
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly active Cu/ZnO–Al catalyst for methanol synthesis: effect of aging on its structure and activity
N. Mota,
R. Guil-Lopez,
B. G. Pawelec,
J. L. G. Fierro and
R. M. Navarro *
*
Instituto de Catálisis y Petroleoquímica-CSIC, Calle de Marie Curie, 2, Cantoblanco 28049, Madrid, Spain. E-mail: r.navarro@icp.csic.es
First published on 5th June 2018
Abstract
The influence of aging of precipitates on the physical and catalytic properties of a copper/zinc oxide-aluminium (Cu/ZnO–Al) catalyst with an optimized composition (low Al concentration, Cu/Zn/Al = 68/29/3) prepared using co-precipitation has been investigated in detail. The change in the structure of precipitates with aging (from amorphous zincian georgeite to crystalline zincian malachite) strongly influences the micro- and nano-structure (Cu and ZnO crystallite size, exposed copper surface area, Cu–ZnO interactions and stability of ZnO) of the final Cu/ZnO–Al catalysts obtained after calcination and reduction of the precipitates. The results of catalytic activity in methanol synthesis from syngas show the higher intrinsic activity of the catalysts derived from aged zincian malachite precipitates as consequence of the increase in the exposed copper surface area and the Cu–ZnO contacts. The stability of catalysts under the reaction conditions was also improved in the catalysts derived from precipitates aged after crystallization of malachite. The catalyst derived from the precipitate removed close to the point of crystallization of malachite shows very poor activity in the methanol synthesis as consequence of its segregated large Cu crystallites in low contact with ZnO derived from the absence of carbonate retention after calcination of the precipitate and the presence of sodium species after conventional washing which favour the strong sintering and crystallization of Cu during reduction.
1. Introduction
Copper/zinc oxide (Cu/ZnO) catalysts promoted by aluminium (Al) are widely studied for the synthesis of methanol (CH3OH) as well as in the water gas shift reaction. The Cu/ZnO–Al catalysts are usually prepared using co-precipitation of metal nitrate solutions with sodium or ammonium carbonate to generate mixed hydroxycarbonates followed by calcination and reduction to generate the active Cu/ZnO–Al catalysts.1 The preparation using co-precipitation of Cu–ZnO–Al catalysts has been the subject of an extensive optimization in research reported literature because the synthesis is complex and very sensitive to synthesis parameters (pH, temperature, stirring, aging and so on)2–4 and because the final activity of the Cu/ZnO–Al catalysts is determined by the nature and structure of the precursors formed during the precipitation.1,5Aging of precipitates in the mother liquor is an important step to achieve precursors which lead to the formation of Cu/ZnO–Al catalysts with high activity. The initially formed solids from co-precipitation of Cu, Zn and Al are amorphous hydroxycarbonates and they evolve during aging in contact with the mother liquor to crystalline forms with different composition, morphology and structure as a result of Ostwald ripening, dissolution/re-precipitation or agglomeration.1,6–9 Most of the published works about aging focused on binary Cu/ZnO as model systems representative of industrial formulations (Cu/Zn around 70/30) reported the slow transformation of the initially amorphous zincian georgeite [(Cu,Zn)7(OH)4(CO3)5] towards crystalline zincian malachite [(Cu,Zn)2(OH)2CO3] and aurichalcite [(Cu,Zn)5(OH)6(CO3)2] in different phase mixtures depending on the precipitation conditions. The point of crystallization of the precipitates correlates with a change in the pH during aging and is usually related to a sudden increase in the activity of the resulting Cu/ZnO catalyst. Some authors have identified zincian malachite as the relevant precursor phase1 whereas others10 have indicated that aurichalcite is a precursor of highly active Cu/ZnO catalysts. The efficient incorporation of Zn into crystalline malachite or aurichalcite is regarded as crucial in the synthesis of highly active CH3OH catalysts because atomic distribution of Cu and Zn in the precipitate will lead to porous copper(II) oxide (CuO) and ZnO uniformly mixed and distributed after the thermal decomposition of the precipitate.11 The uniform and porous mixture of CuO and ZnO ensures that catalysts with large accessible Cu surface area and highly dispersed metallic copper particles will be in close contact with ZnO with suitable reduction conditions. These Cu–ZnO contacts play an important role in the improvement of the intrinsic activity of the Cu active sites. The origin of the Cu–ZnO synergy has been investigated thoroughly in the last few decades and several models have been proposed in the literature to explain the role of Zn on the improvement of the Cu activity. These include defects and lattice strain in Cu particles caused by Zn,12,13 coverage of the Cu surface with metallic Zn atoms,14 preferential exposition of Cu surfaces on ZnO,15 stabilization of Cu+/Cu-species, and so on. In spite of the fact that the origin of the Cu–ZnO synergy has not yet been unequivocally established, the empirical studies performed up to now have revealed the importance of control in the composition and structure of the Cu/Zn precipitates as a way to optimize the activity of the catalysts.
Recently there have been attempts to rationalize the relationship between synthesis and structural properties of Cu–Zn hydroxycarbonates in the systematic works by Behrens et al.7,11,16 who have identified the zincian malachite as an essential precursor phase for highly active Cu/ZnO catalysts. According to these works, the crystallization of zincian malachite ensures the formation of the proper porous meso-structure of catalysts whereas the efficient incorporation of Zn into the malachite structure is responsible for a favourable nano-structure, which leads to Cu particles highly dispersed in close contact with ZnO.
Addition of Al (5–10 mol%) to Cu/ZnO catalysts has usually been considered to promote both catalyst activity and stability.17,18 However, recent studies also associate the promotion of Al in Cu/ZnO catalysts with structural and electronic effects.16 These studies demonstrated that low amounts of Al (2–4 wt%) promote the incorporation of Zn into zincian malachite during aging, thus, reducing the formation of aurichalcite and improving the formation of uniformly mixed CuO and ZnO. In parallel, the Al also acts as dopant in ZnO promoting the formation of defects in the ZnO in contact with Cu. The strong influence of aging of precipitates on the catalytic activity of the resulting catalysts has been widely described in the literature for the binary Cu/ZnO system but the detailed study of aging on the structure of precipitates for optimized catalyst compositions with low Al concentration (Cu/Zn = 70–0 wt%, Al = 2–4 wt%) does not appear so far, to have been studied systematically. In spite of the fact that the low amount of Al (3 wt%) does not change the global evolution from amorphous to crystalline precipitates during the aging observed for binary Cu/ZnO systems,19 the Al ions can have an influence on the kinetics and chemistry of precursors during aging. Thus, the purpose of this research is to investigate the transformations happening on precipitates during aging and their impact on the structural properties and activity corresponding to the optimized composition of Cu/Zn/Al catalysts according to the most recent developments in this field (Cu/Zn = 70–0 wt%, Al = 2–4 wt%). Precipitates aged at different times were characterized well and relationships between the properties of precipitates and the micro- and nano-structure of catalysts were established and related to the activity of the final catalysts for CH3OH synthesis from syngas.
2. Experimental
2.1 Preparation of precursors and catalysts
On the basis of the most recent developments in the chemistry of precipitation of mixed Cu/Zn hydroxycarbonate precursors for highly active Cu/ZnO–Al catalysts an optimized composition (Cu/Zn/Al = 68/29/3) was selected and prepared following the methodology recently proposed as the benchmark system for Cu-based catalysts.19 Hydroxycarbonate precursors were synthesized using co-precipitation from Cu, Zn, Al aqueous nitrate solutions, for example, copper(II) nitrate hemi-pentahydrate [Cu(NO3)2·2.5H2O, 1 M, Sigma-Aldrich >98%], zinc nitrate hexahydrate [Zn(NO3)2·6H2O, Sigma-Aldrich >99%] and aluminium nitrate nonahydrate [Al(NO3)3·9H2O >99% Fluka] with an aqueous solution of sodium carbonate (Na2CO3, 1.6 M, 99.999% PanReac AppliChem) as precipitating agent under controlled conditions (pH = 6.5, 65 °C). Both solutions were dosed into a continuously stirred flask with Milli-Q water (40 mL, 65 °C, pH = 6.5) to maintain and control the pH and temperature during the co-precipitation process. After complete addition of the precipitating solutions the precipitates were aged in a mother liquor under continuous stirring at 65 °C and samples of the precipitates were removed at 30, 70, 100 and 160 min. Evolution of pH and change in colour from blue to greenish were observed during aging with a minimum pH occurring at 50 min. Precipitate samples were filtered and washed with Milli-Q water following a conventional washing protocol to remove the residual sodium nitrate (NaNO3) on the precipitates (several washings, total water = 300 mL g−1 precipitate) and dried in air at 70 °C for 12 h. Precipitates were labelled as CZA-t (t = aging time).The catalyst precursors CuO/ZnO–Al were obtained from the thermal decomposition of the CZA-t precipitates. Decomposition of the precipitates was performed under static air at 340 °C for 2 h with a linear heating rate of 2 °C min−1. The catalyst precursors were labelled as CuO/ZnO–Al-t (t = aging time).
