
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of strontium/calcium containing poly(γ-glutamic acid) – organosiloxane fibrous hybrid materials for osteoporotic bone regeneration
Chunxia Gao a,
Ke Zhaoa,
Yaping Wua,
Qiang Gao*ab and
Peizhi Zhu*a
a,
Ke Zhaoa,
Yaping Wua,
Qiang Gao*ab and
Peizhi Zhu*a
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Jiangsu 225002, China. E-mail: qianggao83@gmail.com; pzzhu@yzu.edu.cn
bKey Laboratory of Eco-Textiles, Ministry of Education, Jiangnan University, Wuxi 214122, China
First published on 18th July 2018
Abstract
Recent researches have proved that combination of several therapeutic metal ions, such as silicate (Si), calcium (Ca), strontium (Sr) and so on, with biomaterials may have promising effects for stimulating bone regeneration. In the present study, the Sr/Ca containing silicate hybrid materials (Sr/Ca-γ-PGA-silica) with a mimetic native extracellular matrix (ECM) structure have been developed by electrospinning. With the aim to promote the solubility of γ-PGA in aqueous-based solution and introduce Sr/Ca elements into the prepared hybrid materials, SrCO3 and CaCO3 were employed due to their nontoxicity and no by-products during chemical reaction between γ-PGA and SrCO3/CaCO3. Results of SEM, EDX and elemental mapping images showed that Sr and Ca have been successfully incorporated into the prepared fibrous hybrid materials with homogeneous dispersion. Results of ICP-AES revealed that there was continuous Si, Sr and Ca ion release behavior of Sr/Ca-γ-PGA-silica hybrid materials in Tris–HCl buffer solution and the Si ions release rate can be tailored by adjusting the molar ratio of Sr to Ca. Immersion of Sr/Ca-γ-PGA-silica hybrid materials in a simulated body fluid (SBF) resulted in the formation of an apatite-like surface layer within 3 days, indicating their excellent bioactivity. In addition, the prepared Sr/Ca-γ-PGA-silica hybrid materials supported the proliferation and alkaline phosphatase (ALP) activity of osteoblast in vitro, showing their good biocompatibility. Altogether, the results indicated that the prepared Sr/Ca-γ-PGA-silica hybrid materials with an adjusted ionic release behavior have great potential for providing an excellent ECM for osteoporotic bone regeneration.
Introduction
Osteoporosis is a progressive bone disease that is characterized by a decrease in bone mass and density which can lead to an increased risk of fractures. It has become a major public health problem, affecting hundreds of millions of people worldwide, especially women age 50 or older.1,2 Considering the increasing proportion of the elderly in the global population, the economic burden of such musculoskeletal diseases will further increase in the near future. At present, there are generally two major pharmacological approaches for the clinical treatment of osteoporosis.3,4 One is for anabolic agents for stimulating bone formation, and another is anti-resorptive agents for inhibiting the bone resorption.5,6 Among all these agents, strontium renalate has received significant attention due to its dual effects on stimulating bone formation and inhibiting bone resorption.7 Although strontium renalate can be used for treating osteoporosis and preventing disease progression, it remains difficult to regenerate osteoporosis-related fractures. Recently strontium (Sr) has received considerable attention due to its dual functions in increasing bone formation and decreasing bone resorption. Moreover, introducing Sr into the biomaterials has become a strategy and various Sr-doped biomaterials have been developed for treating osteoporotic bone fractures. Therefore, incorporation of Sr into bioactive materials was expected as a viable method to regenerate the fractures resulting from osteoporosis.In the past decade, a number of research groups have investigated the bone regeneration of Sr-containing bioglasses, bioceramics and bone cement.7–9 Sr doped bone cement have been proved to improve bone formation as well as reduce bone resorption in vivo.10 Although these Sr-containing inorganic biomaterials have been demonstrated to have high mechanical strength and excellent biological properties, their inherent brittleness and slow degradation are still main drawbacks for limiting their further applications. Therefore, there has been a considerable research interest to develop Sr-containing bone substitute materials. To improve the toughness and degradation of these inorganic biomaterials, with the interpenetrating networks of polymers and silica have been developed for bone regeneration.11 Recently, various silica-based hybrid materials have been developed for bone regeneration.12 However, to our knowledge, most of these prepared silica-based hybrid materials have not contained Sr. Therefore, it is necessary to develop the novel Sr/Ca-containing silica-based hybrid materials. In the previous reports, calcium nitrate (Ca(NO3)2) and strontium nitrate (Sr(NO3)2) have been often chosen as the Ca and Sr sources due to their high soluble in the aqueous-based solution.13,14 However, they are not the best choice for fabrication of hybrid materials due to their nitrate by-products are toxic and they need to be removed by thermal treated at above 600 °C which is too high for organic phase in the hybrids.15 Moreover, the homogeneous dispersion and initial burst dissolution behaviours of incorporated Sr and Ca are still challenges. Thus, one of our objectives is to fabricate Sr/Ca-containing silica-based hybrid materials using more suitable precursors for the Sr and Ca sources with homogeneous dispersion and tailorable ions release behaviour.
With this objective, an anionic polypeptide poly(γ-glutamic acid) (H-γ-PGA) was chosen as an organic phase due to their lots of functional carboxylic acid (–COOH) groups which can be easily modified by chemical reactions to yield cross-linked γ-PGA or its derivatives.16 Ho et al. revealed that the free acid form of γ-PGA is tightly compacted helix in aqueous solution and thus strongly hydrophobic, while the salt forms of γ-PGA are random coils and very soluble in water.17 This structural conformation states and the special binding characteristics between the –COOH and the various ions provide a possible for incorporation of Sr and Ca in the γ-PGA and avoiding using the organic solvents. Our previous studies have proved that the strontium carbonate (SrCO3) and calcium carbonate (CaCO3, calcite) can be acted as the effective precursors of Sr and Ca sources due to their nontoxic and no by-products during chemical reactions.8,18 Meanwhile, to introduce the Si resource and simultaneously increase the stability of prepared hybrid scaffolds in physiology environment, a silane coupling agent, 3-glycidoxypropyl trimethoxysilane (GPTMS) was chosen as its epoxy groups are easily activated under acidic conditions to react with the –COOH groups in γ-PGA.19
Of the various fabrication techniques for mimicking the native extracellular matrix (ECM), electrospinning is a well-established technique for producing continuous fibers with diameters ranging from several micrometers down to tens of nanometers. The hypothesis is that the structural conformation of insoluble γ-PGA (H+) could be changed into the soluble γ-PGA salts by the chemical reaction between the –COOH and the various ions which provides a possibility for developing a novel hybrid materials. The microstructure, compositions, mechanical properties, ions release behaviour of the prepared Sr, Ca containing silicate hybrid materials were characterized. Further, in vitro biocompatibility of these fabricated scaffolds was evaluated using mouse preosteoblasts cells (MC3T3-E1) regarding their proliferation and osteogenic differentiation.
