
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlpinumisoflavone attenuates lipopolysaccharide-induced acute lung injury by regulating the effects of anti-oxidation and anti-inflammation both in vitro and in vivo†
Pei-Ying Li‡
a,
Yu-Chia Liang‡b,
Ming-Jyh Sheua,
Shyh-Shyun Huanga,
Che-Yi Chaoc,
Yueh-Hsiung Kuob and
Guan-Jhong Huang *b
*b
aSchool of Pharmacy, College of Pharmacy, China Medical University, Taichung 40402, Taiwan
bDepartment of Chinese Pharmaceutical Sciences and Chinese Medicine Resources, College of Chinese Medicine, China Medical University, Taichung 40402, Taiwan. E-mail: gjhuang@mail.cmu.edu.tw; Tel: +886-4-22053366 ext. 5508
cDepartment of Food Nutrition and Health Biotechnology, Asia University, Taichung 41354, Taiwan
First published on 7th September 2018
Abstract
Alpinumisoflavone (AIF) is a plant-derived pyranoisoflavone that exhibits a number of pharmacological activities, but the protective effects of AIF against pulmonary inflammation are still unknown. This study aimed to investigate the anti-inflammatory effects and possible molecular mechanisms of AIF in both lipopolysaccharide (LPS)-stimulated macrophages and mice. The results revealed that AIF dramatically suppressed the production of pro-inflammatory mediators [including tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β, IL-17, intercellular adhesion molecule-1 (ICAM-1), and nitric oxide (NO)] and increased the levels of anti-oxidative enzymes [including catalase (CAT), heme oxygenase-1 (HO-1), glutathione peroxidase (GPx), and superoxide dismutase (SOD)] both in vitro and in vivo. Additionally, pre-treatment with AIF could not only significantly prevent histopathological changes and neutrophil infiltration but also decreased the expression levels of nuclear factor-kappa B (NF-κB), mitogen-activated protein kinases (MAPKs), and the nucleotide-binding domain-like receptor protein 3 (NLRP3) inflammasome, as well as IL-17 production in LPS-induced lung tissues. The anti-inflammatory effects of AIF were mediated by up-regulating anti-oxidative enzymes and suppressing the NF-κB, MAPK, NLRP3 inflammasome and IL-17 signaling pathways. This is the first study to reveal that AIF has a protective effect against LPS-induced lung injury in mice.
Introduction
AIF is a plant-derived pyranoisoflavone that is found in many plants, such as Erythrina variegate, Erythrina lysistemon and Millettia thonningii and the fruits of Cudrania tricuspidata.1–4 Previous studies have indicated that AIF exhibits various pharmacological activities, such as anti-osteoporotic, anti-bacterial, and anti-metastatic activities, and the promotion of apoptosis in tumors.5–8 Additionally, AIF can not only regulate apoptosis via the NF-κB and MAPK pathways in lung tumor cells but can also exert anti-osteoporotic effects by inhibiting reactive oxygen species (ROS) production and regulating the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) signaling pathway.2,9 Although previous studies have shown various pharmacological activities of AIF, the protective effects of AIF against pulmonary inflammation remain unknown.Inflammation is a highly regulated nonspecific immune reaction that is initiated by a complex process involving the activation of immune cells, such as macrophages, and the release of pro-inflammatory mediators in the affected tissue.10,11 A persistent and uncontrolled inflammatory response is the major provocation in the progression of many chronic diseases in humans, including autoimmune diseases, metabolic diseases, cancers, and organ dysfunction.12,13 Acute lung injury (ALI) is a common clinical disease that involves widespread inflammation in the lung tissues, which is the first step in the development of lung dysfunction.14,15 In the pulmonary inflammatory response, macrophages play a central role in maintaining the balance of immune and host defenses. In the inflammatory response, macrophages are the main source of cytokines in the lungs and play an important role in the pathogenesis of lung injury. Macrophages can induce neutrophil infiltration and more severe inflammatory responses.16 Furthermore, increasing evidence has shown the interaction between inflammation and oxidative stress, which plays an important role in the pathogenesis of ALI.17 Oxidative stress, which is caused by a large number of ROS, myeloperoxidase (MPO), and malondialdehyde (MDA) formation, results in the occurrence of oxidative damage. However, anti-oxidant enzymes, such as CAT, HO-1, GPx, and SOD, can ameliorate this adverse condition.18 These findings suggest that targeting the suppression of inflammation and/or oxidative stress is a potential strategy to improve ALI.
LPS, also known as endotoxin, is the main component of the outer membrane of Gram-negative bacteria.19 When the lungs are heavily exposed to LPS, endothelial and alveolar epithelial cell damage can result, which causes macrophage activation and the leakage of inflammatory cells, especially neutrophils. Therefore, LPS is commonly used to induce ALI.20 The intratracheal instillation of LPS is used in an established mouse model of ALI characterized by alveolar hemorrhagic edema and increased levels of neutrophils, protein content, and cytokines in the bronchoalveolar lavage fluid (BALF). A considerable influx of neutrophils into the lungs not only results in the release of pro-inflammatory cytokines but also causes the production of ROS and oxidative stress.17 Toll-like receptor 4 (TLR-4) is a transmembrane receptor and is an important protein that mediates the communication between LPS and the inside of the cell. The inflammatory processes aroused by LPS have been proven to be mediated by TLR-4 and include the up-regulation of the activation of NF-κB, MAPKs, and NLRP3 inflammasome pathway.21,22 NF-κB, a heterodimer composed of p50 and p65, is a pleiotropic transcription factor that is involved in a variety of processes, such as inflammatory and immune responses and cell growth and survival.23 In unstimulated cells, NF-κB exists in an inactive state due to its interaction with IκB-α and is retained in the cytoplasm.24 Once activated, IκB-α is phosphorylated by IκB kinase (IKK) and undergoes proteasomal degradation, which liberates the p50 and p65 subunits of NF-κB into the nucleus, which in turn, results in the production of inflammatory cytokines. MAPKs, including extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 MAPK, initiate the expression of inflammatory genes via phosphorylation.22 The activation of the NF-κB and MAPK pathways can regulate various inflammatory mediators, including inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), the cytokines TNF-α, IL-6, IL-1β, and IL-17 and ICAM-1.
The NLRP3 inflammasome is multiple protein complexes that is associated with the pathogenesis of many diseases and can recognize a wide range of pathogens and damage-related molecules. The NLRP3 inflammasome can modulate the activation of caspase-1 and lead to the generation of the pro-inflammatory cytokine IL-1β, which results in inflammation. The activation of the NLRP3 inflammasome is mediated by multiple signals, such as various pro-inflammatory factors, including cytokines and TLR promoters, which can regulate the activation of the NF-κB signaling pathway.21
TLR ligands do not directly activate the NLRP3 inflammasome but instead indirectly participate in the activation of this complex. Dual signaling patterns have been proposed for the activation of the NLRP3 inflammasome in macrophages. The first signal (priming) is provided by the microorganism or endogenous molecule that induces the expression of NLRP3 and pro-IL-1β via the activation of IKK/NF-κB, and the second signal (activation) is sparked by ATP, pore-forming toxins, viral RNA or particulate matter, and this response can cause inflammation.25 Indeed, increasing evidence has shown that LPS can activate multiple signal pathways, including the ROS, TLR-4, NF-κB, MAPK, and NLRP3 inflammasome pathways, and all of these pathways are closely related to ALI induced by LPS.18,26
IL-17 cytokines, which are produced by the T helper (Th)-17 subset of CD4+ T cells,27 comprise six members that include IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F, and each of these cytokines has its own specific receptor.28 Notably, IL-17A and IL-17F have the highest amino acid sequence identity (50%) and share the same receptors, i.e., IL-17RA and IL-17RC.13,28 Despite the high homology between IL-17A and IL-17F, these cytokines perform different functions. Previous studies have indicated that the overexpression of IL-17A plays a central role in the host defense against bacterial and fungal infections and is also related to the development of autoimmunity, inflammation, and tumors. In contrast, IL-17F is mainly involved in mucosal host defense mechanisms.13,27 Therefore, IL-17A is a more popular target than the other members of the IL-17 cytokine family and, according to reports, is also associated with the activation of TLR-4, NF-κB, MAPKs, and the NLRP3 inflammasome.28,29
Therefore, the present study aimed to investigate the anti-inflammatory and anti-oxidative effects of AIF in LPS-stimulated RAW264.7 macrophages and a mouse model of LPS-induced ALI.
