
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoration of metal oxide surface with {111} form Au nanoparticles using PEGylation†
Cheon Woo Moon,
Jongseong Park,
Seung-Pyo Hong,
Woonbae Sohn,
Dinsefa Mensur Andoshe,
Mohammadreza Shokouhimehr and
Ho Won Jang*
and
Ho Won Jang*
Department of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hwjang@snu.ac.kr
First published on 21st May 2018
Abstract
The benefit of introducing gold nanoparticles is due to the plasmon relaxation process. The plasmon decay induces various phenomena such as near-field enhancement, hot electron injection, and resonance energy transfer. Shape-controlled octahedral gold nanoparticles can maximize the efficiency of these processes. For practical purposes, a high-coverage decoration method, comparable to physical vapor deposition on a metal oxide semiconductor nanostructure, is indispensable. However, the ligand exchange reaction to attach octahedral gold nanoparticles is limited in aqueous solution due to the inactivity of the gold (111) surface as a result of a densely-packed cetyltrimethylammonium bilayer structure. Herein, we report a controllable high-coverage surface decoration method of octahedral gold nanoparticles on the targeted semiconductor nanostructures via phase transfer by an organic medium with thiolated-polyethylene glycol. Our results deliver an innovative platform for future plasmonic gold nanoparticle applications.
Introduction
Plasmonic nanoparticles (NPs) have opened a wide range of applications in broad research fields including nanomedicine,1,2 engineered nanostructure growth,3,4 optical and chemical sensors,5,6 and water splitting catalysts7,8 due to their exceptional optical response. Gold (Au) has been regarded as the best candidate9 material for plasmonics due to its high chemical stability in harsh environments and tunable plasmon resonance in the visible-to-infrared light region.10 The incoming light energy can be efficiently transferred to a nearby medium from the energy conversion of plasmon relaxation process by Au NPs. The process eventually results in localized heating, near-field enhancement, hot-electron injection, and resonant electron transfer to an adjacent semiconductor.11,12 The efficiencies of these relaxation processes are highly dependent on the morphology and size of Au NPs.13 The plasmon relaxation can be strengthened by size-controlled Au NPs having sharp vertices or edges.14–16 According to the phenomenological plasmonic model, a productive plasmon relaxation process is expected with the shape-controlled Au NPs.17 In previous research, we intensively investigated shape dependence of the plasmon relaxation processes for shape-controlled Au NPs.18,19 Octahedral Au NPs possess much higher activity than quasi-spherical and hemispherical Au NPs in plasmonic solar water splitting. The theoretical simulations and experimental results18,19 indicate that NPs having octahedral shape have the best ability to enhance incident near-fields on semiconductor nanostructures in comparison with NPs of other shapes.In plasmonics, the dielectric property of the medium around Au NPs is also crucial to obtain the preferred characteristics. We cannot expect prolific plasmonic activity of Au NPs near a highly damped medium (dominance of dielectric imaginary part).20 Metal oxide semiconductors are the ideal materials that can optimise the plasmonic properties of a system. For further applications, attachment of octahedral Au NPs on a metal oxide semiconductor with controllable coverage is highly desirable. Such high-coverage Au NP decorations are currently achieved from physical vapor deposition processes,21 precursor reductions16 (thermal or photo) and linker molecules such as PVP (poly(N-vinylpyrrolidone)).22–24 However, these methods distribute shape-uncontrolled Au NPs randomly on the metal oxide semiconductor nanostructures, which impedes their proficiency and practicability. High-coverage decoration of shape-controlled Au NPs on semiconductors has not yet been achieved.
In contrast, citrate reduction and seed-mediated growth procedures can be utilized to synthesize shape-controlled Au NPs. Although the citrate reduction method can provide homogenous quasi-spherical Au NPs over 10 nm in size,25 the synthesis of Au NPs with a well-defined shape from citrate reduction is abstruse. A well-defined shape can be achieved via the seed-mediated growth method using a cetyltrimethylammonium bromide/chloride (CTAB/C) capping agent. CTAB/C mainly determines the morphology of Au NPs and acts as a capping agent to prevent agglomeration. Although CTAB/C is advantageous for the synthesis of shape-controlled Au NPs, the repulsion from the CTA+ surface charge hinders Au NPs from being used in practical applications; in particular, the attachment of Au NPs to a semiconductor using the seed-mediated growth method is limited. The complete removal of the capping agent and the precise Au NP attachment to the semiconductors are very complex because the agglomeration of Au NPs26 and the collapse of the bilayer structure on the Au NP surface are induced below the critical micelle concentration.27 It is essential to design conceivable methods that can achieve the attachment of shape-controlled Au NPs to the semiconductor by using a CTAB/C bilayer structure even though the process is complex. As a result, the Au NP decoration onto semiconductor nanostructures has received great attention.22–24 The successful development of a nanoparticle decoration platform can open up great opportunities for plasmonics using semiconductor nanostructures.
Ligand exchange of colloidal plasmonic nanoparticles is a highly desirable and feasible technique for stability enhancement and surface functionalization. The ligand exchange reaction to attach Au NPs is usually carried out using organic molecules having a thiol (–SH) functional group, owing to high electron affinity of sulfur (S) to Au.28 To attach Au NPs on a semiconductor in the aqueous phase, thiolated-acids (for example, mercaptopropionic acid (MPA); HS–(CH2)2–COOH) are usually selected. Molecule adsorption onto the targeted semiconductor surface is achieved predominantly by dipping29 or dropping.18 However, these methods have restrictions due to the relatively low coverage29,30 and agglomeration of NPs (Fig. S1†) on the surface of the semiconductor. The difficulty arises from the ionic nature of the exchanged thiol-acid ligands (positive sign), which reduces the zeta potential of Au NPs. A non-ionic (covalent) ligand is required to overcome such stability degradation of Au NPs during ligand exchange. The use of such a ligand is required to achieve homogeneous high-coverage Au NP decoration onto the targeted semiconductor nanostructures. Among the possible candidates, thiolated polyethylene glycol (PEG–SH) is a suitable compound for the functionalization of Au NPs due to its stability, solubility in water/organic solvents, commercial availability, biocompatibility, and high probability of attachment to a metal oxide semiconductor due to the hydroxide (–OH) functional group physisorption.31 The molecular adsorption mechanism has been reported in the literature and is also schematically explained in Fig. S2.†28 Hydroxide or carboxylate groups can initiate the adsorption mechanism due to the existence of (–O–) terminal part. The absorbed PEG–SH molecules form a monolayer on the Au surface,32 and the shape can be changed from mushroom to bush according to the number of molecules on the Au surface.33 Even though a simple ligand exchange reaction with PEG–SH for phase transfer has been reported for citrate-capped Au NPs and CTAB/C-capped Au NPs,34,35 octahedral Au NPs obtained from the ligand exchange reaction have not yet been reported.
Herein, we propose an efficient method for preparing coverage-controlled octahedral Au NPs, which are then attached onto a targeted semiconductor nanostructure; this is achieved by a ligand-exchange reaction with PEG–SH via an ethanol–dichloromethane mixture (EtOH–DCM). The advantages of the octahedral Au NPs compared with those of truncated octahedral Au NPs and cuboctahedral Au NPs for near-field enhancement are shown using the finite-difference time-domain (FDTD) simulation. To attach the electric-field-amplifying octahedral Au NPs onto a targeted semiconductor surface, surface functionalization of Au NPs by PEG–SH is conducted in the aqueous phase. However, the ligand exchange reaction does not proceed well enough on the Au (111) surface to provide the attachment of octahedral Au NPs onto the semiconductor surface. To solve the problem, we introduce phase transfer from the aqueous phase to the organic phase through the addition of EtOH–DCM. PEG–SH functionalizes the octahedral Au NPs after removing the CTAB/C capping agent via phase transfer. A particle number density of about 1.65 × 1014 ea cm−2 (about 31.6% coverage) on the Si surface is achieved homogeneously, which is analogous to that of Au NPs generated by physical vapor deposition.18,21 A transparent solution obtained after a few hours represents the complete activation of octahedral Au NPs in solution. Through the results of spectroscopies (Raman and Fourier transform infrared spectroscopy (FTIR)), we schematically visualize the surface CTA+ bilayer structure and PEG–SH activation mechanism at the (100) and (111) surfaces of Au NPs.
