
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated aging and degradation mechanism of LiFePO4/graphite batteries cycled at high discharge rates†
Shun Sun,
Ting Guan,
Xinqun Cheng,
Pengjian Zuo,
Yunzhi Gao,
Chunyu Du and
Geping Yin*
and
Geping Yin*
MIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, No. 92, West Dazhi Street, Harbin 150001, China. E-mail: yingeping@hit.edu.cn; Fax: +86-451-86403807; Tel: +86-451-86413721
First published on 18th July 2018
Abstract
The effects of discharge rates (0.5C, 1.0C, 2.0C, 3.0C, 4.0C and 5.0C) on the aging of LiFePO4/graphite full cells are researched by disassembling the fresh and aged full cells. The capacity degradation mechanism is analyzed via electrochemical performance, surface morphologies and compositions, and the structure of the anode and cathode electrodes. The capacity fade is accelerated with increasing discharge rates. The irreversible loss of active lithium due to the generation of an SEI film is the primary aging factor for the full cells cycled at low discharge rates. However, when the discharge rate is greater than or equal to 4.0C, the performance degradation of the LiFePO4 electrode is distinct due to structure decay, which is caused by quick and repeated intercalation of lithium ions and elevated temperature during discharging. In addition, the SEI film on the anode tends to be unstable after the rapid extraction of lithium ions at high discharge rates, and this enhances the loss of active lithium. Therefore, it is indicated that the degradation mechanism is changed for the full cells aged at 4.0C and 5.0C. Besides, the high discharge rate also increases the internal resistance of the full cell, which is detrimental to high rate discharge performance.
1. Introduction
The lithium ion battery is widely used for portable devices and electric automobiles, and it is considered to be the most promising power source to replace fossil fuels.1 Many novel anode and cathode materials are presented and studied to improve the performances of Li-ion batteries,2–5 such as silicon-based materials,6,7 transition metal oxides,8,9 nanostructured carbons,10,11 or nickel-rich and lithium-rich layered oxide cathodes.12,13 Besides the increasing demand for specific energy and specific power, the lifetime of lithium ion batteries is still the greatest challenge for widespread use, since the service life is even expected to be as long as 10–15 years.14,15The capacity fade of lithium ion batteries is strongly related to the use conditions, such as the charge and discharge rate,16–19 the cut-off voltage,19–21 the depth of discharge (DOD),22–24 the state of charge (SOC)25–27 and the ambient temperature,28–34 which all have influences on the performance and the lifetime of lithium ion batteries. In general, the primary cause of capacity fade is attributed to the loss of active lithium which might result from the generation of solid electrolyte interface (SEI) film on the anode, the deposition of lithium or electrolyte decomposition.19,35 In addition, the performance degradation of electrode materials and the increased internal resistance also have great impact on the aging of lithium ion batteries.36 However, there is a lack of the related systematic research in whether the degradation mechanism is changed under different conditions and the quantitative analysis of the effects of aging factors.
In order to make a rapid evaluation of the lifetime in a short period of time, the lithium ion battery could be tested under accelerated aging conditions. Enhancing the charge and discharge rate is an efficient and maneuverable way to shorten the lifetime of lithium ion battery. In our previous work, the effect of charge/discharge rate on the aging of LiFePO4/graphite batteries was investigated.37 The lithium deposition was generated on the anode surface during the charging process when the charge rate was greater than or equal to 2.0C, which resulted in the change of the mechanism for consumption of active lithium. Therefore, in order to avoid the deposition of metallic lithium during charging, the aging of LiFePO4/graphite batteries was accelerated by enhancing the discharge rate and fixing the charge rate in this work. A kind of commercial LiFePO4/graphite cells were cycled at different discharge rates (0.5C, 1.0C, 2.0C, 3.0C, 4.0C and 5.0C) and the same charge rate of 0.5C. The reasons for the capacity fade of full cells after aging were researched by disassembly analysis, and the effects of discharge rates on LiFePO4/graphite batteries were investigated and compared. If the enhanced discharge rate just shortens the cycle time without altering the degradation mechanism of lithium ion batteries, it would be a rational and effective method to accelerate the capacity fade of full cell.
2. Experimental
2.1 Cycling test
The rated capacity of the commercial LiFePO4/graphite cells (CP523450A cell, HARBIN COSLIGHT POWER CO., LTD) used in this study was 580 mA h, and the dimensions of this prismatic cell were 52 mm × 34 mm × 5 mm. The designed capacity ratio of anode to cathode was about 1.2. Before the cycling tests, the initial capacities of fresh full cells were detected by three full charge/discharge cycles at 0.5C. The full cells were firstly constant current charged to 3.7 V and held at 3.7 V until the current dropped to 0.02C. After a two minutes rest, the full cells were constant current discharged to 2.5 V. Then the full cells were cycled in accordance with the above test procedure at the constant charge rate of 0.5C and the different discharge rates (0.5C, 1.0C, 2.0C, 3.0C, 4.0C and 5.0C) between 2.5 V and 3.7 V respectively. For the conditions of 4.0C and 5.0C, the rest time between two cycles is 10 minutes for the sake of heat dissipation after discharging at high rates. The rest time after discharging is 6 minutes under the test condition of 3.0C, while it is 2 minutes for the relatively low discharge rates. All the charge/discharge cycles were conducted on a LANHE CT2001C multi-channel battery testing system at the temperature of 25 ± 1 °C. In order to record the variation of cell capacity during aging, the cycling process was interrupted every 25 cycles to check the actual capacity at 0.5C with the same procedures of the initial capacity check. When the actual discharge capacity at 0.5C decayed to about 80% of the initial capacity, the aged full cell came to the end of life, and the cycle test was stopped.2.2 Cell disassembly
The fresh and the aged full cells were disassembled in the discharged state (0% SOC) in a glove box filled with pure argon (<1 ppm H2O, <10 ppm O2). After getting rid of the aluminum shell, the anode and the cathode were separated carefully and rinsed with dimethyl carbonate (DMC) to remove the electrolyte.2.3 Analysis after disassembly
In order to evaluate the performance of single electrodes, the active material on one side was wiped off from the current collector with a cotton-based tissue soaked in 1-methyl-2-pyrolidinone (NMP). The LiFePO4/Li and the graphite/Li half-cells were assembled. The specific operation is in ref. 29 Then the capacities of half-cells were tested at 0.02C, 0.1C, 0.2C, 0.5C and 1.0C respectively. For the LiFePO4/Li half-cells, the voltage range is between 2.5 V and 4.2 V, while the voltage range of graphite/Li half-cells is between 0.01 V and 1.5 V. The assembled half-cells were measured on a LANHE CT2001A battery testing system at 25 ± 1 °C.The structures of cathode and anode materials recovered from the fresh and the aged full cells were characterized by X-ray diffraction (XRD, Empyrean, Panalytical). The morphologies of electrodes were recorded and observed by digital camera and scanning electron microscopy (SEM, HELIOS NanoLab 600i, FEI). The chemical constituents on the anode and cathode surface were tested by X-ray photoelectron spectroscopy (XPS, PHI 5700 ESCA System, Physical Electronics). The Raman spectra of graphite anodes were carried out on a confocal Raman spectroscopic system (Renishaw, inVia) using a 532 nm laser.