Materials and methods
Chemicals
Alpinumisoflavone (AIF) was purchased from BJYM Pharmaceutical & Chemical Co., Ltd. (Beijing, People's Republic of China). The purity of AIF used in the present study was >95%. LPS (Escherichia coli 055:B5), dexamethasone (Dex), thiazolyl blue tetrazolium bromide (MTT), 2′,7′-dihydrofluorescein diacetate (DCFH-DA), dimethyl sulfoxide (DMSO) and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against TLR-4, NLRP3, Hsp90α, TRAF6, COX-2, Act1, CAT, phospho-JNK, GPx, SOD, caspase-1, and IL-1β were purchased from Gene Tex (San Antonio, TX, USA). Antibodies against iNOS, NF-κB, IκBα, p38, HO-1, ICAM-1 and β-actin were purchased from Abcam (Cambridge, UK, USA). Antibodies against Phospho-IKKα/β, IKKβ, JNK, Phospho-ERK, ERK, Phospho-p38, and Phospho-IκB-α were purchased from Cell Signaling Technology (Beverly, MA, USA). Polyvinylidene fluoride transfer membranes (Immobilon P) and enhanced chemiluminescence (ECL) reagent were from EMD Millipore (Bedford, MA, USA). The protein assay dye reagent were purchased from Bio-Rad (Hercules, CA, USA).Cell culture
The murine macrophage cell line RAW264.7 (BCRC no. 60001) was obtained from the Bioresources Collection and Research Center (BCRC) of the Food Industry Research and Development Institute (Hsinchu, Taiwan). The cells were grown in DMEM with 10% fetal bovine serum (FBS), 4 mM L-glutamine, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified incubator containing 5% CO2. The RAW264.7 cells (5 × 105 cells per mL) were seeded into plates for 24 h before treatment. Following treatment with different concentrations of AIF (1, 5 and 10 μg mL−1) in the presence or absence of LPS (100 ng mL−1) for 24 h, and the supernatant and cell were collected for further analysis.Cell viability assay
RAW264.7 cells were seeded into 96-well plates at 5 × 105 cells per mL 24 h before treatment. Following treated with different concentrations of AIF (1, 5 and 10 μg mL−1) in the presence or absence of LPS (100 ng mL−1) for 24 h. One-tenth volume of 5 mg mL−1 MTT solution was then added to the culture medium. After a 4 h incubation at 37 °C, equal cell culture volume of 0.04 N HCl in isopropanol was added to dissolve the MTT formazan, and the absorbance value was measured at 570 nm using a microplate reader (Molecular Devices, LLC.).Animals and preparation of ALI model
Adult male ICR mice (6 weeks old) were obtained from BioLASCO (Taipei, Taiwan). All mice were fed with the standard laboratory diet, water ad libitum, and housed in the conditions of cyclic lighting (12 h light/12 h dark) and temperature (21 ± 1 °C). All animal experimental procedures used in this study were approved by the institution of Animal Care and Use Committee in China Medical University and also adhered to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals as well as conducted under the guidelines of the International Association for the Study of Pain (IASP). The mice were divided into the following group: in the normal control group, mice were received normal saline. In the LPS group, mice were intratracheal (i.t.) administrated LPS (5 mg kg−1, 50 μL per mice). In the AIF (1, 5 and 10 mg kg−1) treated group, mice were intraperitoneally (i.p.) administrated 1 h before LPS challenge. In the positive control group, Dex (10 mg kg−1) was i.p. administered 1 h before LPS challenge. All mice were euthanized and collected the bronchoalveolar lavage fluid (BALF) and lung tissues at 6 h after LPS stimulation.Histological study
The lung tissues were fixed in 10% formalin and embedded in paraffin, which were cut into 5 μm sections and stained with hematoxylin and eosin (H&E), then used the light microscopic to examine the pathological changes.Lung wet-to-dry weight (W/D) ratio
The lung tissue was collected and weighed immediately to get the “wet” weight (W), and placed the lung tissues in an over at 80 °C for 48 h in order to obtain the “dry” weight (D). The W/D ratio was calculated to assess tissue edema.BALF and cell counting
After the mice were sacrificed, the lung tissues were lavaged three times with 0.6 mL ice-cold saline through a tracheal cannula, centrifuged at 1000 rpm for 10 min in 4 °C. The supernatant was stored at −80 °C for NO and cytokines analysis. The protein concentration in the cell pellet was reacted with the protein assay dye reagent and determined by a microplate reader.Measurement of MPO activity
The lung tissues were homogenized in ice-cold potassium phosphate buffer (PPB, 50 mM K2HPO4, pH 6.0) and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 10 min in 4 °C. The pellets were re-suspended with PPB containing 0.5% (w/v) hexadecyltrimethylammonium bromide (HETAB) and subjected to three freeze–thaw cycles, then centrifuged at 12000 g for 10 min in 4 °C, and the supernatant was collected and diluted in PPB then froze at −80 °C until further analysis. MPO activity was assessed by measuring the H2O2-dependent oxidation of o-dianisidine chloride, and the absorbance value was measured at 460 nm using a microplate reader. The protein concentrations of samples were reacted with the protein assay dye reagent and determined by the microplate reader. The results of MPO activity in the samples were presented as unit (U) mg−1 of the protein.
