
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of trifluoromethyl-containing isoindolinones from tertiary enamides via a cascade radical addition and cyclization process†
Hui Yu *,
Mingdong Jiao,
Xiaowei Fang and
Pengfei Xuan
*,
Mingdong Jiao,
Xiaowei Fang and
Pengfei Xuan
School of Chemical Science and Engineering, Shanghai Key Lab of Chemical Assessment and Substainability, Tongji University, 1239 Siping Road, Shanghai 200092, P. R. China. E-mail: yuhui@tongji.edu.cn; Fax: +86 21 65981097; Tel: +86 21 65981097
First published on 3rd July 2018
Abstract
A radical trifluoromethylation reaction of tertiary enamides was investigated and trifluoromethyl-containing isoindolinones were prepared under mild conditions. Using TMSCF3 as a radical source, PhI(OAc)2 as an oxidant and KHF2 as an additive, tertiary enamides were converted to isoindolinones via a cascade addition and cyclization process in moderate to good yields.
In recent years, trifluoromethyl-containing azaheterocycles have attracted much attention for their potential application in the fields of pharmaceutical and agricultural chemistry.1 Thus, lots of efforts have been devoted to the synthesis of trifluoromethyl azaheterocycles,2 and among these developed methods, radical cascade addition and cyclization has emerged as a remarkable strategy due to its unique properties such as economy and high efficiency. Unsaturated amides are commonly used substrates for this type of transformation, which could be attacked by a CF3 radical followed by intramolecular C–O, C–N, or C–C bond formation to give different kinds of trifluoromethyl azaheterocycles. Fu reported a metal-free trifluoromethylation of N-allyamides with CF3SO2Na for the synthesis of trifluoromethyl-containing oxazolines via oxytrifluoromethylation.3 In the presence of copper salts, N-acyl-2-allylaniline could be converted to trifluoromethylated indolines in moderate to good yields via aminotrifluoromethylation process.4 With Togni's reagent,5 TMSCF3,6 CF3SO2Na,7 CF3SO2Cl8 and other reagents9 as the CF3 source, α, β-unsaturated amides, tosyl amides, or imides underwent a tandem conversion to give trifluoromethyl-containing oxindoles or isoquinoline-1,3-diones by trifluoromethylation/arylation reaction. On the other hand, as a special type of unsaturated amide containing an active double bond, enamide also exhibited excellent reactivity in radical reactions.10 In fact, trifluoromethylation of enamides has already been investigated, and in most cases trifluoromethylated alkenes were obtained as the main products.11 To the best of our knowledge, the radical trifluoromethylation and cyclization of enamide still remains undeveloped.
Isoindolinones are important N-heterocyclic compounds necessary in organic and pharmaceutical chemistry, and these compounds are used widely as anticoagulants and tranquilizers such as aristolactam, pagoclone, and zopiclone.12 To introduce a CF3 group into isoindolinones, Wang and co-workers explored a convenient way to the synthesis of trifluoromethyl-containing isoindolinones by radical aminotrifluoromethylation (Scheme 1a),13 but this transformation only occurred for N-methoxylbenzamides, and in case of N-alkylbenzamides trifluoromethylated alkenes were obtained as the major products. 1,1-disubstituted terminal alkenes were also not suitable substrates because of the competition between O-trapping and N-trapping process. Thus, development a new method for the synthesis of trifluoromethyl-containing isoindolinones is still in demand. Here in, as a continuation of our efforts on the radical modification of amide derivatives,14 we wish to present our work on the synthesis of trifluoromethyl-containing isoindolinones using enamides as the start materials by radical carbon trifluoromethylation (Scheme 1b).
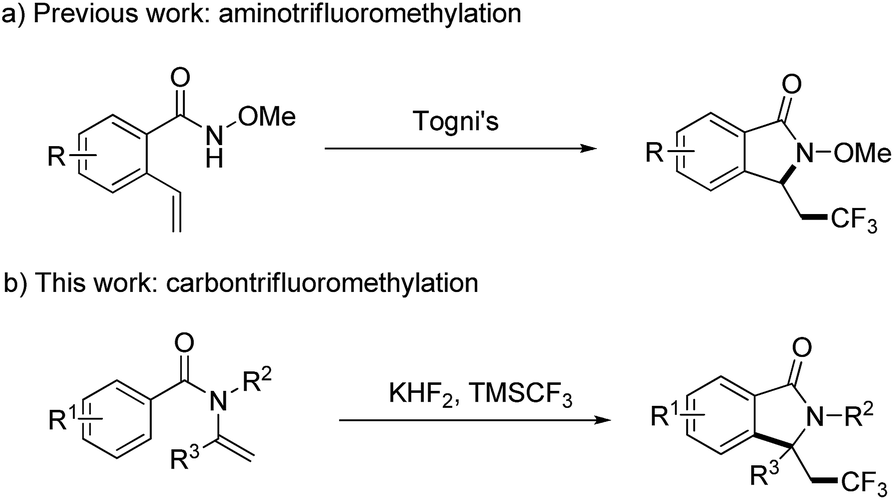 | ||
| Scheme 1 Synthesis of trifluoromethyl-containing isoindolinones. | ||
Initially, N-n-butyl-N-(2-propenyl) benzamide 1a was chosen as the model substrate to optimize the reaction conditions of this radical carbontrifluoromethylation process. As shown in Table 1, the reaction of 1a with TMSCF3 (4.0 equiv.) was firstly examined in CH3CN with PhI(OAc)2 (4.0 equiv.) as the oxidant and NaF (1.0 equiv.) as the additive at 80 °C under N2 atmosphere. 12 hours later, all the start material disappeared monitored by TLC and the desired product 2a was isolated in 15% yield with N-n-butyl benzamide isolated as the main byproduct (Table 1, entry 1). When KF and CsF was used instead of NaF, the desired product 2a was obtained in 35% and 38% yield respectively (Table 1, entries 2 and 3). To our delight, when bifluoride was used as the additive, the yield of 2a could be improved significantly, and KHF2 gave better result than NaHF2 and NH5F2 (Table 1, entries 4–6). Changing the solvent to CH3CN, CH2Cl2 or toluene resulted in the formation of 2a in only 0–32% yields (Table 1, entries 7–9). Increasing the reaction temperature to 100 °C or decreasing the reaction temperature to 60 °C and room temperature led to lower yield of 2a (Table 1, entries 10–12). Another commercial oxidant PhI(OCOCF3)2 (4.0 equiv.) was also tested but to give poor yield of 2a (Table 1, entry 13). When the amount of TMSCF3 and PhI(OAc)2 was reduced to 3.0 equiv., only 62% yield of 2a could be isolated (Table 1, entry 14). The amount of KHF2 was increased to 1.5 equiv. or decreased to 0.5 equiv. also caused lower yield of 2a (Table 1, entry 15). Finally, 1.0 equiv. of NaOAc was added to the reaction but the yield of 2a was not improved (Table 1, entry 16). On the basis of these results, entry 5 represents the best conditions.
|
||||
|---|---|---|---|---|
| Entry | Additive (0.3 equiv.) | Solvent (2 mL) | Temp. (°C) | Yield of 2ab (%) |
| a The reaction was carried out on 0.2 mmol scale in a sealed tube under N2.b Isolated yield.c PhI(OCOCF3)2 (4.0 equiv.) was used as the oxidant.d Reaction carried out with PhI(OAc)2 (3.0 equiv.) and TMSCF3 (3.0 equiv.).e With 1.5 equiv. KHF2.f With 0.5 equiv. KHF2.g 1.0 equiv. NaOAc was added. | ||||
| 1 | NaF | EtOAc | 80 | 15 |
| 2 | KF | EtOAc | 80 | 38 |
| 3 | CsF | EtOAc | 80 | 35 |
| 4 | NaHF2 | EtOAc | 80 | 52 |
| 5 | KHF2 | EtOAc | 80 | 75 |
| 6 | NH5F2 | EtOAc | 80 | 40 |
| 7 | KHF2 | CH3CN | 80 | 21 |
| 8 | KHF2 | CH2Cl2 | 80 | 32 |
| 9 | KHF2 | Toluene | 80 | Trace |
| 10 | KHF2 | EtOAc | 100 | 61 |
| 11 | KHF2 | EtOAc | 60 | 43 |
| 12 | KHF2 | EtOAc | r.t. | NR |
| 13 | KHF2 | EtOAc | 80 | 37c |
| 14 | KHF2 | EtOAc | 80 | 62d |
| 15 | KHF2 | EtOAc | 80 | 58e, 47f |
| 16 | KHF2 | EtOAc | 80 | 73g |

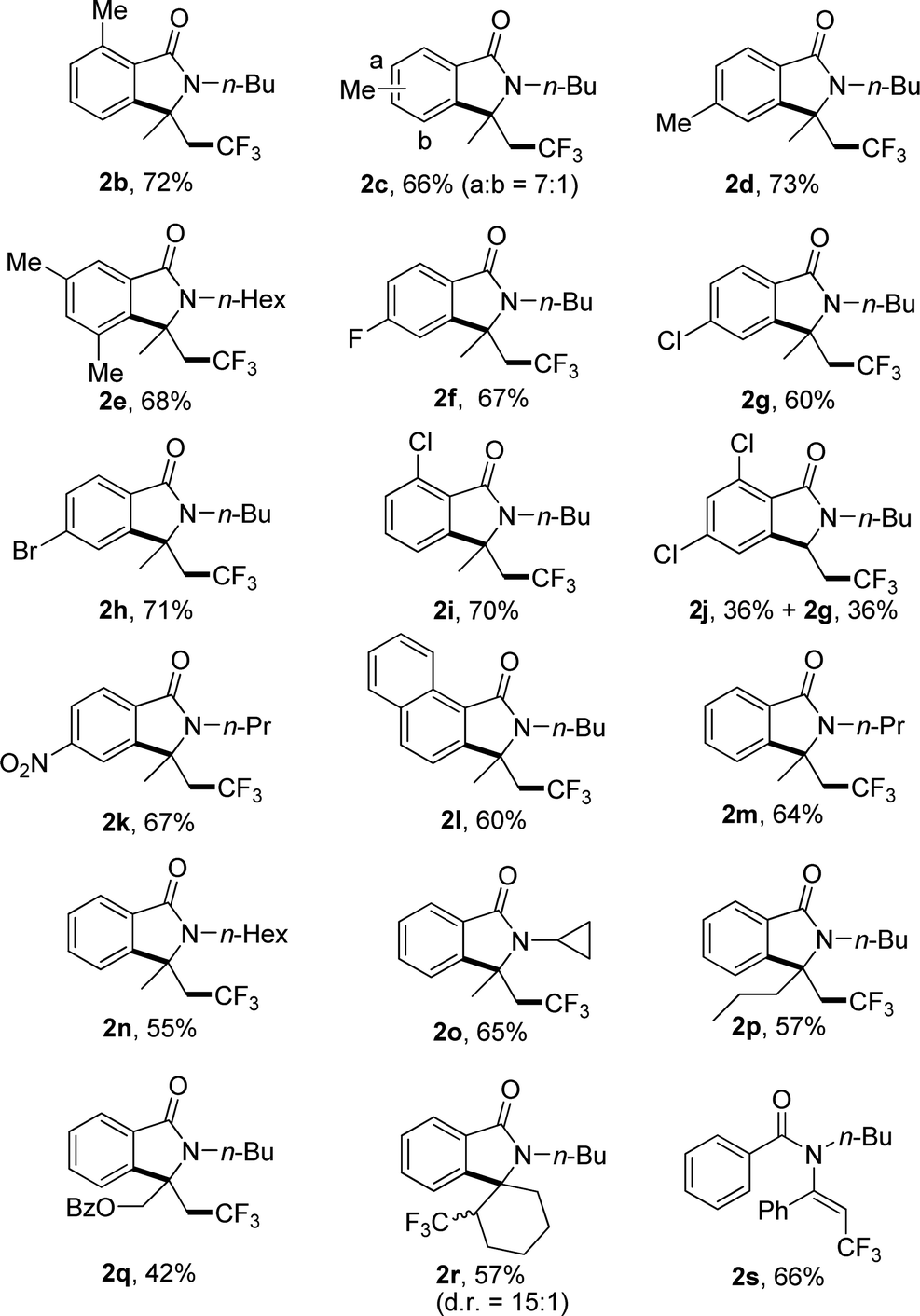
Under the optimized reaction conditions, the scope of substrates was investigated with results summarized in Table 2. For substrates with methyl group substituted at different positions on the benzoyl moiety of 1a, no significant steric hindrance was observed (Table 2, 2b–e). Substrates with electron-withdrawing group also gave satisfied yields (Table 2, 2f–k). Interestingly, 2,4-dichloro benzoyl substrate gave the desired product 2j in 36% yield accompanied with the 2-dechlorination product 2g in the same yield (Table 2, 2j). 1-Naphthoyl substrate underwent a smooth reaction to afford the product in 60% yields (Table 2, 2l). Changing the substituted group on the nitrogen from n-butyl to other alkyl groups also gave the corresponding product in moderate yield (Table 2, 2m–o). Substrates with different substituents on the inner side of double bond also gave the desired products in moderate yields, including a spiro product 2r (Table 2, 2p–r). For substrate with phenyl group on the double bond, no desired product could be found due to the increased steric hindrance, and trifluoromethylated alkene 2s was furnished in 66% yield (Table 2, 2s).

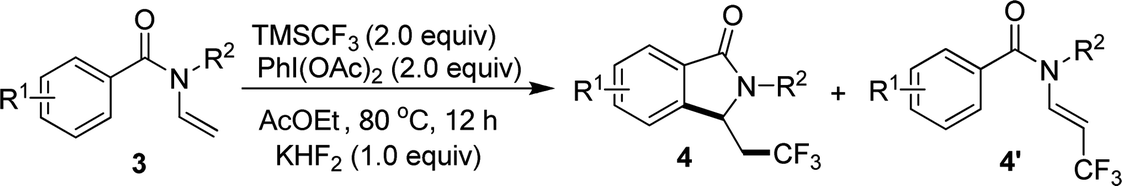
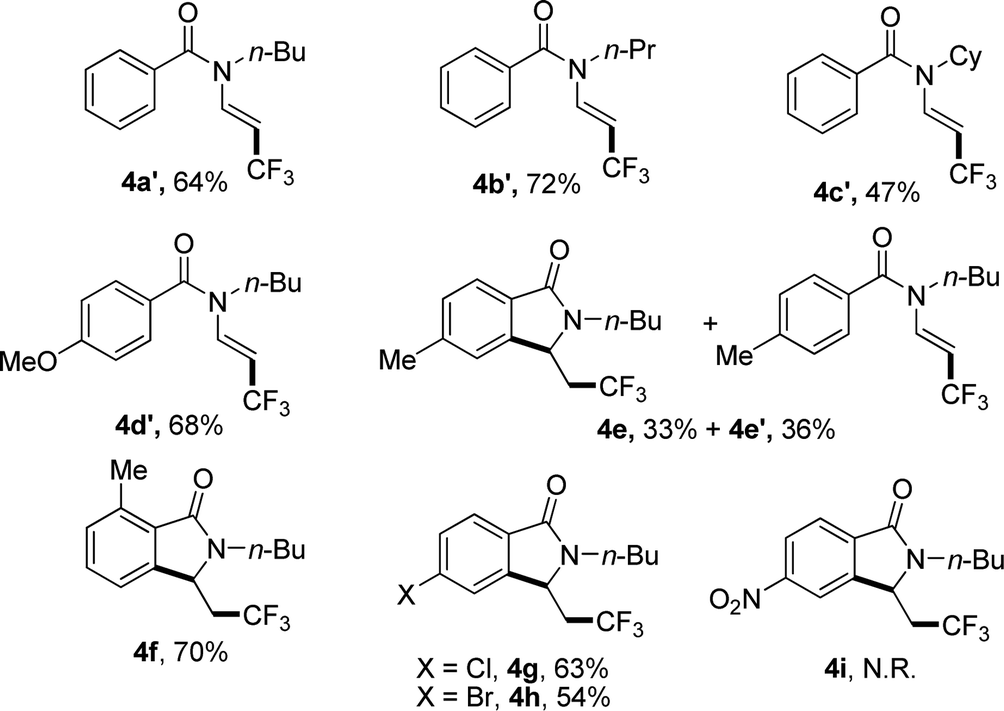
When N-n-butyl-N-(2-vinyl) benzamide 3a was subjected to the reaction conditions, no isoindolinone was observed, and the main product was trifluoromethylated alkene (Table 3, 4a′). Changing the N-protecting group to n-propyl or cyclohexyl also caused the formation of trifluoromethylated alkenes (Table 3, 4b′–4c′). It seemed that the substituents on the benzoyl group had significant influence on the reaction result. For example, substrate with methoxy group on the para position of the benzoyl moiety still gave trifluoromethylated alkene as the main product (Table 3, 4d′), but substrate with methyl group on the para position led to a mixture of trifluoromethylated alkene and isoindolinone (Table 3, 4e/4e′). However, substrate with methyl group on the ortho position or halides on the para position of the benzoyl group gave only isoindolinones as the main products (Table 3, 4f–4h). Substrate with NO2 on the para position displayed low reactivity and no reaction occurred (Table 3, 4i).
Heterocyclic substrate such as 5a and 5b was also examined, but no cyclization product could be found and trifluoromethylated alkene 6a and 6b was obtained as the only product (Scheme 2).
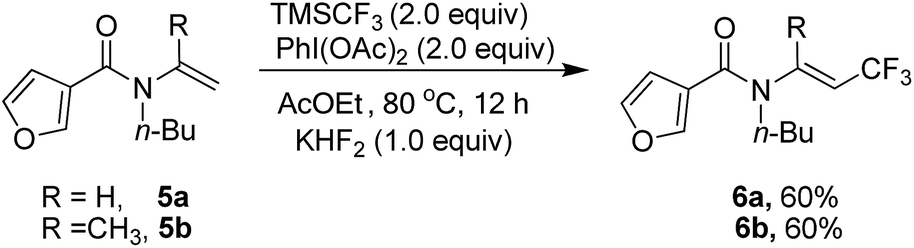 | ||
| Scheme 2 . Results of heterocyclic substrate 5a and 5b. | ||
To gain insights into the reaction mechanism, a control experiment was carried out to elucidate the mechanism. When 1.0 equiv. TEMPO was added to the reaction, the yield of 2a decreased significantly to 15%, which indicated the possibility of a radical pathway. Based on the control experimental result and the previous investigation on aryltrifluoromethylation of alkenes, plausible mechanism for our methodology is proposed in Scheme 2. In the presence of KHF2, TMSCF3 reacted with PhI(OAc)2 to generate CF3 radical, then the CF3 radical attacked enamide 1 or 3 affording radical intermediate A. Depending on the structure of the substrate, intermediate A would be converted to trifluoromethyl-containing isoindolinone or trifluoromethylated alkene according to different pathways as followed: (path a) intramolecular cyclization of A gave the resulting radical B with an aryl ring, which was oxidized to intermediate C then underwent deprotonation to give rise to the final product 2a–r or 4; (path b) A was oxidized to intermediate D then underwent elimination to give trifluoromethylated alkene 2s, 4′ or 6 (Scheme 3).
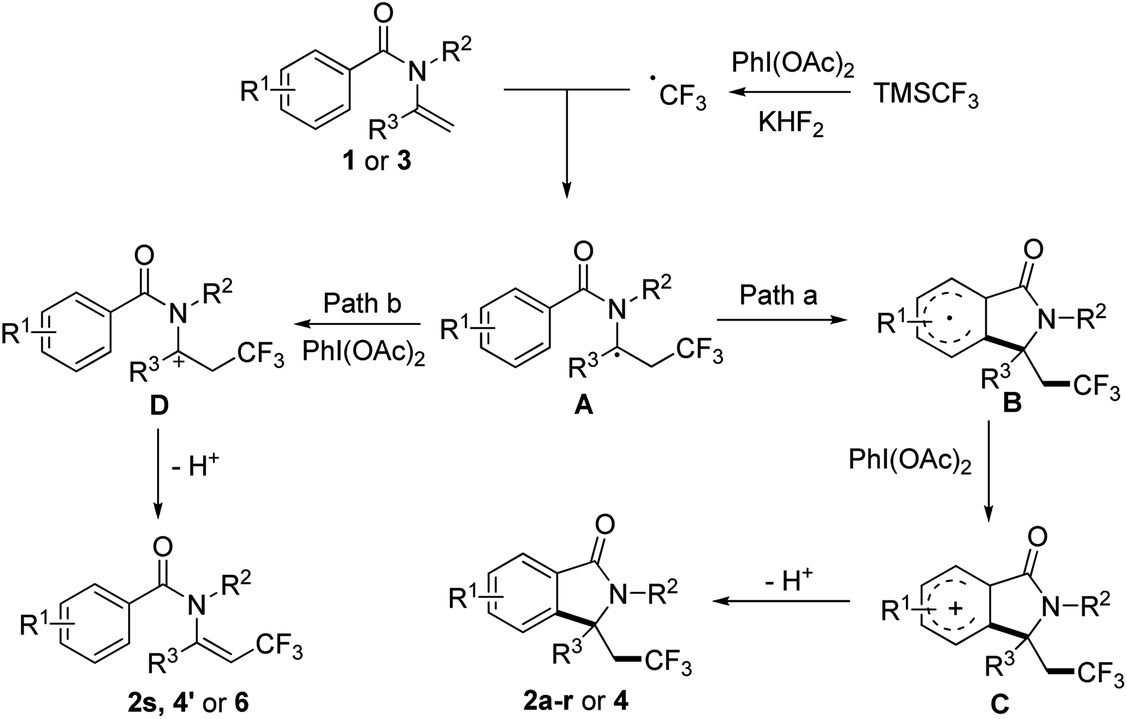 | ||
| Scheme 3 Possible mechanism. | ||
Conclusions
In conclusion, we have demonstrated a simple, facile approach to trifluoromethyl-containing isoindolinones by radical addition and cyclization of enamides with moderate to good yields under mild conditions. KHF2 was found crucial for this cyclization process and further investigation into the mechanism is currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Tongji University (20123231) is gratefully acknowledged.Notes and references
- For reviews, see: (a) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef PubMed; (b) A. A. Gakh and Y. Shermolovich, Curr. Top. Med. Chem., 2014, 14, 952 CrossRef PubMed.
- (a) Y. Zheng and J. A. Ma, Adv. Synth. Catal., 2010, 352, 2745 CrossRef; (b) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef PubMed; (c) T. Koike and M. Akita, Top. Catal., 2014, 57, 967 CrossRef; (d) M. A. Honey, R. Pasceri, W. Lewis and C. J. Moody, J. Org. Chem., 2012, 77, 1396 CrossRef PubMed; (e) J. P. Bouillon, C. Ates, Z. Janousek and H. G. Viehe, Tetrahedron Lett., 1993, 34, 5075 CrossRef.
- J. Yu, H. Yang and H. Fu, Adv. Synth. Catal., 2014, 356, 3669 CrossRef.
- (a) J. S. Lin, X. G. Liu, X. L. Zhu, B. Tan and X. Y. Liu, J. Org. Chem., 2014, 79, 7084 CrossRef PubMed; (b) J. S. Lin, Y. P. Xiong, C. L. Ma, L. J. Zhao, B. Tan and X. Y. Liu, Chem.–Eur. J., 2014, 20, 1332 CrossRef PubMed; (c) H. Y. Zhang, W. Huo, C. Ge, J. Zhao and Y. Zhang, Synlett, 2017, 28, 962 CrossRef.
- (a) P. Xu, J. Xie, Q. Xue, C. Pan, Y. Cheng and C. Zhu, Chem.–Eur. J., 2013, 19, 14039 CrossRef PubMed; (b) H. Egami, R. Shimizu and M. Sodeoka, J. Fluorine Chem., 2013, 152, 51 CrossRef; (c) Y. An, Y. Li and J. Wu, Org. Chem. Front., 2016, 3, 570 RSC; (d) W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480 CrossRef PubMed; (e) N. Yang, Z. Li, L. Ye, B. Tan and X. Y. Liu, Chem. Commun., 2016, 52, 9052–9055 RSC.
- (a) L. Li, M. Deng, S. C. Zheng, Y. P. Xiong, B. Tan and X. Y. Liu, Org. Lett., 2014, 16, 504 CrossRef PubMed; (b) W. Fu, F. Xu, Y. Fu, C. Xu, S. Li and D. Zou, Eur. J. Org. Chem., 2014, 4, 709 CrossRef; (c) X. Mu, T. Wu, H. Wang, Y. Guo and G. Liu, J. Am. Chem. Soc., 2012, 134, 878 CrossRef PubMed; (d) Y. Wang, J. Qiu, D. Kong and F. Chen, Synlett, 2014, 25, 1731 CrossRef.
- (a) L. Shi, X. Yang, Y. Wang, H. Yang and H. Fua, Adv. Synth. Catal., 2014, 356, 1021 CrossRef; (b) L. Zhang, Z. Li and Z. Q. Liu, Org. Lett., 2014, 16, 3688 CrossRef PubMed; (c) W. Wei, J. Wen, D. Yang, X. Liu, M. Guo, R. Dong and H. Wang, J. Org. Chem., 2014, 79, 4225 CrossRef PubMed; (d) R. Sakamoto, H. Kashiwagi, S. Selvakumar, S. A. Moteki and K. Maruoka, Org. Biomol. Chem., 2016, 14, 6417 RSC; (e) J. Liu, S. Zhuang, Q. Gui, X. Chen, Z. Yang and Z. Tan, Eur. J. Org. Chem., 2014, 15, 3196 CrossRef; (f) Q. Lu, C. Liu, P. Peng, Z. Liu, L. Fu, J. Huang and A. Lei, Asian J. Org. Chem., 2014, 3, 273 CrossRef; (g) F. Yang, P. Klumphu, Y. Liang and B. H. Lipshutz, Chem. Commun., 2014, 50, 936 RSC.
- (a) C. Liu, W. Zhao, Y. Huang, H. Wang and B. Zhang, Tetrahedron, 2015, 71, 4344 CrossRef; (b) X. J. Tang, C. S. Thomoson and W. R. Dolbier, Org. Lett., 2014, 16, 4594–4597 CrossRef PubMed; (c) L. Zheng, C. Yang, Z. Xu, F. Gao and W. Xia, J. Org. Chem., 2015, 80, 5730 CrossRef PubMed.
- (a) J. Y. Guo, R. X. Wu, J. K. Jin and S. K. Tian, Org. Lett., 2016, 18, 3850 CrossRef PubMed; (b) J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed; (c) S. Tang, Z. H. Li, M. W. Wang, Z. P. Li and R. L. Sheng, Org. Biomol. Chem., 2015, 13, 5285 RSC.
- For review and selected examples, see: (a) M. X. Wang, Chem. Commun., 2015, 51, 6039 RSC; (b) P. Song, P. Yu, J. S. Lin, Y. Li, N. Y. Yang and X. Y. Liu, Org. Lett., 2017, 19, 1330 CrossRef PubMed; (c) Y. Tang, Y. Zhang, K. Wang, X. Li, X. Xu and X. Du, Org. Biomol. Chem., 2015, 13, 7084 RSC; (d) X. M. Xu, L. Zhao, J. P. Zhu and M. X. Wang, Angew. Chem., Int. Ed., 2016, 55, 3799 CrossRef PubMed; (e) T. Taniguchi, A. Ishita, M. Uchiyama, O. Tamura, O. Muraoka, G. Tanabe and H. Ishibashi, J. Org. Chem., 2005, 70, 1922 CrossRef PubMed; (f) L. He, L. Zhao, D. X. Wang and M. X. Wang, Org. Lett., 2014, 16, 5972 CrossRef PubMed; (g) G. K. Friestad and Y. Wu, Org. Lett., 2009, 11, 819 CrossRef PubMed; (h) C. H. Lei, D. X. Wang, L. Zhao, J. P. Zhu and M. X. Wang, Chem.–Eur. J., 2013, 19, 16981 CrossRef PubMed; (i) M. N. Zhao, Z. H. Ren, D. S. Yang and Z. H. Guan, Org. Lett., 2018, 20, 1287 CrossRef PubMed; (j) C. H. Lei, D. X. Wang, L. Zhao, J. P. Zhu and M. X Wang, J. Am. Chem. Soc., 2013, 135, 4708 CrossRef PubMed.
- (a) R. Ding, Q. Zhang, Y. Xu and T. Loh, Chem. Commun., 2014, 50, 11661 RSC; (b) R. Rey-Rodriguez, P. Retailleau, P. Bonnet and I. Gillaizeau, Chem.–Eur. J., 2015, 21, 3572 CrossRef PubMed; (c) H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang and S. Yu, Chem.–Eur. J., 2012, 18, 15158 CrossRef PubMed.
- For reviews, see: (a) A. Di Mola, L. Palombi and A. Massa, Targets Heterocycl. Syst., 2014, 18, 113 Search PubMed; (b) A. Di Mola, L. Palombi and A. Massa, Curr. Org. Chem., 2012, 16, 2302 CrossRef.
- K. Shen and Q. Wang, Org. Chem. Front., 2016, 3, 222 RSC.
- (a) H. Yu and J. Shen, Org. Lett., 2014, 16, 3204 CrossRef PubMed; (b) H. Yu and J. Shen, RSC Adv., 2015, 5, 9815 RSC; (c) H. Yu, Y. Xu, R. Dong and Y. Fang, Adv. Synth. Catal., 2017, 359, 39 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data for the products. See DOI: 10.1039/c8ra03696a |
| This journal is © The Royal Society of Chemistry 2018 |