-sensitized solar cell applications.34 For Si-corroles, Si-porphyrine derivatives, provides good luminescence properties and they have been applied as a sensor for F− detection.31 Interesting work has been reported recently by Tang et al.; they found that Si-coordinated nitrogen-doped graphene (SiN4G), which is a metal-free catalyst, had catalytic properties that promoted CO oxidation by O2.35 These reports noted that Si atoms doped into 2D materials are a reactive dopant for CO and N2O gases. However, it is unclear why a Si dopant, which is a non-metal atom, was specifically reactive to those gases.
Moreover, to the best of our knowledge, Si doped into N4G for the reduction of N2O by CO has not been studied so far. Thus, we are motivated to ask: How feasible is this metal-free catalyst SiN4G for the reduction of N2O by CO? Is it possible to use this catalyst in a low temperature range? In this work, these questions are answered systemically using a plane-wave-based DFT investigation. The gas adsorption and detailed reaction mechanisms were examined. Moreover, the performance of SiN4G is compared with other catalysts from the literature in order to determine the feasibility of using this catalyst for the reduction of N2O by CO. The results will be valuable for developing low cost catalysts for pollutant gas abatement applications.
2. Method
The gas adsorption and the reaction mechanism of reduction of N2O by CO over SiN4G were examined by using plane-wave-based DFT calculations implemented in the Vienna ab initio simulation package (VASP version 5.4.1).36,37 The projector-augmented wave (PAW)38 with a generalized gradient approximation (GGA) refined by Perdew, Burke and Ernzerhof (PBE)39 was used in this work. Grimme's DFT-D3 was applied for the dispersion contribution.40 The 1 × 10−5 eV per cell and 5 × 10−3 eV Å−1 were set as the energy- and force-convergence parameters, respectively. An energy cutoff of 400 eV and a spin-unrestricted calculation were applied in all cases. The SiN4G slab was constructed by placing one Si at the N4 center of a (5 × 5) slab of graphene with a = 12.4 Å and b = 12.2 Å. The slab was separated by 15 Å of vacuum. This size of the simulated cell is large enough to prevent interfering of adsorption interaction by its replicas. All atoms in the cell were relaxed during the calculations. After the optimized SiN4G was obtained, the simulated cell was fixed during gas adsorption and reaction mechanism calculations. A top view of a center site of SiN4G is illustrated in Fig. 1a. The isolated gas molecule was placed in the 15 Å × 15 Å × 15 Å box. The Monkhorst–Pack grids of 5 × 5 × 1 and 1 × 1 × 1 were applied for slab and isolated gas structures, respectively.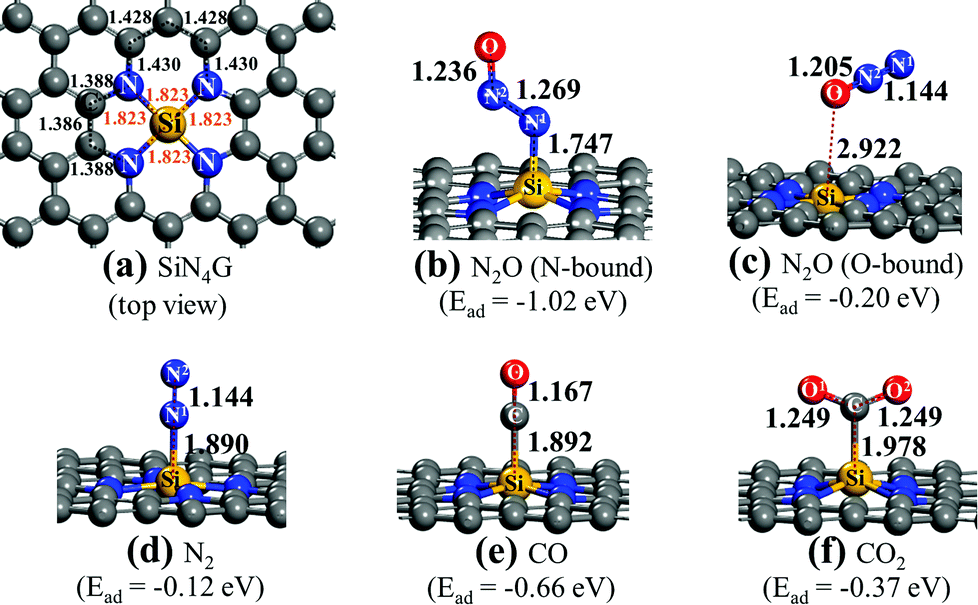 | ||
| Fig. 1 Structures of (a) SiN4G, (b) SiN4G/N2O (N-bound), (c) SiN4G/N2O (O-bound), (d) SiN4G/N2, (e) SiN4G/CO and (f) SiN4G/CO2. Selected bond lengths (Å) and Ead values of adsorbed gas molecules on the catalyst are given. | ||
The transition state (TS) of each elementary step was calculated from the climbing-image nudged elastic band (CI-NEB)41,42 and the dimer method.43,44 The criterion of the force convergence threshold was 0.025 eV Å−1. Each TS structure was confirmed by its single imaginary frequency. The projected density of state (PDOS), electron density difference and Bader charge analyses45,46 were elucidated in this work.