000 g for 10 min in 4 °C. The pellets were re-suspended with PPB containing 0.5% (w/v) hexadecyltrimethylammonium bromide (HETAB) and subjected to three freeze–thaw cycles, then centrifuged at 12000 g for 10 min in 4 °C, and the supernatant was collected and diluted in PPB then froze at −80 °C until further analysis. MPO activity was assessed by measuring the H2O2-dependent oxidation of o-dianisidine chloride, and the absorbance value was measured at 460 nm using a microplate reader. The protein concentrations of samples were reacted with the protein assay dye reagent and determined by the microplate reader. The results of MPO activity in the samples were presented as unit (U) mg−1 of the protein.
Measurement of NO production
The supernatant collected from cell culture or the BALF obtained from mice was mixed with an equal volume of Griess reagent (1% sulphanilamide, 0.1% naphthylethylenediamine dihydrochloride and 5% phosphoric acid), and the absorbance value was measured at 540 nm using an ELISA plate reader.Cytokines analysis
The cytokines (TNF-α, IL-6, IL-1β, and IL-17) yield in the cell supernatants and in BALF were measured by using commercially available ELISA kits (BioLegend, San Diego, CA) according to the manufacturer's instructions.Measurement of ROS generation
The RAW264.7 cells were collected from cell culture for analysis the intercellular ROS generation. Cells were washed with PBS twice then reacted with 10 μM DCFH-DA in the temperature maintained at 37 °C for 45 min. The DCF fluorescence intensities was measured by using Synergy™ HT Multi-Detection Microplate Reader (BioTek Instruments, Inc.) at an excitation and emission wavelength of 485 nm and 528 nm, respectively.Immunofluorescence assay
The treatment of RAW264.7 cells as described above. The supernatant in the plate was removed, following added methanol and wait 10 min then washed with PBS third. The 0.1% Triton X-100 was added and reacted for 10 min, following washed the cells with PBS third, and reacted with 1% bovine serum albumin (BSA) for 1 h. The cells were washed 3 times in PBS and then incubated at 4 °C overnight with primary antibody. The cells were washed 3 times with PBS and incubated with the DyLight™ 488-conjugated goat anti-rabbit IgG antibody (Jackson ImmunoResearch, West Grove, PA) at room temperature for 2 h. After washing the cells with PBS third, the cells were incubated in the presence of 4′,6-diamidino-2-phenylindole (DAPI) for 10 min in the dark. The imaging of cells was acquired by a confocal laser scanning microscope (Leica, TCS SP2, Germany).Western blot analysis
The cell pellet collected from cell culture or the lung tissues obtained from mice were using RIPA buffer (Thermo Fisher Scientific, Waltham, MA) for exhaustive protein extraction. For extracting the protein in the cytoplasmic or nuclear, the nuclear extract kit (Active Motif, Carlsbad, CA, USA) had been used according to the manufacturer's instructions. An equal amount of protein from each sample was separated by SDS-PAGE and then transferred onto PVDF membranes (EMD Millipore, Bedford, MA, USA). The membranes were blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) at room temperature for 1 h then washed the membranes with TBST third and subsequently incubated with primary antibodies at 4 °C overnight. Followed by washes the membranes 3 times with TBST, and incubated the membranes with the HRP-conjugated secondary antibody at room temperature for 1 h, then washes the membranes with TBST third. The blots on the membranes were visualized by ECL reagent and detected by using Kodak Molecular Imaging Software (Version 4.0.5, Eastman Kodak Company, Rochester, NY).Statistical analysis
All values are given as mean ± SD for at least 3 independent experiments. Statistical analysis was executed by using Predictive Analytics Suite Workstation (PASW) Statistical software for Windows, Version 18.0 (SPSS Inc., Chicago, USA) involved one-way ANOVA followed by Scheffe's test. P-value < 0.05 was considered to indicate a statistically significant difference.Results
AIF reduced on production of inflammatory mediators in LPS-stimulated RAW264.7 cells
The release of cytokines into the cell culture medium was measured using ELISAs and western blot analysis. As illustrated in Fig. 1C–G, the treatment of RAW264.7 cells with LPS (100 ng mL−1) alone significantly increased the production of NO, TNF-α, IL-6, IL-1β and ICAM-1, whereas incubating cells with LPS (100 ng mL−1) and AIF at concentrations of 1, 5 and 10 μg mL−1 significantly inhibited the production of NO and cytokines (TNF-α, IL-6 and IL-1β) and ICAM-1 protein expression.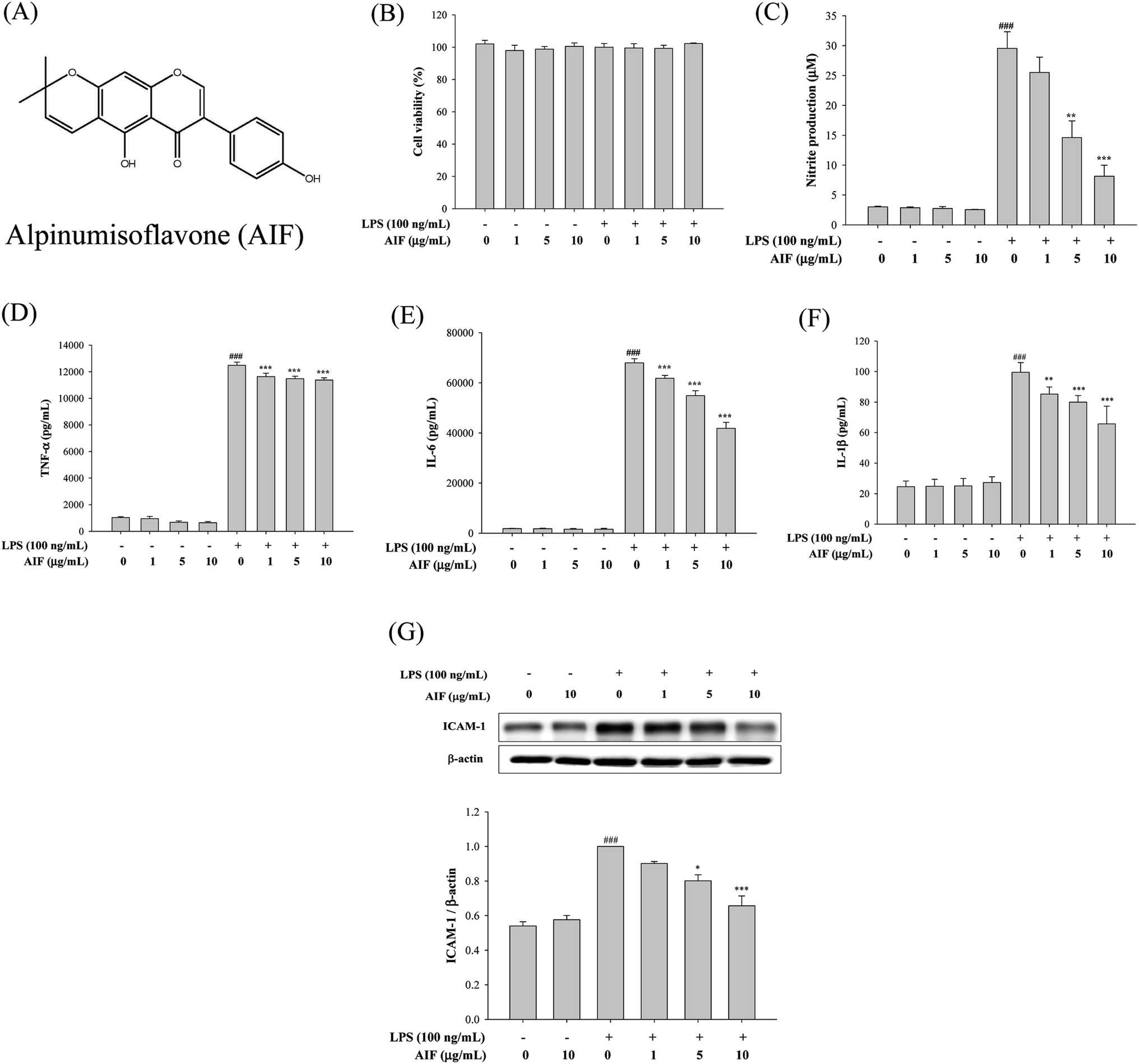 | ||
| Fig. 1 Effects of AIF regarding the inflammatory responses in LPS-induced RAW264.7 cells. (A) The chemical structure of alpinumisoflavone (AIF). (B) Effect of AIF on cell viability were measured using MTT assay. Levels of (C) NO, (D) TNF-α, (E) IL-6, and (F) IL-1β in culture supernatants were quantitated using ELISA. (G) The protein expression of ICAM-1 were measured using western blot. All values represent mean ± S.D. of three or more replicates. ###p < 0.001 compared with normal group; *p < 0.05, **p < 0.01 and ***p < 0.001 compared with LPS treated alone group, respectively. | ||
Effects of AIF decreased LPS-induced activation of NF-κB and MAPKs in RAW264.7 cells