Results and discussion
The finite-difference time-domain (FDTD) method is employed to show the electric-field-amplifying ability of the shape-controlled octahedral Au NPs. Herein, three kinds of NPs (octahedral, truncated octahedral and cuboctahedral) are compared. The heights of these Au NPs are adjusted to 30 nm to fit the scale. The simulation configurations of the three kinds of Au NPs are shown in Fig. 1a. To visually differentiate the facets of these Au NPs, two distinct colors are used according to the facets shown in Fig. 1a. The FDTD simulations obtained at the incident wavelengths of 350, 450, 550 and 650 nm to compare the electric-field-amplifying abilities of the three kinds of Au NPs are shown in Fig. 1b–e. The field intensities are at the highest level near the plasmon resonance frequency of around 550 nm. The overall results show that the incident electric field is significantly enhanced with octahedral Au NPs compared with the results obtained for truncated octahedral or cuboctahedral NPs regardless of the incident light wavelength. The maximum electric field enhancement ratios (|E/E0|) for twelve outputs given in Fig. 1b–e are shown in Fig. S3.† The FDTD simulation results show the excellent electric field-amplifying ability of octahedral Au NPs. In single-crystalline Au NPs without self-assembled structure, the octahedral Au NPs are found to be the best choice to enhance incident near field by both experiment and simulation.18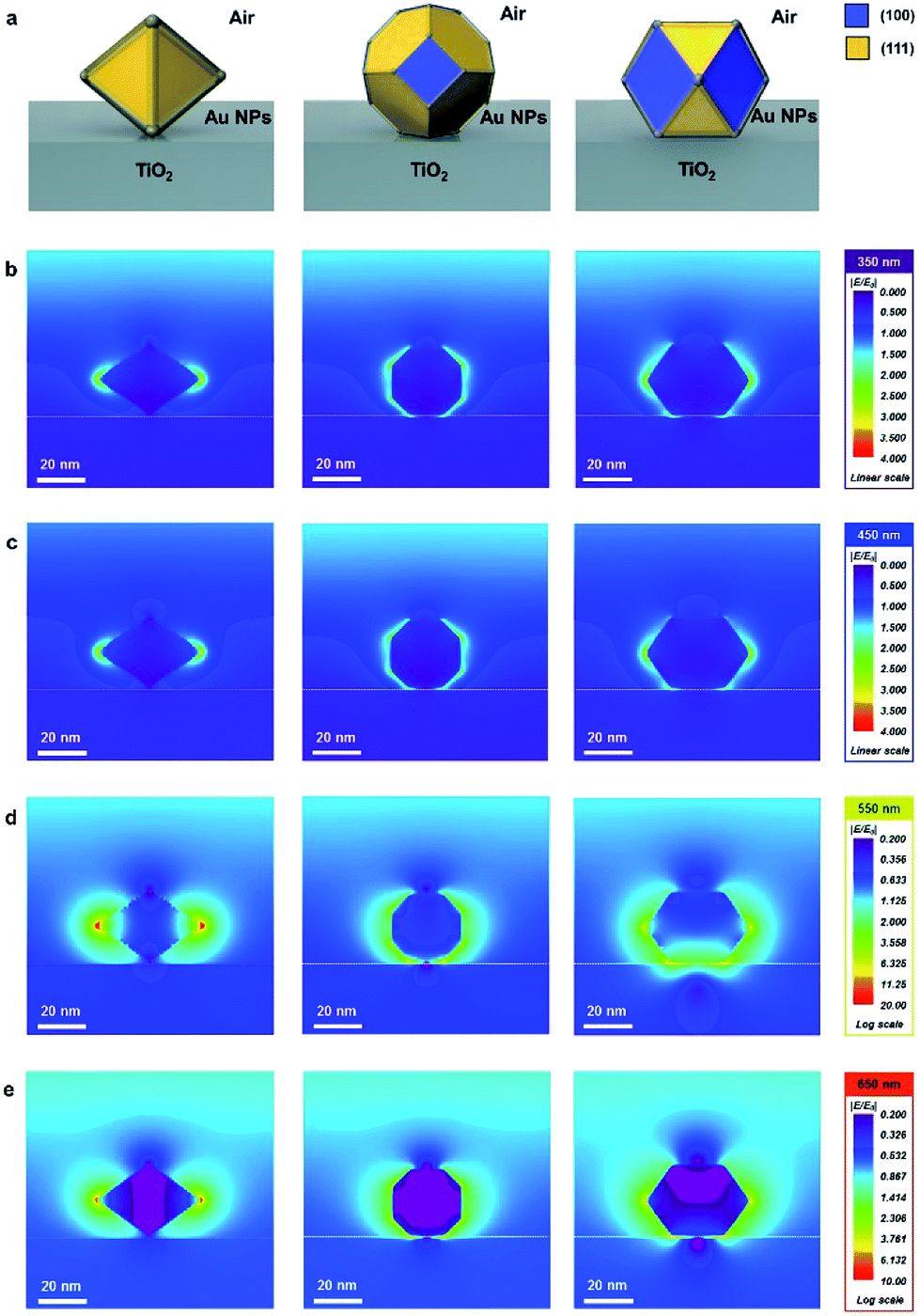 | ||
| Fig. 1 FDTD simulation results (|E/E0|) of the three kinds of Au nanoparticles (octahedral, truncated octahedral and cuboctahedral) at various incident wavelengths. (100) and (111) facets are denoted as yellow and blue, respectively. (a) Schematic configurations of Au nanoparticles. (b) Field enhancement at 350 nm photons. (c) Field enhancement at 450 nm photons. (d) Field enhancement at 550 nm photons. (e) Field enhancement at 650 nm photons. | ||
Fig. 2a–c show the morphologies of the three types of Au NPs (octahedral, truncated octahedral and cuboctahedral) aggregated on a TiO2/Si substrate 40 nm in size. The images were obtained using a field-effect scanning electron microscope (FE-SEM). The transmission electron microscopy (TEM) images of these Au NPs are presented as insets. The 3-dimensional configurations of Au NPs that were reconstructed from the TEM images are also included as an inset. The edge sizes of the three types of Au NPs were estimated to be around 20–25 nm. Fig. S4† shows the proportions of the (111) and (100) facets according to their shapes, as reported36 and calculated. The schematic of the Au NP attachment procedure based on phase transfer via PEGylation34,35 is shown in Fig. 2d. The process consisted of two steps according to their media (in aqueous and organic phases). First, 0.02 mL of PEG–SH aqueous solution (1 mg/1 mL) was added to a glass vial containing an aqueous solution of Au NPs (10 mL). After a while (more than 30 min), an organic solvent mixture of EtOH and DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) was added to the aqueous solution containing Au NPs. Phase transfer automatically occurred upon adding the organic medium (DCM–EtOH). After collecting the Au NP sediments by centrifugation, the concentration of the solution containing Au NPs was adjusted to control the coverage of Au NPs on a targeted semiconductor by adding an organic medium. Au NPs could be simply attached to the substrate via dipping. Finally, after dipping, the targeted substrate was washed with EtOH to remove the residues. Fig. 2e shows the attachment of Au NPs onto the inside of a transparent vial due to PEGylation (PEG–SH adsorption onto the Au NP surface). PEGylated Au NP solution left for 24 h showed that the attachment of Au NPs was the greatest with the cuboctahedral Au NPs and least (almost transparent) with the octahedral Au NPs. This result implied that due to the lack of activity on the (111) facets, PEGylation did not sufficiently proceed on the octahedral Au NPs attached on a targeted semiconductor in the aqueous phase. Fig. 2f shows that without PEG–SH, the phase transfer of octahedral Au NPs from aqueous to the DCM–EtOH medium was not facilitated. Due to the decrease in the micelles of Au NPs with the addition of EtOH, the color of the solution was highly diluted. The phase transfer of Au NPs occurred after the addition of PEG–SH. Within a short time (less than 30 min) of PEGylation, it was observed that the phase transfer did not proceed efficiently. An adequate PEGylation time (over 3 h) was required for sufficient ligand exchange reaction in the aqueous phase. The objective of introducing the EtOH–DCM medium is the removal of the CTA+ bilayer structure to facilitate PEG–SH attachment onto the Au surface. A similar concept was reported in a previous study on a phospholipid bilayer structure.37 To show homogeneous ligand exchange, we obtained an image of the octahedral Au NPs after phase transfer by TEM with the negative staining technique38 (Fig. S5†). TEM images of an isolated octahedral NP (Fig. S5a and b†) and a group of octahedral Au NPs (Fig. S5c†) are shown. For the aqueous phase without PEGylation, the amorphous organic trace around octahedral Au NPs was not observable, as shown in Fig. S5d.†18 To identify the uniformity of Au NPs after PEGylation, we performed dynamic light scattering (DLS) and zeta potential measurements. As shown in Fig. S5e and f,† the average size of the octahedral Au NPs increased from 34.6 to 41.35 nm without changing the polydispersity index (PDI); this implied a homogeneous PEGylation reaction on the Au NP surfaces. PEGylation of Au NPs usually yields a negative zeta potential, as reported in the literature.39,40 A non-negative zeta potential indicates the partial exchange of the CTAB/C bilayer structure on the surface. Fig. 2g shows high-coverage octahedral Au NPs attached onto a glass vial.
