
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBi-functional heterogeneous catalysts for carbon dioxide conversion: enhanced performances at low temperature†
Adrien Comèsa,
Xavier Collarda,
Luca Fusaroa,
Luciano Atzorib,
M. Giorgia Cutrufellob and
Carmela Aprile *a
*a
aUnit of Nanomaterial Chemistry (CNANO), University of Namur (UNAMUR), Department of Chemistry, Rue de Bruxelles 61, 5000 Namur, Belgium. E-mail: carmela.aprile@unamur.be; Tel: +32 81 724514
bDipartimento di Scienze Chimiche e Geologiche, Università di Cagliari, Complesso Universitario di Monserrato, s.s. 554 Bivio per Sestu, 09042 Monserrato, CA, Italy
First published on 16th July 2018
Abstract
Novel heterogeneous bi-functional catalysts bearing tin or zinc inserted as single sites within the silica architecture acting as acid centres and decorated with imidazolium moieties as the nucleophile source were successfully synthesized. The materials were extensively characterized via various techniques including N2 physisorption, solid state nuclear magnetic resonance, X-ray photoelectron spectroscopy, transmission electron microscopy and adsorption microcalorimetry. The solids were tested as catalysts for the conversion of carbon dioxide, selecting the synthesis of styrene carbonate as the target reaction. Both materials exhibited improved performances compared to the analogous solids functionalized with the sole imidazolium salt as well as to other materials reported in the literature. The Sn-based catalyst displayed excellent conversion also in the presence of various epoxides. In all experiments the bi-functional solid allowed reducing the reaction temperature below 150 °C. In the presence of glycidol the temperature was decreased down to 30 °C. The short synthesis protocol of the heterogeneous catalysts, together with the 100% atom economy of the target reaction and the low reaction temperature, make the entire process highly sustainable. Moreover, the Sn-based catalyst was stable under the selected reaction conditions and reusable for multiple catalytic cycles.
Introduction
During the past years, there was a rising interest of the scientific community towards the conversion of carbon dioxide into valuable compounds. This small molecule can be efficiently used as a renewable C1 building block to synthesize a broad range of chemicals such as urea and urethane derivatives, isocyanates, carboxylic acids, and (poly)carbonates.1 However, the elevated thermodynamic and kinetic stability of CO2 makes its transformation still a challenging topic.2,3 In order to transform the energetically unfavourable CO2 conversion into a feasible process, high energy starting materials should be selected in combination with an efficient catalyst. When oxiranes or oxetanes are employed as reactants, the synthesis of the corresponding five or six membered cyclic carbonates can be achieved. Cyclic carbonates are attractive compounds from a sustainable point of view, because of the 100% atom economy of the process as well as for their possible industrial uses.4 They can find application as aprotic polar solvents, electrolytes for batteries and raw-materials for the preparation of polycarbonates.3–7 Amongst the catalysts able to efficiently convert CO2 and epoxides into the corresponding cyclic carbonates, ammonium, phosphonium and imidazolium salts deserve to be mentioned.8 To achieve an easy recovery from the reaction mixture and reuse of the catalyst for multiple cycles, most of these salts were also heterogenized9 on different supports. Silica-based porous materials,10,11 polymers,12,13 single as well as multi-walled carbon nanotubes,14–16 and more recently carbon nanohorns17 among others were already selected for this purpose. The mechanism of the reaction is well known and was already reported in various publications.6 The reaction is initiated by the attack of a nucleophile (usually represented by a halide) to the C1 or the C2 carbon, depending on the substituents, of the epoxide via a SN2 mechanism. The ring opening is followed by CO2 insertion with generation of a cyclic carbonate. However, when a catalyst such as ammonium or imidazolium salts is used in the absence of a co-catalyst, high temperatures (usually higher than 125 °C) are needed.18The high energy input required for the transformation of carbon dioxide can be further reduced employing a species (herein called co-catalyst) able to activate the three-membered ring via coordination of the oxygen with a metal centre acting as Lewis acid or through formation of hydrogen bond.1,18 The first strategy is largely exploited and widely described in the literature. A series of organometallic complexes was already reported by different authors. Various salen-, phthalocyanine-, porphyrins-based complexes bearing Al, Co, Cr or Zn as metal centres were successfully used for this purpose.1 Tangestaninejad and co-workers also proposed a Sn-based porphyrin complex which displayed improved performances due to an increased coordination strength.19 The use of molecules displaying two or three metal centres was also suggested. Kleij and co-workers designed a tri-nuclear Zn-based complex exhibiting bi-functional properties.20 The use of various metal–organic frameworks (MOF) in combination with ammonium or imidazolium salts was also reported.21–23 The majority of the previously reported examples refer to homogeneous catalysts or systems in which one of the two components (catalyst or co-catalyst) was heterogenized. In the broad list of contributions addressing the synthesis of cyclic carbonates from CO2, only a minor number of publications describe the use of a heterogeneous bi-functional catalyst in which both catalyst and Lewis acid co-catalyst are heterogenized on one support and even fewer illustrate the use of inorganic metal centres acting as Lewis acid sites.
Herein, the design of two novel heterogeneous bi-functional catalysts presenting both a metal centre (Zn or Sn) able to bring acid properties and the nucleophile required to promote the opening of the epoxide was successfully achieved. The synthesis procedure involved the initial preparation of a porous support bearing a metal element inserted as single sites within the silica architecture followed by the grafting of an imidazolium salt. The solids were extensively characterized via various techniques including N2 physisorption, X-ray diffraction, 13C and 29Si solid state nuclear magnetic resonance, transmission electron microscopy, combustion chemical analysis and inductively coupled plasma optical emission spectroscopy. The insertion of Sn and Zn as single sites within the silica structure was proved via solid state 119Sn nuclear magnetic resonance and X-ray photoelectron spectroscopy respectively, while the acid properties of the solids were addressed via adsorption microcalorimetry.
The materials were tested under solvent-free conditions in a batch reactor selecting the chemical fixation of CO2 into styrene oxide as target reaction. Both Sn- and Zn-based solids displayed improved performances compared to the analogous silica catalyst functionalized with the sole imidazolium salt as well as to other solids reported in the literature. The versatility of the Sn-based catalyst was also tested with a series of epoxides. In all experiments, the bi-functional solid allowed reducing the reaction temperature below 150 °C.
Results and discussion
The design of the bi-functional heterogeneous solids bearing both imidazolium-based catalysts and co-catalysts with acid properties was based on a two-step procedure.In a first step, mesoporous solid supports bearing Sn and Zn embedded as single site in the silica structure were successfully synthesized optimizing a previously described procedure (see Experimental section).24,25 The selected approach allows obtaining particles with reduced size (XS, below 100 nm) with a short protocol requiring only 60 min synthesis time. This range of size was initially selected because of the improved performances of the catalysts displaying a small particle dimension compared to the large particle analogues.26 For the sake of comparison, a mesoporous all-silica solid with reduced size (XS-MCM-41) was synthesized as well. Transmission electron microscopy investigation confirmed that both Sn- and Zn-based solids (XS-Sn and XS-Zn) as well as XS-MCM-41 display a narrow particle size distribution with a particle diameter comprised between 50 and 100 nm (Fig. S1†). Moreover, at high magnification, the mesoporous arrangement was also clearly distinguished. The characteristic hexagonal organization of pores was confirmed via low-angle XRD measurements (Fig. S2†). All samples display the d100 signal at 2θ ≅ 2°, typical of MCM-41-like architectures. As expected, the main d100 reflection was broader than the one observed for MCM-41 with standard dimensions and the additional d110 and d200 reflections were not clearly visible as a consequence of the reduced size with a consequent decrease of the long range order.27 Analysis of the materials via nitrogen physisorption revealed a high specific surface area and a narrow pore size distribution centred at 2.2 nm (Fig. S3† and Table 1). In view of the selected application, a key parameter is represented by the acidity of the solids, which was investigated by means of adsorption microcalorimetry. In Fig. 1 the differential heat of adsorption, Qdiff, is reported as a function of the amount of NH3 adsorbed for both Sn-XS and Zn-XS samples. For comparison, the analysis of XS-MCM-41 is reported as well. From this investigation emerged that the thermal effect associated with ammonia adsorption on Sn-XS and Zn-XS is much higher than on XS-MCM-41, especially at low NH3 uptake values, indicating that both Sn- and Zn-based solids display acid properties significantly more pronounced than the all-silica sample. On both Sn-XS and Zn-XS Qdiff is initially higher than 200 kJ mol−1, revealing the presence of strong acid sites, and then decreases as the ammonia uptake increases. The point at which the adsorption becomes non-specific can be established taking into account the results obtained on the pure silica XS-MCM-41 sample, for which differential adsorption heat values as high as 67 kJ mol−1 are observed (Fig. 1), in agreement with previous experiments on pure silica samples.28–30 Accordingly, for the Sn- and Zn-based samples the fraction of ammonia uptake corresponding to differential heats below 70 kJ mol−1 was ascribed to non-specific and/or physical adsorption on the pure-silica part of the samples, and thus neglected in calculating the acid sites concentration which resulted 170 and 280 μmol g−1 for Sn-XS and Zn-XS, respectively. The continuous decrease in the differential heat as the ammonia uptake increases on both samples indicates heterogeneity of the acid sites, which can be either Lewis (the Sn or Zn centres) or Brønsted sites (bridged OH groups made acidic by the proximity of the metal site). From the data of ammonia readsorption after overnight evacuation, it was possible to evaluate the differential heat at which the adsorption shifts from irreversible to reversible. It was found that the acid sites able to adsorb ammonia irreversibly are those characterized by Qdiff values higher than 90 kJ mol−1, whose concentration is about 110 and 160 μmol g−1 for Sn-XS and Zn-XS, respectively. From this study emerged that both materials display interesting acid properties in view of their possible application as active supports in the conversion of carbon dioxide.
| Specific surface area (m2 g−1) | Mean pore size (nm) | Total pore volume (cm3 g−1) | Amount of Imia (mmol g−1) | Amount of Mb (mmol g−1) | |
|---|---|---|---|---|---|
| a Range of imidazolium salt determined via combustion chemical analysis on different batches. Amount estimated considering the nitrogen percentage.b Amount of metal (M: Sn or Zn) determined via inductively coupled plasma optical emission spectroscopy (ICP-OES). See Experimental section for more details. | |||||
| XS-MCM-41 | 1090 | 2.1 | 1.24 | — | — |
| XS-MCM-41-Imi | 239 | 0.8 | 0.58 | 1.46 | — |
| XS-Sn | 1086 | 2.3 | 1.43 | — | Not measured |
| XS-Sn-Imi | 234 | 0.8 | 0.71 | 1.25–1.40 | 0.16 |
| XS-Zn | 1125 | 2.2 | 1.37 | — | Not measured |
| XS-Zn-Imi | 298 | 0.8 | 0.60 | 1.30–1.37 | 0.10 |
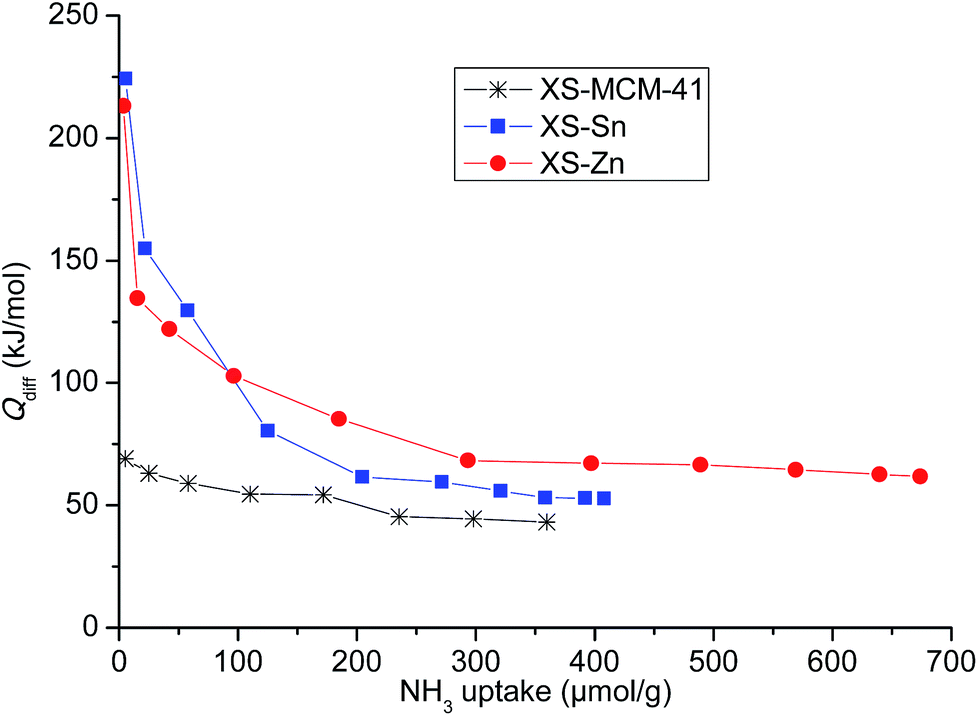 | ||
| Fig. 1 Differential heat of adsorption (Qdiff) as a function of the NH3 uptake for XS-Sn, XS-Zn and XS-MCM-41. | ||
The second step, requiring the functionalization of the surface with an imidazolium-based catalyst, was performed via a one-pot procedure reacting the previously described XS-solids with an equivalent mixture of 3-chloropropyltrimethoxysilane and 1-methyl imidazole as depicted on Scheme 1. This procedure, compared with the standard two-step approach, presents the multiple advantages of a fast functionalization together with a limited number of separation and purification procedures as well as the absence of intermediate characterizations. The amount of organic moieties in the functionalized solids (XS-MCM-41-Imi, XS-Sn-Imi and XS-Zn-Imi) was quantified via combustion chemical analysis (Table 1) while the proof of the presence of imidazolium and not adsorbed imidazole species was obtained via solid state 13C-CP-MAS NMR (Fig. 2 and S4†).31,32 The anchoring of the trimethoxysilane-based linker to the silica surface was addressed via solid state 29Si NMR performed both in cross polarisation and direct excitation.
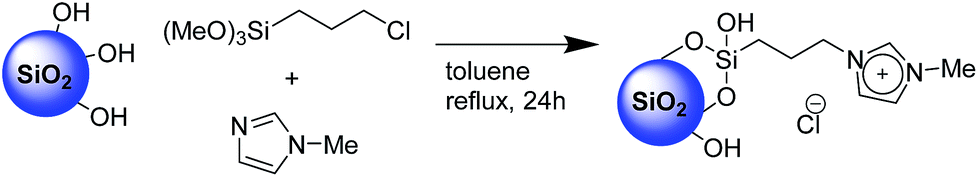 | ||
| Scheme 1 One-pot surface functionalization of silica with imidazolium salt. | ||
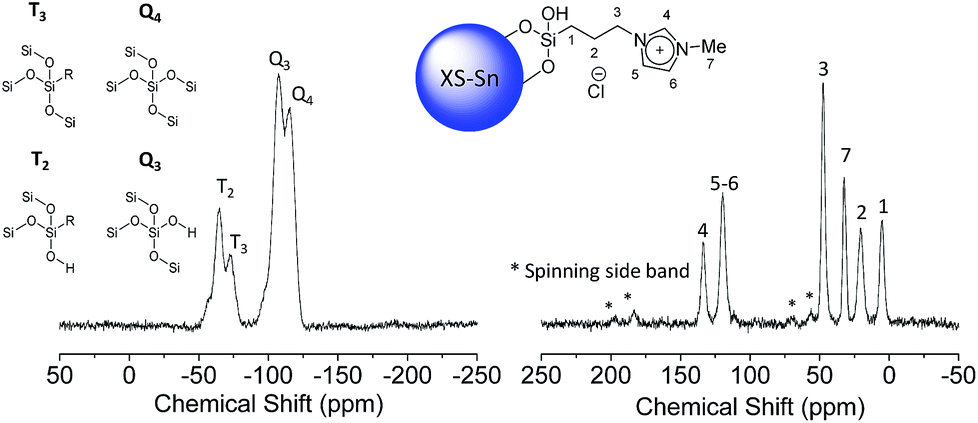 | ||
| Fig. 2 29Si-CP-MAS-NMR (left) and 13C-CP-MAS-NMR (right) of XS-Sn-Imi. | ||
The presence of a combination of T3 and T2 signals in the 29Si-CP-MAS-NMR (Fig. 3 and S4†) confirmed that organic moieties were anchored via a covalent bond and not physisorbed on the silica surface.
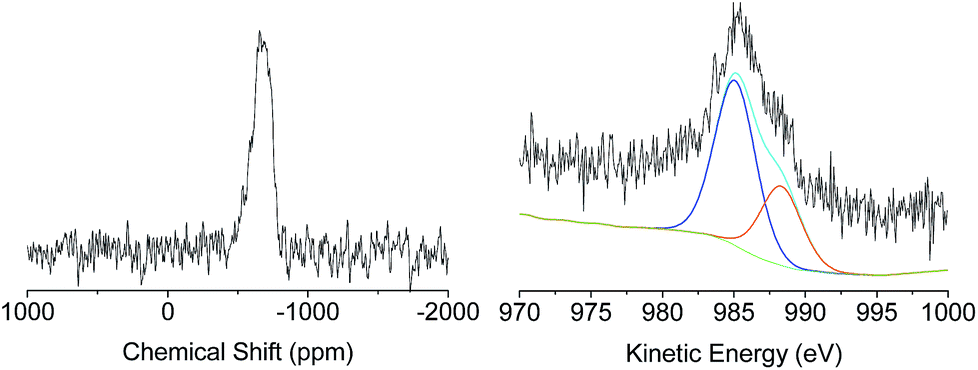 | ||
| Fig. 3 Solid-state 119Sn-NMR of XS-Sn (left) and Zn LMM Auger transition of XS-Zn (right). | ||
A deep investigation on the insertion of Sn and Zn as single site within the silica scaffold was performed as well. Solid-state 119Sn-NMR under static conditions (Fig. 3, left) revealed the presence a broad band centred at −680 ppm which can be attributed to Sn(IV) species in a tetrahedral coordination.33–35 The absence of relevant contribution at higher frequencies (around −600 ppm) allows excluding the formation of a significant amount of SnO2 due to self-condensation of the tin precursors in the reaction medium. The acquisition of solid 67Zn NMR data was hindered by the intrinsic difficulties of this nucleus. It is known that the low natural abundance of 67Zn together with its large quadrupole moment and the small gyromagnetic ratio represent a highly unfavourable combination of features for solid state nuclear magnetic resonance investigation.36 Moreover, the low amount of Zn inserted in the samples (about 2 wt%) reduces also the possibility of efficient use of advanced sensitivity-enhancement techniques or ultrahigh-magnetic-field NMR instruments for a deep investigation of the Zn coordination sphere. In order to overcome these drawbacks, X-ray photoelectron spectroscopy via analysis of the Auger transition (Fig. 3, right) was used to study the insertion of Zn as single site in the MCM-41 solid. The Zn LMM Auger transition can be considered as an overlapping of two different contributions, the first one centred at 988.08 eV corresponds to extra-framework ZnO and the second one at 985.30 eV can be attributed to intra-framework Zn species.24 This result indicates that in the case of XS-Zn-Imi the metal centre is partially present (ca. 30% of the total amount) in the form of ZnO extra-framework species.
The structural and textural properties of the final materials (XS-Sn-Imi and XS-Zn-Imi) were investigated via transmission electron microscopy (TEM), X-ray diffraction (XRD) and nitrogen physisorption. TEM analysis of the samples (Fig. 4) revealed that the post-functionalization did not affect the morphology of the materials with aggregation or sintering phenomena. Both XS-Sn-Imi and XS-Zn-Imi display an almost spherical shape and a narrow particle size distribution similar to the non-functionalized samples. As expected, low angle XRD measurements performed on the imidazolium functionalized solids evidenced a pattern similar to the one already observed for XS-Sn and XS-Zn (Fig. S2†) confirming that the hexagonal order was preserved during the reaction. As expected, nitrogen physisorption analysis (Fig. 5 and S5†) clearly indicated a decrease of the Brunauer–Emmett–Teller (BET) specific surface area as well as of the pore diameter (psd). Due to the shift of the pore size distribution in the microporous region two different methods were applied for the analysis of the data. The standard Barrett–Joyner–Halenda (BJH) was selected to estimate the psd of the non-functionalized XS-solids while the micropore size was evaluated using the method proposed by Horvath and Kawazoe.37,38
 | ||
| Fig. 4 TEM images of XS-Sn-Imi (a) and XS-Zn-Imi (b). | ||
The variation of the pore diameter suggests that the anchoring of the imidazolium moieties occurs not only at the external surface of the particles but also within the pores. It is important to underline that the reduced pore size does not represent a serious drawback in view of the application of these solids in the conversion of carbon dioxide because of the small size of the reactants. As can be seen in Fig. 5 all the materials display a type IV isotherm with an evident hysteresis loop at high relative pressures. This behaviour was previously observed for the materials exhibiting an extra-small size25,26 and can be ascribed to the presence of disordered meso–macro pores as consequence of the inter-particle interactions. Both XS-Sn-Imi and XS-Zn-Imi display promising properties for catalytic applications such as high surface area, relevant amount of metal element inserted as single site which provide the desired acidity and good percentage of imidazolium functionalization. Hence, both solids were tested in the reaction of carbon dioxide with epoxides to give the corresponding cyclic carbonates.
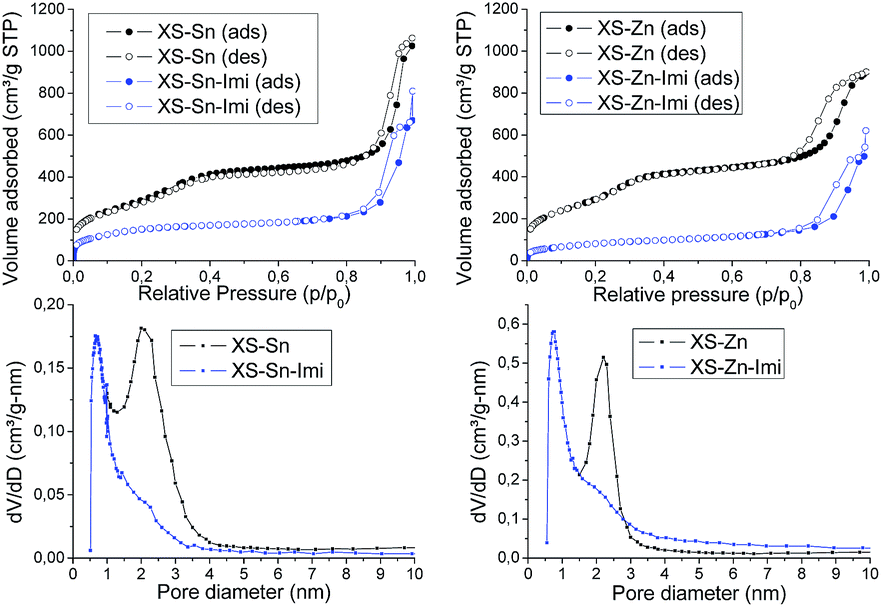 | ||
| Fig. 5 N2 adsorption–desorption isotherms of XS-Sn and XS-Sn-Imi (up-left) and XS-Zn and XS-Zn-Imi (up-right). Pore size distribution of XS-Sn and XS-Sn-Imi (down-left) and XS-Zn and XS-Zn-Imi (down-right). | ||
A challenging substrate as styrene oxide was selected to evaluate the activity of the catalysts and the reaction temperature was decreased to 125 °C compared to the standard value (usually this reaction is performed at 150 °C in absence of co-catalyst).10,16 For comparison, the activity of the all-silica MCM-41 solid functionalized with imidazolium moiety (XS-MCM-41-Imi) was studied as well (see entry 1 in Table 2). Moreover, before starting the investigation in the presence of the bi-functional catalysts an additional catalytic test adding a small amount of ZnCl2 (corresponding approximatively to the quantity inserted in the silica framework) to the reaction mixture was performed in order to evaluate the positive contribution of a salt with Lewis acid properties under homogeneous conditions. In order to allow a meaningful comparison among the solids, as well as with reference catalysts reported in the literature, two different turnover numbers (TON) were calculated. This choice is justified by the presence of two different catalytic active species behaving as catalyst (imidazolium moieties) and co-catalysts (metal inserted as single site) both displaying a synergic role in the reaction. The TON considering the catalyst (TONimi) was defined as moles of epoxide converted/moles of imidazolium salt while the TON based on the co-catalyst (TONM) was defined as moles of epoxide converted/moles of Sn or Zn.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Amount (g) | EtOH (mL) | Conversion (%) | Selectivity (%) | TONimib | TONMc |
| a Reaction conditions: styrene oxide (24.0 mL; 210 mmol), CO2 pressure (40 bar), temperature (125 °C), 500 rpm.b Turnover number (TON) calculated as moles of epoxide converted/moles of imidazolium sites. Amount of imidazolium salt quantified via chemical combustion analysis for each catalyst.c Turnover number (TON) calculated as moles of epoxide converted/moles of Sn or Zn. Amount of metal (Sn or Zn) quantified via inductively coupled plasma optical emission spectroscopy (ICP-OES) for each catalyst.d Reaction performed in presence of ZnCl2 (20 mg, 0.147 mmol). | |||||||
| 1 | XS-MCM-41-Imi | 1 | 1.5 | 54 | >95 | 78 | — |
| 2 | XS-MCM-41-Imid | 1 | 1.5 | 63 | >95 | 91 | 901 |
| 3 | XS-Sn-Imi | 1 | 1.5 | 60 | >95 | 94 | 774 |
| 4 | XS-Sn-Imi | 0.5 | 1.5 | 37 | >95 | 116 | 948 |
| 5 | XS-Zn-Imi | 0.5 | 1.5 | 39 | >95 | 127 | 1489 |
| 6 | XS-Sn-Imi | 0.5 | — | 32 | >95 | 96 | 826 |
| 7 | XS-Zn-Imi | 0.5 | — | 32 | >95 | 98 | 1343 |
As expected, the presence of ZnCl2 caused an improvement of the catalytic performances of the XS-MCM-Imi solid from 54% to 63% conversion (compare entries 1 and 2 in Table 2). Interestingly in the presence of XS-Sn-Imi similar performances compared to the homogeneous Lewis acid catalyst were obtained. The activity of the XS-Sn-Imi solid was preserved after decreasing the amount of catalyst (Table 2, entries 3 and 4). The slightly increase of TONimi obtained under the conditions reported in entry 4 could be attributed to a better dispersion of the solid in the reaction mixture. XS-Zn-Imi catalyst allowed achieving similar conversion (compare entries 4 and 5 in Table 2), thus further proving the positive influence of the insertion of single sites within the silica structure.
All the previously described experiments were performed under the same reaction conditions, hence in presence of a small amount (1.5 mL) of ethanol needed to solubilize the ZnCl2 added in the initial test. However, since it is known that the presence of hydroxyl groups in the reaction mixture may also facilitate the opening of the epoxide via formation of hydrogen bonds, analogous experiments in the absence of alcohol were performed as well.
No relevant differences were observed for both XS-Sn-Imi and XS-Zn-Imi catalysts (compare entries 4 with 6 and 5 with 7), thus proving that the small amount of ethanol was not the main responsible of the increased activity and further confirming the positive active role of the XS-Sn and XS-Zn supports.
The better performances of XS-Zn-Imi in terms of TONM could be attributed to the higher acidity of the support.
The versatility of the catalysts was checked selecting the most challenging (slightly less performant) XS-Sn-imi solid. The data concerning the activity of the Sn-based material in the presence of various epoxides are reported in Table 3. From this investigation emerged that the XS-Sn-Imi displays excellent performances in the presence of various epoxides including the extremely low reactive cyclohexene oxide. Moreover, in the presence of epichlorohydrin and glycidol excellent performances were achieved at the challenging temperatures of 80 °C and 60 °C respectively (entries 7 and 9). All these results confirm the positive synergic effect of the catalyst and co-catalyst.
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Temperature (°C) | Conversion (%) | Selectivity (%) | TONimib |
| a Reaction conditions: epoxide (24.0 mL), catalyst (500 mg), CO2 pressure (40 bar), 500 rpm.b Turnover number (TONimi) calculated as moles of epoxides converted/moles of imidazolium active sites. Amount of imidazolium salts quantified via chemical combustion analysis for each catalyst (not all the catalyst used comes from the same preparation).c Reaction performed for 24 h.d Reaction performed for 6 h with 1 g of catalyst. | |||||
| 1c | Cyclohexene oxide | 150 | 26 | >95 | 90 |
| 2c | Cyclohexene oxide | 125 | 5 | >95 | 17 |
| 3 | Styrene oxide | 125 | 32 | >95 | 96 |
| 4 | Propylene oxide | 125 | 41 | >95 | 196 |
| 5 | Epichlorhydrin | 125 | 53 | >95 | 233 |
| 6 | Epichlorhydrin | 100 | 40 | >95 | 185 |
| 7 | Epichlorhydrin | 80 | 16 | >95 | 78 |
| 8 | Glycidol | 80 | 63 | >95 | 334 |
| 9 | Glycidol | 60 | 35 | >95 | 179 |
| 10d | Glycidol | 30 | 11 | >95 | 32 |
To challenge even more the XS-Sn-Imi, a test with glycidol at 30 °C (entry 10) was performed as well. Also under these conditions, the catalyst showed a good TONimi, higher than other bi-functional catalysts reported in the literature employed under homogeneous39 (TONimi of 26) or heterogeneous conditions in the presence of the same epoxide.
In the recent literature, many efforts were dedicated to the decrease of the working temperature and/or pressure (e.g. using MOF). However, this goal is usually achieved employing imidazolium or more often ammonium salts in homogeneous phase.40–45 It should be mentioned that a direct comparison with the literature is hampered by the huge differences in the reaction conditions employed for the synthesis of cyclic carbonates including presence of solvent, substrate to catalyst molar ratio, nature of nucleophile, use of co-catalyst, temperature, pressure and reaction time. These difficulties can be partially reduced if the catalysts are compared in terms of turnover number or turnover frequency (TOF defined as TON/time). Our solids display better TONimi than some of the most active catalysts previously reported in the literature and used in the absence of co-catalyst. Moreover, they also display excellent TONM or TOFM if compared with other bi-functional heterogeneous catalysts. One of the best comparison can be probably done with similar silica-based porous materials embedding titanium as single site46 and tested in the presence of styrene oxide at 120 °C under solvent free conditions which displayed TONM much lower than the ones achieved with both XS-Sn-Imi and XS-Zn-Imi. Other metal-free catalysts containing only imidazolium-based species can be also selected for comparison to evidence the importance of the co-catalysts. One of the most active silica-supported imidazolium salts displayed a TONimi of 88 in presence of styrene oxide and under conditions similar to those used for the present work. However, it deserves to be mentioned that the counter-anion/nucleophile used was the more active iodide.47
Still considering the synthesis of styrene carbonate, similar TON (e.g. 101 and 106) were achieved with other structured supports such as carbon nanohorns but these catalysts required higher temperature (150 °C).17
When working under heterogeneous conditions, the excellent catalytic performances of a solid are not the only parameter to be considered, the true heterogeneous nature of the catalyst should be also evaluated. In order to exclude the possible leaching of active species in solution and confirm the stability of catalyst under the selected reactions conditions, recycle experiments were performed (Fig. 6). After each cycle, the XS-Sn-Imi catalyst was recovered from the reaction mixture by centrifugation, washed and reused (see Experimental section for more details) without any additional activation treatment. The data reported in Fig. 6 evidence that the catalyst can be used in multiple cycles without any decrease in the catalytic performances.
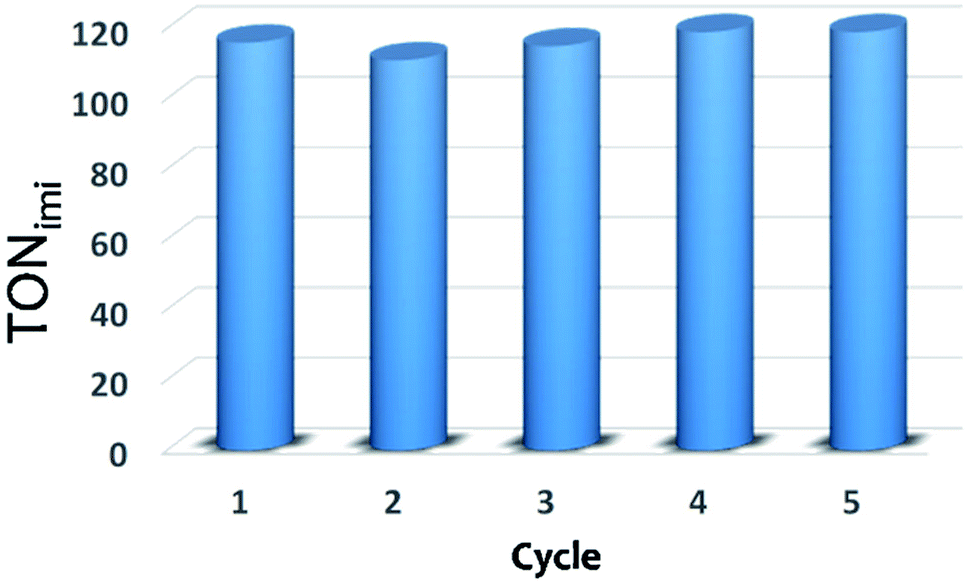 | ||
| Fig. 6 Catalytic performance of XS-Sn-Imi in subsequent reaction cycles. After each cycle, the solid was washed several times with toluene and ethanol, dried and reused without additional treatments. | ||
Experimental section
Materials and methods
Ammonium hydroxide (30%) was purchased from Carl Roth. Cetyltrimethylammonium bromide (CTAB), tetraethylorthosilicate (TEOS), N-methylimidazole, (3-chloropropyl)trimethoxysilane, styrene oxide, epichlorohydrin, cyclohexene oxide, propylene oxide were purchased from TCI. SnCl4·5H2O, Zn(NO3)2·6H2O, glycidol were purchased from Sigma-Aldrich. All chemicals were used as received without any further purification.Transmission electron microscopy (TEM) images were obtained using a Philips Tecnai 10 microscope operating at 80 kV. Samples were prepared by dispersion of a small quantity of material in absolute ethanol and deposited onto a copper grid. Nitrogen adsorption–desorption analyses were carried out at 77 K with a volumetric adsorption analyser (Micromeritics Tristar 3000 and ASAP 2000). Prior to the analysis, the samples were pre-treated at 150 °C for 16 h under reduced pressure (0.1 mbar). The Brunauer–Emmett–Teller (BET) method was applied in the 0.05–0.30 p/p0 range to calculate the specific surface area, while the pore size distributions were calculated from the adsorption isotherm using the Barrett–Joyner–Halenda (BJH) method. Powder X-ray diffraction (XRD) patterns were recorded on a PANalytical X'pert diffractometer with Cu Kα radiation (λ = 1.54178 Å). A Tian-Calvet heat flow calorimeter (Setaram) equipped with a volumetric vacuum line was used for microcalorimetric measurements. Each sample was pre-treated overnight at 250 °C under vacuum (10−3 Pa). Ammonia adsorption was carried out at 80 °C by admitting successive doses of the probe gas and recording the corresponding thermal effect. The run was stopped at a final pressure of 133.3 Pa. After overnight outgassing at 80 °C, a second adsorption run was carried out. The Si environment and the coordination of the Sn atoms were studied by 29Si Magic Angle Spinning (MAS) and static 119Sn Nuclear Magnetic Resonance (NMR). 29Si and 13C NMR spectra were recorded at room temperature on a Bruker Avance-500 spectrometer operating at 11.7 T (99.3 MHz for 29Si and 125.7 MHz for 13C) using a 4 mm CP-MAS Bruker probe. The sample was packed in a 4 mm zirconia rotor and measured with a spinning frequency of 8000 Hz. Cross polarization CP-MAS spectra were recorded using a 5 s relaxation delay and 5 ms contact time for 29Si and 2 ms for 13C. The processing comprised exponential multiplication of the FID with a line broadening factor of 30 Hz for 29Si or 10 Hz for 13C, zerofilling, Fourier transform, phase and baseline corrections. The chemical shift scale was calibrated at room temperature with respect to a sample of solid 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) (0.0 ppm). 119Sn NMR spectra were recorded at room temperature on a Varian VNMRS-400 spectrometer operating at 9.4 T (149 MHz for 119Sn) using a 10 mm wideline probe. The sample was packed in a 10 mm Kel-F container and studied in static condition. 119Sn spectra were recorded using the Hahn echo pulse sequence and following acquisition parameters: 60 s relaxation delay, 3.0 μs (90°) excitation pulse, 5 ms acquisition time. The processing comprised exponential multiplication of the FID with a line broadening factor of 1000 Hz, zero-filling, Fourier transform, phase and baseline corrections. The chemical shift scale was calibrated at room temperature with respect to the isotropic shift of SnO2 to −603 ppm.48 1H NMR spectroscopy of liquid samples was performed on a JEOL ECA operating at 9.4 T (400 MHz). The X-ray photoelectron spectroscopy (XPS) analyses were performed on a ThermoFisher ESCALAB 250Xi instrument. Inductively coupled plasma optical emission spectroscopy (ICP-OES) employed to determine the chemical composition of the materials was performed an Optima 8000 ICP-OES Spectrometer.
Chemical combustion analyses were performed on a Perkin-Elemer 2400 Serie 2 analyser.
Catalysts preparation
Catalytic tests
All catalytic tests were performed in a Cambridge Design Bullfrog batch reactor with individual control of the temperature and the stirring. During the entire experiment in addition to temperature and stirring, the evolution of pressure can be monitored as well. Prior to any experiment, the catalyst was dried overnight at 100 °C. In a typical test, the catalyst was added to the epoxide (24.0 mL) in a Teflon vial under solvent-free conditions. After closing the reactor, the mixture was stirred at 500 rpm. The system was purged with a gentle flow of N2 before the addition of CO2. After a short period of pressure stabilisation at room temperature, the system was heated to the desired temperature with a rate of 1 °C min−1. The reaction was allowed to run at the desired temperature for a variable time (in function of the selected epoxide). After this time, the reactor was cooled down to room temperature, the pressure was released and the reactor was opened. The reaction mixture was submitted to centrifugation for 10 min at 4500 rpm. The supernatant was removed and analysed by 1H-NMR using CDCl3 as solvent.For recycling experiment, the solid was added in the centrifugation tube in presence of toluene (50 mL) and it was sonicated for 15 minutes, centrifuged at 4500 rpm for 10 min and the supernatant removed. This washing operation was repeated 2 times with toluene and 2 times with ethanol. Then the solid was dried at 100 °C overnight prior to next catalytic run.
Conclusion
Two bi-functional silica-based materials bearing tin or zinc inserted as single sites within the silica architecture acting as acid centres and decorated with imidazolium moieties as the nucleophile source (XS-Sn-Imi and XS-Zn-Imi) were successfully synthesized. The solids display promising features for catalytic applications including high surface area, reduced particle size, good amount of imidazolium salts and acid properties estimated via microcalorimetry analysis. Both XS-Sn-Imi and XS-Zn-Imi were used as catalyst for the chemical fixation of carbon dioxide onto epoxides to give the corresponding cyclic carbonates, displaying excellent performances. The versatility of the Sn-based catalyst was proved in the presence of various epoxides. In all experiments, the bi-functional solids allow reduction of the reaction temperature below 150 °C. Moreover, with glycidol the temperature was decreased down to 30 °C. The short synthesis protocol of the heterogeneous catalysts, together with the 100% atom economy of the target reaction and the low reaction temperature make the entire process highly sustainable. The XS-Sn-Imi catalyst was stable under the selected reaction conditions and reusable for multiple catalytic cycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Comès acknowledges the University of Namur for a PhD fellowship. The authors acknowledge FNRS for supporting this work. This research used resources of the “Plateforme Technologique Physico-Chemical Characterisation” – PC2. This research also used resources of the Electron Microscopy Service located at the University of Namur. This Service is member of the “Plateforme Technologique Morphologie – Imagerie”.Notes and references
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- D. Darensbourg, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- F. Jutz, J. M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef PubMed.
- C. Aprile, H. Garcia and P. P. Pescarmona, in Catalytic Methods in Asymmetric Synthesis, John Wiley & Sons, Inc., 2011, pp. 177–208, DOI:10.1002/9781118087992.ch3.
- M. Buaki-Sogo, H. Garcia and C. Aprile, Catal. Sci. Technol., 2015, 5, 1222–1230 RSC.
- L. A. Bivona, O. Fichera, L. Fusaro, F. Giacalone, M. Buaki-Sogo, M. Gruttadauria and C. Aprile, Catal. Sci. Technol., 2015, 5, 5000–5007 RSC.
- H. Zhong, Y. Su, X. Chen, X. Li and R. Wang, ChemSusChem, 2017, 10, 4855–4863 CrossRef PubMed.
- T. Wang, W. Wang, Y. Lyu, X. Chen, C. Li, Y. Zhang, X. Song and Y. Ding, RSC Adv., 2017, 7, 2836–2841 RSC.
- S. Baj, T. Krawczyk, K. Jasiak, A. Siewniak and M. Pawlyta, Appl. Catal., A, 2014, 488, 96–102 CrossRef.
- L. Han, H. Li, S.-J. Choi, M.-S. Park, S.-M. Lee, Y.-J. Kim and D.-W. Park, Appl. Catal., A, 2012, 429–430, 67–72 CrossRef.
- M. Buaki-Sogó, A. Vivian, L. A. Bivona, H. García, M. Gruttadauria and C. Aprile, Catal. Sci. Technol., 2016, 6, 8418–8427 RSC.
- C. Calabrese, L. F. Liotta, E. Carbonell, F. Giacalone, M. Gruttadauria and C. Aprile, ChemSusChem, 2017, 10, 1202–1209 CrossRef PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef PubMed.
- F. Ahmadi, S. Tangestaninejad, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork and A. R. Khosropour, Inorg. Chem. Commun., 2011, 14, 1489–1493 CrossRef.
- M. V. Escarcega-Bobadilla, M. Martinez Belmonte, E. Martin, E. C. Escudero-Adan and A. W. Kleij, Chemistry, 2013, 19, 2641–2648 CrossRef PubMed.
- W. Y. Gao, Y. Chen, Y. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. Cai, Y. S. Chen and S. Ma, Angew. Chem., Int. Ed. Engl., 2014, 53, 2615–2619 CrossRef PubMed.
- J. Liang, Y. Q. Xie, Q. Wu, X. Y. Wang, T. T. Liu, H. F. Li, Y. B. Huang and R. Cao, Inorg. Chem., 2018, 57, 2584–2593 CrossRef PubMed.
- M. Ding and H.-L. Jiang, ACS Catal., 2018, 3194–3201, DOI:10.1021/acscatal.7b03404.
- X. Collard, P. Louette, S. Fiorilli and C. Aprile, Phys. Chem. Chem. Phys., 2015, 17, 26756–26765 RSC.
- L. Li, X. Collard, A. Bertrand, B. F. Sels, P. P. Pescarmona and C. Aprile, J. Catal., 2014, 314, 56–65 CrossRef.
- X. Collard, L. Li, W. Lueangchaichaweng, A. Bertrand, C. Aprile and P. P. Pescarmona, Catal. Today, 2014, 235, 184–192 CrossRef.
- N. Godard, X. Collard, A. Vivian, L. A. Bivona, S. Fiorilli, L. Fusaro and C. Aprile, Appl. Catal., A, 2018, 556, 73–80 CrossRef.
- N. Cardona-Martinez and J. A. Dumesic, J. Catal., 1990, 125, 427–444 CrossRef.
- G. Colón, I. Ferino, E. Rombi, E. Selli, L. Forni, P. Magnoux and M. Guisnet, Appl. Catal., A, 1998, 168, 81–92 CrossRef.
- D. Meloni, D. Perra, R. Monaci, M. G. Cutrufello, E. Rombi and I. Ferino, Appl. Catal., B, 2016, 184, 163–173 CrossRef.
- N. Fattori, C. M. Maroneze, L. P. da Costa, M. Strauss, F. A. Sigoli, I. O. Mazali and Y. Gushikem, Langmuir, 2012, 28, 10281–10288 CrossRef PubMed.
- L. Wang and E. Y. X. Chen, ACS Catal., 2015, 5, 6907–6917 CrossRef.
- N. Godard, A. Vivian, L. Fusaro, L. Cannavicci, C. Aprile and D. P. Debecker, ChemCatChem, 2017, 9, 2211–2218 CrossRef.
- P. Shah, A. V. Ramaswamy, K. Lazar and V. Ramaswamy, Microporous Mesoporous Mater., 2007, 100, 210–226 CrossRef.
- E. V. Beletskiy, X. Hou, Z. Shen, J. R. Gallagher, J. T. Miller, Y. Wu, T. Li, M. C. Kung and H. H. Kung, J. Am. Chem. Soc., 2016, 138, 4294–4297 CrossRef PubMed.
- S. E. Ashbrook and S. Sneddon, J. Am. Chem. Soc., 2014, 136, 15440–15456 CrossRef PubMed.
- G. Horvath and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470–475 CrossRef.
- P. L. Llewellyn, E. Bloch and S. Bourrelly, in Characterization of Solid Materials and Heterogeneous Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 853–879, DOI:10.1002/9783527645329.ch19.
- J. Melendez, M. North and P. Villuendas, Chem. Commun., 2009, 2577–2579, 10.1039/b900180h.
- M. Taherimehr, B. Van de Voorde, L. H. Wee, J. A. Martens, D. E. De Vos and P. P. Pescarmona, ChemSusChem, 2017, 10, 1283–1291 CrossRef PubMed.
- J. Lan, M. Liu, X. Lu, X. Zhang and J. Sun, ACS Sustainable Chem. Eng., 2018, 6, 8727–8735 CrossRef.
- X. Guo, Z. Zhou, C. Chen, J. Bai, C. He and C. Duan, ACS Appl. Mater. Interfaces, 2016, 8, 31746–31756 CrossRef PubMed.
- Z. Xue, J. Jiang, M.-G. Ma, M.-F. Li and T. Mu, ACS Sustainable Chem. Eng., 2017, 5, 2623–2631 CrossRef.
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031–1036 RSC.
- Y. Ren, Y. Shi, J. Chen, S. Yang, C. Qi and H. Jiang, RSC Adv., 2013, 3, 2167–2170 RSC.
- D. Srinivas and P. Ratnasamy, Microporous Mesoporous Mater., 2007, 105, 170–180 CrossRef.
- P. Agrigento, S. M. Al-Amsyar, B. Sorée, M. Taherimehr, M. Gruttadauria, C. Aprile and P. P. Pescarmona, Catal. Sci. Technol., 2014, 4, 1598–1607 RSC.
- C. Cossement, J. Darville, J.-M. Gilles, J. B. Nagy, C. Fernandez and J.-P. Amoureux, Magn. Reson. Chem., 1992, 30, 263–270 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03878c |
| This journal is © The Royal Society of Chemistry 2018 |