When cells are activated with LPS, p-IKK and the degradation of IκB-α are known to be involved in the process of activating NF-κB, which leads to the translocation of NF-κB into the nucleus, thereby initiating the transcription of various inflammatory genes.23 Because the NF-κB and MAPK pathways play crucial roles in LPS-induced macrophage activation, regulating the transcription of a number of genes that are involved in inflammation, we studied the effects of AIF on LPS-induced NF-κB nuclear translocation, which is required for NF-κB-dependent transcription following LPS-stimulation. To determine the effect of AIF on the NF-κB pathway, the cytosolic levels of p-IKK, NF-κB and IκB-α and the nuclear level of NF-κB were assessed using western blot analysis. The results suggested that the AIF treatment effectively reduced the phosphorylation of IKK and NF-κB and blocked IκBα phosphorylation and degradation in the LPS-induced RAW264.7 cells (Fig. 2A). To further verify these results, an immunofluorescence assay was performed to examine the NF-κB localization in RAW264.7 cells. As illustrated in Fig. 2B, compared with the unstimulated cells (controls), intracellular NF-κB clearly translocated from the cytoplasm into the nucleus of the cells that were stimulated with LPS alone. However, treatment with AIF (10 μg mL−1) and LPS (100 ng mL−1) for 30 min significantly decreased the level of NF-κB localized in the nucleus. Furthermore, to determine the effect of AIF on the MAPK pathway, we performed a western blot analysis. The results suggested that the AIF treatments (5 and 10 μg mL−1) effectively reduced the phosphorylation of ERK, JNK and p38 in the LPS-induced RAW264.7 cells (Fig. 2C).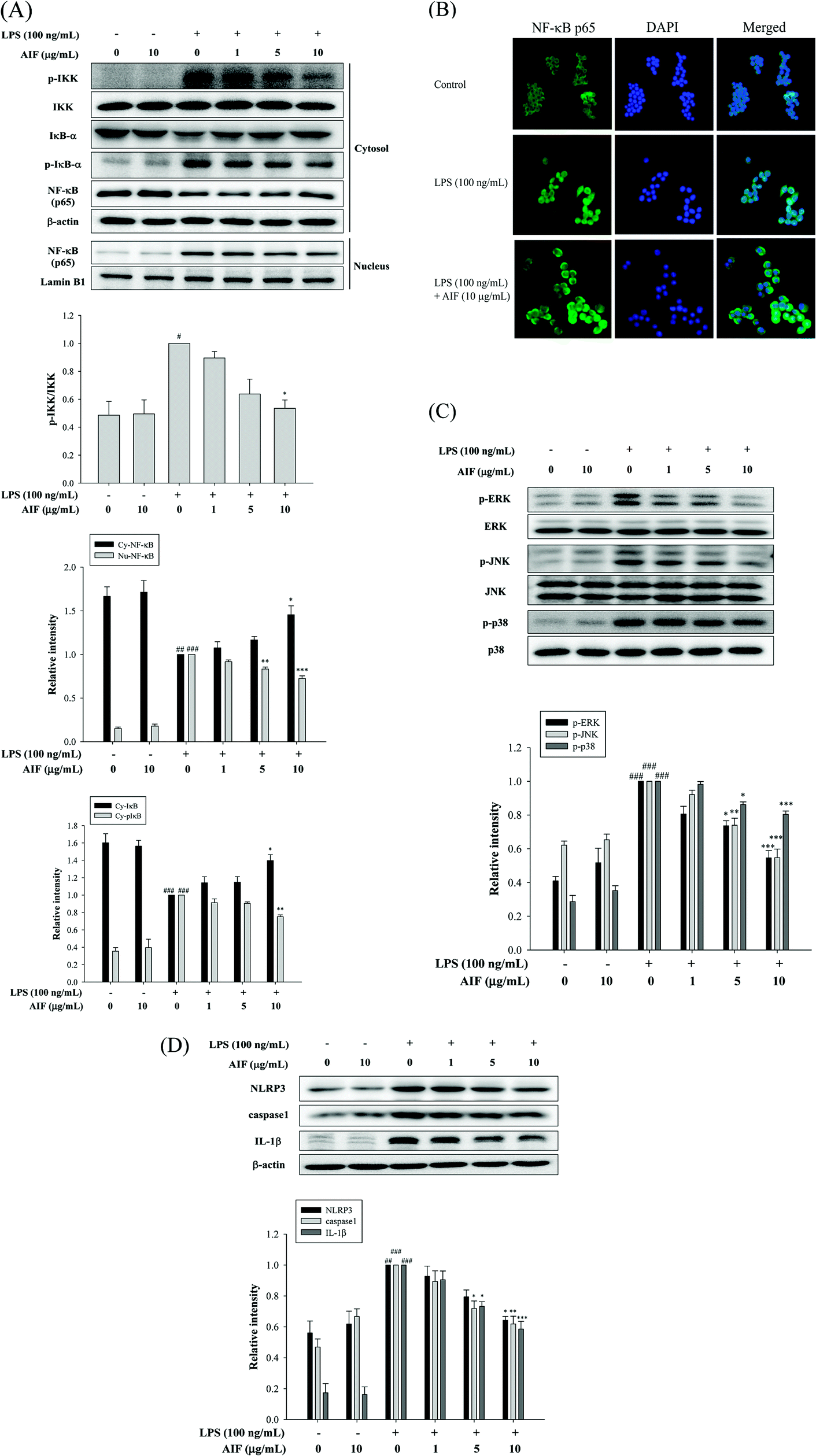 | ||
| Fig. 2 Effects of AIF regarding the activation of NF-κB, MAPKs, and NLRP3 inflammasome in LPS-induced RAW264.7 cells. (A) Effect of AIF on IKK, IκB-α phosphorylation and NF-κB activation were measured using western blot. (B) Effect of AIF on NF-κB nuclear translocation was detected by immunofluorescence assay using an antibody for p65 subunit. (C) Effect of AIF on MAPKs activation were measured using western blot. (D) The protein expression of NLRP3, caspase-1, and IL-1β were measured using western blot. All values represent mean ± S.D. of three or more replicates. #p < 0.05, ##p < 0.01 and ###p < 0.001 compared with normal group, respectively; *p < 0.05, **p < 0.01 and ***p < 0.001 compared with LPS treated alone group, respectively. | ||
AIF reduced the expression of the NLRP3 inflammasome in LPS-stimulated RAW264.7 cells
In macrophages, the level of NLRP3 inflammasomes is dependent on NF-κB-dependent transcriptional priming that is sufficient to facilitate inflammasome activation.30 The NLRP3 inflammasome is an upstream factor of caspase-1, and activated caspase-1 can convert pro-IL-1β into mature IL-1β and cause inflammation.31 Therefore, inhibiting the upstream molecule, the NLRP3 inflammasome, reduces caspase-1 expression, thereby reducing inflammation. To further confirm the effects of AIF on LPS-induced NLRP3 inflammasome expression in macrophages, we used LPS-stimulated RAW264.7 cells. The results suggested that LPS treatment caused significant increases in the expression levels of the NLRP3, caspase-1 and IL-1β proteins. Our results indicated that AIF treatment markedly inhibited the LPS-induced activation of the NLRP3 inflammasome, caspase-1, and IL-1β proteins, especially when a high dose was applied (Fig. 2D).Effects of AIF against LPS-induced IL-17 pathway activation in RAW264.7 cells
The IL-17A-induced Act1/Hsp90 interaction is TRAF6-dependent and can activate NF-κB, which leads to the production of multiple pro-inflammatory molecules that trigger inflammation.13,32 Our findings showed that the treatment of macrophages with LPS caused significant increases in the levels of the Act1, Hsp90, and TRAF6 proteins and the IL-17A cytokine. However, co-treatment with LPS (100 ng mL−1) and AIF (5 or 10 μg mL−1) led to significantly reduced protein and cytokine IL-17A expression (Fig. 3).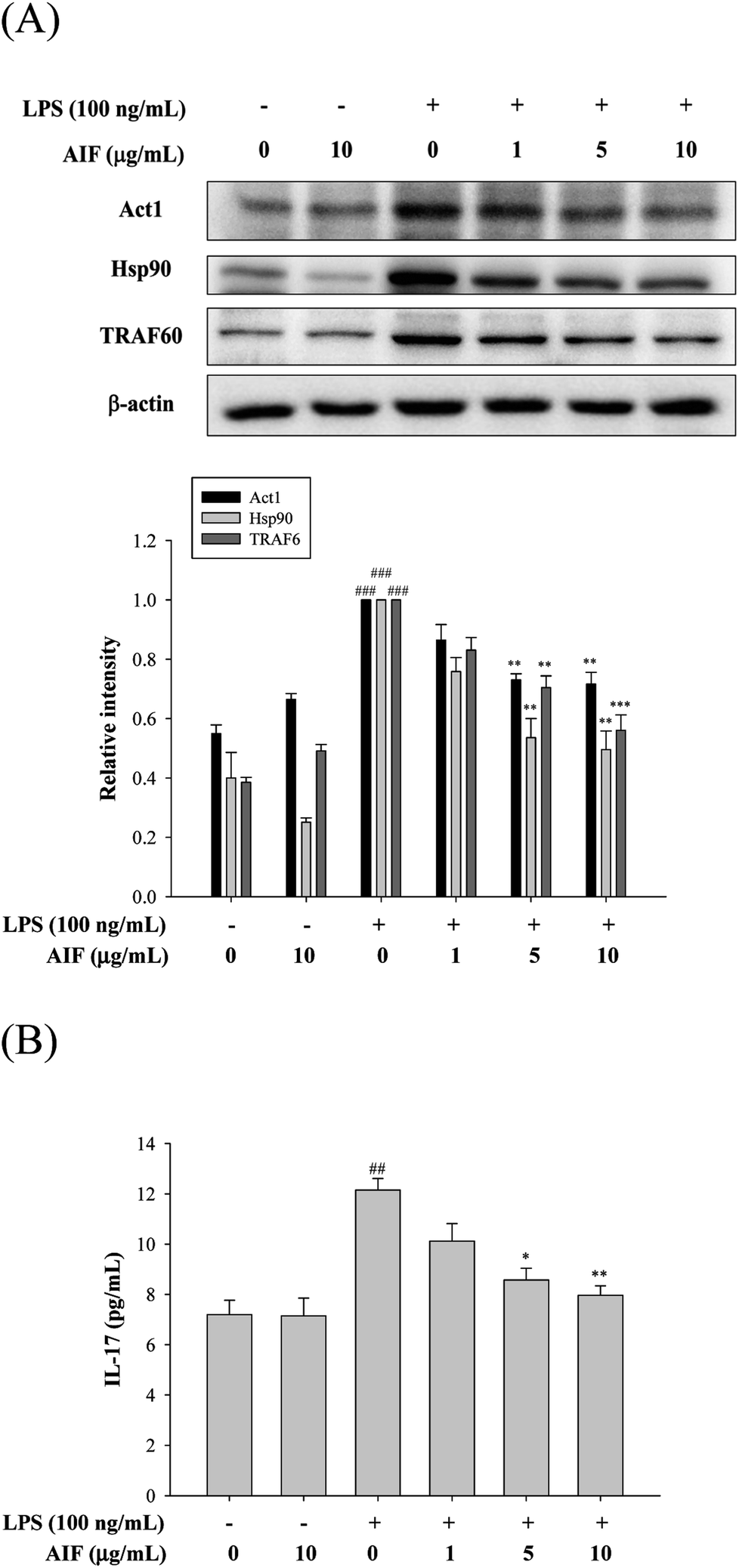 | ||
| Fig. 3 Effects of AIF regarding the activation of IL-17 signaling pathway in LPS-induced RAW264.7 cells. (A) The protein expression of Act1, Hsp90, and TRAF6 were measured using western blot. (B) Levels of IL-17 in culture supernatants were quantitated using ELISA. All values represent mean ± S.D. of three or more replicates. ##p < 0.01 and ###p < 0.001 compared with normal group, respectively; *p < 0.05, **p < 0.01 and ***p < 0.001 compared with LPS treated alone group, respectively. | ||
Effects of AIF regarding the expression of anti-oxidant enzymes, TLR-4, iNOS, and COX-2 in LPS-stimulated RAW264.7 cells
LPS activates TLR-4, which is expressed on pro-inflammatory cells. Moreover, the activation of TLR-4 leads to the promotion of ROS production, which activates a number of intracellular signaling proteins, including NF-κB, MAPKs, iNOS, and COX-2. Our findings revealed that the LPS treatment of RAW264.7 cells caused significant increases in the expression levels of the TLR-4, iNOS, and COX-2 proteins. We observed a loss of LPS-mediated TLR-4 protein induction at a dose of 10 μg mL−1. Co-treatment with LPS (100 ng mL−1) and AIF (1, 5, 10 μg mL−1) also significantly reduced iNOS protein expression. However, AIF did not block COX-2 induction by LPS (Fig. 4A). Furthermore, we also found AIF can significantly increase the expression of antioxidant enzymes in a dose-dependent manner (Fig. 4B). Next, we determined the effect of AIF on intracellular ROS, which were measured using the fluorescent probe DCFH-DA. Fig. 4C shows the effect of AIF on intracellular ROS production in the LPS-stimulated RAW264.7 cells. The treatment of these cells with AIF (5 and 10 μg mL−1) significantly inhibited the LPS induction of intracellular ROS generation.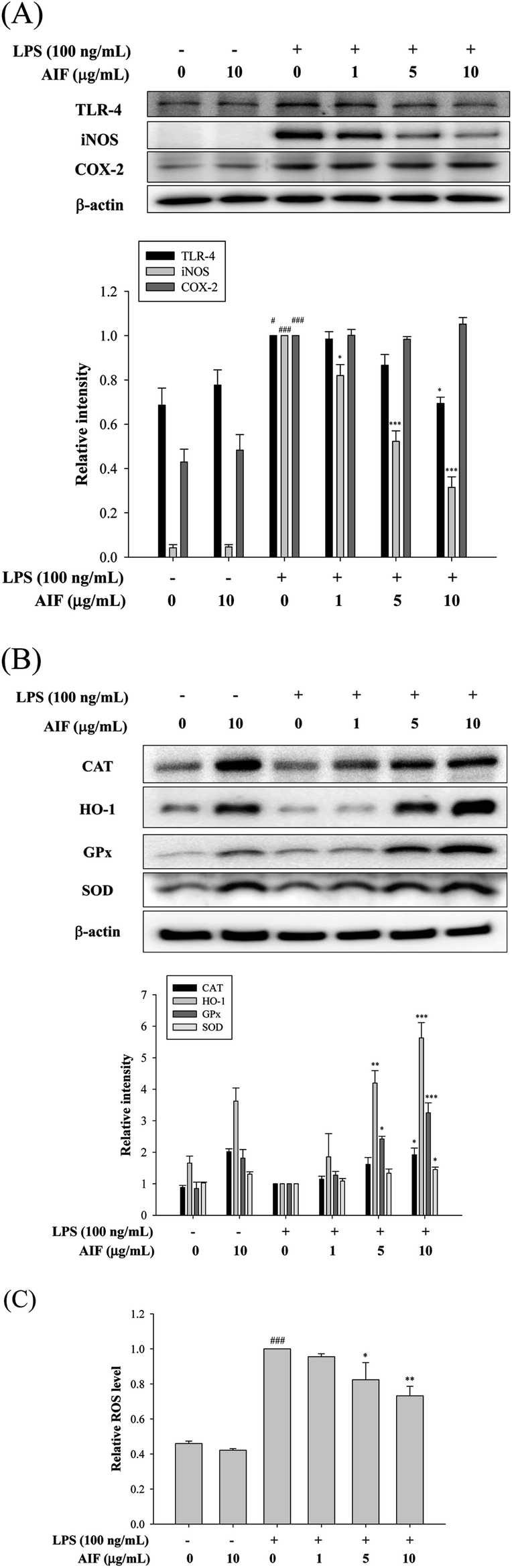 | ||