Materials and methods
Materials
Poly(γ-glutamic acid) (H-γ-PGA, Mw = 1500–2500 kDa) was purchased from Wako Pure Chemicals Industries, Ltd. (Osaka, Japan). Calcium carbonate (CaCO3, calcite, 99%) and strontium carbonate (SrCO3, 99%) was purchased from Alfa Aesar (Tianjin, China). (3-Glycidoxypropyl)trimethoxysilane (GPTMS) and 1 N hydrochloric acid (HCl) were purchased from Sigma-Aldrich (St. Louis, Mo, USA). Milli-Q water (18.7 MΩ) was used throughout the experiments. All of the reagents were used as received without treatment or further purification.Preparation of Sr/Ca-containing silica-based hybrid materials
Three groups of hybrid materials were prepared, (1) Sr containing γ-PGA-silica hybrid material (Sr-γ-PGA-silica); (2) Sr/Ca-containing γ-PGA-silica hybrid material (Sr/Ca-γ-PGA-silica), (3) Ca-containing γ-PGA-silica hybrid material (Ca-γ-PGA-silica), group (1) and (3) acted as control groups. To prepare the electrospinning solution, γ-PGA was firstly mixed with SrCO3 and CaCO3 (Sr2+/Ca2+ = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0; 1:1; 0:1) with a molar ratio of –COOH/(Sr2+ + Ca2+) is 2:1 and then added Milli-Q water to prepare the ∼10 wt% γ-PGA aqueous solution under vigorous stirring at room temperature. There were lots of bubbles produced when the water was added and the solution became complete clear after about 2 hours. Then, predetermined GPTMS at a weight ratio of 1 to γ-PGA was added into this homogeneous mixture followed by further stirring for ∼1 h. The concentration of γ-PGA in total mixture solution was regulated to ∼8.5 wt%. An electrospinning unit (NEU, Kato Tech Co, Japan) was used to electrospin the prepared blend solutions. A high electric field of 21 kV was applied to the needle where the tip of the needle was positioned 14 cm from a grounded rotating drum that was used as the fiber collector. The as-spun hybrid materials were then transferred to an oven and heat treated at 60 °C for ∼12 h.
:0; 1:1; 0:1) with a molar ratio of –COOH/(Sr2+ + Ca2+) is 2:1 and then added Milli-Q water to prepare the ∼10 wt% γ-PGA aqueous solution under vigorous stirring at room temperature. There were lots of bubbles produced when the water was added and the solution became complete clear after about 2 hours. Then, predetermined GPTMS at a weight ratio of 1 to γ-PGA was added into this homogeneous mixture followed by further stirring for ∼1 h. The concentration of γ-PGA in total mixture solution was regulated to ∼8.5 wt%. An electrospinning unit (NEU, Kato Tech Co, Japan) was used to electrospin the prepared blend solutions. A high electric field of 21 kV was applied to the needle where the tip of the needle was positioned 14 cm from a grounded rotating drum that was used as the fiber collector. The as-spun hybrid materials were then transferred to an oven and heat treated at 60 °C for ∼12 h.
Microstructural characterizations
The surface morphology and elements of the scaffolds were analysed by Scanning Electron Microscopy (SEM) equipped with an Energy-Dispersive X-ray Spectroscope (EDX) (JSM-6301F, JEOL, Japan). Prior to the SEM and EDX observation, all of the hybrid materials were pre-coated by Au–Pd coating (SC7620, Quorum Technologies) and then observed by SEM with 15 kV of accelerating voltage and 15 mm of working distance. ImageJ software was used to determine the average fiber diameter and standard deviation by measuring at least 100 fibers.Composition analysis of the prepared scaffolds was performed using Fourier transform infrared (FT-IR) spectroscopy (IRPrestige-21, Shimadzu, Japan). FT-IR was performed in the wave-number range of 400–4000 cm−1, each FT-IR spectrum was obtained from 40 scans at a resolution of 2 cm−1. Measurements were performed in transmission mode using pellets, a mixture of 3 mg sample and 297 mg spectroscopic-grade KBr. Wide-angle X-ray diffraction (XRD, X′ pert-MPD, Philips) was used to determine the presence of any crystalline phases in all of these samples. The XRD analysis was performed using Ni-filtered CuKα radiation (λ = 1.5402 Å) in a step-scan mode (2° per minute) in the 2θ range 5–60°.
Mechanical behaviour evaluation
Mechanical testing of prepared samples was performed in a tensile testing machine (AGS-G, Shimadzu, Japan) at a constant deformation rate of 1 mm min−1. The specimens were 20 mm long, 5 mm width, and ∼55 μm thickness. Prior to testing, the thickness and width of the specimens were measured at three locations along the sample length using a micrometer, and the average values were taken. Six samples per group were tested.In vitro degradation evaluation
To characterize the Si, Sr and Ca ions released behavior from the electrospun hybrid materials, 10 mg samples were immersed in 20 ml Tris–HCl buffer solution (0.05 M, pH = 7.4) at 37 °C for 50 days. The concentration of the released Si, Sr and Ca ions from the fibrous hybrid scaffolds was monitored by inductively coupled plasma atomic emission spectroscopy (ICP-AES, ICPS-7000, Shimadzu, Japan). Three samples of each sample were tested at each time point.In vitro activity evaluation
In vitro bioactivity of the prepared hybrid scaffolds was evaluated from their reactions in the simulate body fluid (SBF) solution. The SBF solution with a pH of 7.4 was prepared by dissolving reagent-grade NaCl, KCl, NaHCO3, MgCl2·6H2O, CaCl2, and KH2PO4 in distilled water at 37 °C and buffering with TRIS (trishydroxymethyl aminomethane) and 1 N HCl solution according to a method described elsewhere.20,21 Constructs with the shape of thin discs (22 mm in diameter × ∼55 μm thick) were placed individually in a static 12-well plate containing 3 ml of SBF per well, and the system was kept at 37 °C in 5% CO2 atmosphere. After incubated for 3 days, the samples were removed from the SBF, rinsed 3 times with distilled water and freeze-dried. The morphology of the samples was investigated by SEM using the procedures described previously.In vitro biocompatibility
In vitro biocompatibility of the prepared samples was evaluated using a mouse pre-osteoblastic MC3T3-E1 cells line, obtained from the Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). Prior to seeding with cells, discs (15 mm in diameter × ∼55 μm thickness) were cut from electrospun hybrid materials. The samples were sterilized using ethylene oxide gas and then placed in 24-well culture plates. MC3T3-E1 cells were seeded onto samples with a density of 50000 cells per well (n = 3). Alpha-modified minimum essential medium with L-glutamine and phenol red (α-MEM, Gibco, NY, USA), supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco™, Invitrogen) was used as the culturing medium. A Thermanox® culture plastic plate (TCP) was used as the control group. The cultures were incubated at 37 °C in a humidified atmosphere of 95% air and 5% CO2, with the medium changed every 2 days.
After incubation for 1, 3, 5, 7 and 14 days, cell proliferation was examined using Cell Counting Kit-8 (CCK-8, Dojindo, Japan), in accordance with the manufacturer's instruction. Briefly, samples were removed after each incubation, rinsed two times with PBS and added 1 ml fresh culture media. Then, 100 μL of CCK-8 reagent was added to each sample and cultured in an incubator at 37 °C with 5% CO2 for an additional 2 h. After 2 h incubation, 100 μL of media were transferred to each well of a 96-well plate to measure absorption value at a wavelength of 450 nm using a microplate reader (SUNRISE Remote, TECAN, Switzerland).