3. Results and discussion
3.1 Cycling tests
Fig. 1a and b show the capacity loss of LiFePO4/graphite full cells aged at different discharge rates (0.5C, 1.0C, 2.0C, 3.0C, 4.0C and 5.0C) with the cycle time and the cycle number respectively. The charging and discharging time under different discharge rates during the whole cycle are presented in Fig. 1c. The time for resting and capacity check is excluded in the statistics. With the increasing discharge rate, the proportion of discharging time declines, and the capacity fade of full cell is accelerated. The acceleration is enhanced obviously as the discharge rate is raised from 0.5C to 3.0C. However, when the discharge rate is higher than 3.0C, the accelerating effect on the capacity fade is not evident, as well as the change of discharging time. Generally, if the capacity loss of LiFePO4/graphite full cell is mainly caused by the consumption of active lithium owing to the generation and development of SEI film on the anode, the square root of lifetime has a negative correlation with the discharge rates,29,38 and the relationship is shown in Fig. 1d. It seems that the relation becomes different after the discharge rate is higher than 3.0C, which indicates that the degradation mechanism of full cell capacity might be changed.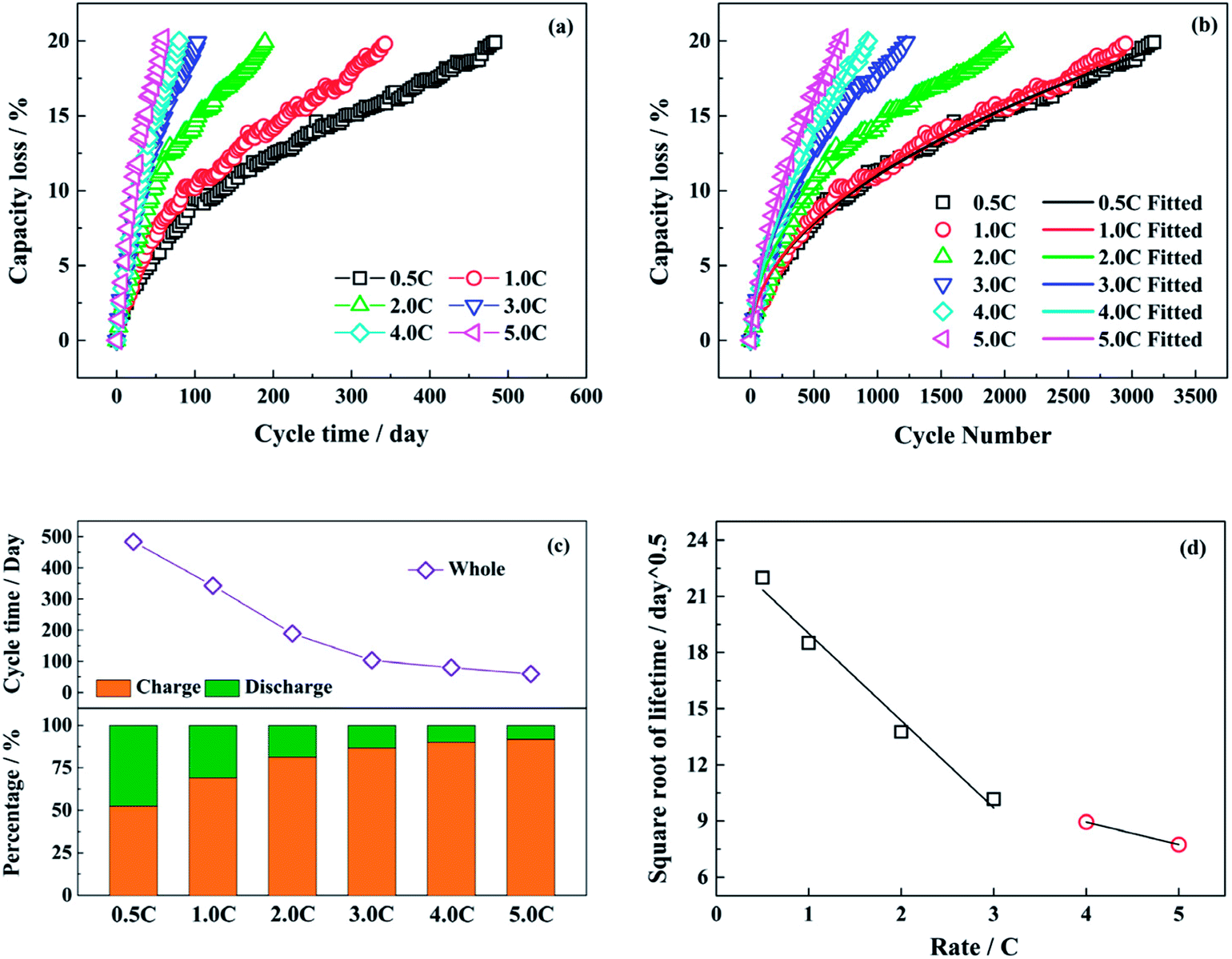 | ||
| Fig. 1 The capacity loss of the LiFePO4/graphite full cells aged at different discharge rates with (a) the cycle time and (b) the cycle number; (c) the percentages of the charging and discharging time as well as the whole cycle time under different discharge rates; (d) the relation of the square root of lifetime with the discharge rates. | ||
In general, the relationship between the capacity loss of lithium ion battery and the number of cycles can be presented as formula (1).39
| Qloss (n) = C × (n)z | (1) |
The fitted values of C and z in formula (1) are presented in Table 1. The exponential term is approximately equal to 0.5 for the relatively low discharge rates, while the values of z become larger at the discharge rates of 4.0C and 5.0C.
| Fitted values | 0.5C | 1.0C | 2.0C | 3.0C | 4.0C | 5.0C |
|---|---|---|---|---|---|---|
| C | 0.37 | 0.36 | 0.46 | 0.39 | 0.27 | 0.32 |
| z | 0.49 | 0.50 | 0.50 | 0.56 | 0.63 | 0.63 |
Therefore, the capacity fade rate for each cycle decreases with the cycle number in a power function with a negative exponent. The calculated data and the fitted lines are shown in Fig. 2a. At the beginning of the cycling, the capacity fade rate drops significantly, then experiences a gradual decline. In the end, the rate of decay tends to be stable after a long cycle. From the fitted curves under different test conditions, the fitted decay rate of full cell capacity becomes higher with the increasing discharge rate as a whole. Whereas, there is little difference between the test conditions of 0.5C and 1.0C.