The final catalysts were obtained from the thermal reduction of CuO/ZnO–Al precursors under diluted hydrogen flow (2.21 vol% H2/Ar) following the temperature programme: heating to 150 °C (2 °C min−1) with subsequent heating up to 200 °C (1 °C min−1) and then holding at 200 °C for 2 h. The final temperature was fixed at 320 °C for the reduction of the sample of CuO/ZnO–Al-70 taking into account its lower reducibility. The catalysts were labelled as Cu/ZnO–Al-t (t = aging time).
2.2 Physicochemical characterization
X-ray powder diffraction (XRD) patterns of precipitates, calcined precursors and reduced catalysts were recorded using an X'Pert Pro PANalytical polycrystal diffractometer with an X'Celerator real time multiple strip (RTMS) detector and nickel filtered Cu Kα1 radiation (λ = 0.15406 nm, 45 kV, 40 mA). For each sample, Bragg angles between 5 and 90 (2θ) were scanned with a step size of 0.0335. Volume averaged crystallite sizes were determined using applying the Debye–Scherrer equation from the line broadening (βr) of the characteristic (020) malachite, (111) CuO and the (111) Cu-metal peaks after subtracting the instrument effect (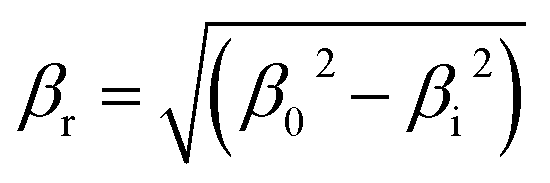 ; β0 = total line broadening, βi = instrumental broadening = 0.058 rad).
; β0 = total line broadening, βi = instrumental broadening = 0.058 rad).
Gravimetric analysis during the thermal decomposition of CZA-t precipitates was studied from 25 °C to 600 °C (at a heating rate of 10 °C min−1) under an oxygen/nitrogen (O2/N2) flow (20 vol%, 25 mL min−1) using a Mettler Toledo TGA/SDTA 851e thermo-gravimetric analyser.
The N2 adsorption–desorption isotherms of precipitates and calcined precursors were obtained at −196 °C under a whole range of relative pressures, using a Micromeritics ASAP 2100 automatic device on samples previously degassed at 120 °C for 12 h to remove all the gases adsorbed on their surfaces. The specific surface area values were calculated by applying the Brunauer–Emmett–Teller (BET) equation to the N2 adsorption isotherm within the relative pressures 0.05 < P/P0 < 0.30. Desorption data were used to determinate the pore size distribution by applying the Barrett–Joyner–Halenda (BJH) method, assuming a cylindrical pore model.
The surface morphology and size of precipitates were observed using field emission scanning electron microscopy (FE-SEM) with a Philips XL30 S-FEG microscope.
The nano-morphological characteristics of the reduced Cu/ZnO–Al-t catalysts were obtained using transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) with a TEM/STEM Jeol JEM-2100F operating at 200 kV accelerating voltage with a field emission gun (FEG), obtaining a point resolution of 0.19 nm.
Infrared spectroscopy was performed on the precipitates and calcined precursors using an attenuated total reflection accessory (ATR) on a JASCO FT/IR-6300 Fourier-transform infrared spectrometer (FTIR). The ATR accessory had a diamond plate and a zinc selenide crystal. The system was purged with N2 during the experiments. The background and the sample IR spectra were recorded with a MCT_M detector with 200 accumulations, a resolution of 4 cm−1 and a scan range of 4000–400 cm−1.
Temperature programmed reduction experiments were carried out using a semi-automatic PID Eng & Tech apparatus equipped with a U-shaped quartz reactor and a thermal conductivity (TC) detector. Before reduction experiments the calcined samples (20 mg) were pre-treated under a helium (He) flow (30 mL min−1) at 120 °C for 20 min to remove water and other adsorbed contaminants and then cooled down to room temperature. Temperature programmed reduction (TPR) profiles were obtained by heating the samples in a 10 vol% H2/Ar flow (50 mL min−1) from room temperature to 300 °C at a linearly programmed rate of 2 °C min−1. The effluent gas was passed through a cold trap to remove water before measuring the amount of H2 consumed during reduction by the TC detector.
Specific Cu surface areas in reduced catalysts were determined using nitrous oxide (N2O) chemisorption after TPR experiments. The reduced catalysts were passivated with a flow of diluted N2O/Ar (2 vol%, 7.5 mL min−1) at room temperature for 15 min. After that the samples were flushed with He for 30 min to remove the residual N2O and subsequently reduced by a H2/Ar flow (10 vol%, 50 mL min−1) up to 280 °C for 5 min at a heating rate of 10 °C min−1. The H2 consumption corresponding to the reduction of passivated samples was equivalent to the amount of N2O consumed during surface passivation. The Cu0 surface area was calculated assuming a molar stoichiometry of Cu(s)/N2O = 2 and an average value of 1.47 × 1019 copper atoms per m2 for the surface density of the copper metal.20 The values were normalized to the weight of the calcined precursor loaded in the reactor.
2.3 Methanol synthesis tests
Catalytic activity tests were performed in a fixed-bed continuous flow stainless steel reactor (8.9 mm id) with a coaxially centred thermocouple in contact with the catalytic bed. The reactor was loaded with 0.2 g of calcined precursor diluted with silicon carbide (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 vol) to avoid adverse thermal effects. Prior to the catalytic measurement, the calcined precursors were reduced in a diluted H2 flow (2.21 vol% H2/Ar, 50 mL min−1) following the temperature programme: firstly heating to 150 °C (2 °C min−1) with a subsequent heating to 200 °C (320 °C for the CuO/ZnO–Al-70 sample) (1 °C min−1) and then holding for 2 h. The pre-treatment gases were flushed from the reactor with N2 before the reaction mixture was added. After reduction a syngas mixture with a composition (vol%) of 4.5% carbon dioxide (CO2), 22.0% carbon monoxide (CO), 58.8% H2 and 14.7% N2 was introduced in the reactor (75 mL min−1) and the pressure was raised to 30 bar. Activity was measured at 250 °C and the reaction was maintained for 8 h. The reaction gas products were regularly determined on-line using gas chromatography (GC) with a Varian 450-GC with TCD equipped with a QS-bond column (CO2, CH3OH and water) and molecular sieve 5A (H2, O2, N2, CO) capillary columns connected in series, using He as carrier gas. Activity data were reported as the CH3OH production rate normalized to the weight of calcined precursor loaded in the reactor (μmol CH3OH/min gcat).
:3 vol) to avoid adverse thermal effects. Prior to the catalytic measurement, the calcined precursors were reduced in a diluted H2 flow (2.21 vol% H2/Ar, 50 mL min−1) following the temperature programme: firstly heating to 150 °C (2 °C min−1) with a subsequent heating to 200 °C (320 °C for the CuO/ZnO–Al-70 sample) (1 °C min−1) and then holding for 2 h. The pre-treatment gases were flushed from the reactor with N2 before the reaction mixture was added. After reduction a syngas mixture with a composition (vol%) of 4.5% carbon dioxide (CO2), 22.0% carbon monoxide (CO), 58.8% H2 and 14.7% N2 was introduced in the reactor (75 mL min−1) and the pressure was raised to 30 bar. Activity was measured at 250 °C and the reaction was maintained for 8 h. The reaction gas products were regularly determined on-line using gas chromatography (GC) with a Varian 450-GC with TCD equipped with a QS-bond column (CO2, CH3OH and water) and molecular sieve 5A (H2, O2, N2, CO) capillary columns connected in series, using He as carrier gas. Activity data were reported as the CH3OH production rate normalized to the weight of calcined precursor loaded in the reactor (μmol CH3OH/min gcat).