Alkaline phosphatase (ALP) activity, an early indicator of osteoblastic differentiation, was measured using an ALP substrate kit (p-nitrophenyl phosphate tablets, Sigma-Aldrich Cor., USA). Scaffolds and TCP controls were seeded with MC3T3-E1 cells (50000 cells per well) as described previously, and incubated in the modified culture medium which prepared by adding 0.025 g of ascorbic acid, 1.5756 g of β-glycerophosphate and 1 ml of dexamethasone (100 nM) into the 500 ml of normal α-MEM medium. These regents were purchased from Wako, Japan. In accordance with the manufacture's instruction, ALP activity was examined by measuring the absorbance of resulting medium at 405 nm after incubation for 5, 10, 14 and 21 days.
The morphologies of the attached cells on the fibrous hybrid scaffolds after 1, 3 and 14 days of culture were examined by SEM. After incubation, samples with attached cells were washed three times with PBS and fixed in 2.5% glutaraldehyde in PBS for 2 h at 4 °C. The samples were washed 3 times with PBS and dehydrated through a graded series of ethanol (50–99%). After the final washing with 99% ethanol, the scaffolds were treated twice with hexamethyldisilazane for 5 min each. Finally, the samples were sputter-coated with Au–Pd and examined by SEM, using the procedure described previously in Section 2.3.
Statistical analysis
All biological experiments were (3 samples in each group) were run in duplicate. The data are presented as the mean ± standard deviation (SD). Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey's honestly significant difference as post hoc test (p < 0.05).Results and discussion
Morphology, compositions and microstructure of fibrous hybrid materials
SEM image (Fig. 1a) showed that the electrospun Sr/Ca-γ-PGA-silica hybrid materials were composed of randomly distributed fibers with an average diameter 236 ± 50 nm, which were free from bead-like defects. The elemental compositions of prepared materials were analyzed by EDX as shown in Fig. 1b, which indicated that the Sr and Ca elements have been successfully incorporated into the electrospun Sr/Ca-γ-PGA-silica hybrid materials. Elemental mapping images of Si, Sr and Ca were shown in Fig. 1c–e, which further confirmed the successfully incorporation of Si, Sr and Ca elements and these elements have homogeneous dispersion in the resulting hybrid materials. | ||
| Fig. 1 Morphological and compositions of prepared Sr/Ca-γ-PGA-silica hybrid materials. (a) SEM images. (b) EDX elemental spectrum. Elemental mapping images of the silicon (c), strontium (d) and calcium (e). | ||
Si, Sr and Ca ions have been proved as potential therapeutic agents with the ability to enhance bone regeneration by their stimulating effects on osteogenesis and angiogenesis.22,23 Therefore, incorporation Sr/Ca ions with the silicate-based biomaterials with controlled ions released behavior should be beneficial for stimulating bone regeneration. In this study, to introduce the Sr/Ca sources and convert the γ-PGA from the insoluble helical structure to the soluble random-like salt structure, CaCO3 and SrCO3 were chosen as effective additives due to its known nontoxicity and no by-products during chemical reaction between γ-PGA and SrCO3/CaCO3. Further, to introduce the Si source and simultaneously increase the stability of prepared fibrous materials in physiology environment, a silane coupling agent, GPTMS was chosen as its epoxy groups are easily activated under acidic conditions to react with the –COOH groups in γ-PGA.19,24,25 According to Fig. 1, Sr/Ca-γ-PGA-silica hybrid materials with homogeneous Si, Sr and Ca elements dispersion have been successfully prepared by electrospinning an aqueous-based γ-PGA/GPTMS solution.
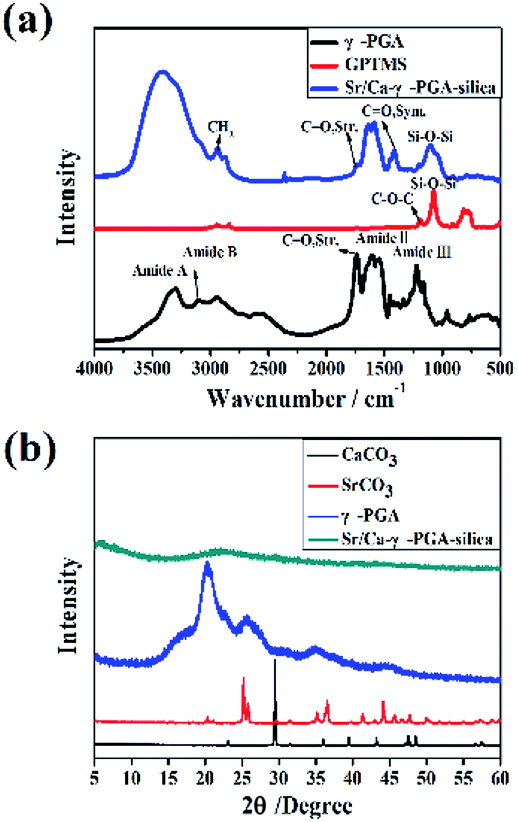
Fig. 2a showed the FTIR spectra of protonated γ-PGA (H-γ-PGA, raw material), GPTMS (dried powder after sol–gel process) and Sr/Ca-γ-PGA-silica hybrid materials. The main characteristic bands observed for protonated γ-PGA were as follows: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1736 cm−1 for the amide I, N–H bending vibration at 1600–1500 cm−1 for the amide II and at 1220 cm−1 for amide III, N–H stretch vibration at 3310 cm−1 for the amide A which overlaps the O–H stretch in the same region, and N–H bending vibration at 2952 cm−1 for the amide B, as shown in Fig. 2a. According to the spectrum of GPTMS, the main characteristic bands were Si–O–Si asymmetric stretching at 1110 cm−1 and Si–OH asymmetric stretching at 814 cm−1. FTIR spectrum of Sr/Ca-γ-PGA-silica hybrid materials contained the main characteristic bands of γ-PGA and GPTMS, indicated that the γ-PGA and GPTMS were incorporated into the hybrid fibrous materials.26 Moreover, we noticed that the band of CO stretching vibration at 1736 cm−1 which corresponds to the free carboxylic acid (COOH) greatly decreased and new symmetric stretching band at 1419 cm−1 which are assigned to the characteristic bands of carboxyl anion (COO–) appeared, as shown in Fig. 2a. This indicated that the γ-PGA stay as its deprotonated phase. The appearance of new shoulder peaks at 1419 cm−1 confirmed the formation of esterification reaction between the –OH of the GPTMS and the –COO– of the γ-PGA.27 Meanwhile, it was obvious to observe that the band at 814 cm−1 in the spectra of GPTMS, which corresponds to Si–O- or Si–OH stretching (non-bridging oxygen bonds), disappeared in the hybrid materials. And the resonances at 1110 cm−1 in hybrid scaffold were attributed to the presence of Si–O–Si bonds (Si–O stretch) in cyclic network configuration. These results suggested that the condensation reaction in GPTMS preceded in the hybrid materials after electrospinning and dried.26 Therefore, GPTMS was successful in providing a cross-linking between γ-PGA polymer chains through the chemical reactions between carboxyl group on the side chain of γ-PGA and epoxy terminated group of GPTMS. The silica was formed by the condensation reaction in GPTMS.