| Fig. 4 Effects of AIF regarding the expression of TLR-4, iNOS, COX-2, and anti-oxidant enzymes in LPS-induced RAW264.7 cells. (A) The protein expression of TLR-4, iNOS and COX-2 were measured using western blot. (B) The protein expression of anti-oxidative enzymes (CAT, HO-1, GPx, and SOD) were measured using western blot. (C) Levels of ROS production in RAW264.7 cells were determined using ELISA. All values represent mean ± S.D. of three or more replicates. #p < 0.05 and ###p < 0.001 compared with normal group, respectively; *p < 0.05, **p < 0.01 and ***p < 0.001 compared with LPS treated alone group, respectively. | ||
AIF attenuated the inflammatory response in LPS-induced ALI mice
To evaluate the effect of AIF on LPS-induced lung inflammation-related histological changes, lung sections were subjected to hematoxylin and eosin staining. Compared with the lungs of the normal control group, those of the LPS group exhibited marked inflammatory responses that were characterized by the presence of interstitial edema, hemorrhages, the thickening of the alveolar walls and the infiltration of inflammatory cells. However, pretreatment with AIF improved the LPS-induced histopathological damage, similarly to pretreatment with Dex (Fig. 5A). To assess the severity of the formation of edema, the W/D ratios of the lungs and the total protein concentration in the BALF were examined. As shown in Fig. 5B and D, an LPS challenge caused significant increases in the W/D ratio and total protein concentration compared with the normal control group. In contrast, pretreatment with AIF (10 mg kg−1) and Dex (10 mg kg−1) remarkably decreased the lung edema and total protein concentration. Additionally, increased MPO activity represents the accumulation of polymorphonuclear neutrophils in the lungs. LPS irritation caused significant increases in the lung MPO activity compared with that in the normal control group. However, pretreatment with AIF (10 mg kg−1) and Dex (10 mg kg−1) significantly decreased the MPO activity in the lung (Fig. 5C). Because TNF-α, IL-6, IL-1β and ICAM-1 are typical pro-inflammatory mediators in various infectious disorders, the effects of AIF on the LPS-induced ALI inflammatory reaction were examined by determining the levels of these cytokines in the BALF via ELISA and western blot analyses. As illustrated in Fig. 5E–H, the levels of the pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) and ICAM-1 were significantly higher in the LPS-stimulated group than in the normal control group. In contrast, pretreatment with AIF (5 and 10 mg kg−1) and Dex (10 mg kg−1) remarkably decreased the levels of pro-inflammatory cytokines in the ALI mice (Fig. 5E–H).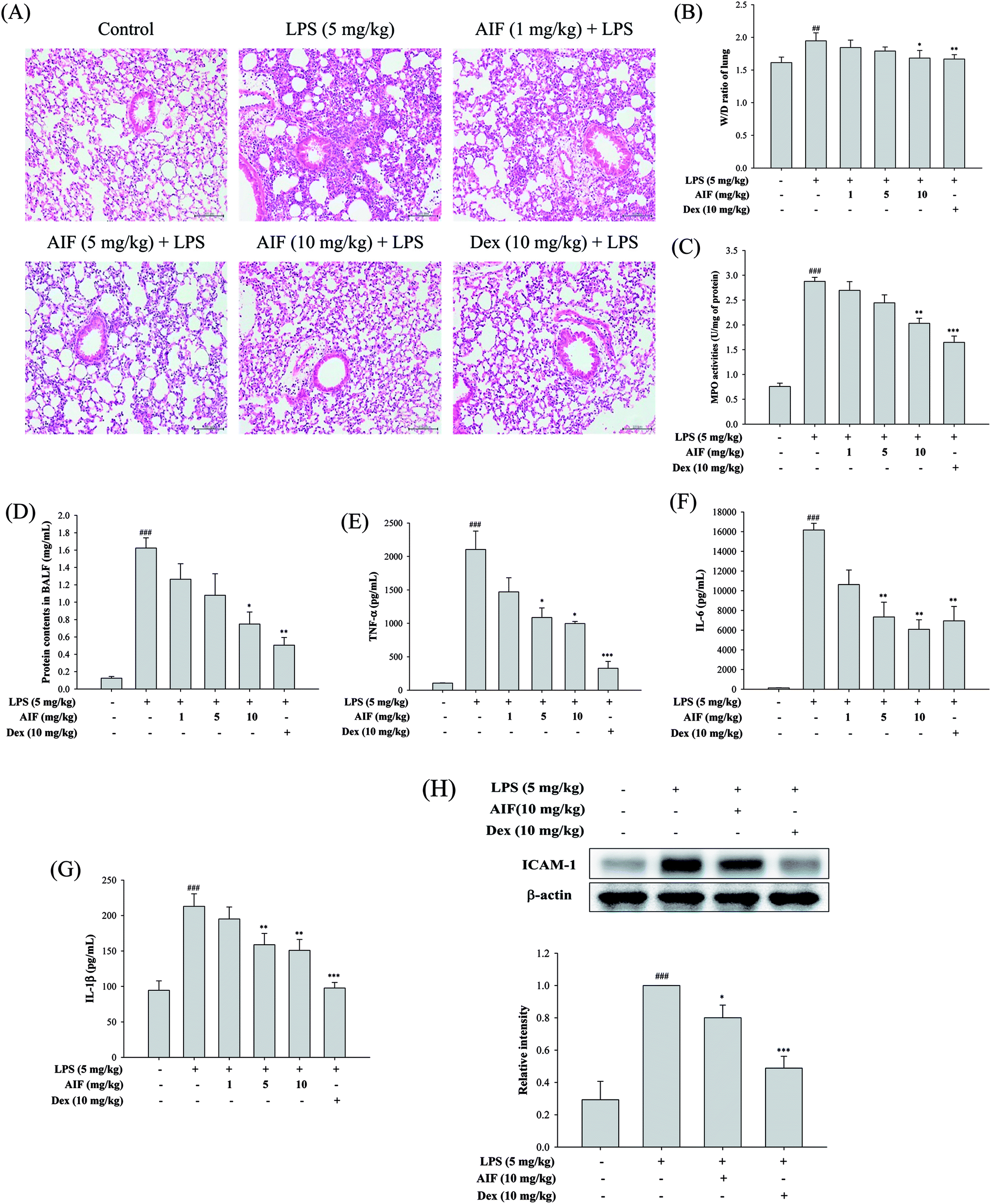 | ||
| Fig. 5 Effects of AIF regarding the inflammatory response in LPS-stimulated ALI mice. (A) Pathological examinations on lung tissues in LPS-induced ALI mice. Lung sections were stained with hematoxylin and eosin (H&E). Original magnification: 200×. Scale bar represents 100 μm. Effects of AIF on the (B) wet-to-dry weight (W/D) ratio and (C) MPO activity in lung tissues of LPS-induced ALI. BALF was collected to measure the (D) protein contents using Bradford assay, and the levels of (E) TNF-α, (F) IL-6, and (G) IL-1β using ELISA. (H) The protein expression of ICAM-1 in lung tissues were measured using western blot. All values represent mean ± S.D. of three or more replicates. ##p < 0.01 and ###p < 0.001 compared with normal group, respectively; *p < 0.05, **p < 0.01 and ***p < 0.001 compared with LPS treated alone group, respectively. | ||
Effects of AIF decreased LPS-induced NF-κB activation in LPS-stimulated ALI mice
The activation of NF-κB and MAPKs prompts the transcription of most pro-inflammatory cytokines, thereby causing greater pathogenesis in cases of ALI.18,33 To investigate the signaling pathway through which AIF inhibited pro-inflammatory cytokine production, western blot analyses were performed to evaluate the expression levels of the proteins in the NF-κB and MAPK pathways. The results indicated that the LPS-induced NF-κB translocation, IκBα phosphorylation, and activation of MAPKs were inhibited by pretreatment with AIF in the ALI mice (Fig. 6A–B).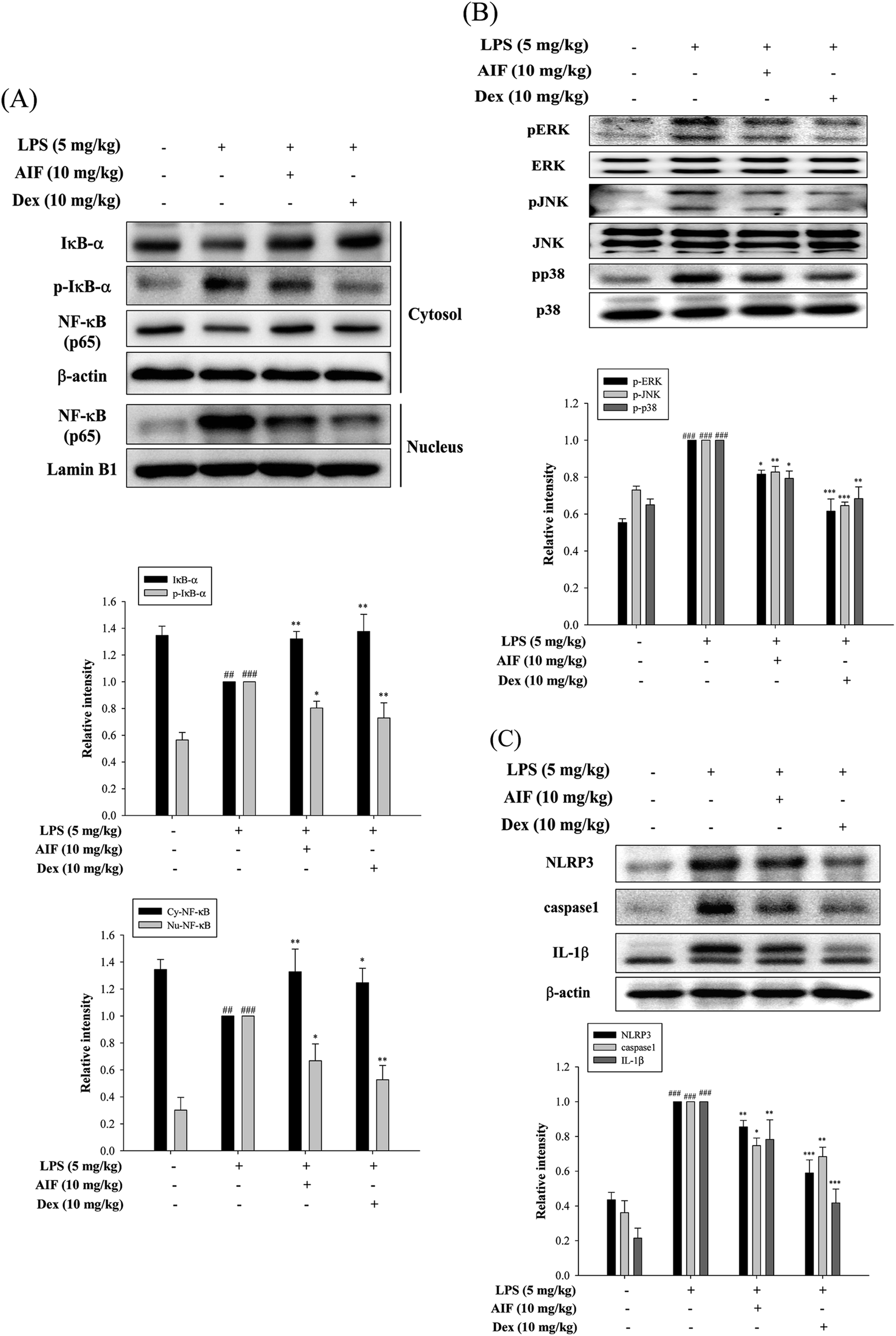 | ||
| Fig. 6 Effects of AIF regarding the activation of NF-κB, MAPKs, and NLRP3 inflammasome in ALI mice. Effect of AIF on (A) IκB-α phosphorylation, NF-κB activation, (B) MAPKs (ERK, p38, and JNK) activation, (C) NLRP3, caspase-1, and IL-1β in lung tissues were measured using western blot. All values represent mean ± S.D. of three or more replicates. ##p < 0.01 and ###p < 0.001 compared with normal group, respectively; *p < 0.05, **p < 0.01 and ***p < 0.001 compared with LPS treated alone group, respectively. | ||
AIF ameliorated the expression of the NLRP3 inflammasome in LPS-stimulated ALI mice
The NLRP3 inflammasome activates caspase-1, which results in the maturation of pro-IL-1β.31 High levels of NLRP3 mRNA are expressed in alveolar macrophages, and for the above reason, the NLRP3 inflammasome has also been found to influence ALI.30 In this experiment, AIF pretreatment reduced the expression levels of the NLRP3 and caspase-1 proteins in the ALI mice and thereby reduced the production of the IL-1β protein (Fig. 6C).Effects of AIF regarding LPS-induced IL-17 pathway activation in ALI mice
To further observe the protective mechanism of AIF pretreatment against LPS-induced ALI, the levels of Act1, Hsp90, TRAF6, and IL-17, which are responsible for increasing lung injury, were analyzed using western blot and ELISA analyses. The results indicated that both AIF (10 mg kg−1) and Dex (10 mg kg−1) significantly decreased the Act1, Hsp90 and TRAF6 protein levels and the level of IL-17 in the BALF of the ALI mice (Fig. 7).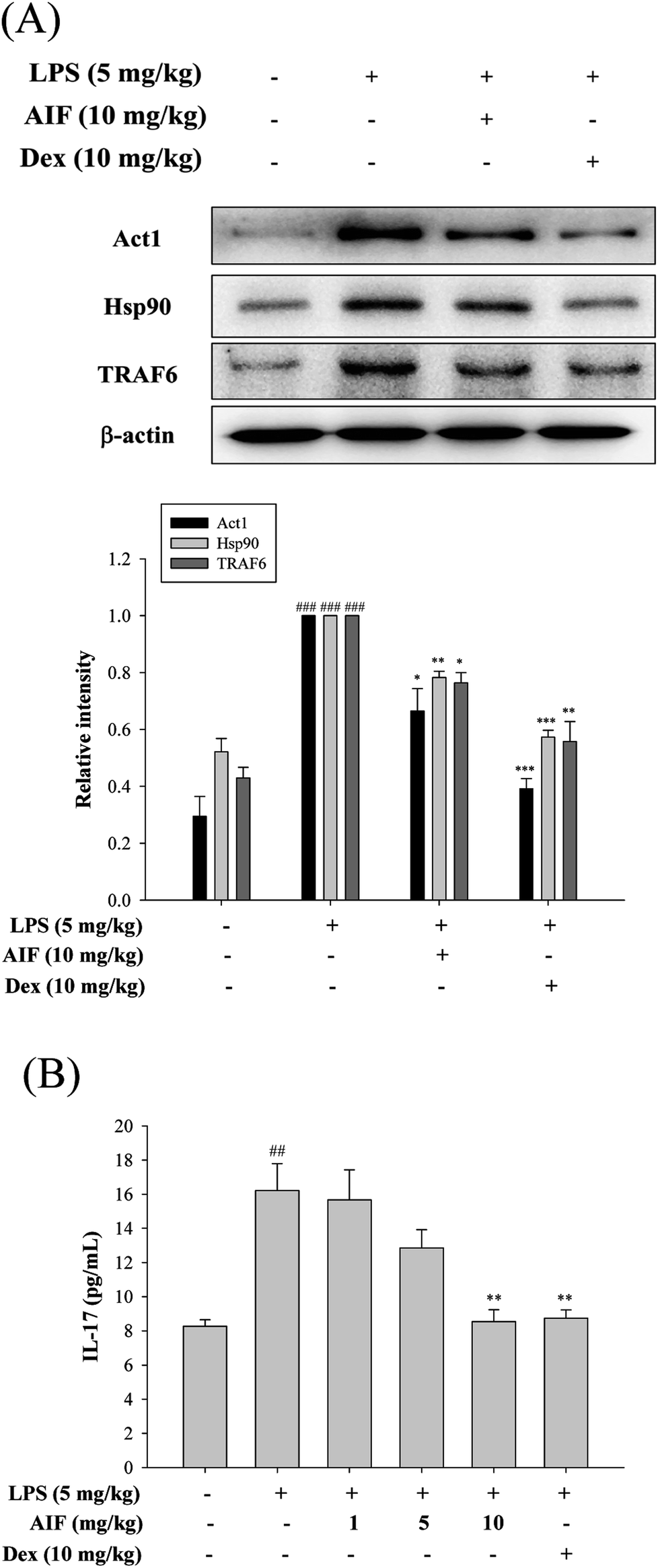 | ||
| Fig. 7 Effects of AIF regarding the activation of IL-17 signaling pathway in ALI mice. (A) The protein expression of Act1, Hsp90, and TRAF6 in the lung tissues were measured by western blot analysis. (B) Levels of IL-17 in the BALF were quantitated using ELISA. All values represent mean ± S.D. of three or more replicates. ##p < 0.01 and ###p < 0.001 compared with normal group, respectively; *p < 0.05, **p < 0.01 and ***p < 0.001 compared with LPS treated alone group, respectively. | ||
Effects of AIF concerning the expression of anti-oxidant enzymes, TLR-4, iNOS, and COX-2 in LPS-stimulated ALI mice
Oxidative damage plays an important role in LPS-induced ALI in mice. Therefore, we investigated whether pretreatment with AIF enhanced the anti-oxidant enzyme levels in the ALI mouse model. In this experiment, AIF pretreatment not only significantly increased the protein expression levels of anti-oxidant enzymes but also obviously reduced TLR-4 protein expression, thereby reducing the production of the iNOS protein in the ALI mice (Fig. 8A–B). However, AIF pretreatment had no significant effect on the COX-2 protein (Fig. 8A).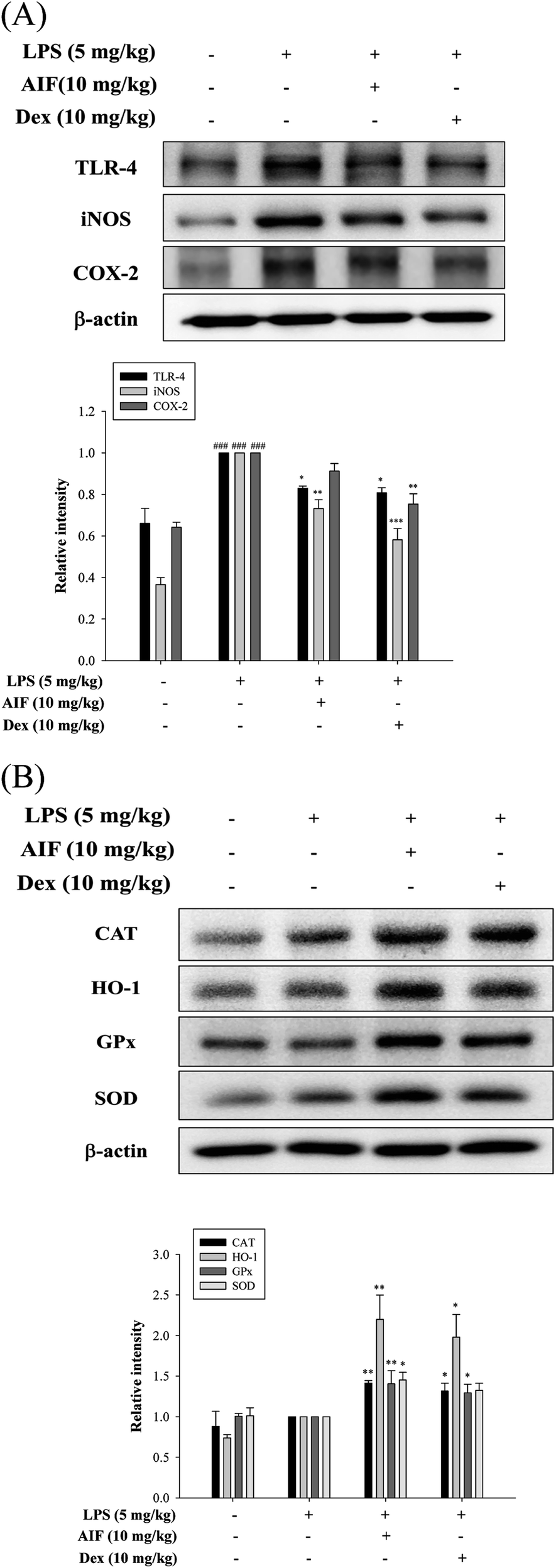 | ||
| Fig. 8 Effects of AIF regarding the expression of TLR-4, iNOS, COX-2, and anti-oxidant enzymes in ALI mice. (A) The protein expression of TLR-4, iNOS, COX-2, and (B) anti-oxidative enzymes (CAT, HO-1, GPx, and SOD) in the lung tissues were measured by western blot. All values represent mean ± S.D. of three or more replicates. ###p < 0.001 compared with normal group. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with LPS treated alone group, respectively. | ||
Discussion
It is well known that the lungs are susceptible to the air in the environment and are often exposed to the risk of direct or indirect injury due to various harmful elements, such as infectious microorganisms.34,35 ALI is a clinical disease that involves widespread inflammation in the lungs and is characterized by diffuse alveolar injury, the formation of lung edema, neutrophil infiltration, surfactant dysfunction, etc.14,15 ALI is commonly caused by Gram-negative bacteria that are present in serious lung infections.36 LPS is the major component of the cell walls of Gram-negative bacteria and is a potent stimulator of macrophages. TLR-4 recognizes and binds LPS, which initiates the activation of monocytes and macrophages, thereby resulting in the release of pro-inflammatory mediators.37 Various signal transduction pathways are triggered in LPS-stimulated macrophages and subsequently produce a variety of pro-inflammatory mediators to counteract the infectious agents.38 The mouse model of LPS-induced lung inflammation is a simple model that enables the study of ALI, which is characterized by alveolar hemorrhagic edema and increased levels of neutrophils, proteins and cytokines in the BALF.12,33 Long-term changes in pro-inflammatory proteins (such as iNOS and COX-2), pro-inflammatory cytokines (such as TNF-α, IL-6, IL-1β, and IL-17) and the adhesion molecule ICAM-1 are closely related to the development of inflammatory diseases. MPO is the most abundant peroxidase enzyme in neutrophils and has been found to be related to tissue damage in many diseases, especially those caused by inflammation.39 MPO activity is an important index of neutrophil infiltration into the lung tissues.36 Moreover, such inflammatory mediators are the hallmarks of LPS-induced ALI.18In the present study, we found that treatment with AIF not only effectively alleviated LPS-induced lung tissue lesions and pulmonary edema and reduced the protein content and MPO activity but also significantly reduced the secretion of TNF-α, IL-6, and IL-1β and suppressed the expression of the TLR-4, iNOS, and ICAM-1 proteins in both LPS-stimulated macrophages and ALI mice. However, AIF surprisingly had no effect on the expression of the COX-2 protein. To assess the anti-inflammatory mechanism of AIF in LPS-induced ALI, we investigated the effects of AIF on NF-κB, MAPKs, and NLRP3 inflammasome activation in lung tissues. The IKK complex contains two catalytic subunits, IKKα and IKKβ, and ample evidence indicates that IKKα/β regulates NF-κB, which plays a pivotal role in the inflammatory response.40 The translocation of NF-κB to the nucleus is required for the transcriptional activation of pro-inflammatory genes. The inhibition of the NF-κB and MAPK pathway could ameliorate ALI and reduce the expression levels of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β.18 Therefore, we investigated the effects of AIF on NF-κB nuclear translocation and MAPK activation. The results revealed that AIF significantly inhibited the phosphorylation of IKK, IκB-α and MAPKs and inhibited the translocation of NF-κB from the cytoplasm to the nucleus, thereby inhibiting the production of downstream inflammatory cytokines. The NLRP3 inflammasome is a multiprotein complex that is necessary for the initiation of acute inflammation.31 Previous research has indicated that NLRP3 expression is induced by TLR agonists in an NF-κB-dependent manner. Moreover increasing evidence proves that the inhibition of both the NLRP3 inflammasome and NF-κB expression can improve ALI.41 Therefore, we investigated the inhibitory effects of AIF on the NLRP3 inflammasome and caspase-1 in LPS-induced RAW264.7 cells and ALI mice. The results revealed that AIF dramatically prevented the LPS-induced activation of the NLRP3 inflammasome and caspase-1 and thereby decreased the expression of the IL-1β protein.
IL-17A, one of the pro-inflammatory cytokines that participate in the pathogenesis of various inflammatory processes, has recently received increasing attention. According to previous studies, many cells (including macrophages, neutrophils, and epithelial cells) increase the production of pro-inflammatory cytokines (such as IL-6, IL-8, GM-CSF, and G-CSF), chemokines, and metalloproteases via the activation of IL-17A, and this process is related to the development of various chronic inflammatory conditions, tumors, allergies, and autoimmune diseases.27,42 Some studies have reported that after induction, Act1 and Hsp90 can assemble together in a receptor-proximal platform to initiate an IL-17A-mediated cascade of TRAF6-dependent pathways and trigger the activation of NF-κB and MAPKs.43 Additionally, several studies have reported that IL-17A can increase the number of neutrophils in LPS-stimulated airways, and this process may be associated with a synergy among the TLR ligands, TLR-4, the NLRP3 inflammasome, IL-1β, and TNF-α, resulting in an enhanced inflammatory response.43,44 Therefore, we further explored whether AIF could inhibit the activation of the IL-17A pathway. The results showed that IL-17A and the expression of its related proteins (i.e., Hsp90, Actl, and TRAF6) were significantly increased in LPS-induced ALI. However, pretreatment with AIF markedly reduced the activation of IL-17A and down-regulated the expression levels of Hsp90, Act1 and TRAF6, which dramatically reduced lung inflammation. These findings indicate that AIF can improve LPS-induced ALI by inhibiting the IL-17A signaling pathway.
Free radicals cause excessive or sustained inflammatory reactions, which are associated with the development of many diseases, such as rheumatoid arthritis, asthma, atopic dermatitis, diabetes, atherosclerosis, cancer, and especially ALI.39 Previous studies have suggested that excessive oxidative stress and/or excessive inflammation play central roles in the pathogenesis of ALI.18,39 Oxidative stress interacts with NF-κB, MAPKs, and the NLRP3 inflammasome.18,40 To further explore the molecular mechanisms of the protective effect of AIF against LPS-induced ALI, we investigated the anti-inflammatory and anti-oxidation activity of AIF. The results revealed that while LPS obviously induced enhanced NF-κB and NLRP3 inflammasome activation by increasing the levels of the p-IKK, NLRP3, caspase-1, and IL-1β proteins and increasing ROS production, these LPS-induced effects were blocked via treatment with AIF. In addition, we also found that treatment with AIF alone increased the levels of oxidative enzymes in unstimulated macrophages. Notably, when AIF was administered to LPS-induced macrophages, the expression levels of oxidative enzymes were significantly enhanced compared with those observed in normal macrophages, and similar results were found in LPS-stimulated ALI. These results provide strong support for the notion that AIF is crucial for the inhibition of oxidative stress and inflammatory damage in ALI, and these effects may be associated with the activation of anti-oxidative enzymes.
Collectively, as illustrated in Fig. 9, the present study demonstrated that AIF significantly ameliorated LPS-induced ALI by reducing oxidative stress and inflammatory damage; these effects were primarily dependent on increases in the expression levels of anti-oxidative enzymes and the suppression of the activations of the NF-κB, MAPK, NLRP3 inflammasome and IL-17 signaling pathways. In summary, this research provides evidence supporting the exploitation of the protective effects of AIF for the treatment inflammation- and oxidative stress-associated diseases, especially bacteria-induced ALI.
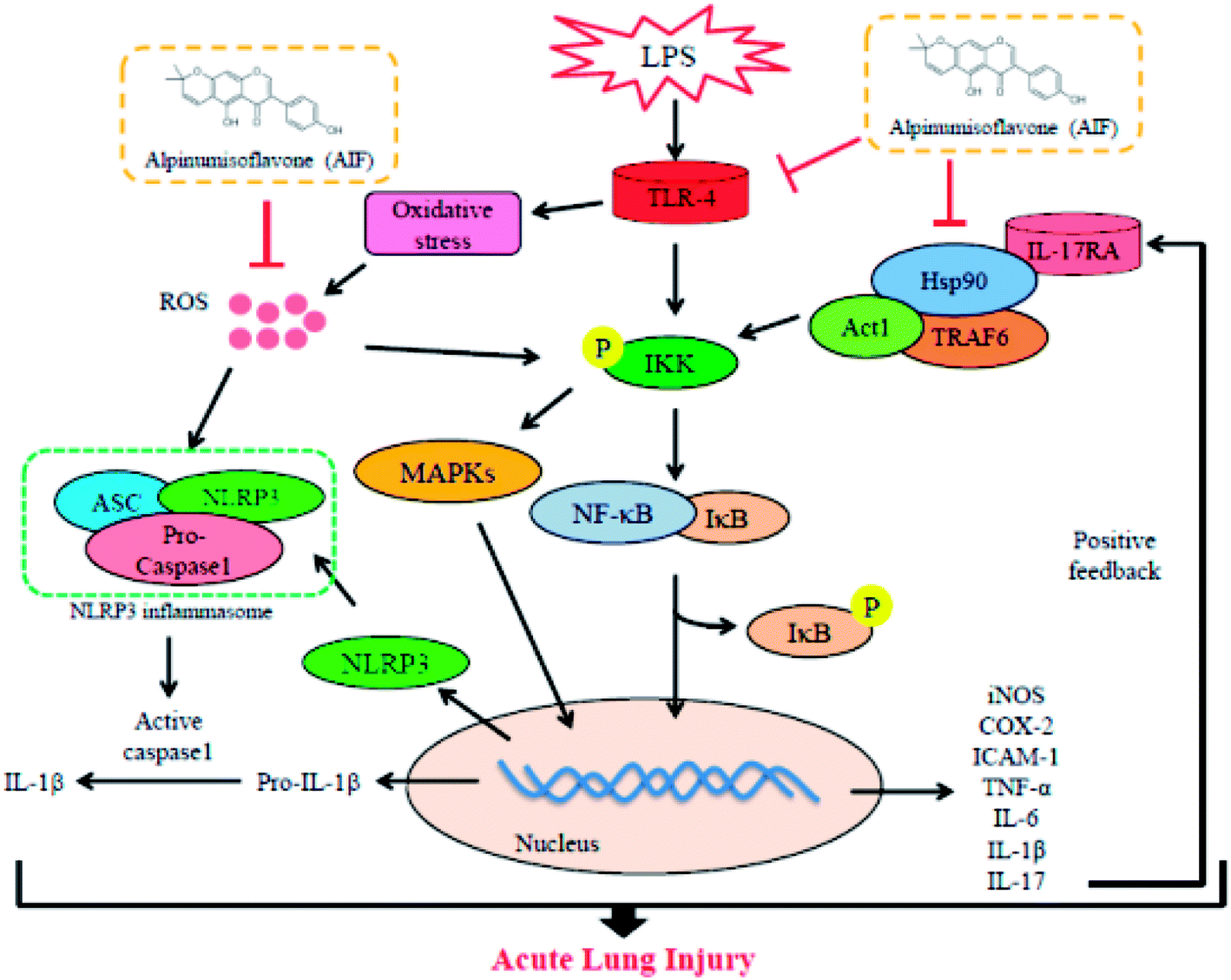 | ||
| Fig. 9 Scheme of the mechanisms in the protective effect of alpinumisoflavone on LPS-induced ALI. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank the financial supports from the National Science Council (MOST 105-2320-B-039-046- and MOST 106-2320-B-039-045-), China Medical University (CMU) and Asia University (ASIA104-CMUH-06 and CMU105-ASIA-23).References
- N. Baskar, B. P. Devi and R. M. Kumar, Anti-cancer activity of methanol extract of root bark of Erythrina variegata Linn, International Journal of Toxicological and Pharmacological Research, 2010, 2, 74–76 Search PubMed
.
- S. Namkoong, T. J. Kim, I. S. Jang, K. W. Kang, W. K. Oh and J. Park, Alpinumisoflavone induces apoptosis and suppresses extracellular signal-regulated kinases/mitogen activated protein kinase and nuclear factor-κB pathways in lung tumor cells, Biol. Pharm. Bull., 2011, 34, 203–208 CrossRef
- R. Kingsford-Adaboh, B. Dittrich, C. B. Hübschle, W. S. Gbewonyo, H. Okamoto, M. Kimura and H. Ishida, Invariom structure refinement, electrostatic potential and toxicity of 4-O-methylalpinumisoflavone, O,O-dimethylalpinumisoflavone and 5-O-methyl-4-O-(3-methylbut-2-en-1-yl)alpinumisoflavone, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 843–849 CrossRef PubMed
- X. H. Han, S. S. Hong, J. S. Hwang, S. H. Jeong, J. H. Hwang, M. H. Lee, M. K. Lee, D. Lee, J. S. Ro and B. Y. Hwang, Monoamine Oxidase Inhibitory Constituents from the Fruits of Cudrania tricuspidata, Arch. Pharmacal Res., 2005, 28, 1324–1327 CrossRef
- W. Cong, C. Zhou and J. Yin, Alpinumisoflavone inhibits osteoclast differentiation and exerts anti-osteoporotic effect in ovariectomized mice, Biomed. Pharmacother., 2017, 93, 344–351 CrossRef PubMed
- J. C. Chukwujekwu, F. R. Van Heerden and J. Van Staden, Antibacterial Activity of Flavonoids from the Stem Bark of Erythrina caffra Thunb, Phytother. Res., 2011, 25, 46–48 CrossRef PubMed
- T. Wang, Y. Jiang, L. Chu, T. Wu and J. You, Alpinumisoflavone suppresses tumour growth and metastasis of clear-cell renal cell carcinoma, Am. J. Cancer Res., 2017, 7, 999–1015 Search PubMed
- B. Zhang, X. Fan, Z. Wang, W. Zhu and J. Li, Alpinumisoflavone radiosensitizes esophageal squamous cell carcinoma through inducing apoptosis and cell cycle arrest, Biomed. Pharmacother., 2017, 95, 199–206 CrossRef PubMed
- Y. Wang, J. Liu, Q. Pang and D. Tao, Alpinumisoflavone protects against glucocorticoid-induced osteoporosis through suppressing the apoptosis of osteoblastic and osteocytic cells, Biomed. Pharmacother., 2017, 96, 993–999 CrossRef PubMed
- H. Bae, R. Kim, Y. Kim, E. Lee, H. Jin Kim, Y. Pyo Jang, S. K. Jung and J. Kim, Effects of Schisandra chinensis Baillon (Schizandraceae) on lipopolysaccharide induced lung inflammation in mice, J. Ethnopharmacol., 2012, 142, 41–47 CrossRef PubMed
- T. W. Kim, E. H. Joh, B. Kim and D. H. Kim, Ginsenoside Rg5 ameliorates lung inflammation in mice by inhibiting the binding of LPS to toll-like receptor-4 on macrophages, Int. Immunopharmacol., 2012, 12, 110–116 CrossRef PubMed
- P. Kang, K. Y. Kim, H. S. Lee, S. S. Min and G. H. Seol, Anti-inflammatory effects of anethole in lipopolysaccharide-induced acute lung injury in mice, Life Sci., 2013, 93, 955–961 CrossRef PubMed
- X. Song and Y. Qian, IL-17 family cytokines mediated signaling in the pathogenesis of inflammatory diseases, Cell. Signalling, 2013, 25, 2335–2347 CrossRef PubMed
- M. Bhargava and C. Wendt, Biomarkers in acute lung injury, Transl. Res., 2012, 159, 205–217 CrossRef PubMed
- D. Mokra and P. Kosutova, Biomarkers in acute lung injury, Respir. Physiol. Neurobiol., 2015, 209, 52–58 CrossRef PubMed
- S. Ferretti, O. Bonneau, G. R. Dubois, C. E. Jones and A. Trifilieff, IL-17, Produced by Lymphocytes and Neutrophils, Is Necessary for Lipopolysaccharide-Induced Airway Neutrophilia: IL-15 as a Possible Trigger, J. Immunol., 2003, 170, 2106–2112 CrossRef
- Z. Q. Su, Z. Z. Mo, J. B. Liao, X. X. Feng, Y. Z. Liang, X. Zhang, Y. H. Liu, X. Y. Chen, Z. W. Chen, Z. R. Su and X. P. Lai, Usnic acid protects LPS-induced acute lung injury in mice through attenuating inflammatory responses and oxidative stress, Int. Immunopharmacol., 2014, 22, 371–378 CrossRef PubMed
- C. H. Yeh, J. J. Yang, M. L. Yang, Y. C. Li and Y. H. Kuan, Rutin decreases lipopolysaccharide-induced acute lung injury via inhibition of oxidative stress and the MAPK-NF-κB pathway, Free Radical Biol. Med., 2014, 69, 249–257 CrossRef PubMed
- Y. Rosenfeld and Y. Shai, Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis, Biochim. Biophys. Acta, 2006, 1758, 1513–1522 CrossRef PubMed
- C. Li, D. Yang, X. Cao, F. Wang, H. Jiang, H. Guo, L. Du, Q. Guo and X. Yin, LFG-500, a newly synthesized flavonoid, attenuates lipopolysaccharide induced acute lung injury and inflammation in mice, Biochem. Pharmacol., 2016, 113, 57–69 CrossRef PubMed
- Y. Qiao, P. Wang, J. Qi, L. Zhang and C. Gao, TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages, FEBS Lett., 2012, 586, 1022–1026 CrossRef PubMed
- S. Y. Cheon, K. S. Chung, E. Jeon, A. Nugroho, H. J. Park and H. J. An, Anti-inflammatory Activity of Saxifragin via Inhibition of NF-κB Involves Caspase-1 Activation, J. Nat. Prod., 2015, 78, 1579–1585 CrossRef PubMed
- T. Zhang, L. Sun, R. Liu, D. Zhang, X. Lan, C. Huang, W. Xin, C. Wang, D. Zhang and G. Du, A Novel Naturally Occurring Salicylic Acid Analogue Acts as an Anti-Inflammatory Agent by Inhibiting Nuclear Factor-kappaB Activity in RAW264.7 Macrophages, Mol. Pharm., 2012, 9, 671–677 CrossRef PubMed
- A. Spooren, R. Kooijman, B. Lintermans, K. Van Craenenbroeck, L. Vermeulen, G. Haegeman and S. Gerlo, Cooperation of NF-κB and CREB to induce synergistic IL-6 expression in astrocytes, Cell. Signalling, 2010, 22, 871–881 CrossRef PubMed
- Y. He, H. Hara and G. Núñez, Mechanism and Regulation of NLRP3 Inflammasome Activation, Trends Biochem. Sci., 2016, 41, 1012–1021 CrossRef PubMed
- L. Jiang, L. Zhang, K. Kang, D. Fei, R. Gong, Y. Cao, S. Pan, M. Zhao and M. Zhao, Resveratrol ameliorates LPS-induced acute lung injury via NLRP3 inflammasome modulation, Biomed. Pharmacother., 2016, 84, 130–138 CrossRef PubMed
- Y. Iwakura, H. Ishigame, S. Saijo and S. Nakae, Functional specialization of interleukin-17 family members, Immunity, 2011, 34, 149–162 CrossRef PubMed
- N. Amatya, A. V. Garg and S. L. Gaffen, IL-17 Signaling: The Yin and the Yang, Trends Immunol., 2017, 38, 310–322 CrossRef PubMed
- A. G. Besnard, D. Togbe, I. Couillin, Z. Tan, S. G. Zheng, F. Erard, M. Le Bert, V. Quesniaux and B. Ryffel, Inflammasome–IL–1–Th17 response in allergic lung inflammation, J. Mol. Cell Biol., 2012, 4, 3–10 CrossRef PubMed
- D. De Nardo, C. M. De Nardo and E. Latz, New Insights into Mechanisms Controlling the NLRP3 Inflammasome and Its Role in Lung Disease, Am. J. Pathol., 2014, 184, 42–54 CrossRef PubMed
- S. Han, W. Cai, X. Yang, Y. Jia, Z. Zheng, H. Wang, J. Li, Y. Li, J. Gao, L. Fan and D. Hu, ROS-Mediated NLRP3 Inflammasome Activity Is Essential for Burn-Induced Acute Lung Injury, Mediators Inflammation, 2015, 2015, 720457 Search PubMed
- C. Wang, L. Wu, K. Bulek, B. N. Martin, J. A. Zepp, Z. Kang, C. Liu, T. Herjan, S. Misra, J. A. Carman, J. Gao, A. Dongre, S. Han, K. D. Bunting, J. S. Ko, H. Xiao, V. K. Kuchroo, W. Ouyang and X. Li, The psoriasis-associated D10N variant of the adaptor Act1 with impaired regulation by the molecular chaperone hsp90, Nat. Immunol., 2013, 14, 72–82 CrossRef PubMed
- Q. Jiang, M. Yi, Q. Guo, C. Wang, H. Wang, S. Meng, C. Liu, Y. Fu, H. Ji and T. Chen, Protective effects of polydatin on lipopolysaccharide-induced acute lung injury through TLR4-MyD88-NF-κB pathway, Int. Immunopharmacol., 2015, 29, 370–376 CrossRef PubMed
- M. A. Kovach and T. J. Standiford, Toll like receptors in diseases of the lung, Int. Immunopharmacol., 2011, 11, 1399–1406 CrossRef PubMed
- S. R. Kim, H. J. Kim, D. I. Kim, K. B. Lee, H. J. Park, J. S. Jeong, S. H. Cho and Y. C. Lee, Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury, Theranostics, 2015, 5, 1343–1362 CrossRef PubMed
- G. Feng, B. Sun and T. Z. Li, Daidzein attenuates lipopolysaccharide-induced acute lung injury via toll-like receptor 4/NF-kappaB pathway, Int. Immunopharmacol., 2015, 26, 392–400 CrossRef PubMed
- K. Tsoyi, H. J. Kim, J. S. Shin, D. H. Kim, H. J. Cho, S. S. Lee, S. K. Ahn, H. S. Yun-Choi, J. H. Lee, H. G. Seo and K. C. Chang, HO-1 and JAK-2/STAT-1 signals are involved in preferential inhibition of iNOS over COX-2 gene expression by newly synthesized tetrahydroisoquinoline alkaloid, CKD712, in cells activated with lipopolysacchride, Cell. Signalling, 2008, 20, 1839–1847 CrossRef PubMed
- S. C. Su, K. F. Hua, H. Lee, L. K. Chao, S. K. Tan, H. Lee, S. F. Yang and H. Y. Hsu, LTA and LPS mediated activation of protein kinases in the regulation of inflammatory cytokines expression in macrophages, Clin. Chim. Acta, 2006, 374, 106–115 CrossRef PubMed
- H. Lv, Q. Liu, Z. Wen, H. Feng, X. Deng and X. Ci, Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3-Nrf2 signal axis, Redox Biol., 2017, 12, 311–324 CrossRef PubMed
- T. Lawrence, M. Bebien, G. Y. Liu, V. Nizet and M. Karin, IKKa limits macrophage NF-κB activation and contributes to the resolution of inflammation, Nature, 2005, 434, 1138–1143 CrossRef PubMed
- S. Yang, Z. Yu, T. Yuan, L. Wang, X. Wang, H. Yang, L. Sun, Y. Wang and G. Du, Therapeutic effect of methyl salicylate 2-O-β-D-lactoside on LPS-induced acute lung injury by inhibiting TAK1/NF-kappaB phosphorylation and NLRP3 expression, Int. Immunopharmacol., 2016, 40, 219–228 CrossRef PubMed
- C. Song, L. Luo, Z. Lei, B. Li, Z. Liang, G. Liu, D. Li, G. Zhang, B. Huang and Z. H. Feng, IL-17-Producing Alveolar Macrophages Mediate Allergic Lung Inflammation Related to Asthma, J. Immunol., 2008, 181, 6117–6124 CrossRef
- C. Gu, L. Wu and X. Li, IL-17 family: Cytokines, receptors and signaling, Cytokine, 2013, 64, 477–485 CrossRef PubMed
- C. Feriotti, E. F. de Araújo, F. V. Loures, T. A. da Costa, N. A. L. Galdino, D. S. Zamboni and V. L. G. Calich, NOD-Like Receptor P3 Inflammasome Controls Protective Th1/Th17 Immunity against Pulmonary Paracoccidioidomycosis, Front. Immunol., 2017, 8, 786 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04098b |
| ‡ Equal contributors. |
| This journal is © The Royal Society of Chemistry 2018 |