3. Results and discussion
3.1 Characterization of precipitates
The evolution of the crystalline structure of precipitates during aging studied using XRD shows the well-known transformation by partial dissolution/re-crystallization of amorphous hydroxycarbonates to crystalline species (Fig. 1). Crystallization was associated with the minimum pH observed at 50 min during aging.6 The XRD pattern of the CZA precipitate removed before the minimum pH (CZA-30, Fig. 1a) revealed an amorphous nature which was consistent with the formation of zincian georgeite and/or Zn hydroxy-rich carbonates because these amorphous forms were usually detected as initial products in the co-precipitation of binary Cu/ZnO systems.21 After the pH drop, the typical pattern of the zincian malachite-like phase [(Cu,Zn)2(OH)2CO3] appeared in the precipitates (CZA-70, CZA-100, CZA-160, Fig. 1b–d).6,22 Diffraction peaks corresponding to additional phases such as hydrotalcite or aurichalcite were not observed in any of the CZA precursors. This is in agreement with results found by Behrens et al.16 who found that low amounts of Al facilitated the incorporation of Zn into the malachite structure, thus inhibiting the formation of crystalline Zn-rich by-product phases. Only in the precipitate examined close to the crystallization point (CZA-70, Fig. 1b) were small diffractions corresponding to crystals of NaNO3 (JCPDS 36-1454) observed. Residual NaNO3 was only observed in this precipitate despite the fact that all the CZA precipitates were subjected to the same intensive washing protocol. This points to the fact that there is a particular difficulty in the removal of Na+ ions in the precipitate removed close to the malachite crystallization point and it could be related to the role of Na+ during the first steps of the crystallization of malachite structures from georgeite as will be explained next. Removal of NaNO3 from the CZA-70 sample required a more aggressive washing protocol but this washing process was discarded because it could act as an additional aging process which could cause rearrangement of the precipitate, and thus, limiting the comparison between the CZA precipitates obtained using the same conventional washing protocol. In this way new insights can be added to the known detrimental effect of Na+ ions on the catalytic performance.24,25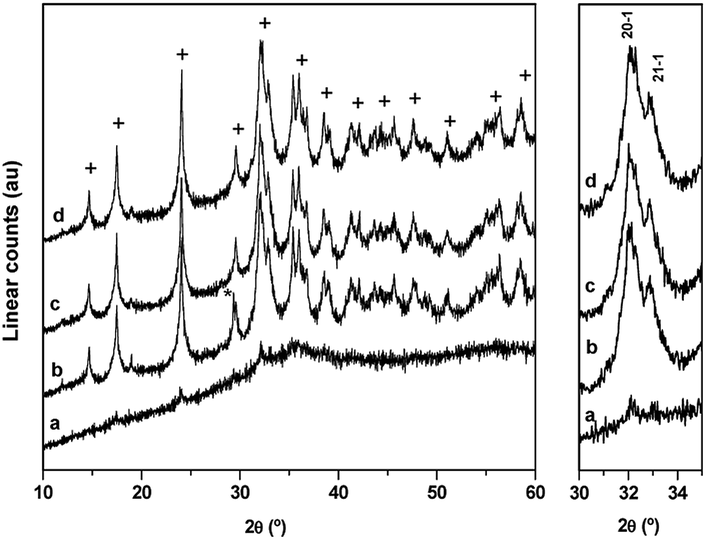 | ||
| Fig. 1 XRD patterns of CZA-t precipitates: (a) CZA-30, (b) CZA-70, (c) CZA-100, (d) CZA-160 (+ zincian-malachite * NaNO3). | ||
Domain sizes of the zincian-malachite phase on CZA precipitates calculated using the Scherrer equation applied to the (020) reflection (Table 1) do not vary during aging. Only the position of the (20−1) reflection of the malachite slightly shifted to higher angles when the precursors were aged. Because the position of the (20−1) reflection is a measure of the degree of Zn incorporation into the zincian malachite-phase,7 the shift observed with aging implied a slight increase in the zinc content in the malachite (Table 1). However, the co-insertion of small amounts of Al3+ ions into the malachite structure could also be possible because it has been reported the favourable incorporation of Al ions for a compositional window similar to that used in this work.16
| (020) size (nm) | (20−1) (2θ) | Zn in zincian malachite (%) | BET (m2 g−1) | Pore diameter (nm) | |
|---|---|---|---|---|---|
| CZA-30 | — | — | — | 31.3 | 20.7 |
| CZA-70 | 28.4 | 31.99 | 18.8 | 74.4 | 13.9 |
| CZA-100 | 28.8 | 32.07 | 21.0 | 86.0 | 16.5 |
| CZA-160 | 28.7 | 32.21 | 24.8 | 75.4 | 17.5 |
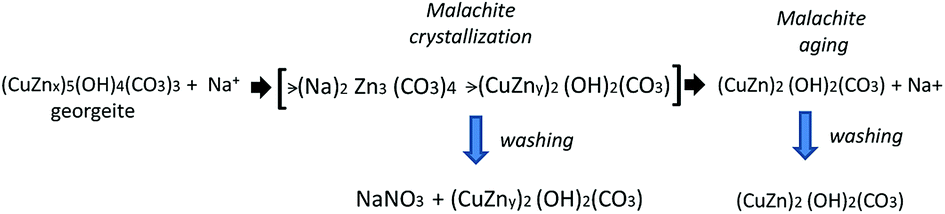
The FT-IR spectrum of the amorphous CZA precipitate (CZA-30, Fig. 2a) shows bands at 1470, 1394 and 831 cm−1 which are characteristic of carbonate vibrations of zincian georgeite [(Cu,Zn)7(OH)4(CO3)5].23,26,30 The FT-IR spectra of the CZA precipitates after crystallization (CZA-70, CZA-100 and CZA-160, Fig. 2b–d) agree well with those of zincian malachite.27,28 In agreement with the data reported in the literature the characteristic bands of carbonates appear at 1515, 1428 and 1390 cm−1 (asymmetric C–O stretching mode ν3), at 820 cm−1 (out-of-plane OCO bending mode), and at 742 cm−1 (asymmetric OCO bending mode).
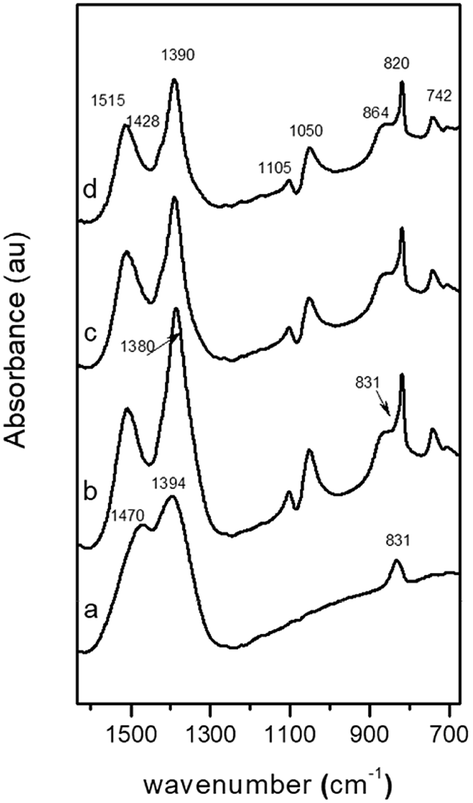 | ||
| Fig. 2 FT-IR spectra (carbonate region) of precipitates: (a) CZA-30, (b) CZA-70, (c) CZA-100, (d) CZA-160. | ||
The three OH deformation bands observed at 1025, 1050 and 864 cm−1 were also in agreement with the Me–OH bending modes assigned to zincian malachite. As observed in Fig. 2, the intensity and positions of the ν3 carbonate bands of zincian malachite in CZA precipitates slightly decrease and shift with aging. These changes agree with the previous XRD patterns which revealed some changes in the Zn content in the malachite-like phase during aging. In the spectrum of the CZA-70 precipitate, additional bands at 1380 and 833 cm−1 characteristic of NO3− vibrations were detected. The presence of aurichalcite in the CZA precipitates could be discarded from the analysis of the fingerprint bands for aurichalcite at 1208 cm−1 (M–OH deformation) and at 970 cm−1 (OH deformation)29 which were absent in all the IR spectra of the CZA precipitates.
Morphological evolution of the CZA precipitates with aging was determined using SEM. SEM images of the CZA precursor removed before crystallization (CZA-30, Fig. 3a) show irregular globular shaped particles which were transformed after crystallization into needle shaped particles (CZA-70 and CZA-100, Fig. 3b and c). This evolution agrees with the microstructure associated with the crystallization of zincian malachite described in the literature.11
 | ||
| Fig. 3 SEM images showing the morphology of precipitates: (a) CZA-30, (b) CZA-70, (c) CZA-100. | ||
The microstructure of CZA precipitates with aging results in variations in their textural properties derived from their N2 adsorption–desorption isotherms (Table 1). The BET surface area of the precipitates increased markedly after crystallization (from 31.3 m2 g−1 in the amorphous precipitate to 73–85 m2 g−1 after crystallization of zincian malachite). The CZA precipitates aged after crystallization (CZA-70, CZA-100 and CZA-160) also showed textural differences between them with a higher surface development in the CZA precipitate aged for 100 min.