:1 volume ratio) was added to the aqueous solution containing Au NPs. Phase transfer automatically occurred upon adding the organic medium (DCM–EtOH). After collecting the Au NP sediments by centrifugation, the concentration of the solution containing Au NPs was adjusted to control the coverage of Au NPs on a targeted semiconductor by adding an organic medium. Au NPs could be simply attached to the substrate via dipping. Finally, after dipping, the targeted substrate was washed with EtOH to remove the residues. Fig. 2e shows the attachment of Au NPs onto the inside of a transparent vial due to PEGylation (PEG–SH adsorption onto the Au NP surface). PEGylated Au NP solution left for 24 h showed that the attachment of Au NPs was the greatest with the cuboctahedral Au NPs and least (almost transparent) with the octahedral Au NPs. This result implied that due to the lack of activity on the (111) facets, PEGylation did not sufficiently proceed on the octahedral Au NPs attached on a targeted semiconductor in the aqueous phase. Fig. 2f shows that without PEG–SH, the phase transfer of octahedral Au NPs from aqueous to the DCM–EtOH medium was not facilitated. Due to the decrease in the micelles of Au NPs with the addition of EtOH, the color of the solution was highly diluted. The phase transfer of Au NPs occurred after the addition of PEG–SH. Within a short time (less than 30 min) of PEGylation, it was observed that the phase transfer did not proceed efficiently. An adequate PEGylation time (over 3 h) was required for sufficient ligand exchange reaction in the aqueous phase. The objective of introducing the EtOH–DCM medium is the removal of the CTA+ bilayer structure to facilitate PEG–SH attachment onto the Au surface. A similar concept was reported in a previous study on a phospholipid bilayer structure.37 To show homogeneous ligand exchange, we obtained an image of the octahedral Au NPs after phase transfer by TEM with the negative staining technique38 (Fig. S5†). TEM images of an isolated octahedral NP (Fig. S5a and b†) and a group of octahedral Au NPs (Fig. S5c†) are shown. For the aqueous phase without PEGylation, the amorphous organic trace around octahedral Au NPs was not observable, as shown in Fig. S5d.†18 To identify the uniformity of Au NPs after PEGylation, we performed dynamic light scattering (DLS) and zeta potential measurements. As shown in Fig. S5e and f,† the average size of the octahedral Au NPs increased from 34.6 to 41.35 nm without changing the polydispersity index (PDI); this implied a homogeneous PEGylation reaction on the Au NP surfaces. PEGylation of Au NPs usually yields a negative zeta potential, as reported in the literature.39,40 A non-negative zeta potential indicates the partial exchange of the CTAB/C bilayer structure on the surface. Fig. 2g shows high-coverage octahedral Au NPs attached onto a glass vial.
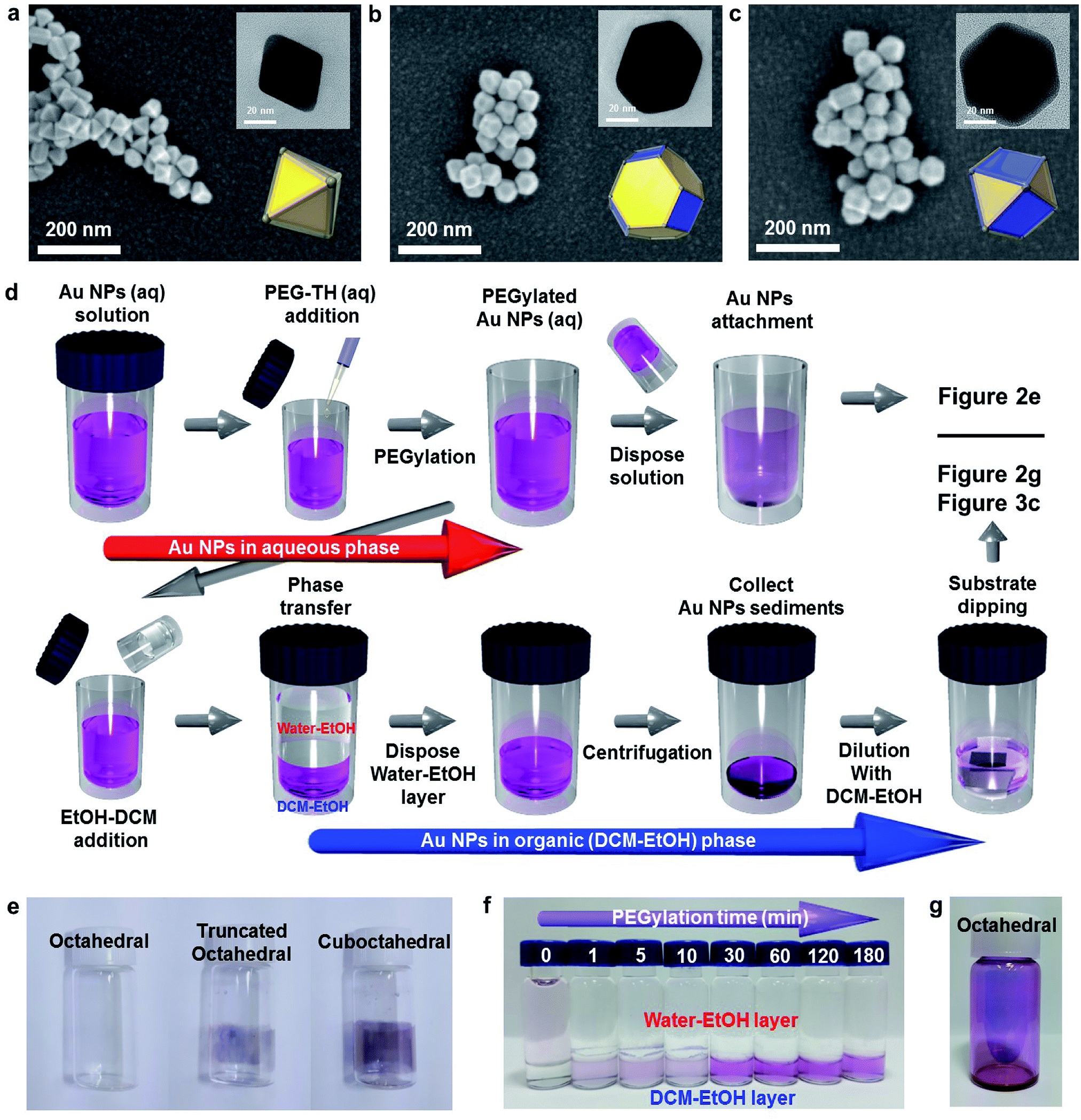 | ||
| Fig. 2 Agglomerated (a) octahedral, (b) truncated octahedral and (c) cuboctahedral Au nanoparticles on TiO2 thin films. (d) A schematic for the process of anchoring Au nanoparticles for the semiconductor nanostructure attachment using PEGylation in aqueous phase and phase transfer to organic medium. (e) A visualization of the Au NP attachment using vials. (f) Phase transfer to organic medium according to the PEGylation time. (g) Image of octahedral Au nanoparticles attached to a vial. | ||
Fig. 3a presents the absorption spectra for the three kinds of Au NP solutions measured and normalized to their resonance peaks from 300 to 800 nm. The resonance peaks for octahedral, truncated octahedral and cuboctahedral Au NPs in aqueous solutions were observed at 541, 538, and 559 nm, respectively. The resonance peaks for truncated octahedral Au NPs and cuboctahedral Au NPs in aqueous solutions were blue-shifted (2 and 3 nm in sequence) after PEGylation due to the change in the dielectric constant of the surroundings, which resulted from the adsorption of PEG–SH onto the Au NP surface.41 Interestingly, the resonance peak shift for octahedral Au NP solution was not observable from the absorbance measurement. The result suggested a sluggish PEGylation process for octahedral Au NPs. For the attachment process, 10 mL of the as-synthesized octahedral Au NP solution was concentrated to 0.5 mL via centrifugation after the phase transfer. The dipping time and solution concentration were controlled to adjust the nanoparticle coverage on the Si substrate. The corresponding results are shown in Fig. 3b (concentration of as-centrifuged Au NP solution is indicated as C). The number of Au NPs on the Si surface increased in proportion to the solution concentration and dipping time. After 1 day, the octahedral Au NP organic solution became transparent (Fig. S6a†) in comparison with the aqueous solution of pristine Au NPs (Fig. S6b†). This result showed the achievement of homogeneous PEGylation via phase transfer to an organic medium (EtOH–DCM). An FE-SEM image of the silicon surface with the highest coverage is shown in Fig. 3c. To show the versatility of the octahedral NPs, they were decorated onto TiO2 nanorods (NRs) (Fig. 3d, S7†) as well as various semiconductor films (Fe2O3, SnO2, WO3, and SiO2; Fig. S8a–d†). In addition, the image of octahedral Au NPs decorated on TiO2 NR/FTO is enclosed as an inset image.