3. Results and discussion
3.1 Gas adsorption on SiN4G
First, the adsorption properties of reactants and products over SiN4G are discussed. The adsorption energy (Ead) is calculated by| Ead = Ecomplexadsorbate–substrate − Eisolatedsubstrate − Eisolatedadsorbate | (1) |
Fig. 1a shows the top view structure of the optimized bare SiN4G sheet. The bond distances around the core site of SiN4G are given. The Si atom is surrounded by two five-membered rings and two six-membered rings. The four Si–N bond lengths are equivalent at 1.823 Å. According the calculated Ead values expressed in Fig. 1, the order of the binding strength of the four gas species on the catalyst is N2O > CO > CO2 > N2. The linear N2O gas strongly adsorbs on the Si atom with the N-bound mode resulting in a bent N–N–O conformation and the calculated Ead is −1.02 eV (see Fig. 1b). Both N–N and N–O bonds are lengthened from 1.146 Å and 1.200 Å (free N2O gas) to 1.269 Å and 1.236 Å, respectively. The formed coordination bond between N and Si is 1.747 Å. In Fig. 1c, the O-bound mode is less favorable for N2O adsorption. In the N2 case, it shows the least binding stability with Ead ∼ −0.12 eV as shown in Fig. 1d. It is worth to note that the attached N2 at Si site in Fig. 1d is slightly less stable than the detached N2 (Ead = −0.17 eV) presented in Fig. S1a of ESI.†
For CO and CO2, their carbon atoms attach to the Si atom and their binding energies are −0.66 and −0.37 eV, respectively. By comparing their Ead values, we can see that the CO adsorption in Fig. 1e is stronger than the CO2 adsorption in Fig. 1f. The end-on configuration of adsorbed CO over SiN4G agrees well with literature.35 The C–O bonds are activated in both CO and CO2 compared with the free molecules. The Ead values from the prior work are −0.55 eV and −0.26 eV for CO and CO2 adsorption, respectively. Our calculated Ead values are slightly stronger than those of the previous calculations because the dispersion correction is included in this work. As a result, the Si site moves along the out-of-plane direction when it chemically adsorbs molecules. On the other hand, the SiN4G sheet is not changed when it binds with gas through physisorption interaction (see Fig. 1c).
A different order of the adsorption energies was observed in the adsorption of these gasses on Si-doped graphene (SiG) studied by Gholizadeh and Yu.15 Their calculated Ead values are small and are in the range of −0.2 eV to −0.15 eV and the adsorption strength follows the order of SiG/CO > SiG/CO2 > SiG/N2O > SiG/N2. Therefore, the coordinated Si with nitrogen at the active center site in the present SiN4G significantly enhances the adsorption ability and changes the order of these gas adsorptions. Due to the distinctly stronger interaction of N2O on SiN4G when compared to CO on SiN4G, the N2O reduction would occur first, rather than the CO oxidation. Therefore, the present results of these adsorption energies also support the claim that the reaction cycle can proceed as the two sequential steps: (1) N2O → N2 + O* and (2) O* + CO → CO2, respectively. The CO2 and N2 products can be easily desorbed due to their low binding strength. This advantage can prevent catalyst poisoning by the products and allows the active site to get ready to react with the reactant in the next cycle.
The projected density of states (PDOSs) of bare SiN4G, and the adsorbed N2O and CO2 on the SiN4G structures, were analyzed and compared in Fig. 2. The Fermi level (EF) is shown by the vertical dashed line at 0 eV. Positive and negative amplitudes of PDOS correspond to the spin-up and spin-down states, respectively. Fig. 2a depicts the PDOS plot of SiN4G. The hybridization of the valence states and bonding can be seen in overlapping peaks. In addition, PDOS analysis of the pyridinic-N embedded in the graphene (N4G) is depicted in Fig. S2 in ESI.† The PDOS of N4G agrees well with those reported in other literatures.47,48 The PDOS peaks of the Si, the four pyridinic-N atoms (4N) and the eight neighbouring carbon atoms around the N atoms (8C) of the bare SiN4G are depicted in Fig. 2a. The right panel of Fig. 2a shows the PDOS of the decomposed s- and p-states of Si around EF, which are relevant for the reaction. SiN4G has symmetrical spin-up and spin-down peaks. The overlapped valence and conduction bands of Si and N atoms, with their adjacent carbons, can be seen in Fig. 2a. In SiN4G, the p-states of Si located close to the EF level can be found in the right panel of Fig. 2a. Furthermore, the PDOS plots of N2O and CO2 adsorbed on SiN4G are also shown in Fig. 2b and c. The chemical bonds between Si and the adsorbed molecules can be observed via the hybridization of their PDOS peaks. To see the variation of PDOS of the adsorbed gas species, PDOS plots of isolated N2O and CO2 molecules are also provided in Fig. 2d and e, respectively. For SiN4G/N2O, we found the coupling between the p-states of Si and the valence p-states the N1 of N2O (g) near EF results in hybridization peaks at EF (right panel of Fig. 2b). This signifies the bonding between N2O and SiN4G. SiN4G donates an electron to N2O resulting in asymmetrical spin-up and spin-down peaks around EF (see Fig. 2b).
 | ||
| Fig. 2 PDOS plots of selected atoms in (a) bare SiN4G, (b) SiN4G/N2O, (c) SiN4G/CO2, (d) isolated N2O gas and (e) isolated CO2 gas. In (a) to (c), the PDOS peaks of Si, 4N, 8C and gas molecules are presented in the left panels, and the s- and p-states of selected Si and atoms of gas molecules are decomposed in the right panels. Positive and negative amplitudes of PDOS indicate the spin-up and spin-down states, respectively. | ||
In contrast to SiN4G/N2O, SiN4G/CO2 shows a symmetrical nature. The broad hybridized peaks can be observed in Fig. 2c. By comparing them with the PDOS of bare SiN4G, the reduction of the occupied valence states can be observed. The PDOS peaks of Si and the gas are decomposed into their s- and p-states and are illustrated in the right panel of Fig. 2c. The overlapping states of Si and CO2 can be seen at EF. The PDOS of only one oxygen atom of CO2 is presented in Fig. 2c, since the two oxygen atoms in CO2 are identical to each other. According to Fig. 2c, the overlapped peaks around EF are the hybridization of the states near EF of Si in bare SiN4G (see Fig. 2a) and the states of isolated CO2 (Fig. 2e).