The thermogravimetric analysis (TGA) curves of the decomposition of CZA precipitates in air are presented in Fig. 4. The decomposition pattern of the amorphous CZA precipitate (CZA-30, Fig. 4a) and its overall mass loss (27.7%) agree fairly well with that reported for zincian georgeite [(Cu,Zn)7(OH)4(CO3)5].30 The decomposition patterns of the CZA precipitates obtained after crystallization were different depending on their aging time. The precipitate removed close the crystallization point (CZA-70 Fig. 4b) decomposed completely in a single step with the maximal weight loss rate around 275 °C. This fact is in agreement with the work of Bems et al.6 which showed the decomposition of malachite precursors at low temperature (250–310 °C) when the precipitates contained NaNO3. The authors explained this effect as inhibition of the decarbonation processes of precursors at high temperature by the nitrate impurities. However, the precipitates aged after the crystallization point (CZA-100 and CZA-160, Fig. 4c and d) decomposed in two steps. According to the literature the first step corresponded to dehydroxylation and decarbonation processes whereas the second decomposition step at high temperatures (400–600 °C) was associated with the decarbonation of an anion modified mixed metal oxides31 or a mixture of carbonates and oxides32 formed from the previous decomposition step.
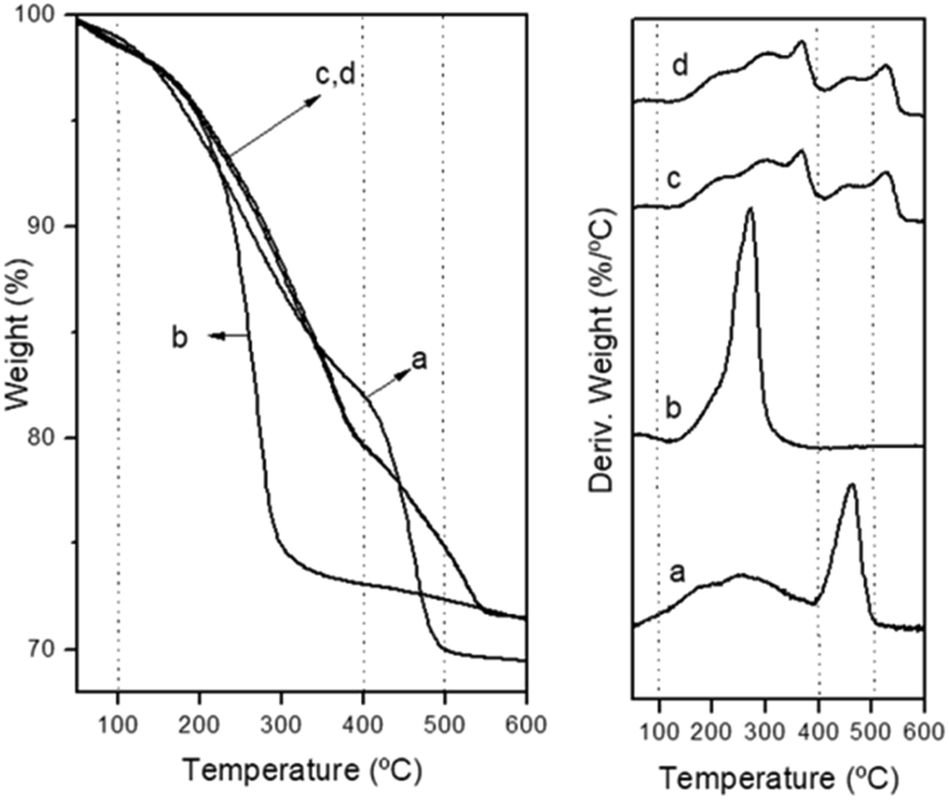 | ||
| Fig. 4 Thermogravimetric and derivative curves corresponding to the decomposition of precipitates: (a) CZA-30, (b) CZA-70, (c) CZA-100, (d) CZA-160. | ||
It is interesting to note that part of the decomposition in these precipitates takes place at very high temperatures (>500 °C) and the decomposition profiles did not correlate with the variations in the incorporation of Zn in the malachite structure observed in the precipitates (Table 1), as usually reported in the literature.5,6 This behaviour could be derived from structures not detected by XRD (in low proportion or amorphous) because differences in the transport conditions during the decomposition can be ruled out because all the crystallized precipitates show a similar texture and shape (Table 1, Fig. 3).
The phase transitions during aging observed in this research were analogous to those described in the well-studied binary Cu–Zn systems6,33 in which the ageing process was differentiated in two sequential steps separated by a period of pH drop and colour change. In our case the CZA precipitate removed before the pH drop was present as amorphous hydroxycarbonate (Fig. 1) with the IR spectra (Fig. 2) and TGA-temperature programmed oxidation (TPO) profile (Fig. 4) characteristic of zincian georgeite whereas the precipitates removed after the pH drop showed crystalline zincian malachite which slowly transformed with ageing. In the observed evolution of the precipitates with aging it is important to note the presence of significant amounts of NaNO3 in the sample removed close to the point of crystallization of malachite (CZA-70). The conventional washing protocol used for the removal of NaNO3 was effective for all CZA precursors except for sample CZA-70, which indicated the particular difficulty for the removal of NaNO3 in this sample. One hypothesis to explain the existence of NaNO3 in the CZA-70 sample could be related to the important role of Na+ ions in the initial crystallization of zincian malachite from amorphous georgeite. It was reported that the initial crystallization of zincian malachite from georgeite can happen faster in the presence of Na+ through the formation of an intermediate zinc sodium salt [Na2Zn3(CO3)4].7,23 In this scenario NaNO3 could be the result of the evolution of the intermediate Zn-sodium salt generated in the initial crystallization step which transformed to more favourable hydroxycarbonates during washing8 (Scheme 1). Upon aging the concentration of the intermediate Zn-sodium salts decreased, incorporating its Zn into the zincian malachite by dissolution and accordingly it could be the reason for the absence of NaNO3 in the precipitates aged after the crystallization of malachite from georgeite. The progressive increase in the insertion of Zn into the zincian malachite structure, from 18.8% to 24.8%, observed in the course of aging after crystallization of malachite was in agreement with the mechanism described previously.
 | ||
| Scheme 1 Evolution of precipitates during the initial crystallization and aging of malachite and the subsequent washing. | ||
3.2 Characterization of calcined precursors and reduced catalysts
The CZA precipitates were calcined at 340 °C to minimize thermal sintering of the CuO phase and considering their TGA profiles they would have different carbonate retention in the catalysts after calcination. The differences in carbonate retention after calcination of the precipitates have important implications in the segregation and crystallization of CuO and ZnO as discussed next. Carbonate retention on calcined CuO/ZnO–Al precursors was studied using FT-IR (Fig. 5). The IR spectrum of the calcined precursor from amorphous georgeite (CuO/ZnO–Al-30, Fig. 5a) shows the same carbonate bands at 1490, 1385 and 824 cm−1 previously observed in the precipitate (Fig. 2a) which indicated that a large proportion of the carbonates remained in the precursor after calcination. However, a marked decrease in the carbonate bands intensity can be seen in the precursor calcined from the precipitate removed close to the crystallization point (CuO/ZnO–Al-70, Fig. 5b). In this sample sharp peaks at 1360 cm−1 and 824 cm−1 were observed which were associated with the NaNO3 previously detected using XRD on the precipitate, because the temperature used in the calcination was below the temperature used for the thermal decomposition of NaNO3 (about 450 °C).34 The calcined samples aged for 100 min and 160 min showed reduction in the carbonate zone when compared to their precipitates and exhibited the main residual carbonate bands at 1482 cm−1 and 1415 cm−1.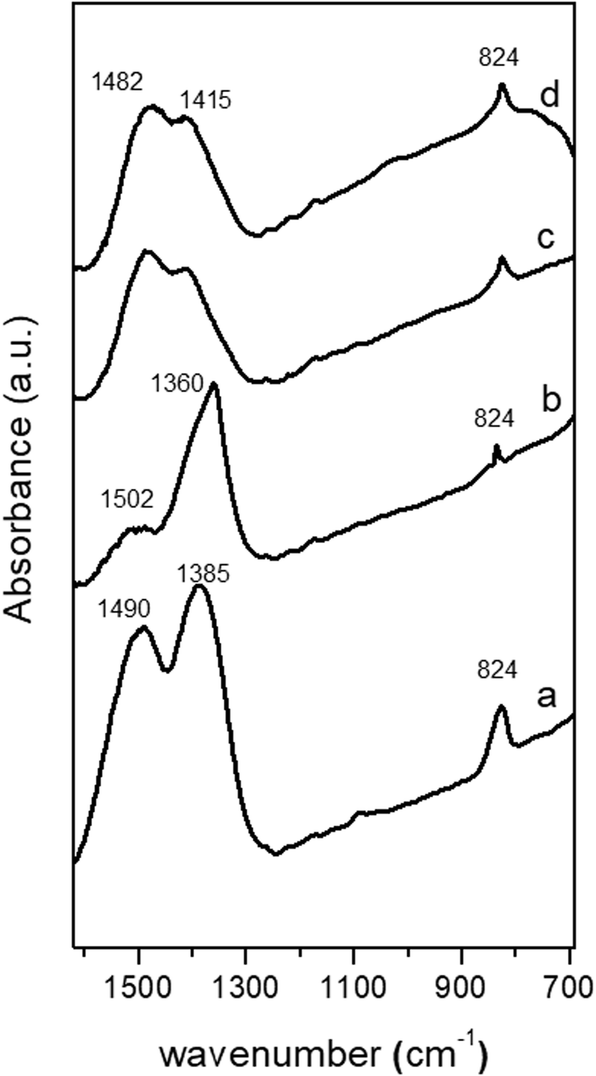 | ||
| Fig. 5 FT-IR spectra (carbonate region) of the calcined precursors: (a) CuO/ZnO–Al-30, (b) CuO/ZnO–Al-70, (c) CuO/ZnO–Al-100, (d) CuO/ZnO–Al-160. | ||