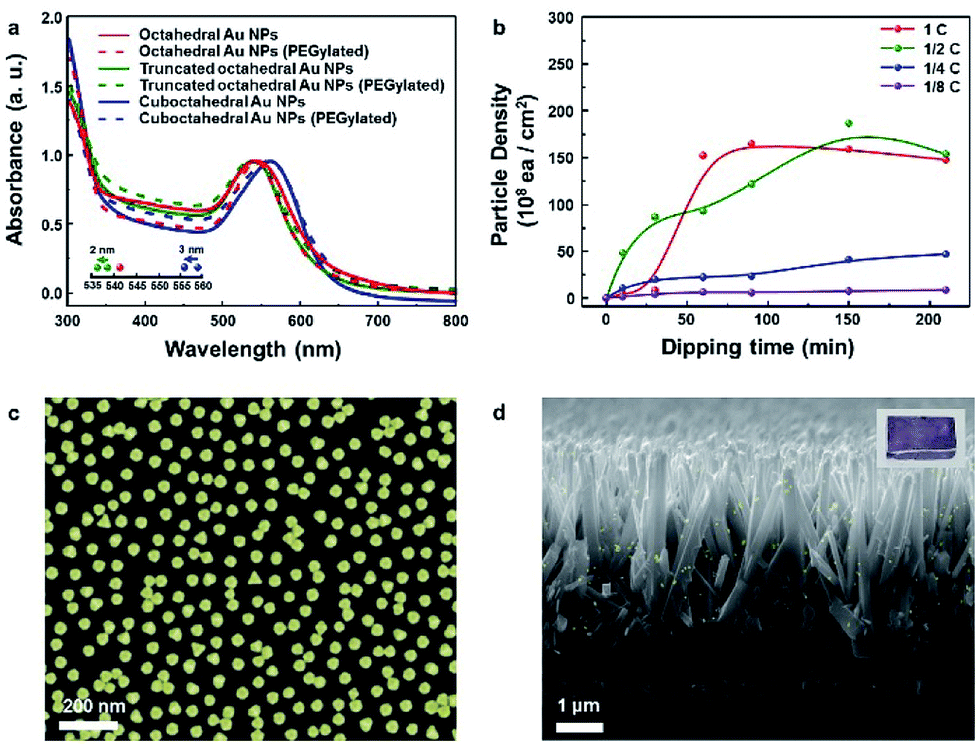 | ||
| Fig. 3 (a) Absorption spectra of octahedral, truncated octahedral and cuboctahedral Au nanoparticles before and after adding PEG–SH in Au nanoparticle aqueous solution. (b) Dependence of particle density on silicon according to the concentration and time (as-centrifuged concentration of Au NP solution from the organic solvent is indicated as C). (c) SEM image showing maximum Au nanoparticle density. (d) SEM image of octahedral Au nanoparticles attached to TiO2 nanorods. | ||
To analyze the results for the shapes of Au NPs, we investigated the CTA+ molecular vibration mode through Raman and FTIR spectroscopy. Fig. 4a shows the main Raman spectra of the CTA+ molecule from 600 to 3000 cm−1 wavenumbers. For comparison, the Raman signal of CTAB 200 mM aqueous solution is also shown in Fig. S9a.† The first domain in Fig. 4a (720 to 800) cm−1 was ascribed to the stretching (ν) vibration of the CTA+ headgroup (trimethylammonium). The vibrational signal at 760 cm−1 was ascribed to the headgroup (C–N+) symmetrical stretching.42 The signal around 730 cm−1 could be ascribed to another head group vibrational mode.43 The vibrational mode at 730 cm−1 is usually unobservable in CTA+-capped Au NPs in a Raman excitation,43,44 and it is only recorded in previous literature with Au nanospheres synthesized from CTA+ growth solution with citrate-reduced seed.45 The phenomenon could be attributed to the facet of a spherical particle that is mainly composed of (111) and a small portion of the (100) facets.46,47 Our result strongly indicates that the CTA+ bilayer structure in octahedral Au NPs is different from those in other NPs. The second domain in Fig. 4a (1000 to 1600 cm−1) originates from the skeletal vibration in the carbon backbone chain of CTA+. The signal at 1085 cm−1 indicates carbon–carbon (CC) stretching (ν) vibration of CTA+ alkyl-chains.45 We can figure out the headgroup vibrational activity compared with that of the backbone from the peak intensity ratio (I1085/(I760 + I730)) of Raman spectroscopy. Summarized data in Table 1 show that octahedral Au NPs have the lowest value. The analysis indicates suppressed backbone vibration and promoted headgroup vibration in CTA+ on the (111) facet. The peaks at 1300 and 1610 cm−1 indicate the CH2 twisting (t) and wagging (w) vibration modes sequentially (out-of-plane vibration) in the hydrocarbon backbone. If the hydrogen moves in the same (or different) vertical direction, it would be wagging (or twisting). The ratio I1300/I1510 shows the vibrational tendency of the backbone chain. As shown in Table 1, hydrogen favors a different vertical directional movement in octahedral Au NPs. The result indicates the presence of strong strain in the CTA+ bilayer structure due to the suppressed atomic vibration of hydrogen. The peak at 1445 cm−1 indicates the CH2 scissoring (δs) vibrational mode (in-plane vibration). The lowest peak intensity ratio (I1445/I1300) of octahedral Au NPs when compared with those of the three Au NPs means suppressed in-plane vibration, which supports the idea of a strong strain in the CTA+ bilayer structure in octahedral Au NPs. The last domain in Fig. 4a (2800 to 3000 cm−1) is attributed to the stretching (ν) vibrational modes of hydrocarbons (CHx).43 The signals at 2850 and 2892 cm−1 indicate stretching (ν) vibrational modes of hydrocarbons in the backbone (CH2) in symmetrical and anti-symmetrical modes, respectively. The two last signals at 2930 and 2973 cm−1 indicate stretching (ν) vibrational modes of hydrocarbons in headgroup (CH3) in symmetrical and anti-symmetrical modes, respectively. The overall comparison of the three intensity ratio results (I2850/I2930, I2892/I2973 and (I2892 + I2890)/(I2973 + I2930)) from Table 1 indicates suppressed CTA+ carbon chain vibration in the octahedral Au NPs. According to the literature, the C–O bonding signs from PEG–SH appear at 1132 and 1160 cm−1;48 however, these peaks are not observed. We think that the reason lies in the low concentration of PEG–SH in the Au NP solution. On the other hand, the Raman signal degradation around 760 and 960 cm−1 is observed, as shown in Fig. S9b.† If a complete ligand exchange with PEG occurs, the CTA+ peaks would be eliminated.49 As per our investigations using DLS and zeta-potential, a partial ligand exchange on the Au NP surface is expected.
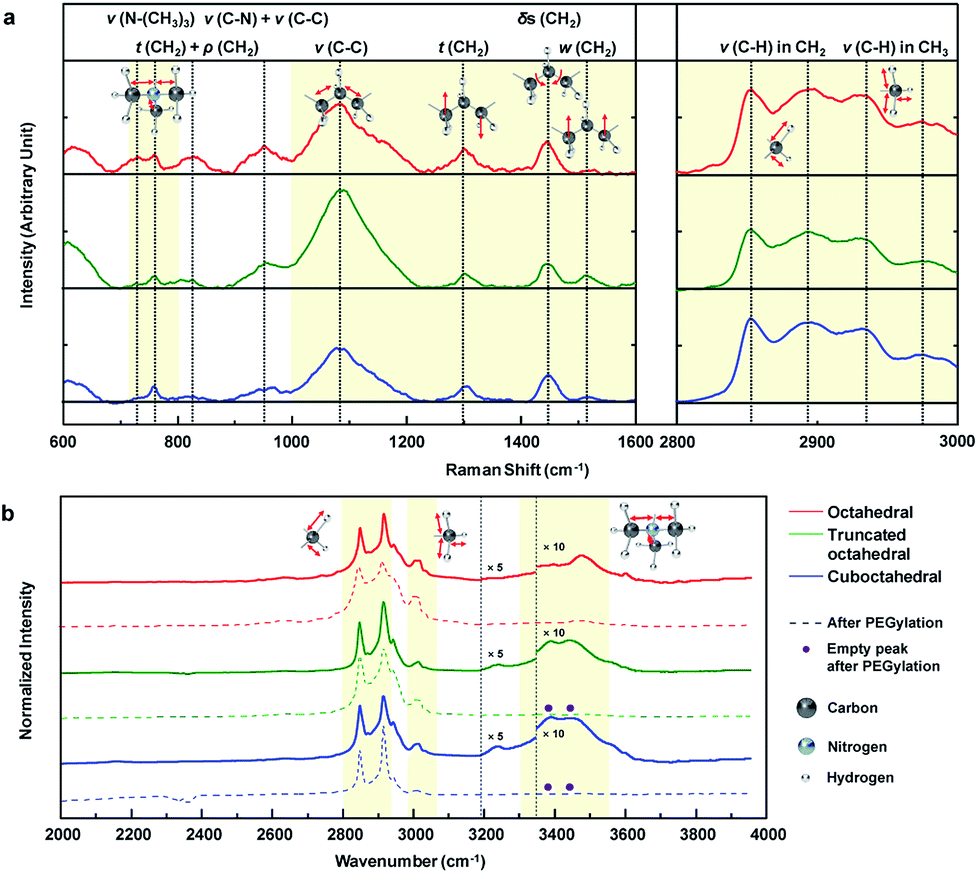 | ||
| Fig. 4 (a) Normalized Raman spectroscopic analysis of the three shape-controlled Au NP solutions from 600 to 3000 cm−1. Smoothed data is also included (black dotted lines (…) indicate peak positions). (b) Normalized FTIR spectroscopic analysis of the three shape-controlled Au NP solutions before and after PEGylation from (2000 to 3950) cm−1. The intensity for FTIR at specific region around 3400 cm−1 is enlarged 10 times for visual assistance. The disappeared peaks after PEGylation are denoted as purple dots (●) (black dotted lines (…) indicate peak positions). | ||
| Intensity ratio | I1085/(I760 + I730)a | I1300/I1510b | I1445/I1300c | I2850/I2930d | I2892/I2973e | (I2892 + I2890)/(I2973 + I2930)f |
|---|---|---|---|---|---|---|
| a (Backbone vibrational activity).b (Out-of-plane vibrational activity (same directional movement)).c (In-plane mode vibrational activity).d (C–H stretching activity in backbone (symmetrical)).e (C–H stretching activity in backbone (anti-symmetrical)).f (C–H stretching activity in backbone (total)). | ||||||
| Octahedral | 1.94 | 7.86 | 1.27 | 1.05 | 1.598 | 1.18 |
| Truncated octahedral | 6.42 | 1.25 | 1.72 | 1.16 | 2.095 | 1.29 |
| Cuboctahedral | 2.56 | 2.98 | 1.76 | 1.10 | 1.669 | 1.22 |
The FTIR absorption spectra of Au NP solution from 2000 to 4000 cm−1 are measured, as shown in Fig. 4b. These data are measured by dropping a small amount of solution onto the glass substrate, and the spectra are normalized according to their highest intensity peak value (2915 cm−1). For reference, the FTIR absorption spectra of CTAB 200 mM solution and PEG–SH aqueous solution (Fig. S10a†) and the designation of the peaks (Fig. S10b†) are included. Several uncommon phenomena are observed in the FTIR results. An unreported peak is observed at 3004 cm−1, which can be attributed to a partial red-shift of the peak at 3015 cm−1 (CH3 stretching (ν) vibration), owing to the strain of the CTA+ bilayer structure. In the literature, the signal around 3239 cm−1 corresponds to the hydroxyl vibration of a surface-absorbed water molecule.50,51 The truncated octahedral and cuboctahedral Au NPs exhibit FTIR signals due to the water molecule adsorption on the Au NP surface, whereas octahedral Au NPs do not show the signal, which means that the water molecule adsorption onto the octahedral Au NP surface is restricted. Therefore, to obtain enhanced hydrophobicity, the CTA+ bilayer in octahedral Au NPs should be more densely-packed than those in the truncated octahedral and cuboctahedral Au NPs.52 Interestingly, the octahedral Au NPs do not show a peak between 3390 and 3440 cm−1, but a new peak at 3474 cm−1 is observed. It is known that this region indicates the stretching vibration due to the adsorption of the CTA+ headgroup.14 The unreported peak at 3474 cm−1 can be regarded as the blue-shifted signal from 3390 and 3440 cm−1 signals owing to the strong binding of the CTA+ headgroup compared to that in NPs of other shapes. Also, strong binding on the Au (111) surface is also expected from computational calculations.53,54 The water solvation frequency of CTA+ around 3600 cm−1 indicates the formation of a CTA+ bilayer structure on the Au NP surface.14 The FTIR signal after PEGlyation shows disappearance of the CTA+ headgroup adsorption signals at 3390 and 3440 cm−1 for truncated octahedral and cuboctahedral Au NPs, whereas the signal at 3474 cm−1 from octahedral Au NPs does not show such an adsorption signal. The overall spectroscopic data suggest that PEGylation on octahedral Au NPs is difficult due to the unique CTA+ bilayer structure (strong binding of headgroup and densely packed structure) on the (111) facet.
To explain the results according to the shape of Au NPs, the facets of Au NPs should be considered. Surface bonding is an indispensable factor for describing selective particle attachment in the aqueous phase. As shown in Fig. 5a, the Au (100) surface atom has 8 neighboring atoms, and 4 additional surface bonds are available. Considering the CTA+ headgroup binding on (100), the Au vacant site and the spectroscopic analysis in Fig. 4, we can observe that the CTA+ headgroup bindings are loose on the (100) surface, and the bilayer density is not high. Sufficient space between the headgroup binding sites can make the CTA+ molecules intersect. The CTA+ bilayer structure on the (100) surface is inferred to be an inter-digitated structure (Fig. 5b), according to our spectroscopy results and the previous report.55 The sparse and thin CTA+ hydrophobic bilayer, in addition to the loose binding of the CTA+ headgroup, allows the PEG–SH molecule to attach successfully to the Au (100) surface (for the truncated octahedral and cuboctahedral Au NPs). Meanwhile, the Au (111) surface atom has 9 neighboring atoms, and 3 additional surface bonds are available, as shown in Fig. 5c. The CTA+ headgroup binding on the (111) Au vacant site and the spectroscopic analysis in Fig. 4 show a dense CTA+ bilayer structure and strong CTA+ headgroup bindings on the (111) surface. The adjacent headgroup binding sites block the intersection of CTA+ molecules. As a result, the CTA+ bilayer structure on the (111) surface is deduced from our spectroscopic result, and the Au (111) facet structure is inferred to be a densely packed structure. The thick and dense CTA+ hydrophobic bilayer, in addition to the strong binding of the CTA+ headgroup, results in the prevention of the PEG–SH binding onto the (111) surface.
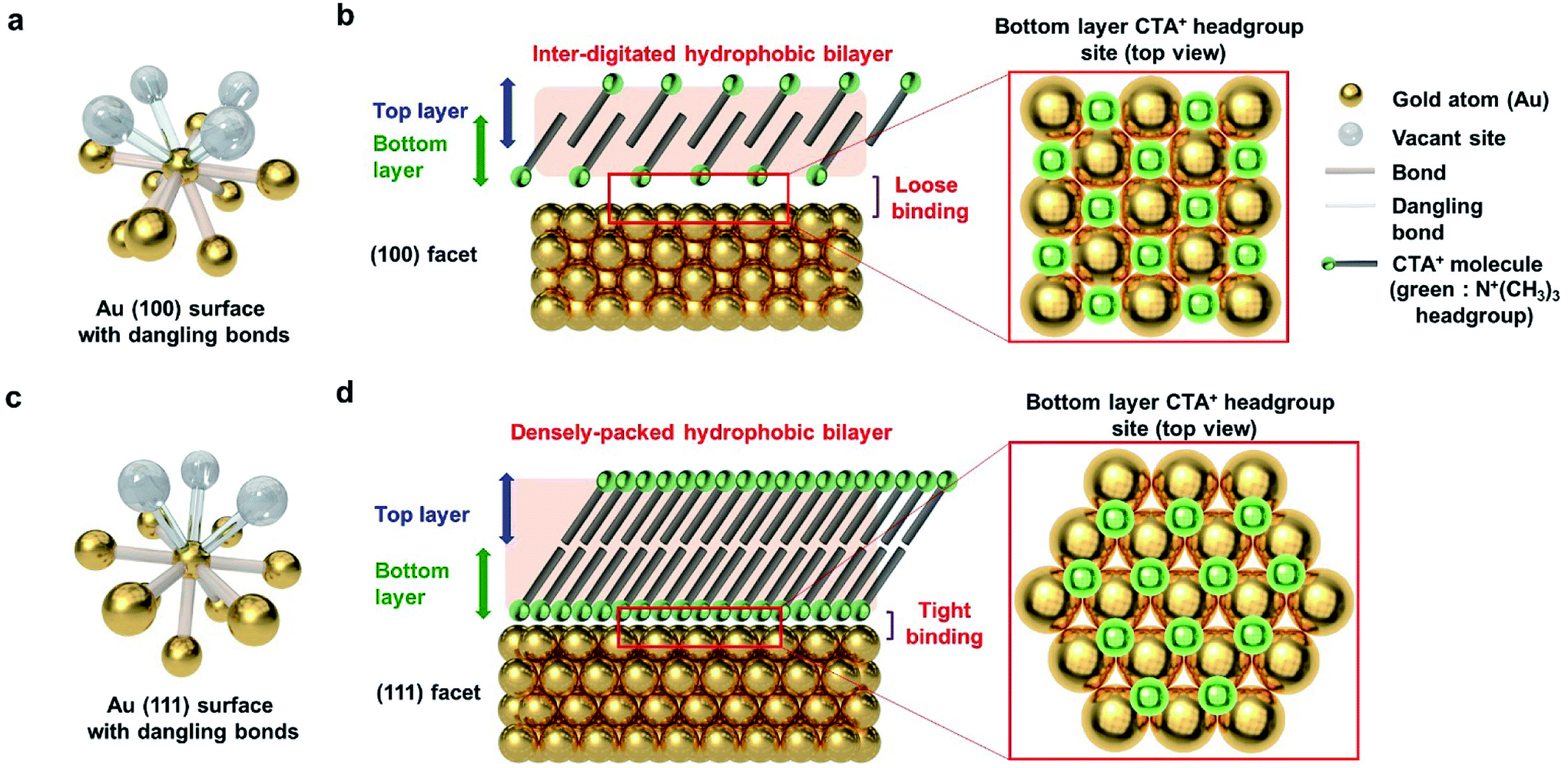 | ||
| Fig. 5 Schematic of the CTA+ bilayer structure according to the facet of Au NPs. (a) One Au (100) surface atom with dangling bonds. (b) Inter-digitated hydrophobic CTA+ bilayer structure on the (100) facet. (c) One Au (111) surface atom with dangling bonds. (d) Densely packed hydrophobic CTA+ bilayer structure on the (111) facet. The CTA+ molecule headgroup adhesion site is denoted in the schematic as green balls. | ||
Conclusions
Through an examination of the Au NP ligand exchange reaction of PEG–SH in water via UV-vis spectroscopy, Raman spectroscopy, FT-IR spectroscopy and the visual observation of a glass vial, we have confirmed that octahedral Au NPs exhibit more decrease in PEGylation in the aqueous phase in comparison with truncated octahedral and cuboctahedral NPs. The PEGylation results for the three shapes of Au NPs indicate that PEG–SH adhesion to the Au surface is more reliable at the Au (100) surface rather than that at the Au (111) surface. We have succeeded in the high-coverage decoration of octahedral Au NPs on a semiconductor surface via removal of the CTAB/C ligand for PEG–SH activation of the inactive Au (111) surface with the EtOH–DCM medium. The controlled attachment of octahedral Au NPs via PEGylation in an organic medium (EtOH–DCM) to the semiconductor with sufficient coverage proves the potential of the proposed method for further plasmonic applications of shape-controlled Au NPs. The covalent bonding property of PEG–SH profoundly reduces the agglomeration of Au NPs. Due to the highly efficient plasmon decay process (including hot electron/heat generation, near-field concentration, and resonant energy transmission) exhibited by shape-controlled Au NPs, we expect that this procedure will facilitate the application of the localized surface plasmon resonance property of Au NPs obtained from their high-coverage attachment.Experimental
Materials
Cetyltrimethylammonium bromide (CTAB, ≥99.9%), L-ascorbic acid (AA, 100%), sodium borohydride (NaBH4, ≥99%), trisodium citrate dihydrate (Na3C6H5O7·2H2O, Na3Cit), gold chloride hydrate (HAuCl4·xH2O, 99.999%) and poly(ethylene glycol) methyl ether thiol (thiolated polyethylene glycol, PEG–SH, average Mn = 2000) were obtained from Sigma-Aldrich. Cetyltrimethylammonium chloride (CTAC, ≥98%) was obtained from Wako. Sodium hydroxide (NaOH, 1 M), anhydrous ethanol (C2H5OH ≥ 99.9%) and dichloromethane (CH2Cl2 ≥99.5%) were obtained from Daejung. 3-Mercaptopropionic acid (MPA ≥ 99%) was purchased from Alfa Aesar. Si wafers and wet oxidized (300 nm) SiO2/Si wafers were purchased from DASOM RMS. These chemicals were used without further purification with deionized water (Pure Water Corporation). Fluorine-doped tin oxide (FTO, 8 Ω □−1) was purchased from Wooyang GMS. Titanium dioxide (TiO2), tungsten oxide (WO3) and hematite (α-Fe2O3) were purchased from Kojundo Chemical (grain shape, 99.9%).Semiconductor thin film preparation from electron beam evaporator (E-beam)
Titanium dioxide (TiO2), tungsten oxide (WO3) and hematite thin film (α-Fe2O3) were deposited on a Si wafer with (100) cut. Film thicknesses were obtained from the thickness monitor. To crystallize the as-deposited film, thermal annealing was performed in ambient air condition in a box furnace (FB 1310M, Thermo Scientific).Au nanoparticle attachment with MPA
We have described this revised method in previously reported studies.18,19 Briefly, 1 mL octahedral-shaped Au nanoparticle solution was added to a scintillation vial. The objective samples were immersed into the octahedral-shaped Au nanoparticle solution. Then, a precipitator solution containing MPA (0.5 M) and NaOH (0.5 M) was added to the octahedral-shaped Au nanoparticle solution. The coverage of the octahedral-shaped Au nanoparticles on the samples was controlled by changing the volume of the precipitator solution. The SEM images in Fig. 2 were achieved via this procedure.Synthesis of CTAC-capped Au seeds
A seed-mediated growth method was used for the synthesis of shape-controlled Au nanoparticles. First, CTAB-capped Au seeds were synthesized from a strong reduction agent (NaBH4). Ice-cold NaBH4 aqueous solution (10 mM, 0.6 mL) was added to 10.0 mL aqueous solution containing 0.25 mM HAuCl4 and 100 mM CTAB. The brownish seed solution was stirred for 2 hours at 30 °C to decompose the remaining NaBH4. Next, CTAC-capped Au seeds were synthesized from the CTAB-capped Au seeds. Then, 100 mM (1.5 mL) AA was added to another aqueous mixture solution containing 5 mM HAuCl4 (0.2 mL), 200 mM CTAC (2 mL) and water (1.8 mL). After the color change of the mixture solution from yellow to transparent, 100 μL CTAB-capped Au seed solution was promptly injected. The solution was left at room temperature until a red wine color was seen. The as-synthesized Au seeds were concentrated by centrifugation and redispersed in 1 mL of 20 mM aqueous CTAC solution.The growth of octahedral Au NPs
The redispersed CTAC seed solution (100 μL) was rapidly injected into the transparent aqueous growth solution containing 5 mM HAuCl4 (0.4 mL), 200 mM CTAB (0.5 mL), 200 mM CTAC (4.5 mL), 100 mM AA (60 μL) and water (4.3 mL). Stirring was vigorously performed during the growth process.Synthesis of truncated octahedral Au NPs
The redispersed CTAC seed solution (100 μL) was rapidly injected into the transparent aqueous growth solution containing 5 mM HAuCl4 (0.4 mL), 200 mM CTAB (0.5 mL), 200 mM CTAC (4.5 mL), 100 mM AA (30 μL) and water (4.3 mL). Stirring was vigorously performed during the growth process.Synthesis of cuboctahedral NPs
The redispersed CTAC seed solution (100 μL) was rapidly injected into the transparent aqueous growth solution containing 5 mM HAuCl4 (0.4 mL), 200 mM CTAB (0.5 mL), 200 mM CTAC (4.48 mL), 100 mM AA (60 μL) and water (4.5 mL). Stirring was vigorously performed during the growth process.FDTD simulations
The FDTD simulations were performed with a program FDTD solution (Lumerical solutions). The near field simulation was conducted with 1.78 fs pulse length. The simulation condition was a perfectly matched layer (PML) boundary condition. The meshing condition was set to the automatic non-uniform mode, and the refinement was set to conformal variant 1. The calculation region was 100 nm × 100 nm, and the mesh size was 0.5 nm × 0.5 nm. The simulation was conducted with titanium dioxide (TiO2) thin film in air.Characterizations
Field emission scanning electron microscopy (FE-SEM, MERLIN compact, Zeiss), high-resolution transmission electron microscopy (TEM, JEM-2100F, JEOL), UV/VIS spectroscopy (V-770, JASCO) and Fourier transform infrared spectroscopy (FT-IR, Nicolet iN10, Thermo Fisher Scientific) were used in this study. For the Raman analysis, a 532 nm laser source was trained on a glass capillary tube containing Au NP solution. For the dynamic light scattering (DLS) analysis, a Malvern Zetasizer was utilized. The instruments were accessed at the Research Institute of Advanced Materials (RIAM, Seoul National University).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study is supported by the Basic Science Research Program (2017R1A2B3009135), the Future Material Discovery Program (2016M3D1A1027666), and the Nano Material Technology Development Program (2016M3A7B4910) through the National Research Foundation of Korea (NRF) and the International Energy Joint R&D Program (20168510011350) of the Korea Institute of Energy Technology Evaluation and Planning (KETEP).References
- E. B. Dickerson, E. C. Dreaden, X. Huang, I. H. El-Sayed, H. Chu, S. Pushpanketh, J. F. McDonald and M. A. El-Sayed, Cancer Lett., 2008, 269, 57–66 CrossRef PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef PubMed.
- F. Lin, J. Yang, S.-H. Lu, K.-Y. Niu, Y. Liu, J. Sun and X.-W. Du, J. Mater. Chem., 2010, 20, 1103–1106 RSC.
- J. Yeo, S. Hong, G. Kim, H. Lee, Y. D. Suh, I. Park, C. P. Grigoropoulos and S. H. Ko, ACS Nano, 2015, 9, 6059–6068 CrossRef PubMed.
- N. Gogurla, A. K. Sinha, S. Santra, S. Manna and S. K. Ray, Sci. Rep., 2014, 4, 6483 CrossRef PubMed.
- W. Xia, C. Mei, X. Zeng, G. Fan, J. Lu, X. Meng and X. Shen, ACS Appl. Mater. Interfaces, 2015, 7, 11824–11832 Search PubMed.
- J. Lee, S. Mubeen, X. Ji, G. D. Stucky and M. Moskovits, Nano Lett., 2012, 12, 5014–5019 CrossRef PubMed.
- Y.-C. Pu, G. Wang, K.-D. Chang, Y. Ling, Y.-K. Lin, B. C. Fitzmorris, C.-M. Liu, X. Lu, Y. Tong, J. Z. Zhang, Y.-J. Hsu and Y. Li, Nano Lett., 2013, 13, 3817–3823 CrossRef PubMed.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238 CrossRef PubMed.
- M. Valenti, M. Jonsson, G. Biskos, A. Schmidt-Ott and W. Smith, J. Mater. Chem. A, 2016, 4, 17891–17912 Search PubMed.
- M. Xiao, R. Jiang, F. Wang, C. Fang, J. Wang and C. Y. Jimmy, J. Mater. Chem. A, 2013, 1, 5790–5805 Search PubMed.
- S. K. Cushing, A. D. Bristow and N. Wu, Phys. Chem. Chem. Phys., 2015, 17, 30013–30022 RSC.
- S. Kundu, J. Mater. Chem. C, 2013, 1, 831–842 RSC.
- P. Sajanlal, C. Subramaniam, P. Sasanpour, B. Rashidian and T. Pradeep, J. Mater. Chem., 2010, 20, 2108–2113 RSC.
- X. Zhang, Y. Liu, S.-T. Lee, S. Yang and Z. Kang, Energy Environ. Sci., 2014, 7, 1409–1419 Search PubMed.
- A. O. Govorov, H. Zhang, H. V. Demir and Y. K. Gun'ko, Nano Today, 2014, 9, 85–101 CrossRef.
- C. W. Moon, S. Y. Lee, W. Sohn, D. M. Andoshe, D. H. Kim, K. Hong and H. W. Jang, Part. Part. Syst. Charact., 2017, 34, 1600340 CrossRef.
- M. G. Lee, C. W. Moon, H. Park, W. Sohn, S. B. Kang, S. Lee, K. J. Choi and H. W. Jang, Small, 2017, 13, 1701644 CrossRef PubMed.
- N. Jiang, X. Zhuo and J. Wang, Chem. Rev., 2018, 6, 3054–3099 CrossRef PubMed.
- X. Shi, K. Ueno, N. Takabayashi and H. Misawa, J. Phys. Chem. C, 2012, 117, 2494–2499 Search PubMed.
- Y.-H. Chiu and Y.-J. Hsu, Nano Energy, 2017, 31, 286–295 CrossRef.
- Y.-H. Chiu, K.-D. Chang and Y.-J. Hsu, J. Mater. Chem. A, 2018, 6, 4286–4296 Search PubMed.
- W.-H. Lin, Y.-H. Chiu, P.-W. Shao and Y.-J. Hsu, ACS Appl. Mater. Interfaces, 2016, 8, 32754–32763 Search PubMed.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098 CrossRef PubMed.
- K. Park, H. Koerner and R. A. Vaia, Nano Lett., 2010, 10, 1433–1439 CrossRef PubMed.
- A. Cifuentes, J. L. Bernal and J. C. Diez-Masa, Anal. Chem., 1997, 69, 4271–4274 CrossRef.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef PubMed.
- Y.-C. Pu, G. Wang, K.-D. Chang, Y. Ling, Y.-K. Lin, B. C. Fitzmorris, C.-M. Liu, X. Lu, Y. Tong and J. Z. Zhang, Nano Lett., 2013, 13, 3817–3823 CrossRef PubMed.
- X. Hu, J. Tian, Y. Xue, Y. Li and H. Cui, ChemCatChem, 2017, 9, 1511–1516 CrossRef.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980–1994 CrossRef PubMed.
- C. S. Levin, S. W. Bishnoi, N. K. Grady and N. J. Halas, Anal. Chem., 2006, 78, 3277–3281 CrossRef PubMed.
- D.-H. Tsai, F. W. DelRio, R. I. MacCuspie, T. J. Cho, M. R. Zachariah and V. A. Hackley, Langmuir, 2010, 26, 10325–10333 CrossRef PubMed.
- J. OckáPark and S. YongáLee, Chem. Commun., 2016, 52, 1625–1628 RSC.
- A. BaniáYaseen, RSC Adv., 2014, 4, 52676–52679 RSC.
- M. Eguchi, D. Mitsui, H.-L. Wu, R. Sato and T. Teranishi, Langmuir, 2012, 28, 9021–9026 CrossRef PubMed.
- S. Parimi, T. J. Barnes and C. A. Prestidge, Langmuir, 2008, 24, 13532–13539 CrossRef PubMed.
- S. C. Boca and S. Astilean, Nanotechnology, 2010, 21, 235601 CrossRef PubMed.
- T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, J. Controlled Release, 2006, 114, 343–347 CrossRef PubMed.
- J. Lipka, M. Semmler-Behnke, R. A. Sperling, A. Wenk, S. Takenaka, C. Schleh, T. Kissel, W. J. Parak and W. G. Kreyling, Biomaterials, 2010, 31, 6574–6581 CrossRef PubMed.
- E. Ringe, J. M. McMahon, K. Sohn, C. Cobley, Y. Xia, J. Huang, G. C. Schatz, L. D. Marks and R. P. Van Duyne, J. Phys. Chem. C, 2010, 114, 12511–12516 Search PubMed.
- S. Lee, L. J. Anderson, C. M. Payne and J. H. Hafner, Langmuir, 2011, 27, 14748–14756 CrossRef PubMed.
- P. B. Santhosh, N. Thomas, S. Sudhakar, A. Chadha and E. Mani, Phys. Chem. Chem. Phys., 2017, 19, 18494–18504 RSC.
- H. Gökce and S. Bahçeli, Opt. Spectrosc., 2013, 115, 632–644 CrossRef.
- M. Tebbe, C. Kuttner, M. Männel, A. Fery and M. Chanana, ACS Appl. Mater. Interfaces, 2015, 7, 5984–5991 Search PubMed.
- C.-H. Kuo, T.-F. Chiang, L.-J. Chen and M. H. Huang, Langmuir, 2004, 20, 7820–7824 CrossRef PubMed.
- A. Dutta, A. Paul and A. Chattopadhyay, RSC Adv., 2016, 6, 82138–82149 RSC.
- D. Yamini, G. D. Venkatasubbu, J. Kumar and V. Ramakrishnan, Spectrochim. Acta, Part A, 2014, 117, 299–303 CrossRef PubMed.
- Z. Zhang and M. Lin, RSC Adv., 2014, 4, 17760–17767 RSC.
- L. Di Landro, M. Pegoraro and L. Bordogma, J. Membr. Sci., 1991, 64, 229–236 CrossRef.
- F. Gu, S. F. Wang, M. K. Lü, Y. X. Qi, G. J. Zhou, D. Xu and D. R. Yuan, Inorg. Chem. Commun., 2003, 6, 882–885 CrossRef.
- F. Schulz, W. Friedrich, K. Hoppe, T. Vossmeyer, H. Weller and H. Lange, Nanoscale, 2016, 8, 7296–7308 RSC.
- J. Feng, R. B. Pandey, R. J. Berry, B. L. Farmer, R. R. Naik and H. Heinz, Soft Matter, 2011, 7, 2113–2120 RSC.
- L. B. Wright, J. P. Palafox-Hernandez, P. M. Rodger, S. Corni and T. R. Walsh, Chem. Sci., 2015, 6, 5204–5214 RSC.
- P.-W. Yang, S. Thoka, P.-C. Lin, C.-J. Su, H.-S. Sheu, M. H. Huang and U.-S. Jeng, Langmuir, 2017, 33, 3253–3261 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03523g |
| This journal is © The Royal Society of Chemistry 2018 |