As a result, the hybridization between the states of Si and the states of N2O and CO2 results in chemical bonds between Si and those gas molecules. In addition, the Bader charge results were also calculated in this work. The changes in the valence electrons (Δe−) of the selected atoms are given in Table S1 in ESI.† Our Bader charge results demonstrate that N2O and CO2 gain more electrons from SiN4G, approximately 1.03|e| and 1.06|e|, respectively. The electronegativity of O, which is greater than those of N, C and Si, also influences the direction of electron transfer in gas adsorbed SiN4G. Therefore, both the PDOS and Bader charge results support the claim that electrons are transferred from SiN4G to those adsorbed gas molecules.
3.2 Mechanisms
The detailed mechanism of the N2O reduction by CO is discussed in this section. The overall reaction can be described by eqn (2). There are two possible mechanisms for this reaction, which are the concerted- and stepwise-mechanisms. The concerted mechanism presents the reaction that N2O and CO interact at once to produce N2 and CO2. In the stepwise mechanism, the N2O reduction over SiN4G occurs first followed by CO oxidation over SiN4G-O* as represented by eqn (3) and (4), respectively.| N2O (g) + CO (g) → N2 (g) + CO2 (g) | (2) |
| N2O (g) → *N2O → N2 (g) + O* | (3) |
| O* + CO (g) → *CO2 → CO2 (g) | (4) |
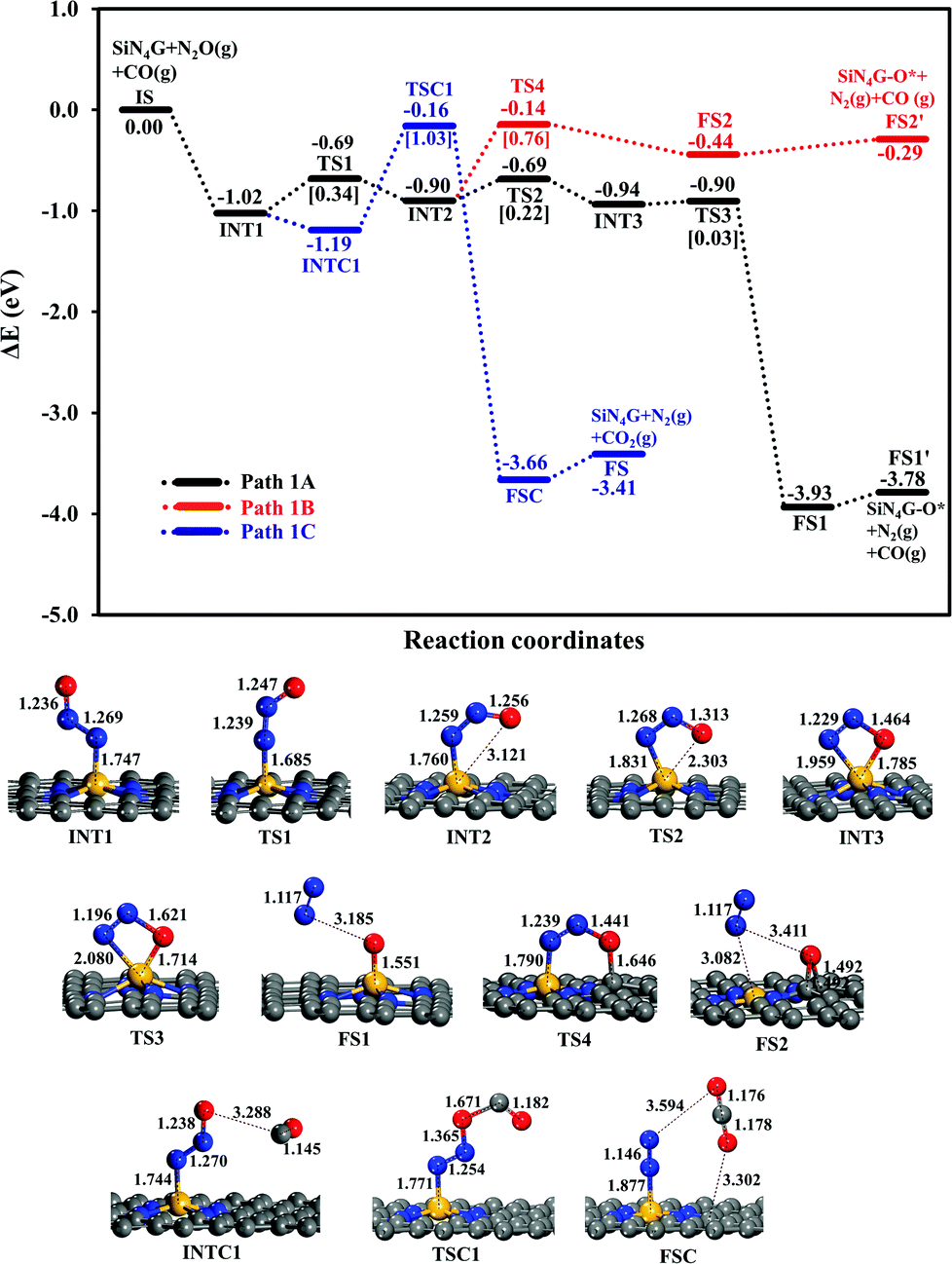 | ||
| Fig. 3 Energy profiles of Path 1A (black line), Path 1B (red line) and Path 1C (blue line) with the corresponding structures. | ||
N2O reduction. Two possible pathways of the N2O dissociation are compared for the stepwise mechanism. The first pathway (Path 1A) represents the conversion of N2O to N2 and leaves its O attached on the Si-site. In another possible pathway (Path 1B), the leaving O atom of N2O is bound at the C–C bridge site of the SiN4G catalyst.