The XRD patterns of the calcined CuO/ZnO–Al precursors are shown in Fig. 6. The XRD pattern of the calcined precursor derived from the amorphous precipitate (CuO/ZnO–Al-30, Fig. 6a) displays ill defined, broad and low intensity diffraction peaks of CuO (JCPDS 48-1548) which implied low crystallinity and size. A significant increase in the crystallinity of CuO and ZnO was observed on the calcined precursor derived from the precipitate removed close to the crystallization point (CuO/ZnO–Al-70, Fig. 6b). This precursor also shows a sharp diffraction peak corresponding to NaNO3, with a similar intensity to that observed in the precipitate which confirms the stability of NaNO3 at the temperature used in the calcination of the precipitate. The calcined precursors aged for 100 min and 160 min (Fig. 6c and d) display a diffraction profile with broad reflections of CuO similar to that observed in the precursor derived from the amorphous precipitate which indicated small crystalline domains and a good intergrowth of the CuO and ZnO phases. No diffraction peaks corresponding to any Cu–Zn carbonates were detected which indicated that residual carbonates on the calcined precursors must be amorphous. Table 2 shows the variation of the domain size of CuO in the calcined CuO/ZnO–Al-t precursors. Because the diffraction of CuO (002) overlapped with that of ZnO (101), the reflection of CuO (111) was used to determine the domain size of CuO. Small crystalline domains of CuO, of about 4 nm, were observed in the calcined precursors derived from the amorphous precipitate (CuO/ZnO–Al-30) as well as in those calcined from the aged precipitates in the form of crystalline zincian malachite (CuO/ZnO–Al-100 and CuO/ZnO–Al-160). The low domain size of CuO agrees with the behaviour which correlates with the generation of CuO of low crystalline size after calcination of zincian georgeite23 or zincian malachite precipitates1,5,35 because these precursors retained the high temperature carbonates after calcination that preserved the segregation and crystallization of CuO. The calcined CuO/ZnO–Al-70 precursor exhibits particles of CuO with a large domain size (7.5 nm) segregated from ZnO. This is in agreement with previous studies found in the literature which showed the negative impact of NaNO3 on the CuO segregation after calcination of the precipitate.35,36 In these studies it was assumed that NaNO3 promoted the CuO agglomeration without indicating the exact cause of this behaviour. In this case it was evident that the crystalline growth of CuO observed in the calcined CuO/ZnO–Al-70 precursor was associated with the absence of carbonate retention after calcination, because as is shown above (Fig. 4b), the nitrate impurities inhibited the decarbonation of the precursor at high temperatures, which facilitated the segregation and crystallization of CuO during the calcination process.
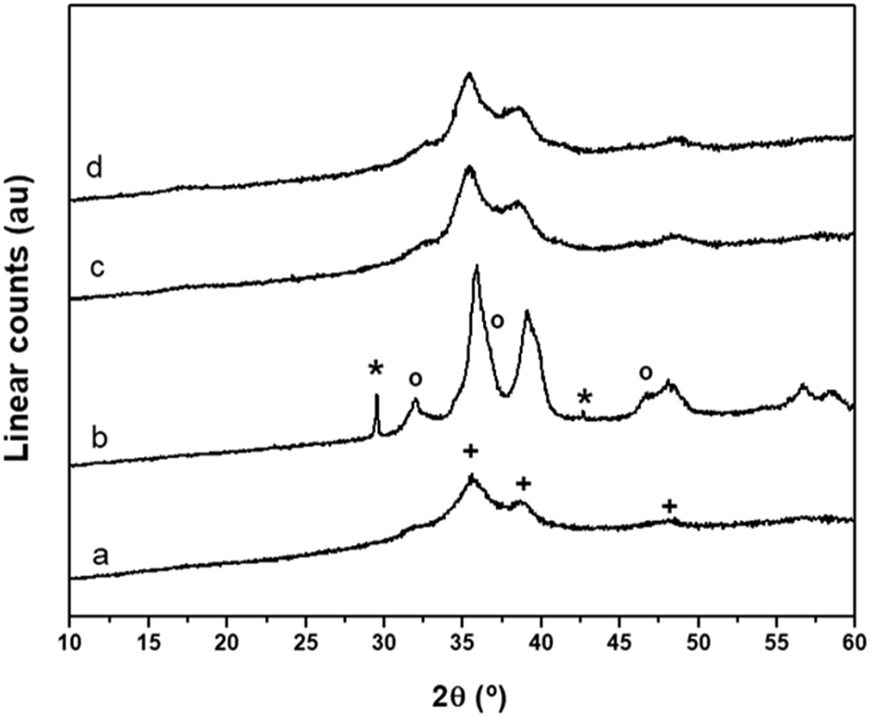 | ||
| Fig. 6 XRD patterns of calcined precursors: (a) CuO/ZnO–Al-30, (b) CuO/ZnO–Al-70, (c) CuO/ZnO–Al-100, (d) CuO/ZnO–Al-160 (+ CuO, o ZnO, * NaNO3). | ||
| BET (m2 g−1) | Pore diameter (nm) | CuO size (nm) | Carbonates (wt%) | |
|---|---|---|---|---|
| CuO/ZnO–Al-30 | 29.5 | 11.2 | 4.1 | 12.5 |
| CuO/ZnO–Al-70 | 40.3 | 17.4 | 7.5 | 1.6 |
| CuO/ZnO–Al-100 | 83.0 | 12.6 | 4.6 | 8.1 |
| CuO/ZnO–Al-160 | 74.5 | 12.4 | 4.5 | 8.1 |
The surface areas of the calcined CuO/ZnO–Al-t precursors showed values (Table 2) similar to those found in the precipitates from which they were derived (Table 1). The specific surface area of the calcined precursor derived from georgeite (CuO/ZnO–Al-30, Table 2) was much lower than those derived from the aged precipitates in the form of crystalline zincian malachite (CuO/ZnO–Al-100 and CuO/ZnO–Al-160) despite the fact that they have a similar CuO domain size. This means that the surface area in the calcined precursors was developed from the meso-structuring of precipitates and not from the nano-structuring of CuO after calcination. The meso-structuring in the calcined precursors was probably related to the residual carbonate structures which remained after calcination.34 Following this reasoning it appeared that the calcined CuO/ZnO–Al-70 precursor which showed a lower surface area (40.3 m2 g−1) compared to its precipitate (74.4 m2 g−1), which was expected because of the absence of residual carbonates after calcination which lead to a drop in the geometric surface area of CuO particles derived from the nitrate impurities which lead to segregation and crystallization of CuO during calcination as previously discussed.
The reduction behaviour of the calcined CuO/ZnO–Al-t precursors was investigated using temperature-programmed reduction (Fig. 7). Calcined CuO/ZnO–Al precursors derived from the zincian malachite precipitates (CuO/ZnO–Al-100 and CuO/ZnO–Al-160) displayed narrow and symmetrical peaks at a low reduction temperature of 161–165 °C, which was typical for the reduction of small CuO nanoparticles in close contact with ZnO.36 A small shoulder before the main reduction peak was also observed and this was because of the reduction of highly dispersed CuO or because of the reduction of carbonate species remaining after calcination leading to defect-rich oxide crystallites.37 The calcined CuO/ZnO–Al-30 precursor also showed a symmetrical reduction peak (Fig. 7a) but at a slightly higher reduction temperature (179 °C) and this was probably associated with the lower accessibility to the CuO particles as consequence of the decrease in the surface area and/or lower Cu dispersion in contact with ZnO.38,39 The CuO/ZnO–Al-70 precursor showed a very different reduction profile with a broad reduction, with the onset of reduction at 225 °C, composed of two overlapped reduction peaks. The first peak includes the reduction of the CuO species, which reduced at a higher temperature as a consequence of their low dispersion and contact with ZnO, whereas the subsequent reduction peak contained the reduction of NaNO3 because the metal Cu could help in the reduction of the nitrate at these temperatures.40 The excess in the amount of hydrogen consumed in this sample also suggests the reduction of the nitrate in the TPR profile of the CuO/ZnO–Al-70 precursor.
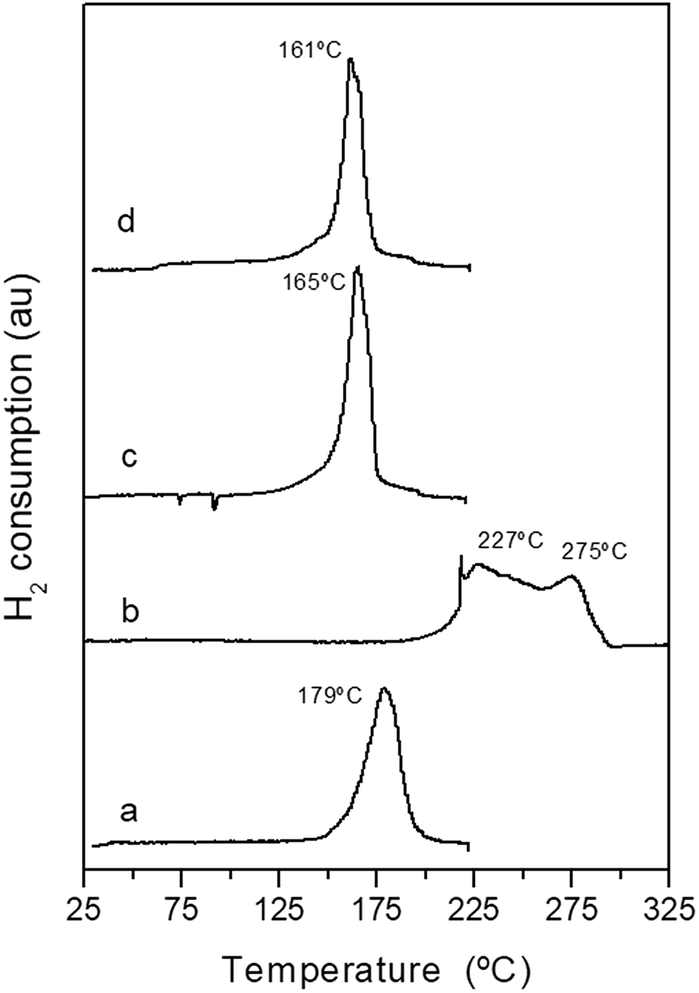 | ||
| Fig. 7 TPR profiles of the calcined precursors: (a) CuO/ZnO–Al-30, (b) CuO/ZnO–Al-70, (c) CuO/ZnO–Al-100, (d) CuO/ZnO–Al-160. | ||
The TEM micrographs of the reduced CuO/ZnO–Al catalysts are depicted in Fig. 8. Reduced catalysts derived from the georgeite precipitate (Cu/ZnO–Al-30, Fig. 8a) and the crystalline zincian malachite (Cu/ZnO–Al-100, Fig. 8c) show metallic Cu and ZnO nanoparticles of a similar size and morphology. Both reduced catalysts show irregular nanoparticles of Cu with a mean particle size of about 8–10 nm in epitaxial contact with small ZnO clusters, and this is in agreement with the nanostructure of the calcined precursors. The high temperature carbonates and the close interaction of CuO–ZnO observed in their calcined precursors may act as growth inhibitors of Cu particles during the reduction.34 However, the Cu particles in the Cu/ZnO–Al-70 reduced catalyst (Fig. 8b) appeared as large round particles, with a mean size interval from 10 nm to 155 nm, which were isolated from irregular and large ZnO particles as expected. Considering the segregation and crystallization of CuO observed in their calcined precursor together with the exothermic reduction of the nitrate impurities both favoured the sintering of Cu particles during reduction.
 | ||
| Fig. 8 Representative TEM images of reduced catalysts: (a) Cu/ZnO–Al-30, (b) Cu/ZnO–Al-70, (c) Cu/ZnO–Al-100. | ||
The specific Cu surface areas of the reduced catalysts determined by N2O chemisorption are shown in Table 3. It is important to note that the N2O chemisorption also titrates, in addition to metallic Cu sites, reduced Zn or ZnOx species in contact with the Cu nanoparticles.41 Therefore the variations in specific Cu surfaces areas listed in Table 3 also include changes in the ZnOx sites. As expected, the differences in the accessible surface area and changes in the Cu particle size resulted in substantial changes in the specific Cu surface area of reduced Cu/ZnO–Al-t catalysts. Obviously the reduced Cu/ZnO–Al-70 catalyst with segregated large Cu crystallites and low contact with ZnO shows a very poor specific Cu surface area. However, the Cu/ZnO–Al-100 and Cu/ZnO–Al-160 catalysts show the higher specific area of Cu, which correlated with their small Cu particles and better surface area development. The contribution of ZnOx species to the values of Cu surface area of Cu/ZnO–Al-100 and Cu/ZnO–Al-100-160 catalysts cannot be discarded because they are promoted by the possible insertion of Al3+ ions into ZnO strongly favoured in the compositional window Al/Cu/Zn used in this work.16 It should be noted that the Cu/ZnO–Al-100 and Cu/ZnO–Al-160 catalysts exhibited very high specific Cu surface areas which agreed well with other published data for highly active CH3OH synthesis catalysts.19
| Cu size (nm) | Cu surface area (m2 g−1) | |
|---|---|---|
| Cu/ZnO–Al-30 | 9 | 21.7 |
| Cu/ZnO–Al-70 | 35 | 7.3 |
| Cu/ZnO–Al-100 | 10 | 38.3 |
| Cu/ZnO–Al-160 | n.m. | 38.5 |
3.3 Methanol synthesis tests
The activity of the catalysts derived from precipitates with different aging was evaluated for CH3OH synthesis at 30 bars and 250 °C and this data are shown in Table 4 and Fig. 9. Analyses of products in all the Cu/ZnO–Al catalysts show CH3OH selectivity beyond 99% without higher alcohol formation. The results of the catalytic assays (Fig. 9) reveal that the Cu/ZnO–Al catalysts present strong differences in their efficiency for CH3OH production, which was expected given the changes in their meso- and nano-structure. The catalyst derived from the precipitate removed close to the crystallization point (Cu/ZnO–Al-70) was almost inactive for the CH3OH synthesis whereas the catalysts derived from crystalline precipitates aged for more than 50 min after the crystallization point (Cu/ZnO–Al-100 and Cu/ZnO–Al-160) were the most active samples with CH3OH rates higher than 970 μmol CH3OH/min gcat. It should be noted the high activity for CH3OH synthesis of the Cu/ZnO–Al-100 and Cu/ZnO–Al-160 catalysts, compared well with the best activity results published in the literature.19 The catalyst derived from the amorphous georgeite (Cu/ZnO–Al-30) showed a CH3OH rate less than half of the level achieved over the most active Cu/ZnO–Al-100 and Cu/ZnO–Al-160 catalysts. This result contrasts with the work published by Kondrat et al.30 which showed that the catalyst derived from zincian georgeite was more active than the catalyst derived from zincian malachite. The difference between the activity found in this study with our work is probably related to the use in the first case of the supercritical antisolvent precipitation with CO2 which leads to stable georgeite with better Cu–Zn mixing than that obtained with our standard co-precipitation method. The georgeite prepared by co-precipitation could suffer from uncontrolled aging during sampling, compared to other hydroxycarbonates resulting in less homogenised Cu–Zn mixing. Zincian georgeite is more stable than pure georgeite but recrystallization to other hydroxycarbonates and malachite can occur in hours at typical catalyst preparation temperatures (60–80 °C).21| COx conversion (%) | CH3OH yield (μmol min−1 g−1) | Turnover frequency (TOF) (s−1) | Relative activity (6 h per initial) | |
|---|---|---|---|---|
| Cu/ZnO–Al-30 | 7.7 | 492.0 | 15.5 | 81.6 |
| Cu/ZnO–Al-70 | 0.01 | 4.2 | 0.34 | — |
| Cu/ZnO–Al-100 | 16.8 | 984.7 | 17.5 | 99.6 |
| Cu/ZnO–Al-160 | 15.1 | 988.5 | 17.5 | 98.1 |
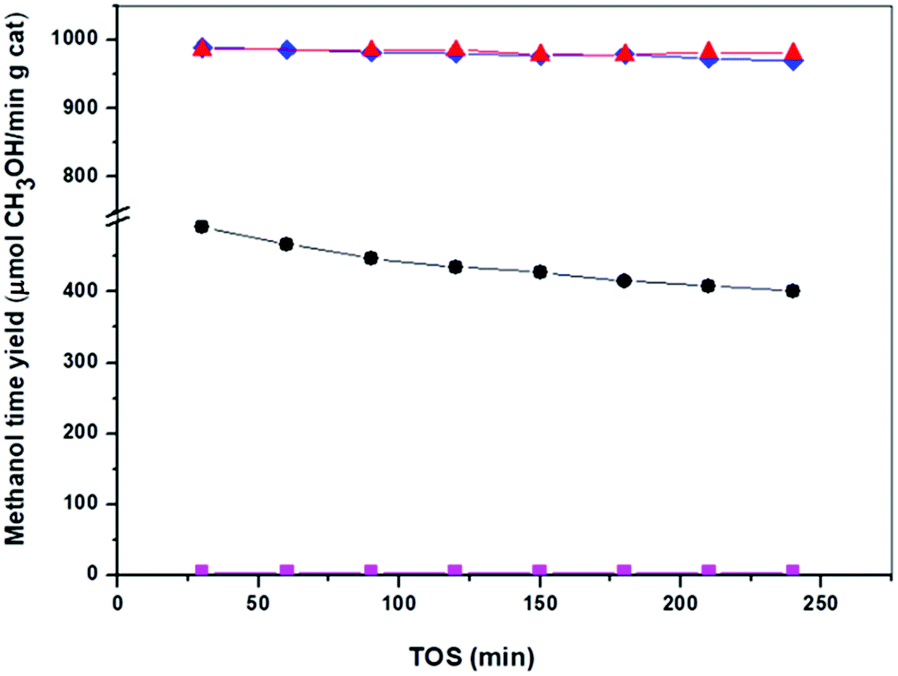 | ||
| Fig. 9 Methanol time-yield from syngas on reduced catalysts: (●) Cu/ZnO–Al-30, (■) Cu/ZnO–Al-70, (▲) Cu/ZnO–Al-100, (*) Cu/ZnO–Al-160, (syngas (% mol): H2 = 58.8, CO = 22, CO2 = 4.5, N2 = 14.7, P = 30 bar, 250 °C, gas hourly space velocity (GHSV) = 32700 h−1). | ||
The initial intrinsic activities of Cu/ZnO–Al catalysts normalized to apparent Cu surface area were calculated and are presented in Table 4. The initial intrinsic activities of Cu/ZnO–Al catalysts were higher for samples derived from precipitates aged after the crystallization of zincian malachite, with the exception of the Cu/ZnO–Al-70 catalyst which showed very poor intrinsic activity, even lower that the catalyst derived from georgeite precipitate. The reduced intrinsic activity of the Cu/ZnO–Al-70 sample correlated well with its low Cu surface area and low development of Cu–ZnO interactions derived from the nitrate impurities present in the precipitate which were difficult to remove using a conventional washing protocol. The presence of Na+ in this sample could also contribute to the low activity because these species can act as poison by increasing the basicity of the catalyst or modifying the Cu–ZnO interactions.23 The initial intrinsic activity of the catalysts derived from aged malachite precipitates, Cu/ZnO-100 and Cu/ZnO-160, was 10% higher than the intrinsic activity of the catalyst derived from the amorphous georgeite. It is widely accepted that CH3OH synthesis on Cu appears to be sensitive to the Cu surface structure42,43 and therefore the differences in the intrinsic activity observed in the catalysts were derived from changes in the “quality” of the Cu surfaces as a result of the different structure of the aged precipitates. Mechanistic models in the literature indicated that the increase in the intrinsic activity of Cu was mainly because of the formation of (partially) reduced Zn species in contact with Cu41 and/or defects in Cu particles induced by their contact with ZnO.13
The results of the characterization of Cu/ZnO–Al catalysts derived from zincian georgeite and zincian malachite show in both cases, that there were small Cu particles in contact with ZnO. However, a slight increase in the reducibility of the Cu particles and their contact with ZnO was observed in the Cu/ZnO–Al-100 and Cu/ZnO–Al-160 catalysts derived from zincian malachite. Thus, the improvement in reducibility and contact with ZnO observed in the Cu particles of the catalysts derived from aged malachite could be the origin of the observed improvement in the intrinsic activity compared to that derived from the amorphous georgeite. Nevertheless, the relatively high initial intrinsic activity of the catalyst derived from georgeite should be emphasized. Georgeite is usually considered as a poor precursor for Cu–Zn catalysts because it suffers uncontrolled ageing after co-precipitation,21 however, the preparation of stable georgeite using supercritical antisolvent precipitation which was found to be a precursor of highly active catalysts for CH3OH synthesis, has been recently reported.30
Differences in the stability of the Cu/ZnO–Al catalysts under different reaction conditions were also observed (Fig. 9). The catalyst derived from the amorphous georgeite (Cu/ZnO–Al-30) lost about 20% of the initial activity after 4 h whereas Cu/ZnO–Al-100 and Cu/ZnO–Al-160 catalysts exhibited more stable behaviour losing less than 2% of their initial activity. It is well-known that Cu sintering is the primary cause of deactivation in Cu/ZnO–Al-based CH3OH synthesis catalysts.44 However, no significant change in the Cu particle size was found in the XRD analysis on used Cu/ZnO–Al-30 and Cu/ZnO–Al-160 catalysts (not shown), which suggested that other phenomena were responsible for the different stability behaviour of catalysts under the reaction conditions used. Changes in ZnO under reaction were also considered as a cause of deactivation45 and therefore, this could be the reason for the different deactivation of Cu/ZnO–Al catalysts. In this regard the XRD analysis of used Cu/ZnO–Al catalysts (not shown) only revealed some crystallization of ZnO for the Cu/ZnO–Al-30 catalyst. The mobility and crystallization of ZnO under reaction observed for the Cu/ZnO–Al-30 sample resulted in a reduction in the Cu/ZnO interactions, therefore lowering the number of active sites. The stability of ZnO in the Cu/ZnO–Al catalysts could be related to differences in their crystallinity, morphology, particle size,46 Al-doping47 and the presence of residual carbonate species34 because these parameters have previously been reported to influence the mobility of ZnO under the reaction conditions used. The precise implication of each parameter in the mobility of ZnO in the Cu/ZnO–Al-30 and Cu/ZnO–Al-100 catalysts will require further investigations but it is tentatively hypothesized that differences in the presence of residual carbonate species and/or the modification of ZnO with Al which could act as stabilizing elements of ZnO in the Cu/ZnO–Al-100 and Cu/ZnO–Al-160 catalysts because these samples show carbonates which decompose at very high temperature (>500 °C, Fig. 4) and because the precipitates of these catalysts show crystallization of zincian malachite which favour the incorporation of Al into ZnO.16
4. Conclusions
The phase transitions of precipitates for an optimized Cu/ZnO–Al catalyst with low Al concentration during aging are analogous to those described for the previously studied binary Cu–Zn systems following the slow transformation of the initially amorphous zincian georgeite towards crystalline zincian malachite with a higher insertion of Zn in the malachite structure as the aging time increases. The precipitate removed close to the point of crystallization of zincian malachite has a different washing behaviour compared to the rest of the precipitates. The conventional washing protocol used for the removal of NaNO3 was effective for all the precipitates except for the sample removed close to the crystallization of the malachite phase which could be related to the important role of Na+ ions in the initial crystallization of malachite from amorphous georgeite. The precipitates show different thermal decomposition profiles in which the extent of carbonation differs after calcination. The nature of the precipitate determines the meso-structuring and interactions of CuO and ZnO in the precursors obtained from the calcination of precipitates. Small crystalline domains of CuO of similar size were observed in the calcined precursors derived from both amorphous georgeite and crystalline malachite precipitates because these precipitates retain carbonates after calcination, which preserve the segregation and crystallization of CuO. The exception was the sample removed close to the crystallization of malachite that exhibits crystalline growth of CuO segregated from ZnO because the nitrate impurities inhibit the presence of carbonates after calcination. The surface area of the calcined precursors was similar to that developed for precipitates indicating that the final surface area in the catalysts was developed from the mesostructuring of precipitates and not from the nanostructuring of CuO and ZnO after calcination. Nevertheless, the residual carbonates remaining after calcination play a role in the maintenance of the surface area because the precipitated sample removed close to the crystallization of malachite does not have any residual carbonates after calcination and it shows a dramatic loss in surface area. The differences in the accessible surface area and Cu particle size lead to variations in the specific Cu surface areas of reduced catalysts giving the catalysts derived from aged zincian malachite the higher Cu surface area (almost doubling the area developed by the sample derived from the amorphous georgeite). However, the catalyst obtained from the precipitate removed close to the crystallization point of malachite has a very poor Cu surface area as a result of the segregation and crystallization of CuO observed in their calcined precursor together with the exothermic reduction of the nitrate impurities both favouring the sintering of Cu particles during reduction. The results of catalytic activity in CH3OH synthesis from syngas showed higher intrinsic activity of the catalysts derived from aged malachite precipitates compared to that derived from the georgeite counterpart as a consequence of the increase in the contacts Cu–ZnO developed in the former. Nevertheless, it should be stressed that the relatively high initial intrinsic activity of the catalyst derived from georgeite despite co-precipitation does not ensure a good initial Cu–Zn mixing in georgeite. The sample removed close to the crystallization of malachite was almost inactive as consequence of its low Cu surface area and low Cu–ZnO interactions. The stability of catalysts under reaction conditions was also improved in the catalysts derived from precipitates aged after the crystallization of the malachite. The catalyst derived from amorphous georgeite shows deactivation derived from the crystallization of ZnO under the reaction conditions used. The catalysts derived from the precipitates aged after crystallization of malachite show better stability, which can be related to the presence of residual carbonate species and/or modification of ZnO by Al, which could act as stabilizing elements of ZnO under the reaction conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work was performed within the research program CTQ2016-76505-C3-1 supported by the Spanish Ministry of Economy, Industry and Competitiveness (MINECO).Notes and references
- D. Waller, D. Striling, F. S. Stone and M. S. Spencer, Faraday Discuss. Chem. Soc., 1989, 87, 107–120 RSC.
- C. Baltes, S. Vukojevic and F. Schüth, J. Catal., 2008, 258, 334–344 CrossRef.
- B. V. Farahani, F. H. Rajabi, M. Bahmani, M. Ghelichkhani and S. Shebdelfar, Appl. Catal., A, 2014, 482, 237–244 CrossRef.
- M. Behrens and R. Schlögl, Z. Anorg. Allg. Chem., 2013, 639, 2683–2695 CrossRef.
- G. J. Millar, I. H. Holm, P. J. R. Uwins and J. Drennan, J. Chem. Soc., Faraday Trans., 1998, 94, 593–600 RSC.
- B. Bems, M. Schur, A. Dassenoy, H. Junkes, D. Herein and R. Schlögl, Chem.–Eur. J., 2003, 9, 2039–2052 CrossRef PubMed.
- S. Zander, B. Seidlhofer and M. Behrens, Dalton Trans., 2012, 41, 13413–13422 RSC.
- D. M. Whittle, A. A. Mirzaei, J. S. J. Hargreaves, R. W. Joyner, C. H. J. Diely, S. H. Taylor and G. J. Hutchings, Phys. Chem. Chem. Phys., 2002, 4, 5915–5920 RSC.
- S. H. Taylor, G. J. Hutchings and A. A. Mirazaei, Chem. Commun., 1999, 1373–1374 RSC.
- T. Fujitani and J. Nakamura, Catal. Lett., 1998, 56, 119–124 CrossRef.
- M. Behrens, J. Catal., 2009, 267, 24–29 CrossRef.
- M. M. Günter, T. Ressler, B. Bems, C. Büscher, T. Genger, O. Hinrichsen, M. Muhler and R. Schlögl, Catal. Lett., 2001, 71, 37–44 CrossRef.
- M. Behnrens, F. Studt, I. Kasatkin, S. Kühl, M. Häwecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, M. Tovar, R. W. Ficher, J. K. Norskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef PubMed.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjaer, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef PubMed.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef PubMed.
- M. Behrens, S. Zander, P. Kurr, N. Jacobsen, J. Senker, G. Koch, T. Ressler, R. V. Fischer and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 6061–6068 CrossRef PubMed.
- H. Wilmer, T. Genger and O. Hinrichsen, J. Catal., 2003, 215, 188–198 CrossRef.
- M. Kurtz, H. Wilmer, T. Genger, O. Hinrichsen and M. Muhler, Catal. Lett., 2003, 86, 77–80 CrossRef.
- J. Schumann, T. Lunkenbein, A. Tarasov, N. Thomas, R. Schlögl and M. Behrens, ChemCatChem, 2014, 6, 2889–2897 CrossRef.
- G. C. Bond and S. N. Namijo, J. Catal., 1989, 118, 507–510 CrossRef.
- A. M. Pollard, M. S. Spencer, R. G. Thomas, P. A. Williams, J. Holt and J. R. Jennings, Appl. Catal., A, 1992, 85, 1–11 CrossRef.
- M. Behrens and F. Girgsdies, Z. Anorg. Allg. Chem., 2010, 636, 919–927 CrossRef.
- S. A. Kondrat, P. J. Smith, J. H. Carter, J. S. Hayward, G. J. Pudge, G. Shaw, M. S. Spencer, J. H. Bartley, S. H. Taylor and G. J. Hutchings, Faraday Discuss., 2017, 197, 287–307 RSC.
- G. Prieto, K. P. de Jong and P. E. de Jongh, Catal. Today, 2013, 215, 142–151 CrossRef.
- K.-W. Jun, W.-J. Shen, K. S. Rama Rao and K.-W. Lee, Appl. Catal., A, 1998, 174, 231–238 CrossRef.
- P. J. Bridge, J. Just and M. H. Hey, Mineral. Mag., 1979, 43, 97–98 Search PubMed.
- M. Schmidt and H. D. Lutz, Phys. Chem. Miner., 1993, 20, 27–32 CrossRef.
- D. Stoilova, V. Koleva and V. Vassileva, Spectrochim. Acta, Part A, 2002, 58, 2051–2059 CrossRef.
- M. Behrens, F. Girgsdies, A. Trunschke and R. Schlögl, Eur. J. Inorg. Chem., 2009, 1347–1357 CrossRef.
- S. A. Kondrat, P. J. Smith, P. P. Wells, P. A. Chater, J. H. Carter, D. J. Morgan, E. M. Fiordaliso, J. B. Wagner, T. E. Davies, L. Lu, J. K. Bartley, S. H. Taylor, M. S. Spencer, C. J. Kiely, G. J. Kelly, C. W. Park, M. J. Rosseinsky and G. J. Hutchings, Nature, 2016, 53183–53187 Search PubMed.
- T. M. Yurieva, React. Kinet. Catal. Lett., 1995, 55, 513–521 CrossRef.
- R. L. Frost, A. J. Locke, M. C. Hales and W. N. Martens, J. Therm. Anal. Calorim., 2008, 94, 203–208 CrossRef.
- D. M. Whittle, A. A. Mirzaei, J. S. J. Hargreaves, R. W. Joyner, C. J. Kiely, S. H. Taylor and G. J. Hutchings, Phys. Chem. Chem. Phys., 2002, 4, 5915–5920 RSC.
- P. Gimenez and S. Fereres, Energy Procedia, 2015, 69, 654–662 CrossRef.
- J. Schumann, A. Tarasov, N. Thomas and R. Schlögl, Appl. Catal., A, 2016, 516, 117–126 CrossRef.
- J. Sloczynsky, R. Grabowski, A. Kozlowska, P. K. Olszewski and J. Stoch, Phys. Chem. Chem. Phys., 2003, 5, 4631–4640 RSC.
- J. P. Breen and J. R. H. Ross, Catal. Today, 1999, 51, 521–533 CrossRef.
- P. Gao, F. -Li, F. K. Xiao, N. Zhao, N. N. Sun, W. Wei, L. S. Zhong and Y. H. Sun, Catal. Sci. Technol., 2012, 2, 1447–1454 Search PubMed.
- J. Agrell, K. Hasselbo, K. Jansson, S. G. Järås and M. Boutonnet, Appl. Catal., A, 2001, 211, 239–250 CrossRef.
- S. Yuvaraj, L. Fan-Yuan, C. Tsong-Huei and Y. Chuin-Tih, J. Phys. Chem. B, 2003, 107(4), 1044–1047 CrossRef.
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Angew. Chem., 2014, 126, 6051–6055 CrossRef.
- J. Yoshihara and C. T. Campbell, J. Catal., 1996, 161, 776–781 CrossRef.
- R. Van den Berg, G. Prieto, G. Korpershoek, L. I. van der Wal, A. J. Van Bunningen, S. Laegsgaard-Jorgensen, P. E. de Jongh and K. P. de Jong, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- B. S. Clausen and H. Topsoe, Catal. Today, 1991, 9, 189–196 CrossRef.
- M. B. Fitchtl, D. Schlereth, N. Jacobsen, I. Kasatkin, J. Schumann, M. Behrens, R. Schlögl and O. Hinrichsen, Appl. Catal., A, 2015, 502, 262–270 CrossRef.
- T. K. Gupta and R. L. Coble, J. Am. Ceram. Soc., 1968, 51, 521–525 CrossRef.
- J. Han, P. Q. Mantas and A. M. R. Senos, J. Mater. Res., 2001, 16, 459–468 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |