
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDietary polyphenol canolol from rapeseed oil attenuates oxidative stress-induced cell damage through the modulation of the p38 signaling pathway†
Xiaoyang Xiaa,
Xia Xiang *a,
Fenghong Huang*a,
Mingming Zhenga,
Renhuai Congb,
Ling Hana and
Zhen Zhanga
*a,
Fenghong Huang*a,
Mingming Zhenga,
Renhuai Congb,
Ling Hana and
Zhen Zhanga
aOil Crops Research Institute of the Chinese Academy of Agricultural Sciences, Oil Crops and Lipids Process Technology National & Local Joint Engineering Laboratory, Key Laboratory of Oilseeds Processing, Hubei, Key Laboratory of Lipid Chemistry and Nutrition, Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture, Wuhan 430062, China. E-mail: xiangxia0130@163.com; jiagongzx@oilcrops.cn; Tel: +86-27-86711526
bInfinitus (China) Company. Ltd, China
First published on 5th July 2018
Abstract
Canolol (CAO) is a main phenolic compound with remarkable antioxidative properties that is generated in rapeseed oil during microwave pressing. The objective of this study was to identify the protective effect of CAO in hydrogen peroxide (H2O2)-triggered oxidative stress and reveal the role of the p38 MAPK pathway during the protective process. CAO treatment showed an observable cytoprotective effect. Results showed that CAO significantly improved H2O2-stimulated cell death, and diminished ROS production and malondialdehyde (MDA) level. Moreover, CAO increased glutathione (GSH) content and promoted the activities of superoxide dismutase (SOD) and catalase (CAT). As a result, apoptosis was ameliorated and depletion of the mitochondrial membrane potential was restored. Western blotting analysis demonstrated CAO downregulated the expression of caspase-3 and decreased the ratio of Bax/Bcl-2. Notably, the phosphorylation of p38 MAPK was inhibited by CAO in H2O2-induced apoptosis, which was confirmed by its inhibitor (SB203580). Taken together, our study demonstrated the pivotal role of the p38 MAPK pathway in the cytoprotective effect of CAO on oxidative stress-induced cell damage, suggesting CAO is a promising antioxidant in food and health-related fields.
1 Introduction
Oxidative stress, as mediated by reactive oxygen species (ROS), is a major factor in cell damage and chronic diseases, such as diabetes,1 cardiovascular2,3 and liver diseases.4 Oxidative stress is also known to activate mitogen-activated protein kinases (MAPKs), including stress-activated p38 mitogen-activated protein kinase (p38 MAPK), extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK). The MAPK pathway plays important roles in cell proliferation and apoptosis induced by various stimuli. Recently, it has been reported that redundant ROS caused by oxidative stress can be effectively eliminated or neutralized by exogenous antioxidants, such as resveratrol, troxerutin, D-limonene, and so on.5,6 The findings suggest that natural antioxidants with effective inhibition effects on oxidative damage have huge prospects in market.Dietary polyphenols are one of the most important antioxidants and receive great attention due to the safety and health benefits, such as anti-oxidant, anti-cancer, and anti-inflammatory, etc. Canolol (CAO), 4-vinyl-2,6-dimethoxyphenol, is an important phenolic compound in rapeseed oil that is produced during microwave pressing. CAO provides a great contribution to oxidation stability of rapeseed oil.7–10 CAO has been previously proven that possess the highest ROO− scavenging activity compared to various known antioxidants including α-tocopherol,10,11 vitamin C,12,13 β-carotene,14 and rutin.15 CAO can suppress the induction of iNOS and various inflammatory cytokines such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and cyclooxygenase-2 (COX-2) in H. pylori-infected Mongolian gerbils.16 These findings suggest CAO has the potential as an anti-oxidant, anti-cancer, and anti-inflammatory. Recently, oxidative stress-induced cell damage has been demonstrated to be prevented by CAO, and the protective mechanism was related to the ERK MAPK mediated pathway.17 However, whether p38 MAPK pathway is related to protective effect of CAO involving oxidative stress-induced cell damage remains unknown.
In the present study, the protective effect of CAO on oxidative stress-induced cell damage was explored by examining the effects of CAO on hydrogen peroxide (H2O2)-triggered loss of cell viability in HepG2 cells, depletion of mitochondrial membrane potential, redox status disturbance and the expression change of apoptotic proteins. More importantly, this study revealed the role of p38 MAPK pathway in the cytoprotective effect of CAO on oxidative damage. All of these may provide an interesting view of the potential application of CAO in food and health-related fields.
2 Materials and methods
2.1 Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco (Life Technologies, Grand Island, NY). Dimethylsulfoxide (DMSO), H2O2 (30%), Trypsin, 2′,7′-dichlorofluorescin diacetate (DCFH-DA), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), propidium iodide (PI) and p38 inhibitor (SB203580) were purchased from Sigma-Aldrich Sigma (St. Louis, MO, USA). 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetra-ethylbenzimidazolcarbocyanine iodide (JC-1) assay kit, lactate dehydrogenase (LDH), malondialdehyde (MDA), reduced glutathione (GSH), superoxide dismutase (SOD) and catalase (CAT) assay kits, Annexin V-FITC/PI kit, protein extraction and quantitation kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Primary antibodies against caspase-3, Bax, Bcl-2, p38 and P-p38 were purchased from Cell Signaling Technology. Antibodies against GAPDH and β-actin were purchased from Abcam Co. Horseradish peroxidase-conjugated goat anti-rabbit/mouse IgG secondary antibody was from Invitrogen Co. Ltd. All chemical reagents were at least of analytical grade.2.2 Preparation and structure determination of CAO
CAO was obtained by preparative chromatography according to our previous report.18 0.5 g rapeseed was prepared for microwave at 800 W and 7 min. The products were crushed and kept in centrifuge tube. Then 500 mL of 70% methanol aqueous solution was added and mixed with vortex finder for 20 min. Finally, the sample was centrifuged at 5000 rpm for 10 min and the supernate was transferred into another centrifuge tube. The above process was repeated three times. The mixture of three extraction solution was concentrated into 20 mL by reduced pressure distillation. The obtained solution was filtrated with 0.22 μm and purified by preparative chromatography. The collected elution solution was frozen and dried to obtain white powder of CAO. The stock solution of CAO in DMSO was kept at −80 °C for use. The structure of CAO was characterized by proton (1H) and carbon (13C) NMR spectra and UPLC chromatogram and the results were shown in Fig. 1A, Fig. S1 and Table S1 (ESI†).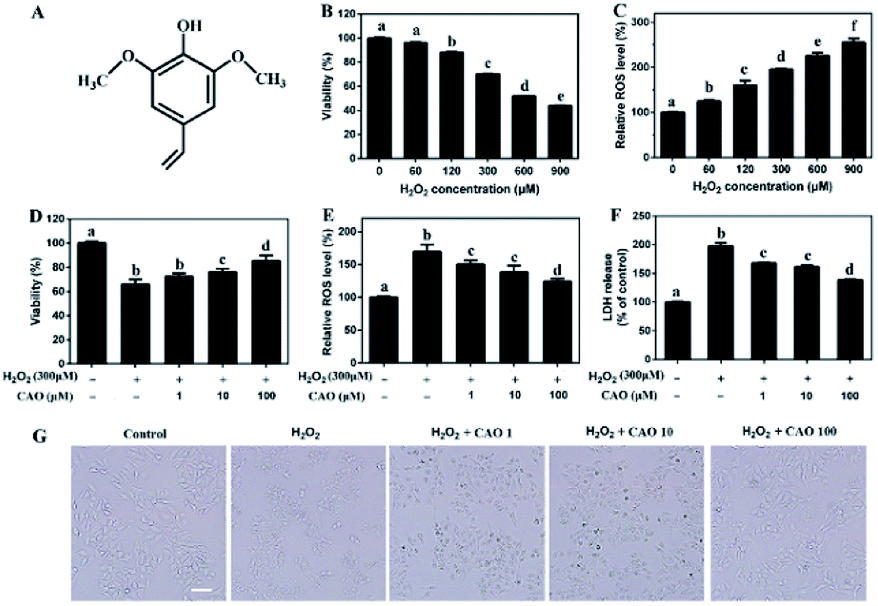 | ||
| Fig. 1 CAO attenuated H2O2-induced loss of cell viability. (A) The structure of canolol. (B) Measurement of HepG2 cell viability incubated with different concentrations of H2O2 (60–900 μM) through MTT assay. (C) Measurement of ROS level induced by H2O2 (60–900 μM) injury through DCFH-DA detection. (D–F) Measurement of cell viability, ROS level and LDH release pretreated with CAO. (G) Images of HepG2 cell morphology (scale bars: 50 μm). Cells were pre-treated with CAO (1, 10, and 100 μM) for 24 h and then incubated with or without 300 μM H2O2 for further 4 h. Data were expressed as mean ± SD, n = 6. One-way ANOVA was performed to test for statistical significance, p < 0.05. | ||
2.3 Cell culture
HepG2 cells was obtained from the American Type Culture Collection (ATCC, Mantissa, VA), and was cultured in DMEM/medium, supplemented with 10% heat-inactivated fetal bovine serum (FBS). The cells were cultured at 5% CO2 and 37 °C. They were seeded in a 25 cm2 culture flask (Falcon, Becton Dickinson) and were counted with a Castell counter (Scharfe System, Reutlingen, Germany) before seeding.2.4 Oxidative stress-induced cell damage in HepG2 cells
HepG2 cells were seeded in culture plates and incubated for 24 h before the experiment. HepG2 cells incubated in medium at 37 °C (control). Subsequently, the HepG2 cells were treated with H2O2 (60, 120, 300 or 900 μM) for another 4 h. The supernate was removed and then washed twice with PBS solution. Subsequently, 100 μL culture medium including 10 μL MTT solutions (5 mg mL−1 in 0.1 M PBS solution) were added to each well for further 4 h incubation. The medium was then replaced with 100 μL DMSO to dissolve formazan crystal. Finally, the absorbance at 490 nm was read by a microplate reader. The relative cell viability was calculated by comparing the absorbance in control wells with culture medium only. The data were presented as the average standard deviation (n = 6).2.5 Experimental groups
HepG2 cells were randomly divided into three experimental groups: (i) control group: HepG2 cells were cultured in medium. (ii) H2O2 group: HepG2 cells were incubated with H2O2 (300 μM) for 4 h. (iii) canolol group (H2O2 + CAO): canolol (1 μM, 10 μM, 50 μM or 100 μM) was administered 24 h prior to H2O2 (300 μM) and run through the following experiment.2.6 Protective effect evaluation of CAO
The protective effect of CAO on H2O2-induced cell death were evaluated by detecting the cell viability using an MTT assay as described previously.19,20 The HepG2 cells were pretreated with different concentrations of canolol at 37 °C for 24 h incubation and followed by 300 μM H2O2 for 4 h treatment. Then the OD values were determined on a microplate reader at 490 nm. Furthermore, multiple viewing fields were photographed under a fluorescence microscope (Olympus, Osaka, Japan).2.7 Measurement of intracellular ROS
2′,7′-dichlorofluorescin diacetate (DCFH-DA) was used as a fluorescence probe to determine the content of intracellular ROS in HepG2 cells. DCFH-DA was added to the cells and further oxidized to fluorescent 2′,7′-dichlorofluorescein (DCF) by ROS. The levels of DCF fluorescence were used to quantitate the generation of ROS. In this work, the HepG2 cells were stained with DCFH-DA (10 μM), which was used to detect the formation of ROS in the cells. After incubation at 37 °C for 30 min, the intoxicated cells were washed thrice with PBS buffer (0.01 M, pH 7.4). Then the photos of cells were taken by an inverted fluorescence microscope. The fluorescence intensity of stained cells were determined at the Ex/Em of 480 nm/525 nm in a microplate reader and detected in flow cytometry (BD LSR II, USA).2.8 Lactate dehydrogenase (LDH) assay
The viability of CAO on H2O2-induced cell death was confirmed by an LDH cytotoxicity assay kit following the manufacturer's instructions (Beyotime Institute of Biotechnology). In brief, the HepG2 cells were pretreated with different concentrations of canolol at 37 °C for 24 h incubation and followed by 300 μM H2O2 for 4 h treatment. Subsequently, the supernatants were collected after centrifugation, 120 μL of culture medium from each well was transferred to another 96-well plate, 60 μL of reaction solution was then added into each well and the plate was placed on a shaker for 30 min at room temperature. The OD values were determined on a microplate reader at 490 nm. The LDH leakage was calculated by comparing the absorbance in control wells with culture medium only. The data were presented as the average standard deviation (n = 6).2.9 Cell apoptosis assay
Annexin V-FITC/PI apoptosis detection kit was used to measure the apoptosis of HepG2 cells. Briefly, HepG2 cells were seeded in a 6-well culture plate and kept for overnight incubation at 37 °C in 5% CO2. Then, cells were treated with different canolol concentrations (CAO: 1 μM, 10 μM, 50 μM and 100 μM). At 24 h post-treatment, cells were followed by H2O2 for 4 h treatment, and then were harvested, washed with PBS, and resuspended in binding buffer in tubes with treatment by Annexin-FITC and PI and kept at 4 °C for 20 min under dark conditions. Stained cells were analyzed using Flow cytometry with Cell-Quest software.2.10 Measurement of mitochondrial membrane potential (Δψm)
The level of Δψm was measured according to the instructions of JC-1 mitochondrial membrane potential assay kit.22 The lipophilic cationic probe JC-1 existed as a green fluorescent monomer at low membrane potentials or as a red fluorescent J-aggregate at high membrane potentials in living cells. The ratio of red/green JC-1 fluorescence depended on the MMP of cells. Briefly, the treatment HepG2 cells were washed twice with PBS and then incubated with 500 μL of JC-1 working solution (20 μM) After 20 min of staining at 37 °C, the cells were washed twice, and the images of cells were observed under an inverted fluorescence microscope. For signal quantification, the intensity of red/green fluorescence was calculated by flow cytometry and the values were normalized to the control group.2.11 Measurement of CAT, SOD, MDA and GSH
CAT, SOD, MDA and GSH play an important role in cell signaling against oxidative stress.21 The treated cells were harvested and sonicated at 4 °C. The supernatants were collected for the analysis of CAT, SOD, MDA, and GSH level by commercial kits following the manufacturer's instructions.2.12 Western blotting
Protein extract was prepared from HepG2 cells and western blotting was performed as described.23 Briefly, cells were lysed with ice-cold RIPA lysis buffer and the protein concentration was detected with a BCA protein assay kit. Equal amounts of protein samples were electrophoresed on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then were transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). The membranes were blocked in 5% skimmed milk (Millipore, Billerica, MA, USA) for 1 h at room temperature, and probed with appropriate primary antibodies against caspase-3, Bax, Bcl-2, P-p38, and p38 at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000 dilutions overnight at 4 °C. The PVDF membranes were washed thrice with TBST (0.05% Tween-20 in 1× PBS solution) and then incubated with secondary antibodies at 1:3000 dilutions for 1.5 h at room temperature. Immunolabeled proteins were detected using the enhanced chemiluminescence (ECL) reagents (Millipore, Billerica, MA, USA) and the images were scanned by Molecular Imager® ChemiDoc™ XRS+ with Image LABTM Software.
:2000 dilutions overnight at 4 °C. The PVDF membranes were washed thrice with TBST (0.05% Tween-20 in 1× PBS solution) and then incubated with secondary antibodies at 1:3000 dilutions for 1.5 h at room temperature. Immunolabeled proteins were detected using the enhanced chemiluminescence (ECL) reagents (Millipore, Billerica, MA, USA) and the images were scanned by Molecular Imager® ChemiDoc™ XRS+ with Image LABTM Software.
2.13 Data analysis
Data were presented as mean ± standard deviation (SD) from at least triplicate analyses for each sample. The evaluation of statistical significance of observed differences was computed by one-way analysis of variance (One-way ANOVA),24 the differences between means were determined using Tukey's multiple comparison test, and the statistical significance was defined at p < 0.05 using SPSS software 18.0 (SPSS Inc., Chicago, IL, USA).3 Results
3.1 CAO attenuated H2O2-induced loss of cell viability
H2O2 has been extensively used for studying the protective effects of natural compounds and plant extracts against oxidative stress-induced cell injury.25 To construct oxidative stress model, the cytotoxicity of H2O2 on HepG2 cells was first tested by MTT assay. As shown in Fig. 1B and C, HepG2 cells underwent an obvious decrease in cell viability and a notable increase in ROS value after H2O2 insult in a dose-dependent manner. When the concentrations of H2O2 ranged from 60 μM to 900 μM with incubation, the cell viabilities decreased from 98.51 ± 0.077% to 43.85 ± 0.23% and ROS values increased from 125.08 ± 3.58% to 255.05 ± 8.84%, which suggested that exposure of HepG2 cells to H2O2 triggered the generation of ROS and cytotoxicity. Due to approximate 35% cell death, 300 μM H2O2 was selected in following experiments.The protective effect of CAO against H2O2-induced cell injury was evaluated. The HepG2 cells treated with 300 μM H2O2 caused significant loss of cell viability to 65.9%. Whereas CAO pretreatment enhanced the cell viability from 72.4% to 86.2% in the concentration range between 1 and 100 μM (Fig. 1D). ROS level detection (Fig. 1E) showed that CAO prevented the elevation of H2O2-induced ROS level in a dose-dependent manner. The LDH assay (Fig. 1F) exhibited that CAO markedly reduced H2O2-induced LDH leakage from 198% to 138% in the concentration range from 1 to 100 μM. Cellular imaging demonstrated that CAO definitely inhibited H2O2-induced morphological damage (Fig. 1G). Thus, H2O2-induced loss of cell viability can be reversed by CAO pretreatment.
3.2 CAO ameliorated H2O2-induced apoptosis and restored mitochondrial membrane potential in HepG2 cells
Apoptosis detection using a flow cytometer (Fig. 2) by Annexin V-FITC/PI double staining method displayed that H2O2 treatment caused significant increase in cell apoptosis compared to the control group. By contrast, CAO pretreatment diminished the apoptosis in a dose-dependent manner and reduced the percentage from 30.8% to 13.5%, which was nearly close to the control group. The results had same tendency with the MTT assay. Additionally, the mitochondrial membrane potential was found to be reduced by H2O2 both qualitatively and quantitatively by JC-1 staining (Fig. 3). However, the results were restored by CAO pretreatment. The decline of Δψm was remarkably alleviated from 45.5% to 77.5% by CAO. These results indicated that CAO not only inhibited and reversed apoptosis induced by H2O2, but also protected mitochondrial functions.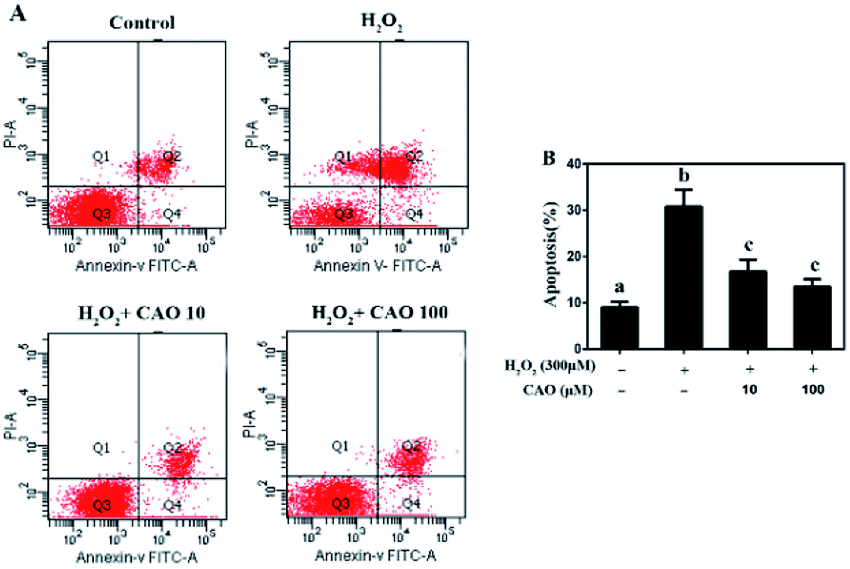 | ||
| Fig. 2 CAO suppressed H2O2-induced apoptosis. HepG2 cells were pre-incubated with CAO (10 and 100 μM) for 24 h, and then incubated with or without 300 μM H2O2 for another 4 h. (A) The apoptosis cells were detected by staining with annexin V-FITC positive and PI negative (AV+/PI−). (B) The number of apoptotic cells was counted using Flow cytometry. Data were expressed as mean ± SD, n = 6. One-way ANOVA was performed to test for statistical significance, p < 0.05. | ||
 | ||
| Fig. 3 CAO attenuated the depletion of H2O2-induced mitochondrial membrane potential (Δψm). After pre-treatment with CAO (10 and 100 μM) for 24 h, HepG2 cells were then incubated with or without 300 μM H2O2 for another 4 h. The Δψm levels in the mitochondrial membrane were detected by the JC-1 staining. (A) Images of the HepG2 cells stained with JC-1(scale bars: 20 μm). (B) The ratio of red/green fluorescent positive cells using Flow cytometry. Data were expressed as mean ± SD, n = 6. One-way ANOVA was performed to test for statistical significance, p < 0.05. | ||
3.3 CAO attenuated H2O2-induced cellular redox status imbalance
Considering that the loss of mitochondrial membrane potential is an important indicator of cell apoptosis, we further evaluated the cellular redox status, including the GSH and MDA content and activities of intracellular enzymes (CAT and SOD), and cellular ROS level. After HepG2 cells subjected to H2O2 injury, significant reduction in CAT activity, SOD activity and GSH content were observed (Fig. 4A, C and D) in comparison with H2O2 control group. By contrast, a marked increase in MDA content (Fig. 4B) was obtained. As expected, CAO pretreatment caused significant increase in CAT activity, SOD activity and GSH content, and decrease in MDA level. Cellular oxidative stress was determined by the specific ROS fluorescence probe indicated that H2O2-induced ROS could be largely eliminated by CAO pretreatment in a concentration-dependent manner (Fig. 4E). The flow cytometry results quantitatively confirmed that CAO significantly reduced intracellular ROS production from 139% to 107% (Fig. 4F). These results implied that the protection of CAO may maintain the cellular redox balance against H2O2-induced oxidative stress in HepG2 cells. | ||
| Fig. 4 CAO attenuated H2O2-induced cellular redox status imbalance. After pre-treatment with CAO (10 and 100 μM) for 24 h, HepG2 cells were then incubated with or without 300 μM H2O2 for another 4 h. (A) CAT activity in cells. (B) MDA level in cells. (C) SOD activity in cells. (D) GSH level in cells. (E) Images of ROS generation by 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) staining (scale bars: 50 μm). (F) Measurement of intracellular ROS level using flow cytometry. Data were expressed as mean ± SD, n = 6. One-way ANOVA was performed to test for statistical significance, p < 0.05. | ||
3.4 CAO inhibited the expression of caspase-3 and regulated the expression of Bcl-2 family proteins in HepG2 cells
The possible mechanism that CAO prevented H2O2-induced apoptosis was investigated. Western blotting assay showed (Fig. 5) that the protein expression levels of proapoptotic Bax and capsase-3 were upregulated in H2O2-treated group compared to the control untreated group, whereas the protein expression level of Bcl-2 was downregulated. CAO pretreatment prevented the H2O2-induced upregulation of Bax and capsase-3, as well as downregulation of Bcl-2. The Bax/Bcl-2 ratio also confirmed the above results. These findings indicated that CAO inhibited HepG2 apoptosis through a mechanism that utilized the pathways involving Bcl-2, Bax, and caspase-3. | ||
| Fig. 5 CAO inhibited H2O2-induced caspase-3 and modulated the express of Bcl-2 family proteins in HepG2 cells. After pre-treatment with CAO (10 and 100 μM) for 24 h, HepG2 cells were then incubated with or without 300 μM H2O2 for another 4 h. (A) The protein expression levels of caspase-3, Bax, and Bcl-2 were analyzed by western blot analysis. (B) Quantitative analysis of caspase-3 was performed by measuring intensity relative to the untreated control. (C) The ratio of Bax/Bcl-2. Each value represents the mean ± SD for three independent experiments. One-way ANOVA was performed to test for statistical significance, p < 0.05. | ||
3.5 CAO inhibited the phosphorylation of p38 in HepG2 cells in response to H2O2
It is widely acknowledged that p38 pathway plays a crucial biochemical role in cell survival and apoptosis.26 Therefore, it was explored whether p38 pathway was involved in the protective effect of CAO on H2O2-induced cell damage. As shown in Fig. 6, although there was no change in the total p38 protein level, CAO-pretreated cells significantly inhibited p38 phosphorylation induced by H2O2. Furthermore, pretreatment for 2 h with 10 μM SB203580 (a specific inhibitor of p38 MAP kinase), obviously blocked the p38 phosphorylation induced by H2O2. These results indicated that the pretreatment with CAO and SB203580 inhibited H2O2-induced p38 MAPK activity. The p38 MAPK pathway mediated and played a key role in the cytoprotective effect of CAO on H2O2-induced oxidative stress damage. | ||
| Fig. 6 Effect of CAO on the expression of P-p38 in HepG2 cells in response to H2O2. The cells were incubated with 100 μM CAO for 24 h with or without pretreatment with 10 μM SB203580 for 2 h before 300 μM H2O2 incubation for 4 h. (A) Effects of p38 inhibitor SB203580 on the cell viability. (B) Measurement of protein phosphorylation by western blot analysis. (C) Quantitative analysis was performed by measuring intensity relative to the untreated control. Each value represents the mean ± SD for three independent experiments. One-way ANOVA was performed to test for statistical significance, p < 0.05. | ||
4 Discussion
Oxidative stress is widely recognized to play an important role in the development of diseases. Oxidative stress induced by H2O2 is believed to be a key cause of cell dysfunction.27,28 H2O2 contains active oxygen, can permeate cellular membrane and enter the cell to cause additional membrane damage.29,30 Currently, natural antioxidants from plants have attracted more attention due to the huge benefit nutrition and safety. Many reports indicate that CAO possess powerful antioxidate, anti-inflammatory, and antitumorigenic properties.30–32 Cell model studies explore that CAO can be quickly uptaken and provide protective effects for cell damage.17,30 Although the protective effect of CAO have been reported, very little is known about its antioxidant activity and underlying mechanism in relation to H2O2-induced apoptosis.Our study suggests that CAO extracted from rapeseed oil attenuated oxidative stress-induced apoptosis and cellular redox status imbalance by inhibition of p38 phosphorylation (Fig. 7). In our present experiments, we found that H2O2 caused cytotoxicity to HepG2 cells and showed a concentration-dependent manner (Fig. 1B), which was in line with other reports. The cell morphology was shown in shrinkage and turned small with H2O2 exposure (Fig. 1G). In addition, annexin V-FITC/PI staining assay showed that H2O2 also induced the apoptosis of HepG2 cells (Fig. 2). The CAT activity, SOD activity and GSH content were reduced (Fig. 4A–C), and MDA content were significantly increased (Fig. 4D). When cells pretreated with CAO, the cell viability of HepG2 cells incubated with H2O2 was markedly increased in the concentration range from 1 to 100 μM (Fig. 1D). The morphological damage induced by H2O2 was restored by CAO (Fig. 1G). CAO also attenuated the H2O2-induced apoptosis in HepG2 cells. The reduction in CAT activity, SOD activity and reduced GSH content with H2O2 exposure was prevented, and the increased in MDA content was also reversed. These imply that CAO confers protective effect in HepG2 cells with H2O2 insult and maintains the cellular redox balance.
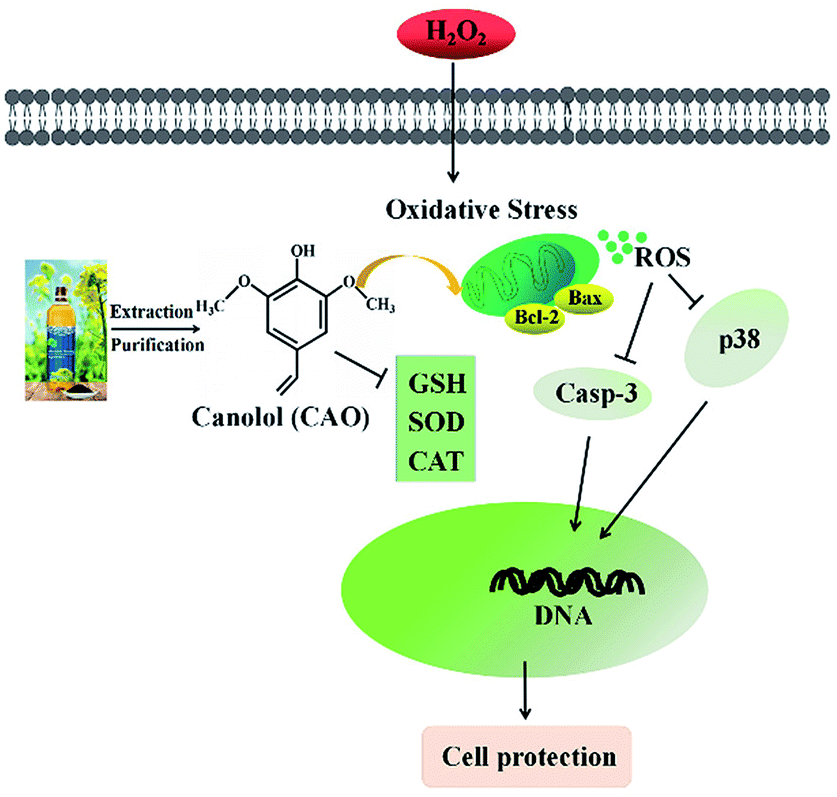 | ||
| Fig. 7 Simplified scheme of the proposed mechanism of CAO underlying H2O2 toxicity in HepG2 cells. | ||
It's widely accepted that ROS generation as an indicator of oxidative stress is associated with a large array of chronic diseases.10,31 Our previous study showed that H2O2 increased the oxidative stress injury in AGS cells through elevating ROS production and CAO significantly attenuated H2O2-induced ROS accumulation.30 Here, we found that H2O2 resulted in the collapse of the Δψm and increase of ROS in HepG2 cells, while pretreatment with CAO significantly suppress these abnormal changes in HepG2 cells with concentration-dependent manner (Fig. 3 and 4E and F). The decrease of cell viability and the increase of LDH release caused by H2O2 were significantly suppressed by CAO (Fig. 1D and F), suggesting that CAO offers distinct protection against oxidative stress.
The loss of mitochondrial membrane potential is a key factor to evaluate the mitochondrial function in cells and plays a crucial role in apoptosis.32 It has been reported that strategies to block mitochondrial apoptotic pathway are potential therapeutics to prevent cells death.33,34 In this study, our results showed the increase of intracellular ROS induced by H2O2 led to the damage of mitochondrial membrane, and resulted in the collapse of the Δψm and activation of the apoptosis (Fig. 2, 3 and 4E). However, the H2O2-induced loss of Δψm was suppressed by pretreatment with CAO (Fig. 3). It has been reported that suppressing ROS production prevents apoptosis.35 Consistent with the results, we also found in our study that H2O2-induced apoptosis was accompanied by an increase in the expression level of caspase-3. However, pre-treatment with CAO suppressed the activation of caspase 3 (Fig. 5).
Bcl-2 family members including antiapoptotic (Bcl-2, Bcl-XL, etc.) and proapoptotic (Bax, Bak, etc.) play an important role in regulating apoptosis.36–38 The ratio of Bax to Bcl-2 proteins is vital for confirming the occurrence of apoptosis. An increase in this ratio promotes the release of cytochrome from the mitochondria into the cytosol, leading to the subsequent activation of caspase-3.39 In this study, it was found that the ratio of Bax to Bcl-2 was obviously higher in cells treated with H2O2, which was inhibited by pretreating cells with CAO (Fig. 6). In a word, these results indicate that Bcl-2 family proteins may play an important role in apoptosis in HepG2 cells induced by H2O2 and CAO protects cells from the H2O2-induced apoptosis by regulating the expression levels of Bax and Bcl-2 proteins.
Oxidative stress can modulate phosphorylated mitogen-activated protein kinase (MAPK) levels, which have been shown to play roles in many diseases.40–42 It has been reported that CAO reduces oxidative stress-induced cell damage by activating the ERK cascade as one of the major MAPK pathways.17 Multiple members of the kinase families in this pathway can be activated by protein phosphorylation and p38 MAPK plays key roles in cellular apoptosis and death.40 To detect the role of p38 pathway that is involved in the cytoprotective effect of CAO against oxidative stress, we first examined the expression of this major signalling protein in the MAPK pathway. Our findings showed that H2O2-induced apoptosis was mainly mediated through the activation of p38 MAPK and CAO inhibited the phosphorylation of p38 MAPK to prevent oxidative damage (Fig. 6). To further elucidate the effects of CAO on p38 MAPKs pathways, SB203580 (the specific inhibitor of p38 MAPKs) was used in this test. We found that pretreatment cells with SB203580 effectively abolished the p38 phosphorylation induced by H2O2. Therefore, these results provide mechanistic evidence that p38 MAPK pathway plays pivotal role in the cytoprotective effect of CAO on oxidative stress-induced cell injury.
5 Conclusions
In this study, our findings demonstrate that CAO is able to reduce H2O2-induced insult in HepG2 cells through the regulation of multiple mechanisms including (1) restoring the cell viability; (2) suppressing the LDH release and MDA content; (3) increasing the levels of CAT, SOD and GSH content; (4) restraining the production of intracellular ROS and suppressing apoptosis; (5) modulating Δψm and caspase 3 dependent pathway; (6) regulating the expression levels of Bax and Bcl-2; (7) inhibiting activation of p38 signaling. These findings provide new insights into the protective effect of CAO in HepG2 cells and reveal the pivotal role of p38 MAPK pathway in cytoprotective effect of CAO. As a main phenolic compound in rapeseed oil, our results offer a support for the potential development of CAO as novel antioxidant application to food-related fields although the effects of CAO in vivo required further investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (31601438, 31471620, 31671820), the Agricultural Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2013-OCRI), the Earmarked Fund for China Agriculture Research System (CARS-13), National Key R&D Program of China (2016YFD0401401), the Director Fund of Oil Crops Research Institute (1610172016003) and the Major Program of Technology Innovation Program of Hubei Province (2017ABA144).References
- J. Bertoncelj, U. Dobersek, M. Jamnik and T. Golob, Food Chem., 2007, 105, 822–828 CrossRef.
- S. Liu and H. Huang, J. Funct. Foods, 2015, 18, 1095–1105 CrossRef.
- H. Wan, D. Liu, X. Yu, H. Sun and Y. Li, Food Chem., 2015, 175, 601–608 CrossRef PubMed.
- K. L. Wolfe and H. L. Rui, J. Agric. Food Chem., 2010, 58, 6621 CrossRef PubMed.
- R. Guo, W. Li, B. Liu, S. Li, B. Zhang and Y. Xu, Med. Sci. Monit. Basic Res., 2014, 20, 82–92 CrossRef PubMed.
- N. A. Panat, D. K. Maurya, S. S. Ghaskadbi and S. K. Sandur, Food Chem., 2016, 194, 32–45 CrossRef PubMed.
- A. Koski, S. Pekkarinen, A. Hopia, K. Wahala and M. Heinonen, Eur. Food Res. Technol., 2003, 217, 110–114 CrossRef.
- H. Kuwahara, A. Kanazawa, D. Wakamatu, S. Morimura, K. Kida, T. Akaike and H. Maeda, J. Agric. Food Chem., 2004, 52, 4380–4387 CrossRef PubMed.
- A. S. M. Satu Vuorela and M. Heinonen, J. Agric. Food Chem., 2004, 52, 8202–8207 CrossRef PubMed.
- D. Wakamatsu, S. Morimura, T. Sawa, K. Kida, C. Nakai and H. Maeda, Biosci., Biotechnol., Biochem., 2005, 69, 1568–1574 CrossRef PubMed.
- R. Guitard, J. F. Paul, V. Nardello-Rataj and J. M. Aubry, Food Chem., 2016, 213, 284 CrossRef PubMed.
- J. P. Spencer, M. M. Abdelmohsen and C. Riceevans, Arch. Biochem. Biophys., 2004, 423, 148–161 CrossRef PubMed.
- E. Passerieux, M. Hayot, A. Jaussent, G. Carnac, F. Gouzi, F. Pillard, M. C. Picot, K. Böcker, G. Hugon and J. Pincemail, Free Radicals Biol. Med., 2015, 81, 158 CrossRef PubMed.
- R. H. Liu, J. Nutr., 2004, 134, 3479–3485 CrossRef PubMed.
- S. Knasmüller, W. Parzefall, R. Sanyal, S. Ecker, C. Schwab, M. Uhl, V. Mersch-Sundermann, G. Williamson, G. Hietsch and T. Langer, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1998, 402, 185–202 CrossRef.
- X. Cao, T. Tsukamoto, T. Seki, H. Tanaka, S. Morimura, L. Cao, T. Mizoshita, H. Ban, T. Toyoda and H. Maeda, Int. J. Cancer, 2008, 122, 1445–1454 CrossRef PubMed.
- X. Dong, Z. Li, W. Wang, W. Zhang, S. Liu and X. Zhang, Mol. Vision, 2011, 17, 2040–2048 Search PubMed.
- M. Yang, C. Zheng, Q. Zhou, C. Liu, W. Li and F. Huang, J. Agric. Food Chem., 2014, 62, 1956–1963 CrossRef PubMed.
- D. Bedoya-Ramirez, A. Cilla, J. Contreras-Calderon and A. Alegria-Toran, Food Chem., 2017, 219, 364–372 CrossRef PubMed.
- H. Gwak, S. Kim, D. N. Dhanasekaran and Y. S. Song, Cancer Lett., 2015, 371, 347–353 CrossRef PubMed.
- J. Duan, G. Wei, C. Guo, J. Cui, J. Yan, Y. Yin, Y. Guan, Y. Weng, Y. Zhu and X. Wu, Am. J. Chin. Med., 2015, 43, 1159–1175 CrossRef PubMed.
- E. Thomas, V. Gopalakrishnan, R. R. Somasagara, B. Choudhary and S. C. Raghavan, Sci. Rep., 2016, 6, 23255 CrossRef PubMed.
- M. Srivastava, M. Hegde, K. K. Chiruvella, J. Koroth, S. Bhattacharya, B. Choudhary and S. C. Raghavan, Sci. Rep., 2014, 4, 6147 CrossRef PubMed.
- K. K. Adom and R. H. Liu, J. Agric. Food Chem., 2005, 53, 6572–6580 CrossRef PubMed.
- G. Cao, H. M. Alessio and R. G. Cutler, Free Radicals Biol. Med., 1993, 14, 303–311 CrossRef PubMed.
- G. Yang, S. Li, L. Yuan, Y. Yang and M. Pan, Food Funct., 2017, 8, 4668–4674 RSC.
- G. Qi, Y. Mi, Y. Wang, R. Li, S. Huang, X. Li and X. Liu, Food Funct., 2017, 8, 4421–4432 RSC.
- J. B. Park, Food Funct., 2013, 4, 1632–1638 RSC.
- Q. Hu, D. Wang, J. Yu, G. Ma, F. Pei and W. Yang, J. Funct. Foods, 2017, 37, 586–593 CrossRef.
- L. Han, X. Xia, X. Xiang, F. Huang and Z. Zhang, RSC Adv., 2017, 7, 42826–42832 RSC.
- H. C. Ting, Y. W. Hsu, C. F. Tsai, F. J. Lu, M. C. Chou and W. K. Chen, Food Chem., 2011, 125, 652–659 CrossRef.
- C. Li, D. Wang, W. Wu, W. Yang, S. S. Ali, Y. Zhao, Y. Duan, L. Wang, X. Zhou and D. Zhao, Aging Cell, 2018, 17, e12693 CrossRef PubMed.
- L. L. Wang, Q. L. Yu, L. Han, X. L. Ma, R. D. Song, S. N. Zhao and W. H. Zhang, Food Chem., 2018, 244, 394–402 CrossRef PubMed.
- H. L. Hung, S. Y. Cheng, Y. T. Cheung, S. Wuwongse, N. Q. Zhang, Y. S. Ho, M. Y. Lee and C. C. Chang, Redox Biol., 2017, 14, 7–19 CrossRef PubMed.
- B. Jie, Z. Yi, W. Gang and L. Ping, Oxid. Med. Cell. Longevity, 2015, 2016, 1–12 Search PubMed.
- M. G. Vander Heiden, N. S. Chandel, E. K. Williamson, P. T. Schumacker and C. B. Thompson, Cell, 1997, 91, 627–637 CrossRef PubMed.
- U. Anilkumar and J. H. M. Prehn, Front. Cell. Neurosci., 2014, 8, 281 Search PubMed.
- B. Antonsson, Mol. Cell. Biochem., 2004, 256–257, 141–155 CrossRef PubMed.
- N. A. Thornberry and Y. Lazebnik, Science, 1998, 281, 1312–1316 CrossRef PubMed.
- D. S. Kim, J. H. Kim, G. H. Lee, H. T. Kim, J. M. Lim, S. W. Chae, H. J. Chae and H. R. Kim, Biol. Pharm. Bull., 2010, 33, 545–549 CrossRef.
- X. Wang, B. Wang, Z. Li, G. Zhu, L. Heng, X. Zhu, Q. Yang, J. Ma and G. Gao, RSC Adv., 2016, 6, 49187–49197 RSC.
- Z. Xia, M. Dickens, J. Raingeaud, R. J. Davis and M. E. Greenberg, Science, 1995, 270, 1326 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Proton (1H) and carbon (13C) NMR spectra and UPLC chromatogram of canolol. See DOI: 10.1039/c8ra04130j |
| This journal is © The Royal Society of Chemistry 2018 |