The relative energy profile of Path 1A with structures and selected bond distances are shown as the black profile in Fig. 3. The total energies of the bare SiN4G, isolated N2O and isolated CO are used as a reference to calculate the relative energy (ΔE). The Ea value of each transition state is given in the bracket. This pathway starts from the N2O adsorption intermediates of INT1 and INT2 with an Ead of approximately −1 eV. The transformation of INT1 to INT2 needs energy barrier of 0.34 eV at the TS1 state. The N2O molecule is bent and distorted on SiN4G, in particular for INT2 with the terminal O atom directed to the Si center. For the INT2 → TS2 → INT3 steps, a small Ea of 0.22 eV is required to enable the bonding between Si and O atoms; at TS2, the S–O distance decreases to 2.303 Å to form a bond and is 1.785 Å at INT3. The N–O bond is then broken simultaneously with a negligible Ea of 0.03 eV at TS3. Finally, N2 is produced and the O atom remains at the Si site at the final state (FS1) of this elementary step. The Si–O bond length of FS1 is 1.551 Å. The energy difference between the FS1 and FS1′ describes that the N2 is easily desorbed from SiN4G-O* with very little energy, approximately 0.15 eV. In addition, Path 1A is a highly exothermic reaction with a reaction energy of −3.78 eV.
Another possible route (Path 1B) is examined, and its energy profile with structures is depicted by the red profile in Fig. 3. This pathway proceeds through the INT2 → TS4 → FS2 steps. An Ea of 0.76 eV is required to surmount the energy barrier at TS4. Both N–O and N–Si bond lengths are elongated at TS4 in order to release N2 as a product. For the TS4 to FS2 step, the dissociated O atom binds on atop C site at TS4. Then N2 is simultaneously desorbed from Si and the dissociated O atom moves simultaneously towards the C–C bridge site, which is more energetically favorable than the top C site, at the final state (FS2). According to the relative energies, the SiN4G-O* structure of FS2, in which the O atom is attached to the C–C bridge site, is less stable than that of FS1, where the O atom binds with the Si site. Unlike Path 1A, this pathway is an endothermic reaction due to the less stable FS2.
In summary, Path 1A is more kinetically and thermodynamically favorable than Path 1B. The activation barrier of N2O dissociation on SiN4G for the N2O reduction is less than that on SiG (Ea ∼ 0.5 eV).15 Thus, the surrounding N atoms at the Si center not only improve the adsorption ability of N2O adsorption but also enhance the activity of catalyst in the N2O dissociation process.
CO oxidation. The second step is the CO oxidation, in which CO is oxidized by the pre-adsorbed oxygen from the previous step to produce CO2, as outlined in eqn (4). The energy profile of this step and the structures are displayed in Fig. 4. Two competitive pathways, Path 2A and Path 2B, are considered. Both of these pathways start from INT4 where the CO molecule weakly interacts with SiN4G-O*. In Path 2A, CO interacts with O* and then forms the CO2 product attached on the Si site with C atom. The energy barrier of the INT4 → TS5 → FS3 step is 0.66 eV and this step is slightly endothermic by +0.01 eV relative to IS2. Then CO2 is desorbed directly from FS3 to FS3′. This step requires energy approximately 0.37 eV. The PES and corresponding configurations of the direct desorption CO2 desorption process, the FS3 to FS3′ step, are shown in Fig. S5 in ESI.†
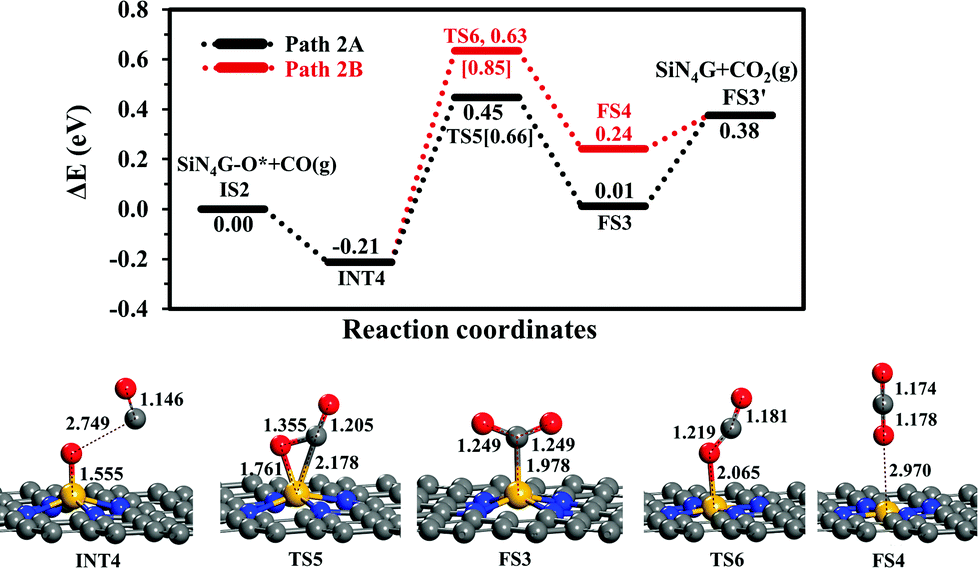 | ||
| Fig. 4 Energy profiles of Path 2A and 2B with the corresponding structures. | ||
In Path 2B, CO interacts with O* and release CO2 from Si site simultaneously (see FS4 in Fig. 4). The energy barrier to surmount TS6 is 0.85 eV. For this state, CO2 needs 0.14 eV to desorb from the catalyst in FS3′. Similar to Path 2A, the CO2 desorption energy is less than the energy of breaking the O–CO bond. This step is also endothermic. Consequently, Path 2A is more favorable than Path 2B in view of energetics. The present Ea value of Path 2A is lower by approximately 0.1 eV than the reported one by Tang et al. in the study of CO oxidation by O2.35 In summary, the CO oxidation step is less thermodynamically and kinetically preferable than the N2O reduction step, however, this reaction is feasible at low temperatures, which is supported by the calculated activation energy barriers and reaction energy. Finally, SiN4G is completely regenerated and ready for the next N2O reduction.