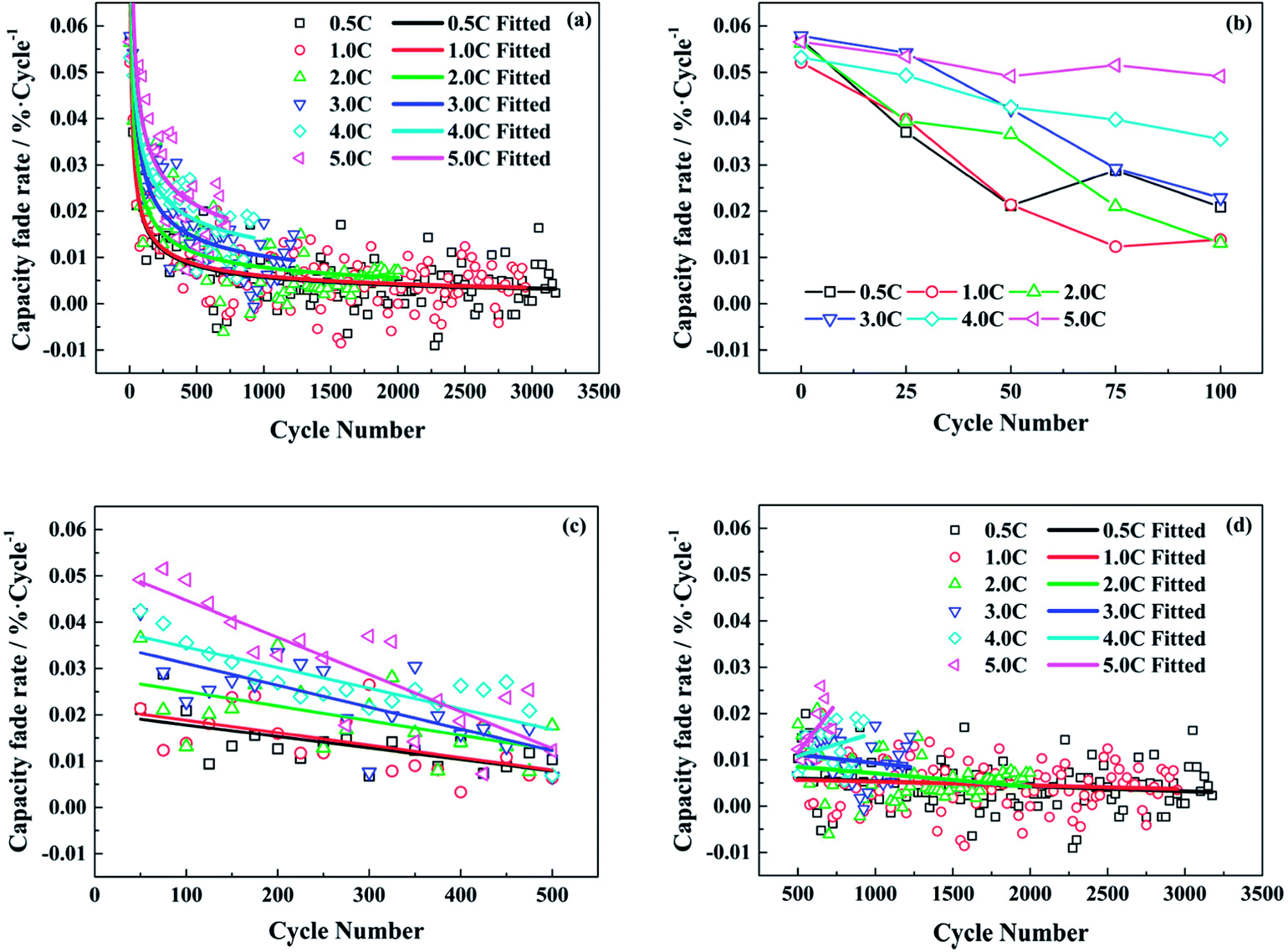 | ||
| Fig. 2 The capacity fade rate per cycle of the LiFePO4/graphite full cells aged at different discharge rates: (a) the whole aging process; (b) the first 100 cycles; (c) the 50th to the 500th cycles; (d) the 500th cycle to the end. | ||
In order to analyze the change rule of capacity fade rate in depth, it is inspected at different stages and the results are shown in Fig. 2b–d. In Fig. 2b, the capacity fade rates are high for all the test conditions in the very beginning. When the discharge rate is less than 4.0C, the capacity fade per cycle drops to be not more than 0.03% after 50 cycles. However, the fade rates decrease slowly with the number of cycles at the discharge rates of 4.0C and 5.0C. In Fig. 2c, the capacity fade rates under different test conditions all present a downward trend from the 50th cycle to the 500th. Afterwards, the situation is changed in Fig. 2d. For the relatively low discharge rates, the rates of decay decline gradually and stabilize around 0.005% per cycle till the end of cycle life exactly as the fitted curves in Fig. 2a. Conversely, the capacity fade rates start to increase under the conditions of 4.0C and 5.0C.
According to the above analysis, it is supposed that the SEI film on the anode is incomplete or nonuniform at the very beginning of cycle life. Therefore, the rapid capacity loss during the first few cycles is caused by the heavy consumption of active lithium due to the generation of SEI film. When the discharge rates are relatively low, the capacity fade rates drop significantly and then decrease gently with the number of cycles, as the SEI film becomes comparatively stable. However, when the discharge rates are high, a mass of lithium ions are simultaneously extracted from the graphite electrode in the discharging process, which can result in a broken and unstable SEI film on the anode. Hence, though the decay rate of capacity continues to decline after the start of cycling at the discharge rate of 4.0C or 5.0C, it still maintains a high level. As for the increased decay rate of capacity in the later period of cycling, it might be attributed to the performance degradation of electrode active materials after the repeated discharging at high rates.
Fig. 3 shows the normalized charge and discharge profiles at different discharge rates after the first and the last capacity check respectively. Since the discharge capacities in the capacity check are all tested at 0.5C, the next charging curves after the first or the last capacity check are similar in spite of the different test conditions. From Fig. 3a, in the case of the initial cycle, the discharge voltage platform drops with the increasing discharge rate. However, the discharge capacity does not decrease accordingly. The discharge capacities at 1.0C, 2.0C and 3.0C are about 98.8%, 96.4% and 97.4% of the corresponding charge capacity severally, while the discharge capacities at 4.0C and 5.0C are both close to 100% of the charge capacity. As the full cell is discharging at high rates, the heat is unceasingly produced and accumulated. The surface temperature of full cells can be higher than 43 °C in the end of discharging at 4.0C. This facilitates the diffusion of lithium ions inside the full cell, and counteracts the effects of the polarization due to the large current. In addition, the coulombic efficiencies at different discharge rates during cycling are all closed to 100%, except the first cycle after every capacity check.
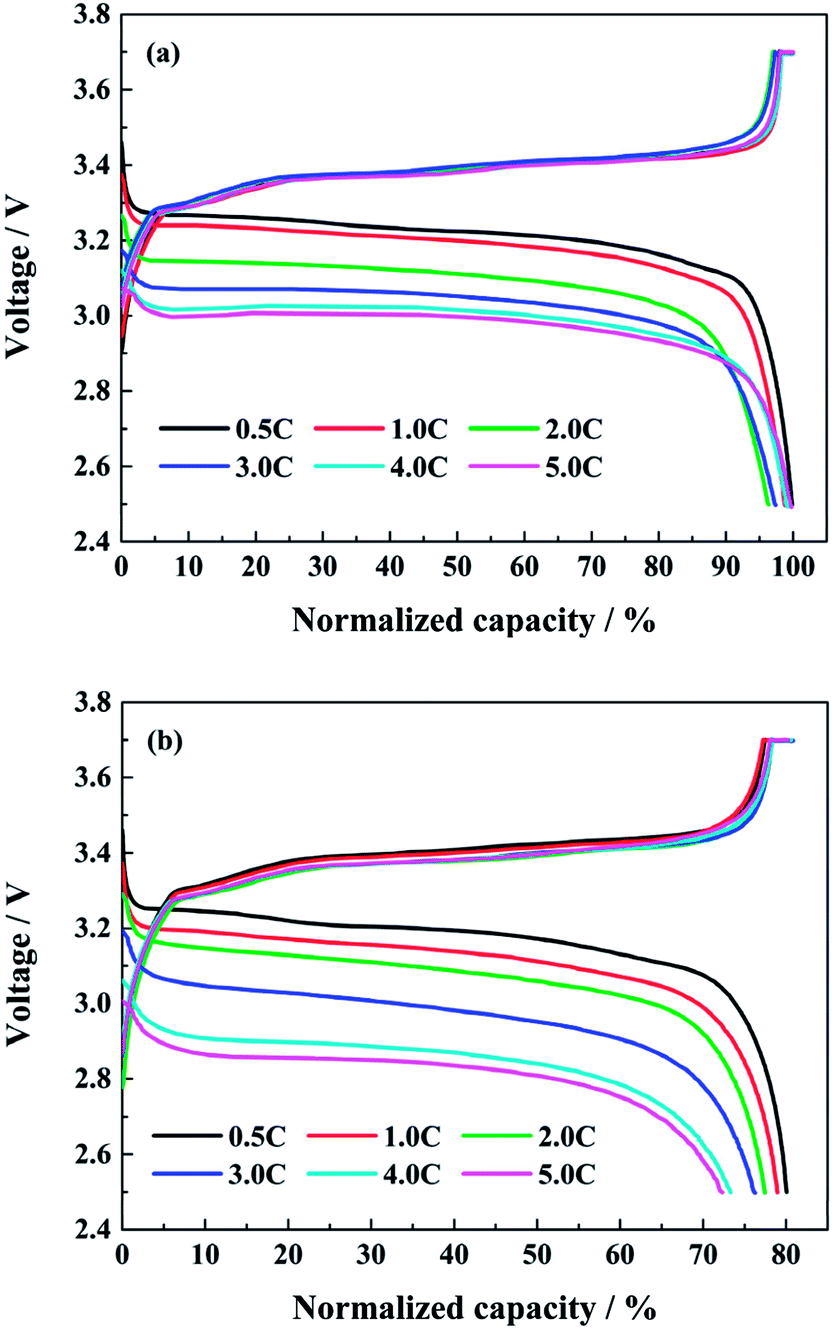 | ||
| Fig. 3 The normalized charge and discharge profiles at different discharge rates after (a) the first and (b) the last capacity check. | ||
When the full cells reach to the end of life, the normalized charge and discharge profiles at different discharge rates after the last capacity check are shown in Fig. 3b. The discharge voltage platform drops more obviously as the discharge rate rises. The discharge capacities at 1.0C, 2.0C, 3.0C, 4.0C and 5.0C are 98.7%, 96.5%, 93.7%, 89.5% and 89.2% of the normalized charge capacity at 0.5C, respectively. Compared with the corresponding values in Fig. 3a, it can be indicated that the high rate discharge performance of lithium ion battery declines, and the internal resistance of the full cells increases evidently after aging at high discharge rates.
3.2 The performance of single electrodes
In general, the performance of the single electrode is mainly affected by the material structure and the diffusion barrier due to the surface film.29,30 In order to evaluate the intrinsic capacity of electrode materials, the LiFePO4/Li and the graphite/Li half-cells were tested at 0.02C, for the effect of polarization due to the test current can be basically ignored. The change in capacities tested at 0.02C are mainly attributed to the structure degradation. When the test rate is elevated, the effect of the polarization caused by the surface film on the electrode performance is also increased.Fig. 4a and b show the specific capacities at 0.02C and 0.1C of the LiFePO4 and the graphite electrode materials, respectively, harvested from the fresh and the aged full cells tested at different discharge rates. For the LiFePO4 cathode material recovered from the fresh cell, the specific capacities at 0.02C and 0.1C are 150.6 mA h g−1 and 149.3 mA h g−1 respectively. Since the material structure is undamaged and the polarization effect is ignored, the capacity at 0.02C of the fresh electrode is regarded as the comparison standard for calculating the extent of the effects caused by the structure and the surface film. The effect of structure degradation on the capacity loss is quantified by the ratio of the change value of capacities at 0.02C to the comparison standard. The difference between the capacities at 0.02C and at 0.1C is considered to be the adverse impact of the surface film on the electrode performance, and the ratio of the difference value to the comparison standard represents for the effect of the surface film.
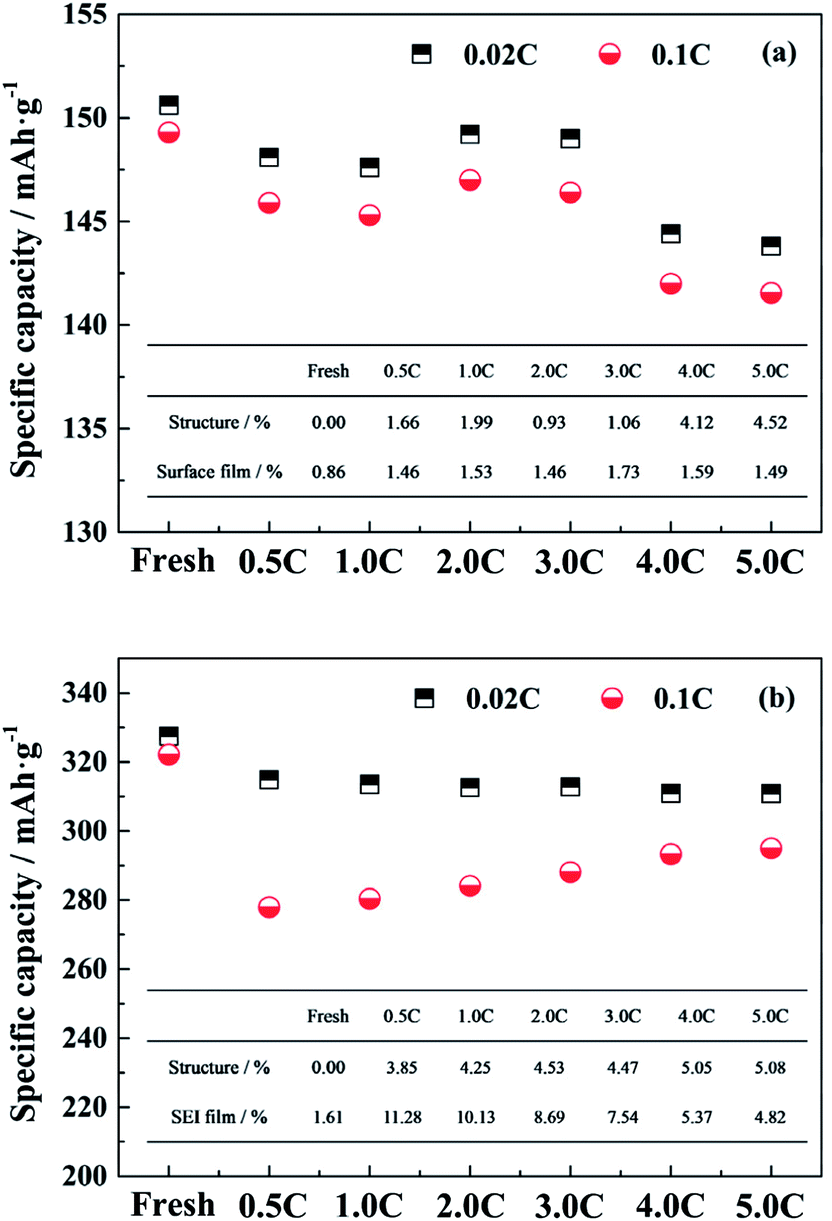 | ||
| Fig. 4 The specific capacities at 0.02C and 0.1C of (a) the LiFePO4 and (b) the graphite electrode materials for the fresh and the aged full cells tested at different discharge rates. Illustration is the extent of the effects caused by the structure and the surface film on the performance of single electrodes. | ||
After cycling, the specific capacities at 0.02C of aged LiFePO4 electrodes are decreased compared to the fresh electrode especially for the full cells aged at 4.0C and 5.0C discharge rates. It is demonstrated that the high discharge rate has a negative effect on the cathode structure. The specific capacities tested at 0.1C exhibit the same trend with the variation of the specific capacities at 0.02C, and the difference values between the two show little significant changes, which indicates that the effect of surface film on the cathode performance is not sensitive to the different discharge rates. The detailed values of the extent of the effects caused by the structure and the surface film for the LiFePO4 electrode are calculated and presented in the illustration of Fig. 4a. It is suggested that the change in the performance of aged LiFePO4 electrodes is mainly attributed to the structure decay in cathode.
The same method is used to analyze the effects of the structure and the SEI film on the performance of graphite electrodes. The reversible capacities at 0.02C of aged graphite electrodes are decreasing, but the values are similar to each other. It is indicated that the influence of the decay in anode structure has no obvious difference for the full cells aged at different discharge rates. When the graphite/Li half-cell is tested at 0.1C, the reversible capacity is reduced compared to that at 0.02C, and follows a trend of decline as the discharge rate reduces. The detailed values of the extent of the effects caused by the structure and the SEI film on the performance of graphite electrodes are calculated and shown in the illustration of Fig. 4b. As the discharge rate is increased, the effect of the SEI film on the anode is reduced. When the discharge rate reaches to 4.0C, the effect degrees of the structure decay and the polarization caused by SEI film are almost equal. However, the generation of SEI film on the anode dominates the performance of graphite electrodes when the full cells aged at relatively low rates. Since the SEI film is mostly generated during the charging process, the long cycle time leads that plenty of active lithium is consumed to form a thick SEI film on the anode surface, which has a great impact on the capacity fade of full cell.
The rate capabilities of the cathodes and the anodes recovered from the fresh and the aged full cells are shown in Fig. S1a and b.† Through the comparison between the two sets of data, the high rate performance of graphite electrodes is quite poor, especially for the anodes aged at high discharge rates. Therefore, the high rate discharge ability of full cell is primarily determined by the performance of graphite anodes.
3.3 The morphologies analysis
The macroscopic feature and morphologies of the LiFePO4 electrodes have no obvious difference after aging at different discharge rates. However, for the graphite electrodes, there is a change in the macroscopic feature from visual inspection when the discharge rate is greater than or equal to 4.0C. As shown in Fig. 5a–d, some active materials fall off from the current collector and adhere to the separator. This may be caused by the fast deintercalation of lithium ions from the graphitic layers and the expansion of interlayer spacing under elevated temperature.29 | ||
| Fig. 5 The digital photographs of the graphite electrodes and separators aged at (a and b) 4.0C and (c and d) 5.0C discharge rates. The SEM morphologies of the graphite electrodes harvested from the aged full cells tested at different discharge rates: (e) 0.5C, (f) 1.0C, (g) 2.0C, (h) 3.0C, (i) 4.0C and (j) 5.0C. | ||
Fig. 5e–j show the SEM morphologies of the graphite electrodes aged at different discharge rates. After aging at relatively low discharge rates, some depositions of lithium compound appear on the graphite anode surface. The depositions normally result from the electrolyte decomposition during charging process, and could decrease the electrode capacity. In addition, there are some fractures in graphite particles when the discharge rate is greater than or equal to 4.0C, which is in agreement with the macroscopic feature. The shedding of active material might cause an adverse effect on the anode rate capability.
3.4 The chemical constituents on electrode surface
The changes of the composition on anode surfaces after aging at 0.5C, 4.0C and 5.0C discharge rates are detected by the XPS test. The C1s, O1s, F1s and P2p spectra of the outermost surface layer on anodes are shown in Fig. 6. The C1s spectra are fitted by four peaks according to the reported binding energies.29,40–42 The O1s spectra, F1s spectra and P2p spectra are all fitted by two peaks separately. The relationship between the fitted peaks at different binding energies and the corresponding bonds is summarized in Table 2.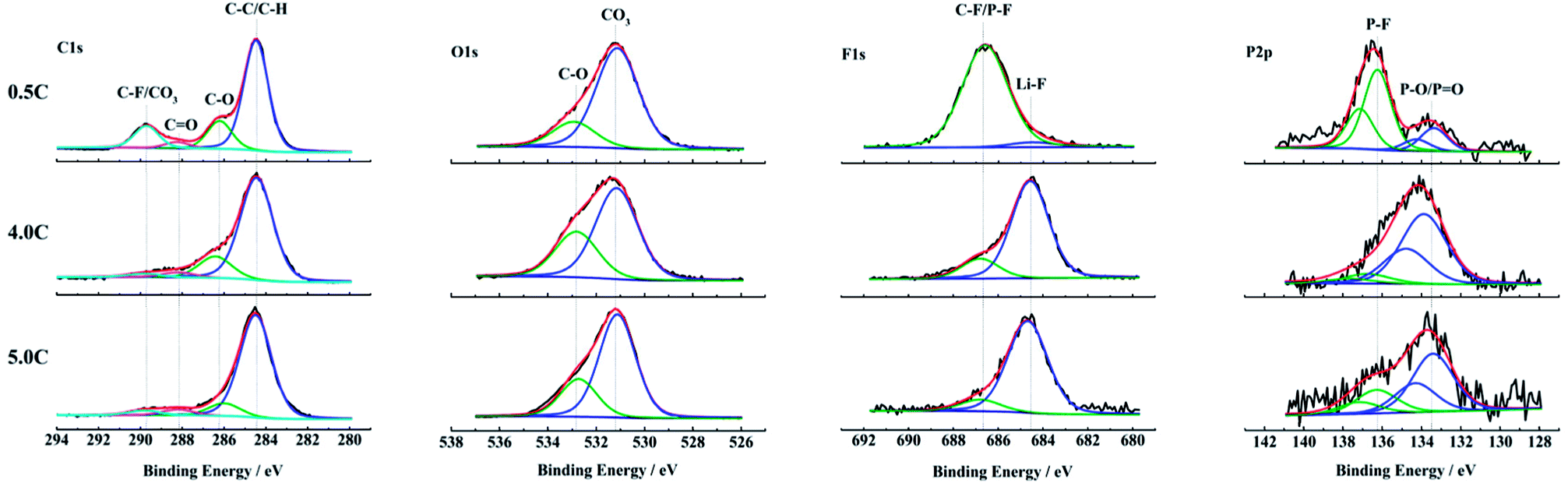 | ||
| Fig. 6 XPS spectra of anode surfaces harvested from the full cells aged at 0.5C, 4.0C and 5.0C discharge rates. | ||
| Element | Bond | Binding energy [eV] |
|---|---|---|
| F1s | C–F/P–F | 686.5–8 |
| LiF | 684.2–7 | |
| O1s | C–O | 532.6–9 |
| CO3 | 530.7–531.2 | |
| C1s | C–F/CO3 | 289.7–290.4 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
287.8–288.3 | |
| C–O | 286.0–4 | |
| C–C/C–H | 284.3–5 | |
| P2p | P–F | 136.2–8 |
| P–O/PO |
133.3–9 |
In C1s spectra, the C–F/CO3 (289.7–290.4 eV) feature in the sample cycle-aged at 0.5C is more pronounced compared to 4.0C and 5.0C discharged rates, and the C–O (286.0–4 eV) as well as the CO (287.8–288.3 eV) species also show the same result.29,40–42 The high content of oxygen containing compounds indicates that there are more decomposition products of electrolyte deposited on the anode surface during aging at 0.5C. This is because that the SEI film is mainly generated during the charging process and its thickness has a positive correlation with the number of cycles.
The O1s spectra exhibit some differences in the relative contents of oxygen containing species. The samples aged at 4.0C and 5.0C discharge rates appear to contain more C–O (532.6–9 eV) compared to the carbonates (530.7–531.2 eV).29,40 This suggests that there are increased amounts of organic components on the anode surfaces aged at 4.0C and 5.0C, which is owing to the rising temperature during the high rate discharge.29,40
According to the F1s and P2p spectra, the samples aged at 4.0C and 5.0C discharge rates show a considerably smaller relative amount of P–F compounds (136.2–8 eV)35,36,43 than the sample aged at 0.5C. In comparison, the relative amounts of LiF (684.2–7 eV)40 and P–O compounds (133.3–9 eV)35,36,43 are apparently larger for the samples aged at high discharge rates. The P–F and P–O species as well as the LiF detected on the anode surfaces are mainly attributed to the decomposition of LiPF6 salt.44,45 With the proceeding of decomposition reactions, the P–F species are gradually replaced by the P–O species and LiF.44,46 It is indicated that the LiPF6 salt experiences a more complete decomposition reaction under the condition of high discharge rates. The SEI film on the anode aged at high discharge rates contains a relatively large quantity of LiF that has an adverse effect on the lithium ion diffusion in the interface layer.47 So, it causes that the internal resistance of full cell increases, and the polarization voltage rises, which results in a reduced ability of high rate discharge performance. This result is in accordance with the poor rate capability of graphite materials in Fig. S1b† and the obvious decline of discharge voltage platform in Fig. 3b after aging at high discharge rates.
The XPS spectra of cathode electrodes recovered from the full cells decayed to the capacity retention of 80% at 0.5C, 4.0C and 5.0C discharge rates are compared in Fig. S2,† and the binding energy assignments for different fitted peaks are also presented in Table 2. After a general comparison among the three samples, except the slight difference in the O1s spectra, it is concluded that there is no other obvious change for the chemical constituents on cathode surfaces aged at different discharge rates.
3.5 The structures analysis
According to the analysis of half-cell tests, the decay in structure has more serious influence on the performance of single electrodes when the full cells are cycle-aged at high discharge rates. In order to confirm the change in structure, the powders of LiFePO4 and graphite materials scraped from the electrodes are tested by XRD respectively. The XRD patterns of the LiFePO4 powders recovered from the fresh and the aged full cells tested at different discharge rates are shown in Fig. 7. As shown in Fig. 7a, the intensity of peaks for the FePO4 phase is increased after aging, which is indicated that some lithium ions cannot come back to the cathode when the full cell is discharged to 2.5 V. Generally, this part of active lithium is consumed irreversibly in the anode due to the formation of SEI film or other side reactions.16,29 In addition, the peaks of (200), (020) and (002) shift to low angle direction when the full cells decay to the end of life, especially for the high discharge rates (Fig. 7b). It is indicated that the distance of interplanar spacing becomes larger after aging.48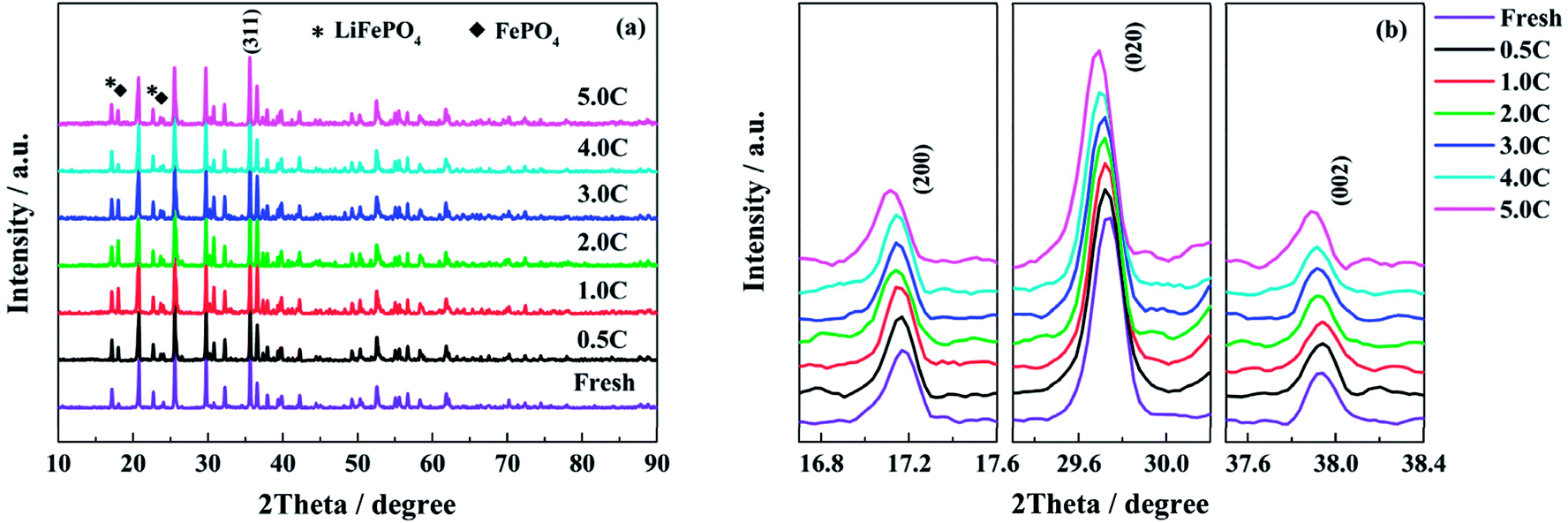 | ||
| Fig. 7 The XRD patterns of the LiFePO4 powders recovered from the fresh and the aged full cells tested at different discharge rates: (a) the full XRD patterns and (b) the magnifications of the (200), (020) and (002) peaks. | ||
Table 3 presents the lattice parameters of LiFePO4 phase. The grain size of LiFePO4 particles is calculated by the radians of full width at half maximum (FWHM) and diffraction angle according to the Debye–Scherrer formula. The values of a, b and c are increased with the elevating discharge rates, while the grain size shows an opposite trend, especially for the discharge rates of 4.0C and 5.0C. The XRD results demonstrate that the high discharge rates have a negative effect on the structure of LiFePO4 materials. The decay in cathode structure might be attributed to the quick and repeated intercalation of lithium ions, the elevated temperature and high utilization ratio of active material during the high rate discharging process.
| LiFePO4 | Fresh | 0.5C | 1.0C | 2.0C | 3.0C | 4.0C | 5.0C |
|---|---|---|---|---|---|---|---|
| 2θ (311) | 35.626 | 35.605 | 35.624 | 35.602 | 35.602 | 35.600 | 35.574 |
| FWHM (311) | 0.154 | 0.160 | 0.157 | 0.158 | 0.154 | 0.164 | 0.168 |
| Grain size/nm | 53.58 | 51.57 | 52.56 | 52.22 | 53.58 | 50.31 | 49.11 |
| a/Å | 10.3139 | 10.3170 | 10.3185 | 10.312 | 10.3197 | 10.3261 | 10.3260 |
| b/Å | 6.0002 | 6.0033 | 6.0032 | 6.0044 | 6.0009 | 6.0057 | 6.0129 |
| c/Å | 4.6949 | 4.6934 | 4.6877 | 4.6913 | 4.6934 | 4.6941 | 4.6980 |
| Vol/Å3 | 290.54 | 290.69 | 290.38 | 290.48 | 290.65 | 291.11 | 291.69 |
The XRD patterns of the graphite powders of the fresh and the aged full cells tested at different discharge rates are shown in Fig. S3,† and the structural parameters are fitted and presented in Table 4. There is no obvious change among the XRD patterns of different graphite powders, and only the graphite peaks are detected. From the inset of Fig. S3,† the peak of (002) has a shift towards the low angle direction for the full cells aged at 4.0C and 5.0C discharge rates, meaning that the layer distance (d002) of the graphite is expanded.49 From Table 4, as the discharge rate increases, the FWHM of the (002) peak is broadened, and the grain size is reduced. For the graphite materials aged at high discharge rates, the increased interlayer spacing and the decrease in grain size indicate that part of active materials is damaged and exfoliated after the long cycle time,49 which coincides with the morphologies analysis of anode electrodes. Fig. S4† shows the Raman spectra of the fresh and the aged graphite electrodes tested at 0.5C, 4.0C and 5.0C discharge rates. After aging, the peaks assigned to the disorder-induced D and D′ bands are obviously enhanced, especially for the high discharge rates, which means that the defect quantity is increased in graphitic materials.50 Comparing with the structure analysis of aged LiFePO4 materials, the high discharge rates are also detrimental to the material structure.
| Graphite | Fresh | 0.5C | 1.0C | 2.0C | 3.0C | 4.0C | 5.0C |
|---|---|---|---|---|---|---|---|
| d002/Å | 3.3670 | 3.3653 | 3.3685 | 3.3686 | 3.3656 | 3.3721 | 3.3721 |
| 2θ | 26.450 | 26.463 | 26.438 | 26.437 | 26.461 | 26.409 | 26.409 |
| FWHM (002) | 0.222 | 0.258 | 0.255 | 0.269 | 0.293 | 0.283 | 0.326 |
| Grain size/nm | 36.35 | 31.28 | 31.65 | 29.99 | 27.54 | 28.51 | 24.75 |
4. Conclusions
In this work, the influence of different discharge rates on the aging of LiFePO4/graphite full cells is investigated. The decay rate of full cell capacity is accelerated by increasing the discharge rate. However, when the discharge rate is higher than 3.0C, the degradation mechanism of full cell is changed.Through disassembling the fresh and the aged full cells, it is concluded that the intrinsic capacity of graphite anode is mainly related to the SEI film on its surface. The irreversible consumption of active lithium due to the formation and development of SEI film is the primary reason for the capacity fade of full cell aged at relatively low discharge rate. When the discharge rates are 4.0C and 5.0C, the capacity fade rate maintains a comparatively high level, for the SEI film on the anode is apt to be broken and unstable after the rapid extraction of lithium ions at high rates. Besides, the decay rate of capacity increases in the later period of cycling, which is attributed to the degradation of active materials. The performance degradation of LiFePO4 electrode and the unstable SEI film indicate that the degradation mechanism is changed when the full cells are aged at high discharge rates. Furthermore, the high discharge rate has adverse effects on the performance of graphite anodes and also can increase the internal resistance of full cell, which is detrimental to the high rate discharge ability.
In conclusion, the high discharge rates can result in the rapid fade of full cell capacity and the changed degradation mechanism. So, when the discharge rate is greater than or equal to 4.0C, it is not suitable to be used for accelerating the aging of the LiFePO4/graphite full cells.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is funded by the National High Technology Research and Development Program (863 Program) of China (No. 2012AA110203) and the National Natural Science Foundation of China (No. 51634003).References
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef.
- J. Pang, A. Bachmatiuk, Y. Yin, B. Trzebicka, L. Zhao, L. Fu, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Adv. Energy Mater., 2018, 8, 1702093–1702135 CrossRef.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- Q. Hao, J. Pang, Y. Zhang, J. Wang, L. Ma and O. G. Schmidt, Adv. Opt. Mater., 2018, 6, 1700984–1700989 CrossRef.
- Y. Xing, S. Wang, B. Fang, Y. Feng and S. Zhang, Microporous Mesoporous Mater., 2018, 261, 237–243 CrossRef.
- J. Pang, R. G. Mendes, P. S. Wrobel, M. D. Wlodarski, H. Q. Ta, L. Zhao, L. Giebeler, B. Trzebicka, T. Gemming, L. Fu, Z. Liu, J. Eckert, A. Bachmatiuk and M. H. Rummeli, ACS Nano, 2017, 11, 1946–1956 CrossRef PubMed.
- A. Soni, L. Zhao, H. Q. Ta, Q. Shi, J. Pang, P. S. Wrobel, T. Gemming, A. Bachmatiuk and M. H. Rummeli, Nano-Struct. Nano-Objects, 2018, 16, 38–44 CrossRef.
- Y. Xing, S. Wang, B. Fang, G. Song, D. P. Wilkinson and S. Zhang, J. Power Sources, 2018, 385, 10–17 CrossRef.
- H. Ta, L. Zhao, D. Pohl, J. Pang, B. Trzebicka, B. Rellinghaus, D. Pribat, T. Gemming, Z. Liu, A. Bachmatiuk and M. Rümmeli, Crystals, 2016, 6, 100–116 CrossRef.
- W. Long, B. Fang, A. Ignaszak, Z. Wu, Y. J. Wang and D. Wilkinson, Chem. Soc. Rev., 2017, 46, 7176–7190 RSC.
- K. Olszowska, J. Pang, P. S. Wrobel, L. Zhao, H. Q. Ta, Z. Liu, B. Trzebicka, A. Bachmatiuk and M. H. Rummeli, Synth. Met., 2017, 234, 53–85 CrossRef.
- A. Manthiram, J. C. Knight, S.-T. Myung, S.-M. Oh and Y.-K. Sun, Adv. Energy Mater., 2016, 6, 1501010–1501032 CrossRef.
- D. H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692–697 CrossRef PubMed.
- A. Barré, B. Deguilhem, S. Grolleau, M. Gérard, F. Suard and D. Riu, J. Power Sources, 2013, 241, 680–689 CrossRef.
- W. Waag, S. Käbitz and D. U. Sauer, Appl. Energy, 2013, 102, 885–897 CrossRef.
- T. Guan, P. Zuo, S. Sun, C. Du, L. Zhang, Y. Cui, L. Yang, Y. Gao, G. Yin and F. Wang, J. Power Sources, 2014, 268, 816–823 CrossRef.
- D. Wong, B. Shrestha, D. A. Wetz and J. M. Heinzel, J. Power Sources, 2015, 280, 363–372 CrossRef.
- J. Groot, M. Swierczynski, A. I. Stan and S. K. Kær, J. Power Sources, 2015, 286, 475–487 CrossRef.
- Y. Gao, J. Jiang, C. Zhang, W. Zhang, Z. Ma and Y. Jiang, J. Power Sources, 2017, 356, 103–114 CrossRef.
- K. Qian, Y. Li, Y.-B. He, D. Liu, Y. Zheng, D. Luo, B. Li and F. Kang, RSC Adv., 2016, 6, 76897–76904 RSC.
- J.-W. Lee, Y. K. Anguchamy and B. N. Popov, J. Power Sources, 2006, 162, 1395–1400 CrossRef.
- S. Saxena, C. Hendricks and M. Pecht, J. Power Sources, 2016, 327, 394–400 CrossRef.
- S. B. Peterson, J. Apt and J. F. Whitacre, J. Power Sources, 2010, 195, 2385–2392 CrossRef.
- W. Gu, Z. Sun, X. Wei and H. Dai, Electrochim. Acta, 2014, 133, 107–116 CrossRef.
- S.-h. Wu and P.-H. Lee, J. Power Sources, 2017, 349, 27–36 CrossRef.
- A. Barai, K. Uddin, J. Chevalier, G. H. Chouchelamane, A. McGordon, J. Low and P. Jennings, Sci. Rep., 2017, 7, 5128–5138 CrossRef PubMed.
- M. Kassem, J. Bernard, R. Revel, S. Pélissier, F. Duclaud and C. Delacourt, J. Power Sources, 2012, 208, 296–305 CrossRef.
- T. Waldmann, M. Wilka, M. Kasper, M. Fleischhammer and M. Wohlfahrt-Mehrens, J. Power Sources, 2014, 262, 129–135 CrossRef.
- S. Sun, T. Guan, B. Shen, K. Leng, Y. Gao, X. Cheng and G. Yin, Electrochim. Acta, 2017, 237, 248–258 CrossRef.
- T. Guan, S. Sun, Y. Gao, C. Du, P. Zuo, Y. Cui, L. Zhang and G. Yin, Appl. Energy, 2016, 177, 1–10 CrossRef.
- M. Ecker, P. Shafiei Sabet and D. U. Sauer, Appl. Energy, 2017, 206, 934–946 CrossRef.
- X. Lin, M. Salari, L. M. Arava, P. M. Ajayan and M. W. Grinstaff, Chem. Soc. Rev., 2016, 45, 5848–5887 RSC.
- V. Ruiz, A. Kriston, I. Adanouj, M. Destro, D. Fontana and A. Pfrang, Electrochim. Acta, 2017, 240, 495–505 CrossRef.
- J. Schmitt, A. Maheshwari, M. Heck, S. Lux and M. Vetter, J. Power Sources, 2017, 353, 183–194 CrossRef.
- M. Klett, R. Eriksson, J. Groot, P. Svens, K. Ciosek Högström, R. W. Lindström, H. Berg, T. Gustafson, G. Lindbergh and K. Edström, J. Power Sources, 2014, 257, 126–137 CrossRef.
- M. Hellqvist Kjell, S. Malmgren, K. Ciosek, M. Behm, K. Edström and G. Lindbergh, J. Power Sources, 2013, 243, 290–298 CrossRef.
- S. Sun, T. Guan, P. Zuo, Y. Gao, X. Cheng, C. Du and G. Yin, ChemElectroChem, 2018 DOI:10.1002/celc.201800326.
- A. J. Smith, J. C. Burns, X. Zhao, D. Xiong and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A447–A452 CrossRef.
- Y. Cui, C. Du, G. Yin, Y. Gao, L. Zhang, T. Guan, L. Yang and F. Wang, J. Power Sources, 2015, 279, 123–132 CrossRef.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef.
- L. Yang, X. Cheng, Y. Gao, Y. Ma, P. Zuo, C. Du, Y. Cui, T. Guan, S. Lou, F. Wang, W. Fei and G. Yin, RSC Adv., 2014, 4, 26335–26341 RSC.
- S. Lou, B. Shen, P. Zuo, G. Yin, L. Yang, Y. Ma, X. Cheng, C. Du and Y. Gao, RSC Adv., 2015, 5, 81235–81242 RSC.
- M. Herstedt, D. P. Abraham, J. B. Kerr and K. Edström, Electrochim. Acta, 2004, 49, 5097–5110 CrossRef.
- L. Terborg, S. Weber, F. Blaske, S. Passerini, M. Winter, U. Karst and S. Nowak, J. Power Sources, 2013, 242, 832–837 CrossRef.
- V. A. Agubra and J. W. Fergus, J. Power Sources, 2014, 268, 153–162 CrossRef.
- G. Gachot, P. Ribiere, D. Mathiron, S. Grugeon, M. Armand, J. B. Leriche, S. Pilard and S. Laruelle, Anal. Chem., 2011, 83, 478–485 CrossRef PubMed.
- D. N. Wong, D. A. Wetz, J. M. Heinzel and A. N. Mansour, J. Power Sources, 2016, 328, 81–90 CrossRef.
- G.-T. Wu, M.-H. Chen, G.-M. Zhu, J.-K. You, Z.-G. Lin and X.-B. Zhang, J. Solid State Electrochem., 2003, 7, 129–133 CrossRef.
- P. Liu, J. Wang, J. Hicks-Garner, E. Sherman, S. Soukiazian, M. Verbrugge, H. Tataria, J. Musser and P. Finamore, J. Electrochem. Soc., 2010, 157, A499–A507 CrossRef.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04074e |
| This journal is © The Royal Society of Chemistry 2018 |