
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of a scalable AuNP catalyst system for plasmon-driven photocatalysis†
Heike Lisa Kerstin Stephanie Stolle *ab,
Frank Garwea,
Robert Müllera,
Thomas Krechc,
Bastian Oberleiterd,
Thomas Rainerd,
Wolfgang Fritzschea and
Achim Stolleb
*ab,
Frank Garwea,
Robert Müllera,
Thomas Krechc,
Bastian Oberleiterd,
Thomas Rainerd,
Wolfgang Fritzschea and
Achim Stolleb
aLeibniz Institute of Photonic Technology (IPHT), Department of Nanobiophotonics, Albert-Einstein-Straße 9, D-07745 Jena, Germany. E-mail: Lisa.Stolle@leibniz-ipht.de
bInstitute of Technical and Environmental Chemistry, Faculty of Chemistry and Earth Sciences, Friedrich-Schiller-University Jena, Philosophenweg 7A, D-07743 Jena, Germany
cJENOPTIK Automatisierungstechnik GmbH, Konrad-Zuse-Straße 9, D-07745 Jena, Germany
dHEGLA boraident GmbH & Co. KG, Köthener Straße 33a, D-06118 Halle/Saale, Germany
First published on 29th August 2018
Abstract
In this work we present a simple, fast and cost-efficient synthesis of a metal nanoparticle catalyst on a glass support for plasmon driven heterogeneous photocatalysis. It is based on efficient mixing of metal salts as particle precursors with porous glass as the supporting material in a mixer ball mill, and the subsequent realization of a complete catalyst system by laser sintering the obtained powder on a glass plate as the support. By this, we could obtain catalyst systems with a high particle proportion and an even spatial particle distribution in a rapid process, which could be applied to various kinds of metal salt resulting in plasmon active metal nanoparticles. Furthermore, the catalyst production process presented here is easily scalable to any size of area that is to be coated. Finally, we demonstrate the catalytic performance of our catalysts by a model reaction of ethanol degradation in a self-designed lab-scale reactor.
1. Introduction
Plasmon active noble metal nanoparticles like silver, gold, platinum or palladium find applications in many fields of research, e.g. in bioanalytical sensing,1–3 surface enhanced Raman spectroscopy (SERS)4,5 and in catalysis.6–11 The synthesis of such particles is possible, in different shapes and sizes, using noble metal salts. Like this, gold nanoparticles have been synthesized, e.g. as colloidal, quasi-spherical particles,12–15 nanorods,16–18 nanocubes19,20 and nanostars.21 For silver nanoparticles, plenty of different shapes are also available, for instance spheres,22,23 nanocubes24,25 and prisms.26–28 However, platinum has mainly been synthesized in non-specific structures29 or as platinum cubes,30 which is similar for palladium nanoparticle synthesis.31–33What makes these particles so interesting for applications is the so called localized surface plasmon resonance (LSPR) effect they show, which is characteristic of such particles.34 In this effect, the valence electrons in the metal, which – under the influence of light – are constantly oscillating against the restoring force of the positively charged nuclei, can be excited by wavelengths resonant to this movement, resulting in a simultaneous oscillation of electrons (plasmon resonance). The wavelength of light necessary to initiate this process is different for each metal but also varies with shape and particle size of the same metal.34–36 By this effect, a strongly enhanced electric field is formed in the direct environment of a particle. This field enhancement can be around six times higher than the field intensity of irradiating photons for gold nanospheres37 and even up to 103 times higher in the cases of other shapes.35 The oscillation can be kept up for times in the fs and ps regions for electron–electron and electron–phonon relaxation, respectively.36,38 Decay is possible in different processes. Thus, the resonant light irradiated can be scattered, which is particularly the case for larger particles above 100 nm,35 or it can form carriers of energetic charge in the case of smaller particles.10 Energetic charge carriers can either relax and form heat at the nanoparticle surface, heating up components in their close environment (plasmonic heating), or they can result in electron–hole-pairs,10,38 meaning electrons can be donated to surrounding molecules or taken from them, with both of these initiating their degradation process.11,35,39
The abundance of possible mechanisms of converting light energy makes metal nanoparticles a desirable material for photocatalysts.39 Photocatalysts in general are becoming more popular, because classical catalysts which are activated with thermal energy require a high energy input, and in order to maintain the temperature for the highest turn-over a constant energy input is required. In the case of photocatalysis, this is not necessary, because in the best cases sunlight, which is abundantly available and makes any other energy sources redundant, can be used to drive reactions.34 Due to this, the catalysis becomes easily switchable by having a light on when energy is required to activate the catalyst, and having the light off when it is not. Gold nanoparticles are especially in demand for implementation as catalysts, as these particles exhibit a rich variety of shapes covering the absorption of almost the entire spectrum of visible light,35,40 and are also very stable.39 Regarding plasmon catalysis, this makes gold a very interesting catalyst material exploiting the energy of the VIS area of (sun) light.34 But combining gold catalysts with particles of other metals like silver,41,42 palladium43–46 or platinum47 and semiconductors like TiO2 (ref. 48–50) is also possible. Such combinations open up new possibilities for plasmon catalysis, as these allow the tuning of the irradiation wavelength needed.41,42
The synthesis of metal nanoparticles of different metals and in different shapes is nowadays state of the art, as described here. Bottom-up techniques range from classical wet-chemical synthesis12–19,21–27,29–33 to microfluidic20,28 and photochemical syntheses51 using metal salts as precursor materials, and to electrochemical synthesis approaches utilizing metal plates as electrodes.52 Classical top-down methods include lithographic approaches and laser ablation, where a metal plate in water is treated with a laser, resulting in monometallic particles,53 but the production of alloys54 or bimetallic particles55 is also possible. Problems regarding these techniques are, however, the low yields of particles as catalysts and the production costs, making them inappropriate for catalyst production and their upscaling.56
Another obstacle is making the particles usable for their application as catalysts. Here, more preparation steps are often required. In (quasi-)homogeneous catalysis, this problem is quite simple to overcome because the particles of the material to be used can stay in the solution where they were synthesized. Afterwards, the reactants are added (as solution) and the catalytic reaction is initiated by light of the required wavelength. This was done, for example, by utilizing Ag and Au nanospheres and AgAu-nanorings in solution to oxidize and degrade methylene blue.41 The case of heterogeneous catalysis (which represents the majority of catalytic applications) is not that simple, as the metal nanoparticles need to be immobilized on a supporting material. Here, mostly particles of different materials are used as a support and are coated with the plasmon active nanoparticles, and are utilized to feed packed bed reactors.6,10,57,58 This involves the use of TiO2 or SiO2 as supportive materials, as shown in the studies from Mukherjee et al. describing the dissociation of H2 mediated by plasmon heating of supported AuNPs prepared in a 3-step process.10,57 Rombi et al. present the oxidation of CO at AuNPs on SiO2, activated by 3-mercaptopropyltrimethoxysilane prior to loading with AuNPs by thermal reduction of a HAuCl4 solution in the presence of citrate.58 Studies from Christopher et al. demonstrated the use of α-Al2O3 particles as a support for AgNPs for plasmon catalyzed oxidation reactions.6 In other systems, like the microfluidic reaction chamber reported by Adleman et al. used for the plasmon-assisted catalysis of ethanol and water in the liquid phase, a catalyst system consisting of gold nanoparticles spin-coated onto a glass object slide was used.8 Hou et al. describe a similar system for gas phase conversion of CO2 to hydrocarbons over TiO2, AuNP/TiO2 or pure AuNP catalysts on a glass support, where gold was introduced by electron beam evaporation.59
The preparation of plasmon-based catalyst systems described so far in the literature is usually based on several working steps, consuming time and also resources, especially regarding the metal precursor salts. Usually the systems described are laid out for classical packed bed reactors and are difficult to scale-up or scale-down for smaller reactor systems. In the present paper, we introduce a new method to design a complete catalyst system consisting of plasmon active nanoparticles on a glass support. In the process, we were able to obtain integrated AuNP catalysts. Therefore, the methodology is very easy and fast to carry out, is cost-efficient regarding the metal precursors used, and is scalable to different sizes of supporting glass plates. This catalyst is intended for use in packed bed reactors with bypass flow, allowing thorough irradiation of the catalyst in comparison to classical packed bed reactors. In total, only two steps are necessary for the catalyst production: the first being the grinding of the metal precursor material HAuCl4 with porous glass, and the second being the laser sintering of the obtained metal precursor/glass mixture on the desired glass plate support, resulting in spherical metal nanoparticles. The process was preliminarily tested for small object slides (coated area 60 × 20 mm), but could be also demonstrated on glass plates of 100 × 100 mm. Finally, the catalytic performance of the realized catalyst systems was evaluated by degradation experiments of ethanol in a self-designed testing plant and a bypass-flow photoreactor.
2. Experimental section
2.1 Chemicals and materials
Ethanol and HAuCl4-trihydrate (49.5% Au content) were obtained commercially from Alfa Aesar, Karlsruhe, Germany and were used without further purification. Porous glass with different pore sizes was delivered by HEGLA boraident (Halle/Saale, Germany). Glass samples with initial pore sizes of 1–2 nm, 50 and 100 nm were delivered in larger pieces, whereas 200 and 370 nm glass samples were obtained preground. As a support material for the coating, Schott BOROFLOAT® plates with a size of 150 × 150 × 1 mm obtained from HEGLA boraident (Halle/Saale, Germany) were used.2.2 Mixing of HAuCl4 with porous glass
For the mixing of the particle precursor, 1 g of porous glass was put into a 35 mL ZrO2-milling beaker and 20 mg of HAuCl4 was added, in order to obtain in total a 1 wt% distribution of metal to glass. Through two slots for milling beakers, the second milling beaker was filled only with porous glass for comparison of the resulting powders. Then, 25 g of 5 mm ZrO2 milling balls was added to each beaker and the grinding process was carried out at 30 Hz for 60 min with a mixer ball mill Retsch 400M.2.3 Characterization of ground mixtures
The grinding mixtures were evaluated by particle size distribution measurements (PSD-measurements by dynamic light scattering, DLS) with a Fritsch Analysette 22 with a wet dispersion unit. Each measurement was carried out with ultrasound. Measurements for the analysis of BET surface areas and total pore volumes were done with a Quantachrome Autosorb iQ3 system. All ten samples (79.5–110.0 mg) were degassed for 1.2 h at temperatures up to 150 °C before nitrogen adsorption measurements were carried out at 77.35 K. For all of the samples, complete adsorption/desorption isotherms were measured. SEM imaging was carried out with a field emission scanning electron microscope, FE-SEM JSM 6300F, JEOL, in BSE contrast after sputtering the samples with graphite. Light microscopy was done with a Zeiss Axio Imager.A1m with Axio Cam MRc5 device.2.4 Laser sintering of obtained mixtures on supporting glass plates
The comminuted mixtures were dispersed in ethanol and homogenized in an ultrasonic bath. Afterwards, the mixture was applied homogeneously onto a 76 × 26 mm object slide in an area of 60 × 20 mm, and onto 150 × 150 mm float glass plates in a square of 100 × 100 mm in the middle of the plates and the ethanol was evaporated under IR radiation. Then, the material was laser sintered with a CO2-laser under mild conditions in a cross-hatch, so that the glass/metal precursor mixture was just superficially sintered. The laser feed rate was 40 mm s−1 and the line distance was 150 μm. The laser power was 0.85 W.UV/VIS measurements of the catalysts on supports were conducted using an Ulbricht sphere with a Perkin Elmer Lambda 900 UV/VIS/NIR Spectrometer. XRD measurements were made with an X’PERT MPD Pro (PANalytical) system using Cu K-alpha-radiation (λ = 0.154 nm). The crystallite size L was calculated according to the Scherrer equation. Raman measurements were carried out using a confocal WITec Raman Microscope alpha300 R setup with a water immersion objective from Nikon with 10× magnification and a numerical aperture (NA) of 0.25. The excitation wavelength was 785 nm by a cw diode laser from Toptica Photonics. Grating was set to 300 grooves per mm and the integration times were 2 s for the pure sample before and after catalysis, and 5 s for the sample with ethanol.
2.5 Catalytic tests
Catalytic tests were carried out in a self-constructed testing plant made of Swagelok® stainless-steel tubes, a temperature-controlled glass saturator and a self-designed bypass-flow-photoreactor. The glass saturator with ethanol was tempered to 273 K by an IKA RC2 Basic thermostat in order to evaporate the alcohol as the component to be degraded. A mass flow controller for nitrogen (Brooks GF series) with a control unit (Brooks 0254) was set to 30 mL min−1, directing the vaporized ethanol towards the reactor. Before reaching the reactor, another mass flow controller for air was set to 300 mL min−1. The pressure of the gas mixture is controlled by a pressure gauge (Keller LEO 2) before and after passing the reactor. Plasmonic catalysis is initiated by irradiation with a Xe arc lamp (LSB530A, obtained from LOT-QuantumDesign GmbH) with a spectral range of 200–900 nm in a LOT Oriel LSH201 housing and control unit. The reaction is monitored with the help of an Agilent gas chromatograph 7890B equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD). A PoraPLOT Q (25 m, 320 μm, 10 μm) was used as the column. The GC was calibrated with different amounts of ethanol in the gas phase and the ethanol concentration was calculated from the peak areas of ethanol. For calculations, a saturated vapor pressure of ethanol at 273.15 K of 1568 Pa (Dortmund Database, DDBST) was used. The measured pressure in the testing plant was 102![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa before and after the reactor chamber. The retention time of CO2 was verified by the injection of pure CO2.
000 Pa before and after the reactor chamber. The retention time of CO2 was verified by the injection of pure CO2.
The calculations of percentual ethanol degradation/CO2 increase are based on the average peak areas measured without light (except outliers) and the respective value measured after the light was switched on. Here, a range of strong ethanol degradation was measured before reaching steady-state conditions. For comparison, the average value of the latter was used.
3. Results and discussion
3.1 Analysis of grinding mixtures
The mixtures obtained from the grinding process – without and with HAuCl4 – are displayed in Fig. 1, left. A change in color can be seen for the sample containing the gold precursor, exhibiting a purple to violet color. This color points to an initial formation of particles already in the grinding step, as mixtures containing only the precursors are expected to be yellow arising from the colour of HAuCl4. SEM images (Fig. 1, right) confirm this assumption, but also show that this happens only occasionally and most of the material is still present as the glass/metal salt bulk. Data for particle size analyses and nitrogen adsorption measurements of the porous glasses with and without addition of HAuCl4 can be found in the ESI (Tables S1 and S2† respectively). | ||
| Fig. 1 Left: Photo of samples ground with no metal salt (white powder), and with HAuCl4 (purple powder). Right: The SEM images of the samples with HAuCl4/glass after grinding show a few gold nanoparticles in the bulk (bright structures). | ||
3.2 Analysis of complete catalyst plates
The formation of metal nanoparticles through laser irradiation without reducing agents present was demonstrated in the literature for AuNPs from HAuCl4 in solution.60 However, for the utilized glass support, tests needed to be done in order to evaluate whether it is possible to obtain a stable and regular coating with nanoparticles through laser impact on mixtures of glass powder and HAuCl4. Therefore, pretests on object slides were done in areas of 20 × 60 mm. The obtained coated glasses exhibit a strong red to deep wine red colour comparable to the powder obtained by grinding, indicating the formation of gold nanoparticles. Also, it can be seen in both cases that the coating is regular, which indicates a uniform coating with the nanoparticles (Fig. 2). | ||
| Fig. 2 Gold particles laser sintered on the object slides (left image; from left to right: HAuCl4 with 1–2, 50, 100, 200 and 370 nm porous glass). A light microscopy close up view with 5× magnification of the coating structure with gold nanoparticles (right image). Here, the course of the laser is visible in the red sintered grids with particles and the white non-sintered spaces containing no particles. | ||
The localization of the catalyst particles on the glass surface is crucial for catalytic applications, in order to enable light activation and to be accessible for the reactants in order to induce catalysis of chosen contaminants. SEM images from the surface of a slide coated with gold (Fig. 3, left) and the fracture plane of such an object slide (Fig. 3, right) show evidence that gold nanoparticles of around 30 nm in size are present on the surface (visible as bright spherical spots), but are also found melted into the glass or in second order pores of sizes up to 3 μm (which were apparently formed by the melting of the glass during the laser sintering process).
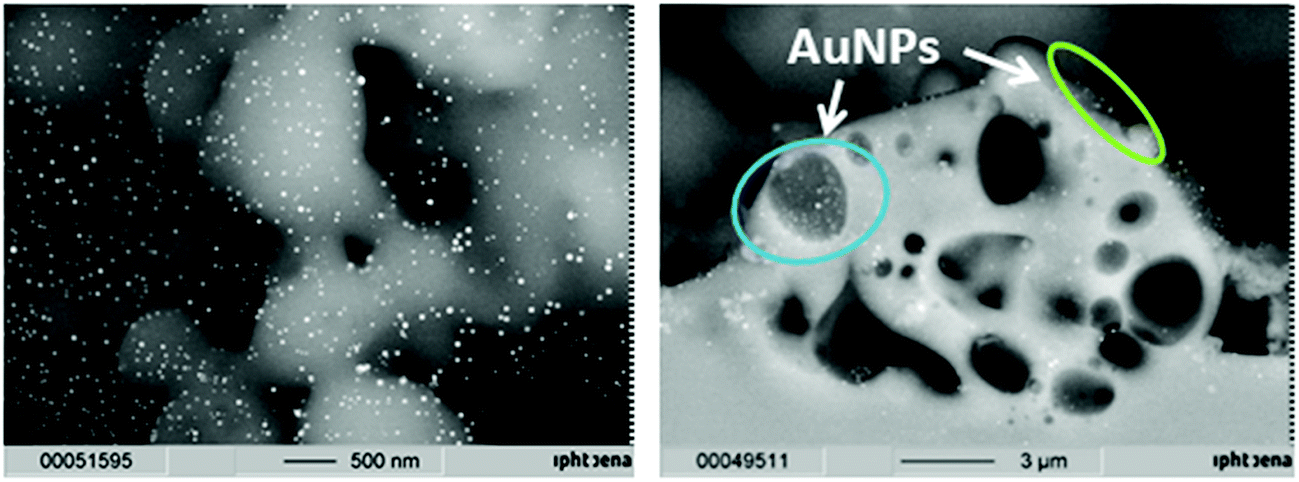 | ||
| Fig. 3 SEM images of gold nanoparticles on a laser sintered glass substrate (top view, left image) and a fracture plane image of an object slide (right image) with gold nanoparticles in pores (blue circle) and on the surface (green circle). | ||
Transmission measurements of the different sample object slides in an Ulbricht-sphere (Fig. 4) revealed a transmission minimum (absorption maximum) of 522 nm for the gold samples with porous glass of 1–2, 50, 100 and 200 nm, and of 528 for the sample with 370 nm porous glass. An absorption peak at 522 nm is typical for 30 nm gold spheres, and at 528 for particles with a size of 42 nm.61 Thus, the Ulbricht sphere measurements support the size found in the SEM images, but also clarify that the particles must be placed at the glass surface.
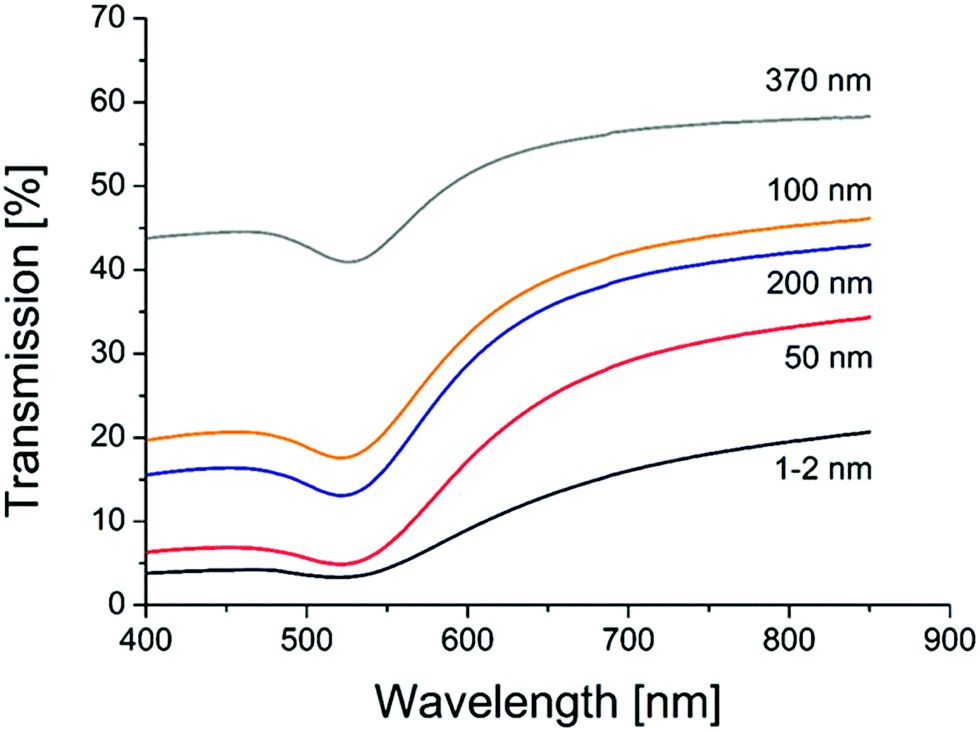 | ||
| Fig. 4 UV/VIS transmission spectra of AuNP catalysts on different porous glasses (pore sizes are given on each curve). | ||
Simulations using Mie-theory revealed a red-shift of the absorption band of gold nanoparticles from air (520 nm) to water (545 nm) and to the glass environment (565 nm; Fig. 5). When comparing these results with the Ulbricht sphere measurements (absorption maxima at 522 and 528 nm; Fig. 4) and knowing that the approximate size of the particles is around 30 nm, it can be concluded that the obtained particles are located on the surface, exposed to air and therefore able to be activated by light and reachable by contaminant molecules. This is also in good agreement with the literature, where similar shifting effects of gold nanoparticles (20 nm size) embedded in glass could be obtained by calculations.62
 | ||
| Fig. 5 Results from Mie-calculations for the absorption wavelengths of 80 nm AuNPs in different surrounding media (air, water and glass). | ||
Besides these UV/VIS measurements using the Ulbricht sphere, XRD was utilized to study the particle size using the Scherrer equation (Fig. 6, Table S3 ESI†). For the measured sample containing gold particles with 200 nm porous glass, the positions of the (111), (200), (220) and (311) features of monocrystalline gold nanoparticles are at positions of 2Θ = 38.2, 44.4, 64.6 and 77.6, which is consistent with the literature.63,64 The calculated crystallite size L resulted in a value of 30 nm for this sample, supporting the previous results from Ulbricht-sphere measurements and from SEM imaging.
 | ||
| Fig. 6 An XRD diffractogram of AuNPs on a layer of porous (200 nm pore size) glass. | ||
The scaling-up of the production process was demonstrated by coating of carrier plates matching the test reactor. These plates are of a 150 × 150 mm size with an area of 100 × 100 mm in the center of the plate to be coated with the catalytic active material. The coating process was therefore carried out exactly as it was for the object slides with the same glass/metal precursor mixture in the pretests. This should result in reproducible products. Indeed, we could also see a uniform, brightly red coloured coating of gold nanoparticles for such a large area.
In total, six plates with a pure gold coating and two with a bimetallic coating were obtained by this process (Fig. 7). This technique also allows estimation of the amount of metal precursor utilized, and therefore the catalyst mass per plate (Table 1). The plates are denoted regarding the pore size of the glass used in the grinding process.
 | ||
| Fig. 7 Catalyst plates with a 150 × 150 mm size and a coated area of 100 × 100 mm each. A = 1–2 nm, B = 50 nm, C = 100 nm, D = 200 nm plate, A, E = 200 nm plate and B, F = 370 nm porous glass. | ||
| Pore size of glass [nm] | Amount of glass/metal precursor per layerb [mg] | Au content per layer [as mg HAuCl4] | Au content in layer [as mg Au] | Costs of HAuCl4 per layer [€] |
|---|---|---|---|---|
| a The Au content of the laser-sintered layers relates to the gold content of the glass/metal precursor and its amount used for laser sintering.b Applied amount of the glass/metal precursor for laser sintering.c For the samples with glass of 200 nm initial porosity, two plates were obtained (displayed in Fig. 7D and E). | ||||
| 1–2 | 439 | 8.91 | 4.41 | 0.76 |
| 50 | 288 | 5.99 | 2.97 | 0.51 |
| 100 | 214 | 4.02 | 1.99 | 0.34 |
| 200 | 299/87 | 6.07/1.76 | 3.00/0.87c | 0.52/0.15 |
| 370 | 159 | 3.14 | 1.55 | 0.27 |
Besides the scalability of the process, the costs of the precursor for the nanoparticles can also be considered as favorable, as they range from 0.15 € in the case of the 200 nm porous glass plate B to 0.76 € for the 1–2 nm porous glass with AuNPs. Through adding more metal precursor to the glass/metal salt mixture, an increase in the number of catalyst particles on the support could be achieved easily, however this consequently results in higher metal salt costs.
3.3 Application of catalyst system in heterogeneous catalysis
The obtained catalysts are intended to be used for plasmon driven heterogeneous catalysis for waste gas cleaning by degradation of relevant contaminants. In the following experiments, the catalytic performance of the constantly irradiated catalysts was validated using ethanol as a model contaminant in the gas phase. In order to investigate the catalytic effect of the produced catalyst system, tests were carried out in a self-constructed laboratory testing plant (Fig. 8).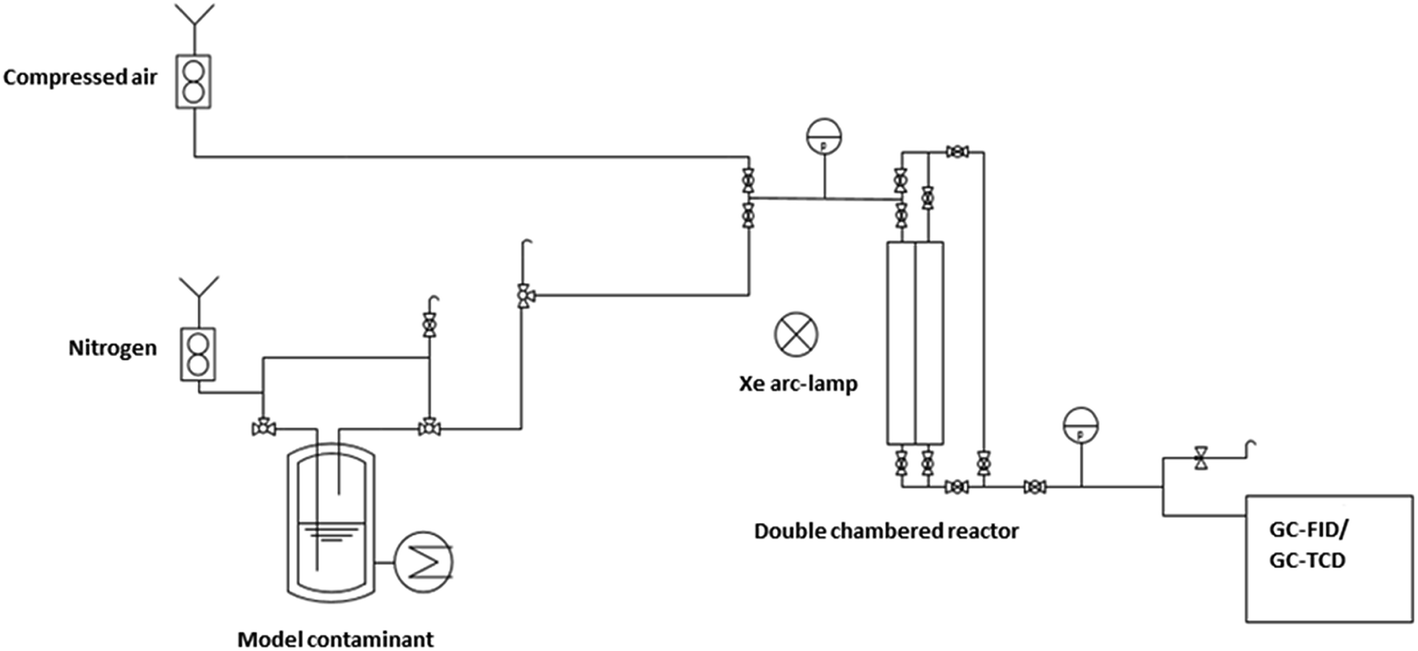 | ||
| Fig. 8 Setup of the laboratory testing plant. | ||
The model contaminant ethanol was placed in a glass saturator and vaporized at 273 K, and was carried into the testing plant by nitrogen in order to inhibit reactions before passing the catalyst. Therefore, oxidant air is mixed with the nitrogen/ethanol gas flow shortly before the reactor. The results of the degradation tests with the 200 nm catalyst plate B are displayed in Fig. 9.
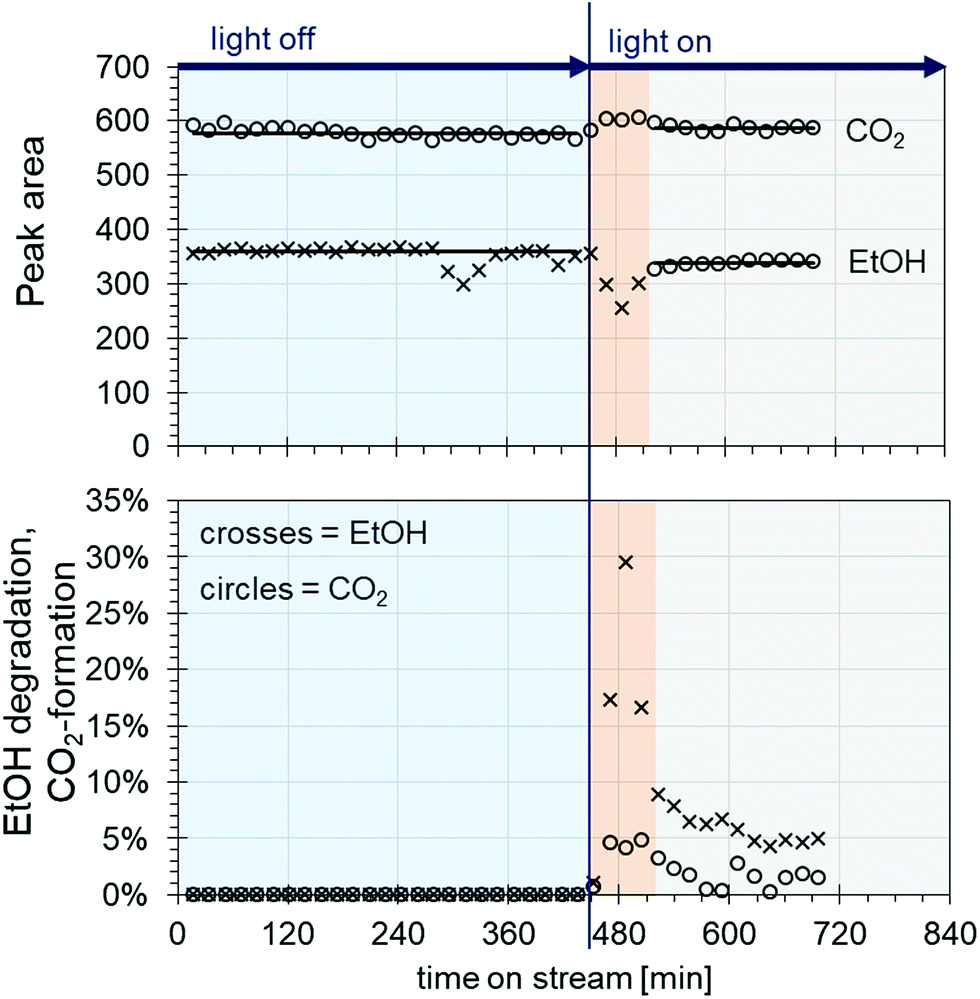 | ||
| Fig. 9 Result of the degradation tests with ethanol at a flow rate of 30 mL min−1 for ethanol/nitrogen and 300 mL min−1 for air at a 273 K saturator temperature regarding evolution of ethanol/CO2 peak areas (top) and the percentage of ethanol degradation/CO2 formation (bottom). | ||
The ethanol peak area measured without light is stable, with the exception of a few outliers (Fig. 9 top, crosses in the blue area). The same applies to the peak areas of CO2 coming from the air flow measured by TCD (Fig. 9 top, circles in the blue area). When the light is switched on, a rapid decrease in the ethanol concentration can be seen along with an increase in CO2, indicating ethanol degradation (Fig. 9 top, crosses and circles in the red area, and see also Fig. S1 ESI†). However, after around 50 min, the ethanol peak area increases again until it reaches steady state. In parallel, the CO2 peak area decreases, and then reaches a plateau (Fig. 9 top, crosses and circles in the grey area). This behaviour could be due to desorption processes of ethanol from the catalyst. As the conditioning and measurements of the initial ethanol concentration without light are carried out by bypass flow through the reactor with the catalyst inside, a certain amount of ethanol could adsorb onto the catalyst surface. When the light is switched on, only the ethanol arriving in the gas flow is degraded, until the catalyst is heated up to the extent that the adsorbed ethanol is also released. This might happen either through the thermal energy coming from the utilized light source or through plasmon heating. The outer temperature at the reactor window was around 45 °C. Later, the catalyst reaches equilibrium of adsorption/desorption and degradation, and a stable ethanol peak area is measured lying beyond the initial ethanol concentration.
Looking at the degradation of ethanol and the CO2 formation by percent (Fig. 9, bottom), an ethanol degradation of up to 30% was achieved along with a CO2 increase of around 5%. After reaching equilibrium, the ethanol degradation was at values of around 5% and the CO2 was around 2%. Along with the degradation rates, it was possible to evaluate the weight hourly space velocity (WHSV) as a catalytic key figure describing the reactor system (Table S4, ESI†). In our case, the WHSV could be calculated as 5.94 h−1.
Regarding the stability of the catalyst plate, it remained stable during the entire catalytic process for 720 min and no change of colour, loss of material, or adsorption of contaminants could be detected. Concerning the last point mentioning adsorption of the model contaminant ethanol on the catalyst, Raman studies were carried out with the used catalyst plate before and after catalysis and with ethanol in order to verify its Raman shift (Fig. S2, ESI†). The measured peak position of ethanol is in good agreement with the literature65,66 and it could be confirmed that no ethanol was adsorbed onto the catalyst plate, guaranteeing still accessible gold nanoparticles after the catalytic process.
4. Conclusion
In the present studies we could demonstrate an elegant, easy and inexpensive way to design a heterogeneous catalyst system on a glass support with different support sizes. The technique requires only two steps; the production of glass/metal precursor powders in a mixer ball mill, and the laser sintering of this material on the glass support with the desired size. Here, we could show that it is possible to obtain catalysts on supports with sizes of 20 × 60 to 100 × 100 mm with AuNPs as plasmon catalytic active particles. Through the grinding and laser sintering processes, the porosity of the glasses mixed with the metal precursors is not preserved, but metal nanoparticles were formed. The sizes of these particles were usually around 30 nm. Thus, it seems to be unimportant whether the utilized glass shows any porosity or which size the pores are, so any kind of glass seems applicable. A very important aspect of this to question is if the formed gold nanoparticles are also located on the glass surface. This could be confirmed, which enables the established catalyst systems to be used for catalytic applications.A key issue regarding the utilization of noble metal catalysts is the costs of such systems, especially for the cost-intensive metal salts used as particle precursors. Calculating the costs of our method, we could obtain prices ranging from a few cents up to 0.76 € for HAuCl4 per 100 cm2 catalyst area, which is a rather inexpensive approach.
Finally, catalytic tests analyzing the degradation of ethanol showed that a decrease in ethanol concentration of up to 30% is possible with the catalyst system displayed. Parallel to the ethanol degradation, CO2 formation could be measured as the resulting degradation product. It turned out, however, that the catalyst requires a certain warm-up time to reach equilibrium of the ethanol adsorption/desorption and degradation, which occurs due to thermal energy input or plasmon heating. This leads to a lowered but stable ethanol degradation of around 5% and a CO2 formation of 2%.
In conclusion, a novel approach for the fabrication of a heterogeneous plasmon-driven photocatalyst system is introduced, based on the laser-assisted sintering of a mixture of ground glass with a metal salt precursor on a glass substrate. The fabrication process is demonstrated, and the resulting catalyst areas are characterized as well as successfully tested using ethanol degradation as a model reaction. The procedure is easily scalable to larger areas and is quite cost-efficient, representing a promising novel approach for heterogeneous photocatalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out within the project #6 of pades Wachstumskern Thüringen (FKZ 03WKCN06). We thank pades Partikeldesign Thüringen and the BMBF (German Federal Ministry of Education and Research) for funding this project. Also we cordially thank Torsten Langer from JENOPTIK Automatisierungstechnik GmbH (Jena) and Robert Bischoff from HEGLA boraident (Halle/Saale). We would also like to express many thanks to Beate Fähndrich from the Institute of Technical Chemistry and Environmental Chemistry, Friedrich-Schiller-University Jena for BET measurements and Andrea Dellith (Leibniz-Institute of Photonic Technology Jena) for SEM imaging, Christa Schmidt (Leibniz-Institute of Photonic Technology Jena) for XRD measurements and Dr Clara Stiebing and Dr Jan Rüger (Leibniz-Institute of Photonic Technology Jena) for Raman measurements. We express our special thanks to the Open Access Fund of the Leibniz Association for the funding of the publication of this article.References
- A. Csáki, M. Thiele, J. Jatschka, A. Dathe, D. Zopf, O. Stranik and W. Fritzsche, Eng. Life Sci., 2015, 15, 266–275 CrossRef.
- K. A. Willets and R. P. V. Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef PubMed.
- S. Szunerits and R. Boukherroub, Chem. Commun., 2012, 48, 8999–9010 RSC.
- A. Campion and P. Kambhampati, Chem. Soc. Rev., 1998, 27, 241–250 RSC.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Chem. Rev., 1999, 99, 2957–2976 CrossRef.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef PubMed.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef PubMed.
- J. R. Adleman, D. A. Boyd, D. G. Goodwin and D. Psaltis, Nano Lett., 2009, 9, 4417–4423 CrossRef PubMed.
- B. Dong, Y. Fang, X. Chen, H. Xu and M. Sun, Langmuir, 2011, 27, 10677–10682 CrossRef PubMed.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef PubMed.
- G. L. Hallett-Tapley, M. J. Silvero, M. González-Béjar, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, J. Phys. Chem. C, 2011, 115, 10784–10790 CrossRef.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- G. Frens, Nature (London), Phys. Sci., 1973, 241, 20–22 CrossRef.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef.
- J. Pérez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzán and P. Mulvaney, Coord. Chem. Rev., 2005, 249, 1870–1901 CrossRef.
- H.-L. Wu, C.-H. Kuo and M. H. Huang, Langmuir, 2010, 26, 12307–12313 CrossRef PubMed.
- M. Thiele, J. Z. E. Soh, A. Knauer, D. Malsch, O. Stranik, R. Müller, A. Csáki, T. Henkel, J. M. Köhler and W. Fritzsche, Chem. Eng. J., 2016, 288, 432–440 CrossRef.
- P. S. Kumar, I. Pastoriza-Santos, B. Rodríguez-González, F. J. García de Abajo and L. M. Liz-Marzán, Nanotechnology, 2008, 19, 015606 CrossRef PubMed.
- A. Pyatenko, M. Yamaguchi and M. Suzuki, J. Phys. Chem. C, 2007, 111, 7910–7917 CrossRef.
- Y. Wan, Z. Guo, X. Jiang, K. Fang, X. Lu, Y. Zhang and N. Gu, J. Colloid Interface Sci., 2013, 394, 263–268 CrossRef PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef PubMed.
- D. Yu and V. W.-W. Yam, J. Am. Chem. Soc., 2004, 126, 13200–13201 CrossRef PubMed.
- I. Pastoriza-Santos and L. M. Liz-Marzán, Nano Lett., 2002, 2, 903–905 CrossRef.
- D. Aherne, D. M. Ledwith, M. Gara and J. M. Kelly, Adv. Funct. Mater., 2008, 18, 2005–2016 CrossRef.
- A. Knauer, A. Csáki, F. Möller, C. Hühn, W. Fritzsche and J. M. Köhler, J. Phys. Chem. C, 2012, 116, 9251–9258 CrossRef.
- T. Teranishi, M. Miyake and M. Hosoe, Adv. Mater., 1997, 9, 65–67 CrossRef.
- T. S. Ahmadi, Z. L. Wang, A. Henglein and M. A. El-Sayed, Chem. Mater., 1996, 8, 1161–1163 CrossRef.
- J. Turkevich and G. Kim, Science, 1970, 169, 873–879 CrossRef PubMed.
- T. Teranishi and M. Miyake, Chem. Mater., 1998, 10, 594–600 CrossRef.
- W. Niu, Z.-Y. Li, L. Shi, X. Liu, H. Li, S. Han, J. Chen and G. Xu, Cryst. Growth Des., 2008, 8, 4440–4444 CrossRef.
- S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814–1833 RSC.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef PubMed.
- J. Li, E. Saydanzad and U. Thumm, Phys. Rev. A, 2016, 94, 051401 CrossRef.
- M. Bernardi, J. Mustafa, J. B. Neaton and S. G. Louie, Nat. Commun., 2015, 6, 7044 CrossRef PubMed.
- M. Gonzalez-Bejar, K. Peters, G. L. Hallett-Tapley, M. Grenier and J. C. Scaiano, Chem. Commun., 2013, 49, 1732–1734 RSC.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- T. S. Rodrigues, A. G. M. da Silva, A. B. L. de Moura, I. G. Freitas and P. H. C. Camargo, RSC Adv., 2016, 6, 62286–62290 RSC.
- H. Wu, P. Wang, H. He and Y. Jin, Nano Res., 2012, 5, 135–144 CrossRef.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2015, 137, 948–957 CrossRef PubMed.
- F. Wang, C. Li, H. Chen, R. Jiang, L.-D. Sun, Q. Li, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef PubMed.
- X. Huang, Y. Li, Y. Chen, H. Zhou, X. Duan and Y. Huang, Angew. Chem., 2013, 125, 6179–6183 CrossRef.
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870–6873 CrossRef PubMed.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- S. A. Ansari, M. M. Khan, M. O. Ansari and M. H. Cho, New J. Chem., 2015, 39, 4708–4715 RSC.
- N. Pugazhenthiran, S. Murugesan, P. Sathishkumar and S. Anandan, Chem. Eng. J., 2014, 241, 401–409 CrossRef.
- F. Kim, J. H. Song and P. Yang, J. Am. Chem. Soc., 2002, 124, 14316–14317 CrossRef PubMed.
- Y.-Y. Yu, S.-S. Chang, C.-L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661–6664 CrossRef.
- V. Amendola, S. Polizzi and M. Meneghetti, J. Phys. Chem. B, 2006, 110, 7232–7237 CrossRef PubMed.
- J. Zhang, D. N. Oko, S. Garbarino, R. Imbeault, M. Chaker, A. C. Tavares, D. Guay and D. Ma, J. Phys. Chem. C, 2012, 116, 13413–13420 CrossRef.
- R. Intartaglia, G. Das, K. Bagga, A. Gopalakrishnan, A. Genovese, M. Povia, E. Di Fabrizio, R. Cingolani, A. Diaspro and F. Brandi, Phys. Chem. Chem. Phys., 2013, 15, 3075–3082 RSC.
- J. Zhang, M. Chaker and D. Ma, J. Colloid Interface Sci., 2017, 489, 138–149 CrossRef PubMed.
- S. Mukherjee, L. Zhou, A. M. Goodman, N. Large, C. Ayala-Orozco, Y. Zhang, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2014, 136, 64–67 CrossRef PubMed.
- E. Rombi, M. G. Cutrufello, R. Monaci, C. Cannas, D. Gazzoli, B. Onida, M. Pavani and I. Ferino, J. Mol. Catal. A: Chem., 2015, 404–405, 83–91 CrossRef.
- W. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. B. Cronin, ACS Catal., 2011, 1, 929–936 CrossRef.
- T. Nakamura, J. Mater. Res., 2008, 23, 968–974 CrossRef.
- W. Haiss, N. T. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef PubMed.
- V. A. G. Rivera, F. A. Ferri and E. Marega Jr, in Plasmonics – Principles and Applications, ed. K. Y. Kim, InTech, Rijeka, 2012, ch. 11, DOI:10.5772/50753.
- S. Krishnamurthy, A. Esterle, N. C. Sharma and S. V. Sahi, Nanoscale Res. Lett., 2014, 9, 627 CrossRef PubMed.
- M. M. H. Khalil, E. H. Ismail and F. El-Magdoub, Arabian J. Chem., 2012, 5, 431–437 CrossRef.
- B. N. Khlebtsov, Z. Liu, J. Ye and N. G. Khlebtsov, J. Quant. Spectrosc. Radiat. Transfer, 2015, 167, 64–75 CrossRef.
- Q. Wang, Z. Li, Z. Ma and L. Liang, Sens. Actuators, B, 2014, 202, 426–432 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03661f |
| This journal is © The Royal Society of Chemistry 2018 |