O stretching vibration at 1736 cm−1 for the amide I, N–H bending vibration at 1600–1500 cm−1 for the amide II and at 1220 cm−1 for amide III, N–H stretch vibration at 3310 cm−1 for the amide A which overlaps the O–H stretch in the same region, and N–H bending vibration at 2952 cm−1 for the amide B, as shown in Fig. 2a. According to the spectrum of GPTMS, the main characteristic bands were Si–O–Si asymmetric stretching at 1110 cm−1 and Si–OH asymmetric stretching at 814 cm−1. FTIR spectrum of Sr/Ca-γ-PGA-silica hybrid materials contained the main characteristic bands of γ-PGA and GPTMS, indicated that the γ-PGA and GPTMS were incorporated into the hybrid fibrous materials.26 Moreover, we noticed that the band of CO stretching vibration at 1736 cm−1 which corresponds to the free carboxylic acid (COOH) greatly decreased and new symmetric stretching band at 1419 cm−1 which are assigned to the characteristic bands of carboxyl anion (COO–) appeared, as shown in Fig. 2a. This indicated that the γ-PGA stay as its deprotonated phase. The appearance of new shoulder peaks at 1419 cm−1 confirmed the formation of esterification reaction between the –OH of the GPTMS and the –COO– of the γ-PGA.27 Meanwhile, it was obvious to observe that the band at 814 cm−1 in the spectra of GPTMS, which corresponds to Si–O- or Si–OH stretching (non-bridging oxygen bonds), disappeared in the hybrid materials. And the resonances at 1110 cm−1 in hybrid scaffold were attributed to the presence of Si–O–Si bonds (Si–O stretch) in cyclic network configuration. These results suggested that the condensation reaction in GPTMS preceded in the hybrid materials after electrospinning and dried.26 Therefore, GPTMS was successful in providing a cross-linking between γ-PGA polymer chains through the chemical reactions between carboxyl group on the side chain of γ-PGA and epoxy terminated group of GPTMS. The silica was formed by the condensation reaction in GPTMS.
 | ||
| Fig. 2 (a) ATR-FTIR spectra of protonated γ-PGA (H-γ-PGA, raw material), GPTMS and prepared Sr/Ca-γ-PGA-silica hybrid materials; (b) XRD of CaCO3 and SrCO3 (raw materials, the spectra of CaCO3 and SrCO3 are shown as references), protonated γ-PGA (H-γ-PGA, raw material) and prepared Sr/Ca-γ-PGA-silica hybrid materials. | ||
Fig. 2b shows the XRD patterns of SrCO3, CaCO3, protonated γ-PGA (which were shown as reference) and prepared Sr/Ca-γ-PGA-silica hybrid materials. Fig. 2b revealed that SrCO3, CaCO3 and γ-PGA were typical crystal with the characteristic diffraction peaks. However, it was obvious to see that there were no measurable peaks in the pattern of the fabricated Sr/Ca-γ-PGA-silica hybrid materials, indicating that the conformation of γ-PGA have been changed from the insoluble α-helix state to the soluble linear random-coil conformation by introducing the Ca2+ or Sr2+ into the γ-PGA.
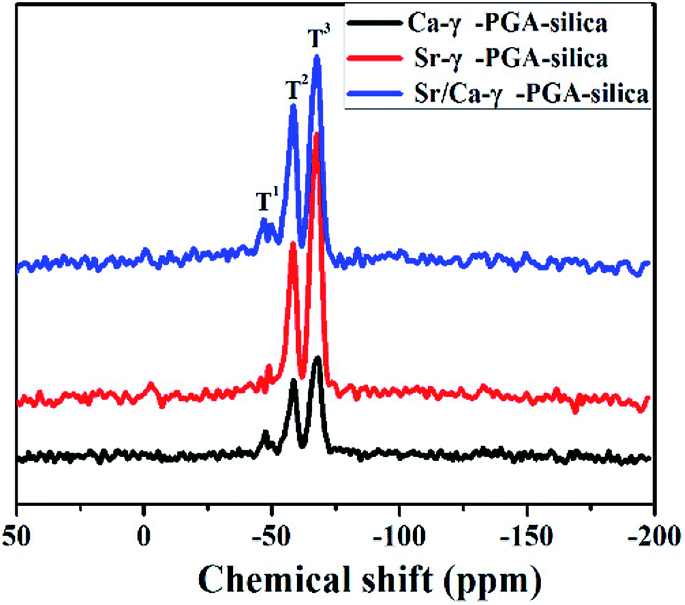
29Si MAS-NMR was employed to clarify the molecular structure of electrospun Sr/Ca-γ-PGA-silica hybrid materials (Ca-γ-PGA-silica, Sr-γ-PGA-silica were shown as references). Fig. 3 shows the 29Si MAS-NMR results of hybrid materials with different ions of the γ-PGA. According to the Fig. 3, two signals could be clearly distinguished with typical shifts of −57 and −67 ppm which corresponding to the T2 and T3.24 And the peak area of T3 was larger than T2 (T1) in all samples. The weak (absence) of T1 and the dominance of T3 suggested that condensation reactions between the silanols from GPTMS alkoxides proceed, agreeing with the results of FTIR in Fig. 2a (the intense peak for Si–O–Si bridging oxygens and the weak peak for Si–O non-bridging oxygens).
 | ||
| Fig. 3 29Si MAS-NMR spectra of electrospun Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials. | ||
Results of SEM, EDX and elemental mapping images revealed that the Si, Sr and Ca have been successfully incorporated into the prepared Sr/Ca-γ-PGA-silica hybrid materials with a homogeneous Si, Sr and Ca dispersion in the resulting hybrid materials. The improved stability of Sr/Ca-γ-PGA-silica hybrid materials was supported by the FTIR analysis through the appeared new shoulder peaks CO at 1736 cm−1 and 1419 cm−1, which confirmed the formation of cross-linking between the –COO groups of γ-PGA and epoxy groups of GPTMS.14 Moreover, 29Si MAS-MNR indicated that the Si–OH non-bridging bond and Si–O–Si bridging bond were formed by hydrolysis and self-condensation of GPTMS, as shown by the presence of T2 and T3 species in all three samples (Fig. 3). The dominance of T3 indicated that the silica matrix condensed well and the GPTMS was successfully condensed into the prepared hybrid materials.