To conclude all the results in this work, the N2O reduction by CO prefers the stepwise mechanism than the concerted mechanism. The energy profile of the most favorable stepwise pathway of the N2O reduction by CO on SiN4G is presented in Fig. 5. A summation of the energies of bare SiN4G, N2O and CO is used as the reference energy. The calculated reaction energy of N2O + CO → N2 + CO2 in this work is −3.41 eV. This value is comparable to −3.5 eV reported by theoretical calculations in Si- and Fe-doped graphene15,49 and −3.8 eV of the direct reaction of CO with N2O from experiment.50 The N2O reduction and CO oxidation prefer Path 1A with an Ea of 0.34 eV and Path 2A with an Ea of 0.66 eV. Thus, the rate-determining step of the overall reaction is the CO oxidation. It is worth mentioning that N2O is adsorbed more strongly than CO2, as seen from their adsorption energies. The small adsorption energies of the N2 and CO2 products in this system indicate that the products easily desorb from the active site, which prevents them from poisoning the catalyst. Hence, the active SiN4G site can be recovered after the reaction is completed and the next reaction cycle can continue further. Overall, the SiN4G catalyst demonstrates a promising performance for the reduction of N2O by CO.
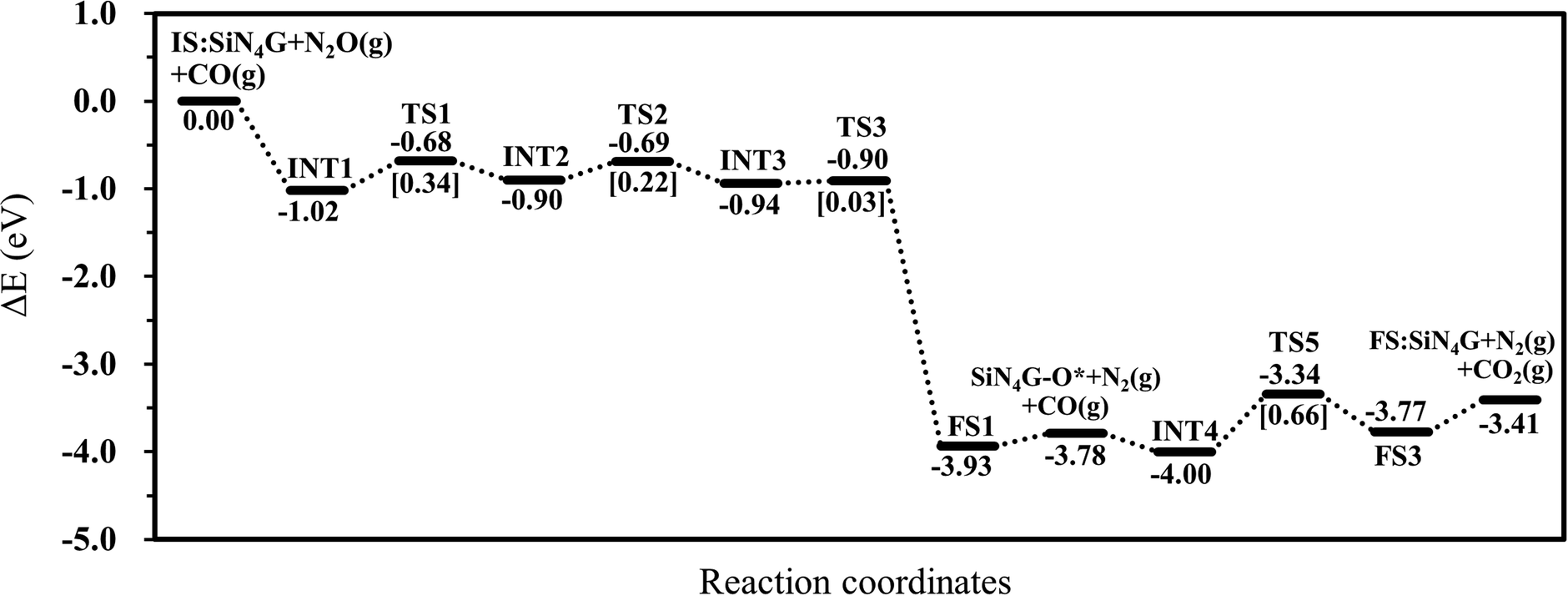 | ||
| Fig. 5 Energy profile of the most favorable pathway of the N2O reduction by CO on SiN4G. | ||
To form a better understanding of the charge property of the systems in the reaction, electron density differences of the intermediates and transition states along the most favorable pathway are also investigated. The results are given in Fig. S3 and S4 in ESI.† For bare SiN4G, the electron density difference is referenced with the isolated Si atom and N4G surface, see Fig. S3 in ESI.† The light blue and yellow regions represent the electron density increment and reduction, respectively. The electron density of Si is depleted when Si is embedded at the porphyrin-like core and is delocalized over N atoms and the Si–N bonds around the di-vacancy site. This result agrees well with the Bader charge analysis in Table S1 in ESI;† the partial charge of Si is about +2.66|e| and the coordinating N atoms show an average negative charge about −1.5|e|. Therefore, these four N atoms strongly coordinate with the embedded Si atom as confirmed by the adsorption energy of Si on N4G of −7.07 eV. This value is comparable with −7.25 eV, reported by Tang et al.35 This strong adsorption stabilizes the catalyst and prevent the leakage of the embedded Si atoms from the catalyst, resulting in increasing the durable operation. When all adsorbed species attached on SiN4G, electrons are obviously transferred from Si and accumulated between Si and the attached atom (see Fig. S4 in ESI†). This electron transfer supports the PDOS result.
3.3 Comparison with other two dimensional catalysts
Herein, the expected catalytic activity of SiN4G for the reduction of N2O by CO is considered in comparison with the Si-based graphenes or nanotubes and other metal-based catalysts from previous reports. The activation energies of the rate-limiting steps of the N2O reduction and the CO oxidation of some catalysts are compared in Table 1. The effect of the coordinating N atoms in the SiN4G on the catalytic activity of the catalyst can be described by comparing SiN4G with SiG.15 SiN4G has a lower Ea in the N2O reduction step but a higher Ea in the CO oxidation step. For the entire N2O and CO2 reaction, the Ea of the rate determining step is 0.5 eV in SiG, which is slightly lower than that of SiN4G. However, as mentioned in Section 3.1, the CO adsorption on SiG is slightly stronger than that of N2O, which indicates that the selectivity of SiG towards the N2O reduction is poorer than SiN4G if CO exists in the system. Thus, the presence of N atoms around the Si does not only enhance the adsorption with the reactants, but it also makes the catalyst become more selective. In SeG, the Ea of the N2O reduction is approximately 1.8 eV which is much higher than the Si doping case.15 Note that for SiN4G, the adsorption CO2 product is not comparative to the adsorption of the CO and N2O reactants. This characteristic is different from the SeG and SiG systems, in which reactants and products show similar adsorption energies. This means that the CO2 product in the case of SeG and SiG may poison the active sites, while in the case of SiN4G the CO2 is readily desorbed to get the active site ready for the next reaction cycle.| Catalyst | Ea (eV) | Ead (eV) | |||
|---|---|---|---|---|---|
| N2O → N2 + O* | CO + O* → CO2 | N2O | CO | CO2 | |
| a BL denotes a barrierless process. | |||||
| SiN4G (this work) | 0.34 | 0.66 | −0.97 | −0.66 | −0.37 |
| SiN4G35 | 0.72 | −0.55 | −0.26 | ||
| Mn-N4 carbon nanotube (MnN4CNT)51 | 1.49 | −2.20 | |||
| Si-doped graphene (SiG)15 | 0.5 | 0.3 | −0.18 | −0.19 | −0.18 |
| Se-doped graphene (SeG)15 | 1.8 | 0.7 | −0.22 | −0.18 | −0.21 |
| Si-doped boron nitride nanotubes (SiB-BNNTs)13 | BLa | 0.08 | N/A | −0.19 | |
| Si-doped boron nitride nanotubes (SiN-BNNTs)13 | BLa | 0.42 | N/A | −0.16 | |
| Silicon carbide nanotubes ((6,0)-SiCNT)17 | 0.71 | 1.01 | −0.64 | −0.38 | |
| Silicon carbide nanosheets (SiCNS)17 | 1.12 | 0.98 | −0.59 | −0.18 | −0.14 |
| Pd-doped graphene (PdG)52 | 0.26 | −1.04 | −0.21 | ||
| Al-doped graphene (AlG)16 | 0.24 | 0.06 | −0.81 | −0.62 | −0.52 |
| Ti-doped graphene (TiG)16 | BLa | 0.16 | N/A | −1.03 | −0.32 |
| Fe-doped graphene (FeG)49 | 0.4 | 0.2 | −0.4 | −1.5 | −0.4 |
| Pd-doped boron nitride (PdBN)53 | 0.23 | −1.07 | −0.06 | ||
| Ag-doped boron nitride (AgBN)54 | 0.17 | −1.04 | −0.36 | ||
| Co-doped boron nitride (AgBN)55 | 0.16 | −1.04 | −0.33 | ||
| Ag6Au7 cluster11 | 1.1 | 0.5 | −0.2 | −0.5 | −0.1 |
| Cu7 cluster1 | BLa | 0.9 | N/A | ||
| Cu12 cluster1 | BLa | 0.8 | N/A | ||
Moreover, the CO oxidation on Si-doped BN nanotubes (Si-BNNTs) and Ti-doped graphene (TiG), and the N2O reduction by CO on Al-doped graphene (AlG) and Fe-doped graphene (FeG) are facile based on their activation energies. In the Fe-doped graphene (FeG), the rate-limiting steps require 0.4 eV and 0.8 eV for the stepwise and concerted mechanisms, respectively. However, the N2O adsorption on FeG (Ead ∼ −0.4 eV) is less favorable than the CO adsorption on FeG (Ead ∼ −1.5 eV).49 Thus, FeG is not selective to the present reaction. As presented in Table 1, many metal-free catalysts were proposed to show better catalytic performance than pure metal cluster catalysts like Cu- and Ag6Au7-clusters.1,11
In conclusion, the results in this work indicate that SiN4G is a promising catalyst because of the following reasons: (1) the durability of the catalyst indicated by the substantial adsorption strength of Si on N4G (−7.07 eV), (2) the high reactivity and selectivity for converting N2O and CO to less harmful products, N2 and CO2, at low temperature, indicated by low activation barriers and large reaction energy, (3) the small adsorption energy of the CO2 product prevents catalyst poisoning, which is a problem with most of the conventional metal catalysts, and (4) a low cost for large-scale reactions in industry.
4. Conclusion
This work investigated the charge, density of state, energetic and structural properties for the reduction of N2O reduction by CO on the Si-coordinated nitrogen-doped graphene catalyst, SiN4G, by employing plane-wave-based DFT calculations. The mechanistic insight of the N2O and CO reaction has been systematically examined. The calculation results show that the four coordinating N atoms of SiN4G increase the reactivity of the Si active site and stabilize the adsorption state by charge transfer. The Si active site is specifically reactive to N2O due to the strong overlap between the Si active site and the N atom of the adsorbed molecule. The reaction mechanism for the reduction of N2O by CO occurs through two consecutive steps: N2O reduction followed CO oxidation. The overall reaction is thermodynamically and kinetically preferable due to its low activation barriers and exothermic characteristic. The N2O reduction occurs easily, with a small energy barrier of 0.34 eV, while the CO oxidation is the rate determining step of the entire reaction, with an Ea of 0.66 eV and slight endothermicity. However, CO2 can be released more easily than it can be dissociated back to CO. The SiN4G is regenerated completely after CO oxidation. Then, a new reaction cycle occurs simultaneously due to the stronger interaction between SiN4G and N2O when compared with the CO product. This catalyst also shows strong reaction activity when compared to proposed catalysts from the literature. In summary, this metal-free catalyst is one of many candidates for eliminating CO and N2O at low temperature, and it is reusable for multiple reaction cycles. The mechanistic insight from this work provides a useful guidance in designing a low-cost metal-free catalyst with great catalytic performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the support from National Nanotechnology Center (NANOTEC), Thailand and the Research Center for Computational Science, Institute for Molecular Science, Nanotechnology Platform program from MEXT, Japan. S. N and A. J acknowledge the Thailand Research Fund (RSA6180080). P. M. thanks the Natural National Science Foundation of China (NSFC) Research Fund for International Young Scientists FY 2016 (21650110450). M. E. acknowledges a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) (JP16H04104, JP16H06511).References
- J. Barabás and T. Höltzl, J. Phys. Chem. A, 2016, 120, 8862–8870 CrossRef PubMed.
- Y. Shi and K. M. Ervin, J. Chem. Phys., 1998, 108, 1757–1760 CrossRef.
- D. K. Böhme and H. Schwarz, Angew. Chem., Int. Ed., 2005, 44, 2336–2354 CrossRef PubMed.
- M. M. Kappes and R. H. Staley, J. Am. Chem. Soc., 1981, 103, 1286–1287 CrossRef.
- V. Blagojevic, G. Orlova and D. K. Bohme, J. Am. Chem. Soc., 2005, 127, 3545–3555 CrossRef PubMed.
- P. A. Hintz and K. M. Ervin, J. Chem. Phys., 1995, 103, 7897–7906 CrossRef.
- A. Yamada, K. Miyajima and F. Mafune, Phys. Chem. Chem. Phys., 2012, 14, 4188–4195 RSC.
- H. Yamamoto, K. Miyajima, T. Yasuike and F. Mafuné, J. Phys. Chem. A, 2013, 117, 12175–12183 CrossRef PubMed.
- O. P. Balaj, I. Balteanu, T. T. J. Roßteuscher, M. K. Beyer and V. E. Bondybey, Angew. Chem., Int. Ed., 2004, 43, 6519–6522 CrossRef PubMed.
- S. Hirabayashi and M. Ichihashi, Phys. Chem. Chem. Phys., 2014, 16, 26500–26505 RSC.
- Y. Wongnongwa, S. Namuangruk, N. Kungwan and S. Jungsuttiwong, Appl. Catal., A, 2017, 538, 99–106 CrossRef.
- X.-L. Xu, E. Yang, J.-Q. Li, Y. Li and W.-K. Chen, ChemCatChem, 2009, 1, 384–392 CrossRef.
- M. D. Esrafili, N. Saeidi and P. Nematollahi, RSC Adv., 2015, 5, 100290–100298 RSC.
- H.-L. Fang, L. Xu, J. Li, B. Wang, Y.-F. Zhang and X. Huang, RSC Adv., 2015, 5, 76651–76659 RSC.
- R. Gholizadeh and Y.-X. Yu, Appl. Surf. Sci., 2015, 357, 1187–1195 CrossRef.
- M. D. Esrafili, F. Mohammadian-Sabet and P. Nematollahi, RSC Adv., 2016, 6, 64832–64840 RSC.
- P. Nematollahi and M. D. Esrafili, RSC Adv., 2016, 6, 59091–59099 RSC.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef PubMed.
- B. Men, Y. Sun, M. Li, C. Hu, M. Zhang, L. Wang, Y. Tang, Y. Chen, P. Wan and J. Pan, ACS Appl. Mater. Interfaces, 2016, 8, 1415–1423 CrossRef PubMed.
- J. Long, X. Xie, J. Xu, Q. Gu, L. Chen and X. Wang, ACS Catal., 2012, 2, 622–631 CrossRef.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef.
- Y.-C. Lin, P.-Y. Teng, C.-H. Yeh, M. Koshino, P.-W. Chiu and K. Suenaga, Nano Lett., 2015, 15, 7408–7413 CrossRef PubMed.
- H. R. Byon, J. Suntivich and Y. Shao-Horn, Chem. Mater., 2011, 23, 3421–3428 CrossRef.
- S. Kattel and G. Wang, J. Mater. Chem. A, 2013, 1, 10790–10797 RSC.
- W. Orellana, J. Phys. Chem. C, 2013, 117, 9812–9818 CrossRef.
- U. I. Kramm, I. Herrmann-Geppert, J. Behrends, K. Lips, S. Fiechter and P. Bogdanoff, J. Am. Chem. Soc., 2016, 138, 635–640 CrossRef PubMed.
- E. Ashori, F. Nazari and F. Illas, Phys. Chem. Chem. Phys., 2017, 19, 3201–3213 RSC.
- L.-L. Liu, C.-P. Chen, L.-S. Zhao, Y. Wang and X.-C. Wang, Carbon, 2017, 115, 773–780 CrossRef.
- M. D. Esrafili, F. Sharifi and P. Nematollahi, Appl. Surf. Sci., 2016, 387, 454–460 CrossRef.
- S. N. Remello, T. Hirano, F. Kuttassery, Y. Nabetani, D. Yamamoto, S. Onuki, H. Tachibana and H. Inoue, J. Photochem. Photobiol., A, 2015, 313, 176–183 CrossRef.
- R. Paolesse, G. Pomarico, D. Monti, M. Bischetti, A. Savoldelli, R. Fronczek Frank, M. Smith Kevin, D. Genovese and L. Prodi, Chem. Eur. J., 2018, 24, 8438–8446 CrossRef PubMed.
- S. N. Remello, F. Kuttassery, T. Hirano, Y. Nabetani, D. Yamamoto, S. Onuki, H. Tachibana and H. Inoue, Dalton Trans., 2015, 44, 20011–20020 RSC.
- S. N. Remello, F. Kuttassery, S. Mathew, A. Thomas, D. Yamamoto, Y. Nabetani, K. Sano, H. Tachibana and H. Inoue, Sustainable Energy Fuels, 2018 10.1039/C8SE00102B , In press..
- J. Liu, X. Yang and L. Sun, ChemComm, 2013, 49, 11785–11787 RSC.
- Y. Tang, W. Chen, Z. Shen, S. Chang, M. Zhao and X. Dai, Carbon, 2017, 111, 448–458 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558–561 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef.
- J. Paier, R. Hirschl, M. Marsman and G. Kresse, J. Chem. Phys., 2005, 122, 234102 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef.
- D. Sheppard, P. Xiao, W. Chemelewski, D. D. Johnson and G. Henkelman, J. Chem. Phys., 2012, 136, 074103 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef.
- J. Kästner and P. Sherwood, J. Chem. Phys., 2008, 128, 014106 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef PubMed.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- Z. Hou, X. Wang, T. Ikeda, K. Terakura, M. Oshima and M.-a. Kakimoto, Phys. Rev. B, 2013, 87, 165401 CrossRef.
- H. S. Kim, H. S. Kim, S. S. Kim and Y.-H. Kim, Nanoscale, 2014, 6, 14911–14918 RSC.
- S. Wannakao, T. Nongnual, P. Khongpracha, T. Maihom and J. Limtrakul, J. Phys. Chem. C, 2012, 116, 16992–16998 CrossRef.
- H. Loirat, F. Caralp, M. Destriau and R. Lesclaux, J. Phys. Chem., 1987, 91, 6538–6542 CrossRef.
- Z. Lu, M. Yang, D. Ma, P. Lv, S. Li and Z. Yang, Appl. Surf. Sci., 2017, 426, 1232–1240 CrossRef.
- G. Xu, R. Wang, F. Yang, D. Ma, Z. Yang and Z. Lu, Carbon, 2017, 118, 35–42 CrossRef.
- Z. Lu, P. Lv, J. Xue, H. Wang, Y. Wang, Y. Huang, C. He, D. Ma and Z. Yang, RSC Adv., 2015, 5, 84381–84388 RSC.
- Z. Lu, P. Lv, Z. Yang, S. Li, D. Ma and R. Wu, Phys. Chem. Chem. Phys., 2017, 19, 16795–16805 RSC.
- Z. Lu, P. Lv, Y. Liang, D. Ma, Y. Zhang, W. Zhang, X. Yang and Z. Yang, Phys. Chem. Chem. Phys., 2016, 18, 21865–21870 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03265c |
| This journal is © The Royal Society of Chemistry 2018 |