Mechanical properties
Fig. 4 shows the typical tensile stress–strain curves of electrospun γ-PGA, Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials. Ultimate tensile strength, the strain to failure, yield stress and yield strain for all samples are summarized in Table 1. For all materials, the stress initially increased rapidly and almost linearly with the elongation. The tensile strength of the γ-PGA fibremats was 1.2 ± 0.5 MPa and the elongation to failure was 3.5 ± 0.9%. In comparison, the tensile strength and the elongation of Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials significantly increased, 5.0 ± 1.2 MPa/40.2 ± 5.2%; 3.2 ± 0.8 MPa/41.0 ± 6.3% and 4.4 ± 0.7 MPa/45.3 ± 3.8%, respectively. Moreover, according to the Fig. 4, we can see that the yield point appeared in the hybrid materials and the value of yield stress/yield strain was not obviously different. Results indicated that the mechanical properties of hybrid materials were mainly determined by the degree of cross-linking (the content of GPTMS).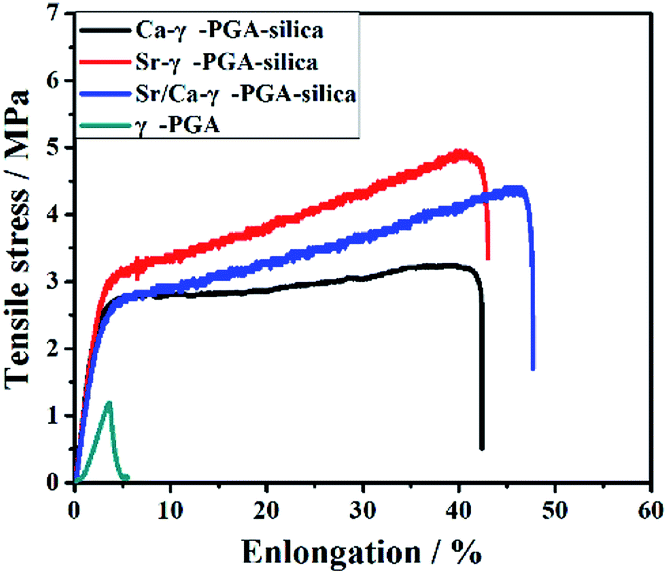 | ||
| Fig. 4 Mechanical response in tension of electrospun γ-PGA, Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials. | ||
| Samples | Ultimate strength (MPa) | Elongation to the failure (%) | Yield stress (MPa) | Yield strain (%) |
|---|---|---|---|---|
| PGA | 1.2 ± 0.5 | 3.5 ± 0.9 | 1.2 ± 0.5 | 3.5 ± 0.9 |
| Ca-γ-PGA-silica | 5.0 ± 1.2 | 40.2 ± 5.2 | 3.1 ± 0.6 | 4.0 ± 1.1 |
| Sr-γ-PGA-silica | 3.2 ± 0.8 | 41.0 ± 6.3 | 2.7 ± 0.4 | 4.3 ± 0.8 |
| Sr/Ca-γ-PGA-silica | 4.4 ± 0.7 | 45.3 ± 3.8 | 2.6 ± 0.5 | 4.1 ± 0.4 |
In vitro bioactivity
In vitro bioactivity of these prepared hybrid materials were evaluated by immersed them in a SBF solution. Results showed that the surface of the electrospun hybrid materials exhibited considerable morphological changes after immersion in SBF for 3 days, as shown in Fig. 5. For the Ca-γ-PGA-silica (Fig. 5a), there were numerous apatite-like spherical particles appeared on the surface of fibers. For the Sr-γ-PGA-silica (Fig. 5b), the fibers exhibited little swelling and there was no new products formed on the surface of fibers. As comparison, for the Sr/Ca-γ-PGA-silica (Fig. 5c), the surface of the fibers was almost completely covered with fine particles, while the porous and fibrous architecture of the materials were still evident. EDX analysis (Fig. 5a′ and c′) showed that Ca-γ-PGA-silica and Sr/Ca-γ-PGA-silica contain the obvious peak at ∼2.1 keV (corresponding to P), and at ∼3.7 keV (corresponding to Ca) after immersed in SBF for 3 days, indicating the reaction products could be apatite-like materials. In comparison, the EDX of Sr-γ-PGA-silica (Fig. 5b′) showed the weak peak of P, Ca and the strong peak of Si, indicating there was little reaction after the sample immersed in SBF solution.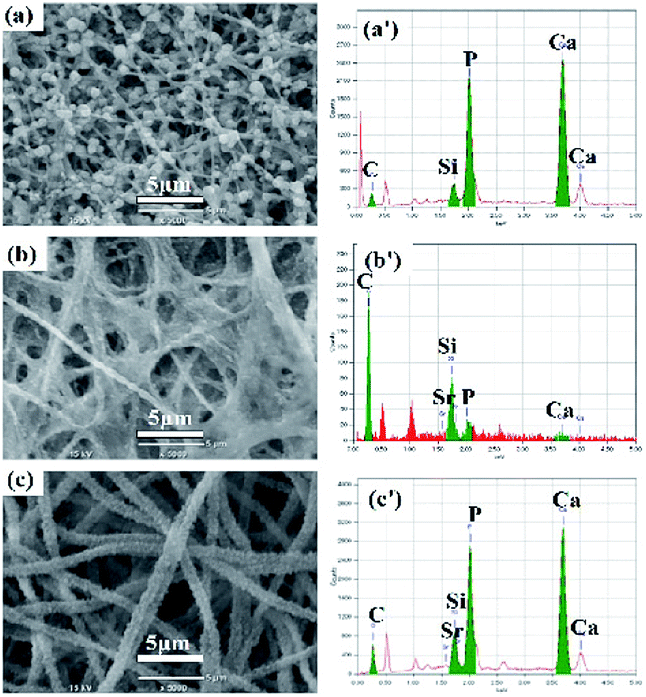 | ||
| Fig. 5 SEM images and EDX spectra of (a and a′) Ca-γ-PGA-silica; (b and b′) Sr-γ-PGA-silica and (c and c′) Sr/Ca-γ-PGA-silica hybrid materials. | ||
Ionic released behavior
Degradation and ionic released behavior of these prepared hybrid materials were monitored by immersed them in a Tris–HCl buffer solution. Fig. 6 showed the amount of released Si, Ca and Sr from the electrospun Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials in the Tris–HCl buffer solution as a function of the soaking time. Firstly, for Si ion release, all samples exhibited the sustained release of soluble silica without any initial burst release, as showed in Fig. 6a. And the percentage of released Si amount of samples were 100%, 71.6% and 37.8%, corresponding to Sr-γ-PGA-silica, Sr/Ca-γ-PGA-silica and Ca-γ-PGA-silica hybrid materials. The Si release indicated that the degradation of the fabricated hybrid materials. The Sr-γ-PGA-silica hybrid materials showed the 100% Si release, and dissolved completely after immersed in buffer solution for 50 days. In comparison, the Ca-γ-PGA-silica showed the 37.8% Si release after immersed in buffer solution for 50 days. Different from the Si release behavior, Ca and Sr showed a rapid release in the first day and then a slow and stable release until 50 days. All these results showed that the Sr, Ca and Si ions can released from these prepared hybrid materials and Sr-γ-PGA-silica hybrid materials have the fastest degradation than the Sr/Ca-γ-PGA-silica and Ca-γ-PGA-silica hybrid materials.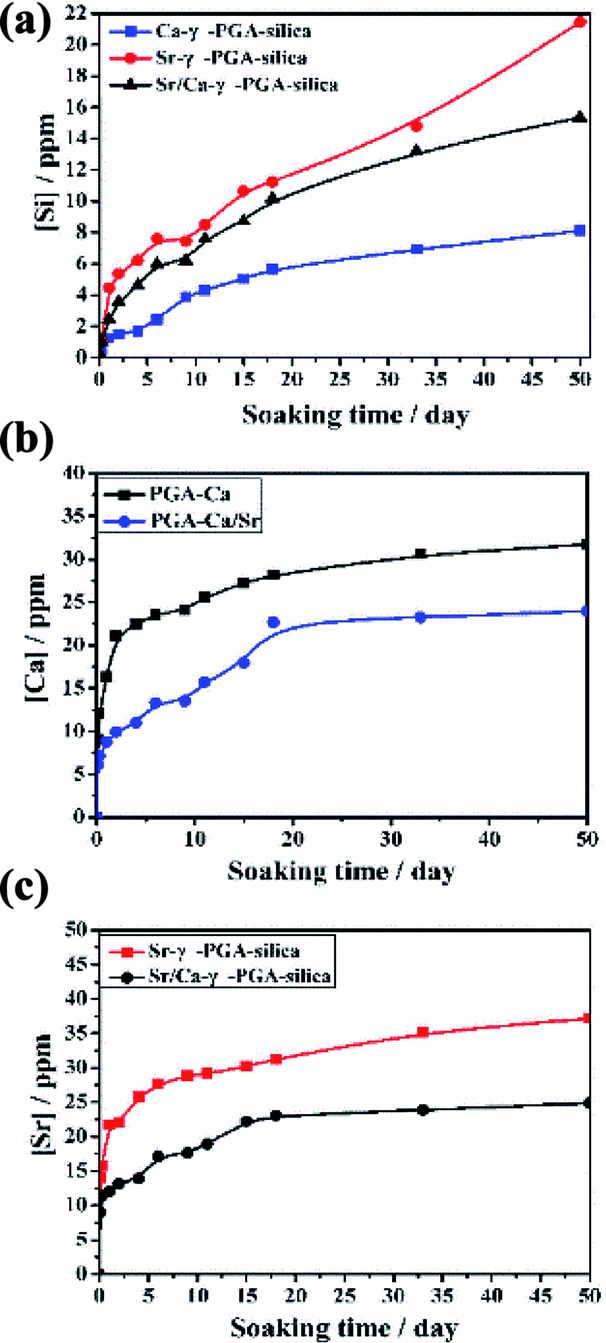 | ||
| Fig. 6 Si, Ca and Sr ions released curves of electrospun Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials (a) Si; (b) Ca and (C) Sr. | ||
The dissolution and release of Si, Sr and Ca ions from the prepared hybrid materials have been seen as a key factor for enhancing their therapeutic effect, and it should be controllable to prevent the possible detrimental effect of high ion concentration. Herein, to understand the effect of the incorporated Sr (Ca), and GPTMS on the ionic dissolution products and degradability of prepared hybrid materials, the Si, Sr and Ca ions release behaviours of prepared hybrid materials in Tris–HCl buffer solution were investigated. Results revealed that all samples exhibited similar trend with a slow and stable release of Si ions (Fig. 6a) due to the similar degree of cross-linking. Meanwhile, the Si ions can be continuously released within 50 days and the amount of released Si increased with the increased Sr content in the hybrid system. Different from the Si ion released profile, Ca and Sr ions exhibited a burst release in the initial stage (within two days), and then showed steadily release until 50 days. Although there was a burst release for Ca and Sr ions, the concentration of released Ca and Sr ions are still within the specified range.
In vitro biocompatibility
Proliferation of MC3T3-E1 cells on the electrospun Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials and the TCP control substrates after incubation times of 1–14 days are shown in Fig. 7a. On the first day, the cell viability on all samples and control were similar. After 3 days, cell activity showed a significantly increase and the Ca-γ-PGA-silica and Sr/Ca-γ-PGA-silica had the better cell viability than the Sr-γ-PGA-silica. A might reason for this different cell viability is the released Si, Ca and Sr ions concentration of these hybrid materials. The in vitro biocompatibility was further evaluated by the ALP assay. ALP is known as a biochemical marker for determining the osteoblast phenotype and is considered as an important factor for assessing bone differentiation and mineralization.28 Results of spectrophotometric measurements of ALP activity of MC3T3-E1 cells cultured on electrospun hybrid materials and on TCP controls are presented in Fig. 7b. The ALP activity reached its highest level at day 10, followed by a reduction at day 21. The increase in the ALP activity is an indication that the cells were able to carry out an osteogenic function on all prepared hybrid materials,29 while the higher ALP activity of the cells on hybrid materials in comparison to the TCP control substrates indicated a greater ability of those materials to support osteoblasts differentiation. In addition, the ALP activity in the sample Sr-γ-PGA-silica was higher than the other samples.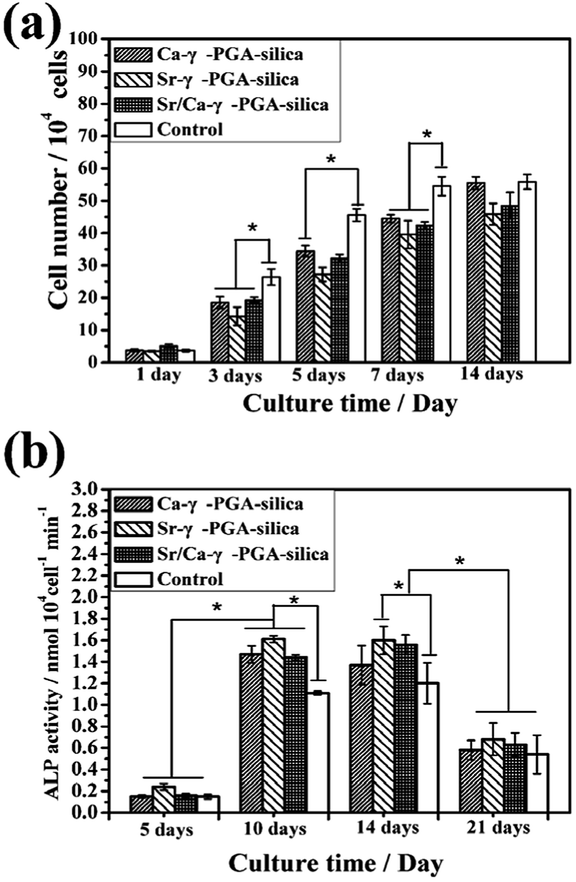 | ||
| Fig. 7 Response of MC3T3-E1 cells to fibrous Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials, and to tissue culture plastic (TCP) control substrates: (a) cell proliferation as a function of incubation time; (b) alkaline phosphatase (ALP) activity of MC3T3-E1 cells on the cell-seeded scaffolds and TCP control after incubation times of 5, 10, 14 and 21 days; *significant difference between pairs of substrates shown (p < 0.05). (Mean ± SD; n = 3). | ||
Osteoblast response to prepared hybrid materials was assessed through CCK-8 assay, ALP activity test and cell morphological observations (Fig. 7, 8 and 9). Results revealed that the Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials were biocompatible. In this study, the enhanced biological properties especially for ALP activity can be attributed to their ECM-like fibrous structure of materials and unique Si, Sr and Ca ions release behavior. Hench et al. suggested that the Si concentrations of 17–21 ppm are critical for up-regulation of several osteogenic genes.30,31 In this study, the Si release concentrations were 1.51, 3.57 and 5.37 ppm within 2 days. Considering refreshing the culture media every 2 days, together with the decrease of release rate, the Si, Ca and Sr concentration released from the materials will be suitable for stimulating osteoblast proliferation and differentiation. Naturally, higher osteogenic differentiation would lead to greater population of cells with osteoblastic phenotype and thus to more secretion of bone.28 Our results showed that Sr/Ca-γ-PGA-silica hybrid materials with the higher Si and Sr released content had higher ALP activity, indicating that material induced osteogenesis occurred.
 | ||
| Fig. 8 SEM images of MC3T3-E1 cells morphology evolutions on the fibrous hybrid materials (a, a′ and a′′) Ca-γ-PGA-silica; (b, b′ and b′′) Sr/Ca-γ-PGA-silica and (c, c′ and c′′) Sr-γ-PGA-silica after incubation for 1 day (a–c); 3 days (a′–c′) and 14 days (a′′–c′′). | ||
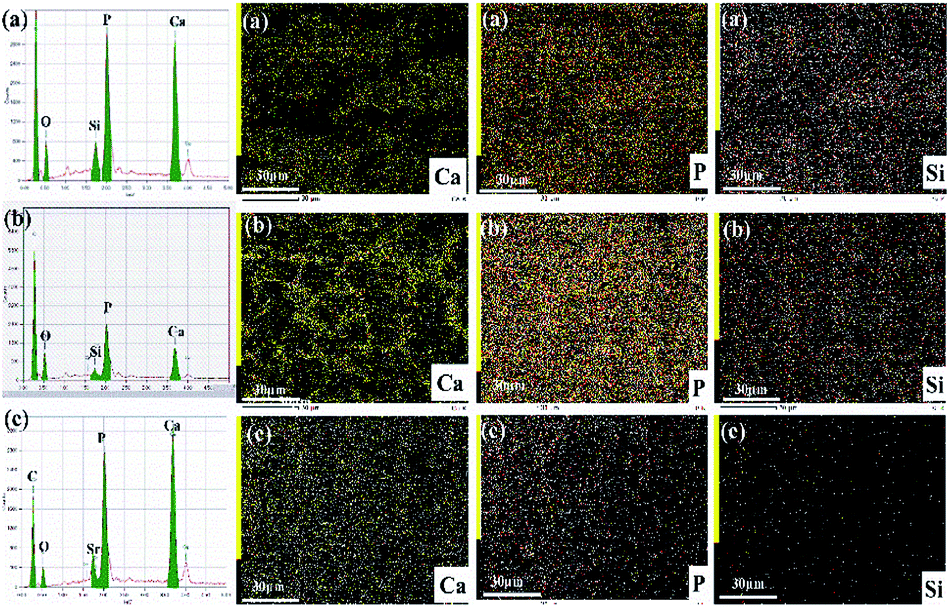 | ||
| Fig. 9 EDX elemental spectra of hybrid materials and elemental mapping images of the silicon, strontium and calcium after incubation of 14 days (a) Ca-γ-PGA-silica; (b) Sr/Ca-γ-PGA-silica and (c) Sr-γ-PGA-silica. | ||
Fig. 8 shows the SEM images of MC3T3-E1 cellular morphological after cultured on the electrospun Ca-γ-PGA-silica, Sr-γ-PGA-silica and Sr/Ca-γ-PGA-silica hybrid materials for 1, 3 and 14 days. Result is compatible with the results of the proliferation and ALP assay that all hybrid materials supported adhesion and growth of MC3T3-E1 cells. As shown in Fig. 8, the size and morphology of cells evidently depended on the culture time. The cells exhibited a typical polygonal morphology with long filopodia on these three samples after 1 day culture (Fig. 8a–c). With the culture time increased to 3 days, cells showed increase in the cell density (Fig. 8a′–c′). Further, the cells were attached to the surface by discrete filopodia and exhibited long and numerous microvilli on their surface. It is interesting to see that these microvilli of cells tended to attach to and integrate with the surrounding fibers to form structured 3D cell-scaffold network (Fig. 8a′–c′). For all samples, cellular density increased and reached confluence after 14 days culture, as shown in Fig. 8a′′–c′′. Moreover, it can be observed that many mineral-like layer presented on the surface of samples. The elemental compositions of these precipitated mineral-like layer of hybrid materials after incubation of 14 days were analyzed by EDX and elemental mapping images, as shown in Fig. 9. Results of EDX confirmed the presence of Ca, P and Si elements. Moreover, elemental mapping images of Ca, P and Si were shown in Fig. 9c–e, which further confirmed the physiological mineralization has occurred as corroborated by the detection of Ca/P ratios close to values found in human bones.
Based on the results described above, the reaction mechanism among the γ-PGA, CaCO3/SrCO3 and GPTMS has been investigated. The possible mechanism for formation of γ-PGA-silica hybrids was shown in eqn (1)–(3). Initially, γ-PGA was ionized by rapid reaction between carboxyl group (–COOH) and CaCO3 and SrCO3 in aqueous solution, as shown in eqn (1); and the mixture solution became homogeneous and transparent within 2 h due to the ionized γ-PGA became water soluble. Then, GPTMS was added to this mixture solution with the predetermined content. As shown in eqn (2), the ring-opening reaction happened in the epoxy groups of GPTMS under the acidic environment, and the methoxysilane groups (Si–OCH3) of GPTMS were hydrolyzed to give silanol (Si–OH) groups; Finally, with the prolonged reaction time, the protonated epoxy group of GPTMS is believed to attack carboxyl side group (–COO–) of the γ-PGA, resulting in the bonding of GPTMS molecules to the γ-PGA chains. Simultaneously, the hydrolyzed silanol (Si–OH) groups can co-condense and formed the Si–O–Si groups, as shown in eqn (3). Finally, this homogeneous mixture solution was then used in the electrospinning step to prepare the fibrous hybrid materials.
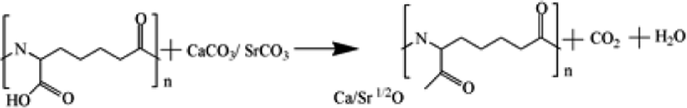 | (1) |
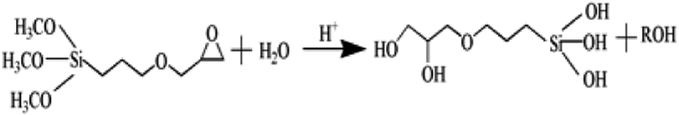 | (2) |
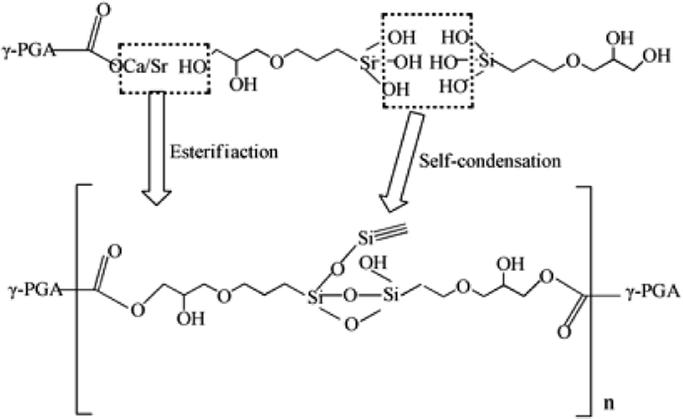 | (3) |
Conclusions
In the present study, a new type of Sr/Ca-γ-PGA-silica with a mimic ECM structure has been fabricated by electrospinning. Results of SEM, EDX and elemental mapping showed that all three prepared hybrid materials presented a continuous and uniform nanofibrous morphology, homogeneous Si and Sr/Ca element dispersion, and enhanced amorphous structure. Results of ICP-AES revealed that the prepared Sr/Ca-γ-PGA-silica hybrid materials can provide a stable and continuous Si, Sr and Ca ions release behavior in Tris–HCl buffer solution until 50 days and the Si release rate can be tailored by adjusting the molar ratio of incorporated Sr and Ca. Immersion of the Sr/Ca-γ-PGA-silica hybrid materials in an SBF resulted in the formation of an apatite-like surface layer within 3 days, indicating its excellent bioactivity. In addition, the prepared Sr/Ca-γ-PGA-silica hybrid materials supported the proliferation and ALP activity of osteogenic MC3T3-E1 cells in vitro, showing their good biocompatibility. Altogether, the results suggested that the prepared Sr/Ca-γ-PGA-silica hybrid materials with an adjusted ionic release behavior have great potential for providing an excellent ECM for repairing and regenerating fractures resulting from osteoporosis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (81601880), the Technology Support Program of Science and Technology Department of Jiangsu Province (BE2017689), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Note and references
- J. A. Kanis, M. L. Rd, C. Christiansen, C. C. Johnston and N. Khaltaev, J. Bone Miner. Res., 1994, 9, 1137–1141 CrossRef PubMed.
- J. A. Kanis, Lancet, 2002, 359, 1929–1936 CrossRef.
- C. Crandall, J. Womens Health Gend. Based Med., 2002, 11, 211–224 CrossRef PubMed.
- K. Akesson, Bull. W. H. O., 2003, 81, 657–664 Search PubMed.
- L. J. Black and G. J. Cullinan, Pharmaceutical compositions containing a bisphosphonate and an anti-resorptive agent for inhibiting bone loss, EP 0693285 A2, 1996.
- A. A. Licata, Clin. Rev. Bone Miner. Metab., 2006, 4, 305–316 CrossRef.
- Y. Xiang and J. Du, Chem. Mater., 2011, 23, 2703–2717 CrossRef.
- C. Gao, H. Liu, Z. P. Luo, Sajilafu, H. Yang and L. Yang, Mater. Sci. Eng., C, 2017, 80, 352 CrossRef PubMed.
- D. Guo, K. Xu, X. Zhao and Y. Han, Biomaterials, 2005, 26, 4073–4083 CrossRef PubMed.
- C. T. Wong, W. W. Lu, W. K. Chan, K. M. C. Cheung, K. D. K. Luk, D. S. Lu, A. B. M. Rabie, L. F. Deng and J. C. Y. Leong, J. Biomed. Mater. Res., Part A, 2004, 68A, 513–521 CrossRef PubMed.
- E. M. Valliant and J. R. Jones, Soft Matter, 2011, 7, 5083–5095 RSC.
- D. Arcos and M. Vallet-Regí, Acta Biomater., 2010, 6, 2874–2888 CrossRef PubMed.
- Y. Y. Özbek, F. E. Baştan and F. Üstel, J. Therm. Anal. Calorim., 2016, 125, 745–750 CrossRef.
- L. Ren, K. Tsuru, S. Hayakawa and A. Osaka, J. Ceram. Soc. Jpn., 2001, 109, 406–411 CrossRef.
- B. Yu, C. A. Turdeanionescu, R. A. Martin, R. J. Newport, J. V. Hanna, M. E. Smith and J. R. Jones, Langmuir, 2012, 28, 17465–17476 CrossRef PubMed.
- I. L. Shih and Y. T. Van, Bioresour. Technol., 2001, 79, 207–225 CrossRef PubMed.
- G. H. Ho, T. I. Ho, K. H. Hsieh, Y. C. Su, P. Y. Lin, J. Yang, K. H. Yang and S. C. Yang, J. Chin. Chem. Soc., 2013, 53, 1363–1384 CrossRef.
- C. Gao, H. Liu, H. Yang and L. Yang, Mater. Technol., 2016, 30, B256–B263 CrossRef.
- C. Gao, S. Ito, A. Obata, T. Mizuno, J. R. Jones and T. Kasuga, Polymer, 2016, 91, 106–117 CrossRef.
- M. Bohner and J. Lemaitre, Biomaterials, 2009, 30, 2175–2179 CrossRef PubMed.
- M. Cerruti, D. Greenspan and K. Powers, Biomaterials, 2005, 26, 4903–4911 CrossRef PubMed.
- M. Xing, Z. Huan, Q. Li, J. Yu and J. Chang, Ceram. Int., 2017, 43, 5156–5163 CrossRef.
- H. Xie, Q. Wang, Q. Ye, C. Wan and L. Li, J. Mater. Sci.: Mater. Med., 2012, 23, 1033–1044 CrossRef PubMed.
- L. S. Connell, L. Gabrielli, O. Mahony, L. Russo, L. Cipolla and J. R. Jones, Polym. Chem., 2017, 8, 1095–1103 RSC.
- M. Vallet-Regã-, M. Colilla and l. B. Gonzã, Chem. Soc. Rev., 2011, 40, 596–607 RSC.
- L. S. Connell, L. Gabrielli, O. Mahony, L. Russo, L. Cipolla and J. R. Jones, Polym. Chem., 2017, 8, 1095–1103 RSC.
- G. Poologasundarampillai, B. Yu, O. Tsigkou, E. Valliant, S. Yue, P. D. Lee, R. W. Hamilton, M. M. Stevens, T. Kasuga and J. R. Jones, Soft Matter, 2012, 8, 4822–4832 RSC.
- A. Wang, X. Ding, S. Sheng and Z. Yao, Yonsei Med. J., 2010, 51, 740–745 CrossRef PubMed.
- M. Sahni, H. L. Guenther, H. Fleisch, P. Collin and T. J. Martin, J. Clin. Invest., 1993, 91, 2004–2011 CrossRef PubMed.
- L. L. Hench, J. Mater. Sci.: Mater. Med., 2006, 17, 967–978 CrossRef PubMed.
- J. R. Jones, Acta Biomater., 2013, 9, 4457 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |