
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrospun metal and metal alloy decorated TiO2 nanofiber photocatalysts for hydrogen generation†
Courtney Ligon a,
Kaniece Latimera,
Zachary D. Hoodbc,
Sanuja Pitigalaa,
Kyle D. Gilroyd and
Keerthi Senevirathne*a
a,
Kaniece Latimera,
Zachary D. Hoodbc,
Sanuja Pitigalaa,
Kyle D. Gilroyd and
Keerthi Senevirathne*a
aDepartment of Chemistry, Florida A&M University, Tallahassee, FL 32307, USA. E-mail: keerthi.senevirathne@famu.edu
bSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA
cCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
dWallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology, Emory University, Atlanta, GA 30332, USA
First published on 24th September 2018
Abstract
Photocatalytic nanofibers of TiO2 decorated with 2% metal (Pt, Pd, and Cu) and metal alloys (Pt2Pd and PtCu) were synthesized by the polymer-assisted electrospinning method, followed by microwave-assisted ethylene glycol reduction. Structurally, nanofibers calcined at 500 °C adopted an anatase phase along with a remnant rutile phase. Morphological, structural, and photocatalytic studies were carried out using scanning and transmission electron microscopy equipped with an energy dispersive spectroscopy attachment, X-ray powder diffraction, X-ray photoelectron spectroscopy, and photocatalytic hydrogen generation under UV-Vis irradiation. The calcined nanofibers were found to have a diameter of 60.0 ± 5.0 nm and length of up to several microns. High resolution TEM imaging suggests that the nanofibers are composed of agglomerated individual TiO2 nanoparticles, which are tightly packed and stacked along the axial direction of the nanofibers. PXRD studies suggest alloy formation, as evident from peak shifting towards higher two-theta values. Surface modification with co-catalysts is shown to contribute considerably to the rate of photocatalytic H2 generation. The amount of H2 generated gradually increases as a function of time. The 2%Pt2Pd/TiO2 catalyst shows the highest rate of H2 generation (4 mmol h−1 gramcatalyst), even higher than that of 2%Pt/TiO2 nanofiber photocatalyst (2.3 mmol h−1 gramcatalyst), while 2%Cu/TiO2 nanofiber photocatalyst shows the least activity among the decorated catalysts (0.04 mmol h−1 gramcatalyst).
Introduction
The current drive towards developing efficient photocatalysts demands the engineering of advanced functional materials. A wide range of inorganic (i.e., metal oxides, nitrides, sulfides, phosphides, and some nonmetal nitrides) catalysts have been explored as photocatalysts for energy conversion and environmental pollution remediation.1–7 The choice of photocatalytic material depends on many variables, including band gap (BG) energy, stability under irradiation, cost effectiveness, and ease of preparation. Additionally, lowering the recombination probability of photogenerated electron–hole carriers and making them readily available for water oxidation and reduction reactions are of high importance. Photocatalytic water splitting using semiconductors is initiated by the direct absorption of incident light, the energy of which is greater than the band gap energy of the photocatalyst. As a result of the photon absorption, electrons and holes (photocarriers) are created. The electrons (e−) excited from valence band (VB) to conduction band (CB) reduce H+ ions into H2 gas (HER, eqn (1)) while holes (h+) oxidize water into O2 (OER, eqn (1)). This process is governed by intrinsic factors of photocatalyst such as the position of energy bands, band gap, crystallinity of material, availability of co-catalysts etc. Most importantly, potentials of the bottom level of CB and the top level of VB are required to be more negative (relative to H+/H2 redox potential) and positive (relative to redox potential of O2/H2O), respectively. Ultimately, the redox potential of water (1.23 eV) needs to be within the band gap energy of the photocatalysts if it is to function as a water splitting catalyst.8| 2H+ + 2e− → H2 (HER) | (1) |
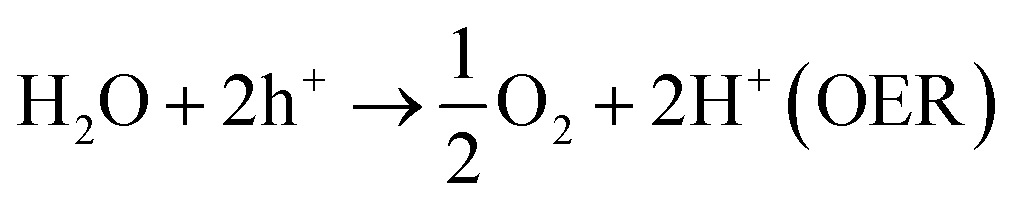 | (2) |
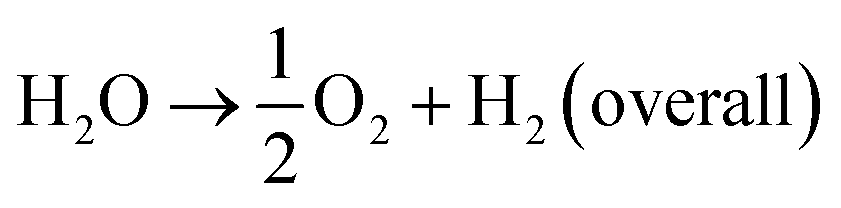 | (3) |
The band gap of the catalyst, i.e., its electronic structure, is strongly associated with the photon energy absorption and electron excitation to create photocarriers. Concurrently, water oxidation and H+ reduction are mostly dependent upon the co-catalysts or promoters loaded on the photocatalyst.3 It is quite challenging to develop low band gap semiconductors with sufficient stability and surface properties that lead to higher activity. Furthermore, preventing the recombination of photocarriers, which is equally responsible for low catalytic activity of photocatalysts, presents its own challenges as well.
Numerous synthesis and fabrication methods have been developed to synthesize one dimensional (1D) nanostructures9 because they are uniquely positioned to be used in various applications. This is due to their high aspect ratio, superior electron survivability,10 and well-defined unidirectional channel for electrical carrier transport.11 For these reasons, 1D nanostructures are presumed to have potential to be used in applications in a wide range of areas, including photocatalysis, solar cells, heterogeneous catalysis, and sensors. The use of nanowires or nanofibers, particularly in photocatalytic water splitting, is advantageous over bulk semiconductor materials due to their reduced radial dimension and increased surface to volume ratio.12 These properties facilitate active and rapid diffusion of photogenerated electron–hole charge carriers to the catalyst surface. Additionally, the nanofiber surface functions as an excellent substrate for secondary materials, in that it can be easily decorated with co-catalysts, which facilitate effective transfer of photogenerated carriers from the catalyst to redox reactions.13–15 It has also been reported that dimensionally unconstrained nanofibers are beneficial in vectorial transport of photogenerated charge carriers through grain boundaries, resulting in enhanced separation of electron–hole pairs compared to nanoparticles.13,16 This process is important for avoiding photogenerated electron–hole recombination.
A wide range of co-catalysts, including noble metals (Pt,17,18 Au,19 Rh,20), core–shell types (Ni@NiO,21,22 Rh@Cr2O3 (ref. 21)), metal oxides (NiO, RuO2 (ref. 3 and 23)), and even doped co-catalysts (Rh2−yCryO3 (ref. 24)) has been studied and has shown significant promise in enhancing photocatalytic activity towards overall water splitting and H2 generation. It has been reported that the physicochemical structure and dispersion of co-catalysts play a vital role in enhancing photocatalytic activity. For instance, (ZnxGa1−x)(OxN1−x) loaded with Rh@Cr2O3 core–shell co-catalyst has produced 6 times more H2 than the same core–shell structure in which the core was replaced with Pt, and 12 times more H2 than a Pd core.21 Even though metal alloys have not been frequently chosen as co-catalysts in photocatalysts, they have been studied as cathode catalysts in oxygen reduction reaction (ORR) in low temperature proton exchange membrane (PEM) fuel cells.25–29 In relation to this study, literature precedence is available in terms of the synthesis of PtxNiy,27,30,31 PtxCoy,26 PtxCuy,25 and PtxPdy32 nanoparticles. Pt3Pd alloy catalysts have shown enhanced catalytic activity and stability, compared to Pt only catalysts in PEM fuel cells, due to better electronic properties of Pd, the alloying effect, and particle size.32
In this study, we discuss the fabrication of TiO2 nanofibers by employing polymer-assisted electrospinning method and decoration of nanofiber surfaces with noble metal and metal alloy co-catalysts. A polymer-assisted electrospinning technique was employed to fabricate TiO2 nanofibers according to methods described elsewhere.33,34 A microwave-assisted polyol reduction method was used to deposit metal and metal alloy co-catalysts. The photocatalytic H2 production activities from methanol–water mixture by TiO2 nanofibers decorated with co-catalysts Pt, Cu, Pd, PtCu, and Pt2Pd have been studied. Numerous analytical and photocatalytic studies were performed to understand and compare photocatalytic behavior of TiO2 nanofiber photocatalysts. From this work, a detailed comparison of photocatalytic activities as a function of the nature of co-catalysts has been made. Herein, we report the physico-chemical characterization of TiO2 nanofiber photocatalysts and the effect of co-catalysts, especially metal alloy co-catalysts toward photocatalytic H2 generation.
Experimental section
Synthesis of TiO2 nanofiber photocatalyst
TiO2 nanofiber photocatalysts were fabricated by electrospinning a solution of titanium isopropoxide, which is a common sol–gel precursor used to synthesize TiO2, as previously reported with slight modifications.35 Briefly, 0.30 g of PVP polymer was dissolved in 7.0 mL of abs. ethanol by stirring. In a separate flask, the precursor solution was prepared by dispersing 4.0 mL of Ti(OiPr)4 (13.0 mmol) in a mixture of 7.5 mL absolute ethanol and 3.0 mL of glacial acetic acid. The spinning solution was prepared by slowly adding the viscous polymer solution into the precursor solution while magnetically stirring. The transparent electrospinning solution was quickly loaded into a 10.0 mL glass syringe, which is attached to a stainless-steel needle (18 gauge) through plastic tubing. The needle was connected to the anode of the DC high-voltage power supply (Gamma High Voltage Research, Ormond Beach, Florida) and a sheet of aluminum foil was used as the sample collector. The electrospinning process was carried out by applying 15 kV and a 1.0 mL h−1 flow rate while maintaining the distance between the tip of the needle and the aluminum foil to be 6 cm. As-prepared PVP/Ti(OiPr)4 composite nanofibers were left overnight for hydrolysis and followed by calcination at 500 °C for 3 hours in air.Metal nanoparticle deposition
TiO2 nanofiber photocatalyst was deposited with 2 wt% of pure metal (Cu, Pt, and Pd) or metal alloy (Pt2Pd and PtCu) co-catalysts by employing a microwave-assisted polyol reduction method. Briefly, 0.40 g of photocatalyst was dispersed in 20.0 mL of ethylene glycol by sonication in order to uniformly disperse the photocatalyst. Next, required amounts of metal precursors were dissolved in ∼2 mL of ethylene glycol and mixed with photocatalyst dispersion by stirring overnight in order to achieve homogeneous distribution of metal precursors. Due to the difficulty of dissolving Pd precursor in pure ethylene glycol, a mixture of water/ethylene glycol was used. The reduction of the metal precursors was carried out under microwave irradiation for 2 minutes with 10 s intervals. The co-catalyst loaded samples were recovered by centrifugation, and a series of washings with de-ionized water was performed afterwards to remove ethylene glycol. The samples were dried in an oven at 70 °C overnight.Physical characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v ratio) and stirred for ∼60 min while purging with argon prior to irradiation. To initiate the photocatalytic reaction, the obtained photocatalyst suspension was then irradiated with a 300 W Xenon lamp without the use of bandpass filters. At a given irradiation time interval, a 50 μL sample of gas was drawn from the headspace of the photocatalytic cell using a gas-tight syringe and analyzed with a Shimadzu GC 8A fitted with a 5 Å mol-sieve column and thermal conductivity detector (TCD).
:5 v/v ratio) and stirred for ∼60 min while purging with argon prior to irradiation. To initiate the photocatalytic reaction, the obtained photocatalyst suspension was then irradiated with a 300 W Xenon lamp without the use of bandpass filters. At a given irradiation time interval, a 50 μL sample of gas was drawn from the headspace of the photocatalytic cell using a gas-tight syringe and analyzed with a Shimadzu GC 8A fitted with a 5 Å mol-sieve column and thermal conductivity detector (TCD).Results and discussion
Herein, we report the synthesis, characterization, and application of TiO2 nanofiber photocatalysts, decorated with noble metal and metal alloy co-catalysts, prepared by electrospinning method combined with microwave-assisted polyol reduction. In electrospinning, when proper conditions are applied, the charged droplet stretches into a stable jet and then forms an elongated fiber, which deposits on the collector as a nanofiber mat. TiO2 nanofibers were synthesized by the spinning of titanium isopropoxide (Ti(OiPr)4) and poly(vinylpyrrolidone) (PVP) polymer mixture dispersed in absolute ethanol. The titanium alkoxide precursor undergoes hydrolysis when it is in contact with moisture and forms a gel. However, the hydrolysis process must be controlled and precipitation needs to be avoided during the solution preparation and spinning process in order to maintain a continuous flow of precursor solution. Addition of glacial acetic acid helps to hinder hydrolysis process and also helps to manipulate porosity of the nanofibers.36 The PVP polymer plays a role as a sacrificial template and also maintains the viscosity in the spinning solution.9 Proper regulation of viscoelastic behavior of the spinning solution is essential in order to produce well-formed nanofibers. It is reported that too low polymer concentrations lead to bead formation as well as non-uniform nanofibers.37 The low viscoelasticity of the spinning solution may cause disruptions in the spinning jet and rather act as electrospray than electrospinning. In contrast, too high polymer concentrations result in non-uniform, thicker fibers as a result of inadequate stretching and thinning process.37,38 Therefore, it is vital to adjust solution viscosity by adjusting the polymer content in order to fabricate uniform nanofibers. The distance from the tip of the spinneret to the substrate is an essential parameter as well and need to be properly adjusted. The distance and the fiber diameter have an inverse relationship (i.e. higher the distance smaller the fiber diameter) possibly because longer distance offers adequate time for thinning process.39 While the reaction mixture must be non-aqueous, the as-spun fibers still must undergo hydrolysis; therefore, environmental moisture was allowed to drive hydrolysis of as-spun fibers left under ambient air overnight before calcining.The conditions applied in the spinning process define the nature of nanofibers produced. It is important to optimize the synthetic parameters, which vary from material to material. The distance from spinneret to the substrate (current collector), applied voltage, and flow rate are some of the key factors that affect the nanofiber morphology and thickness. It is reported that applied voltage higher than that of optimal causes bead formation but does not readily reduce the fiber diameter.37 It is expected to obtain higher yields at a higher flow rate. However, it is evident that higher flow rates significantly increase the fiber diameter. In this study, nanofiber fabrication was carried out by applying 15 kV, 6 cm spinneret to substrate distance, and 1.0 mL h−1 flow rate.
Thermogravimetric analysis (TGA) of uncalcined nanofibers (Fig. 1a) shows first weight loss (≈20%) upon heating up to ∼200 °C in air, which can be attributed to the removal of surface-adsorbed moisture and solvents present due to incomplete drying. The second major weight loss (≈36%) between 300 °C and 500 °C is due to desorption and decomposition of poly(vinylpyrrolidone) polymer (PVP), which is an integral part of the spinning mixture. After 500 °C, the TGA curve levels off, indicating a formation of a thermally stable material and removal of all organic moieties from nanofiber catalyst. Therefore, all as-prepared nanofibers samples were calcined at 500 °C in air in order to remove adsorbed solvents and the PVP polymer and to form a stable crystalline catalyst. In comparison, the TGA weight loss curve recorded for pre-calcined nanofibers indicates little or no weight loss. The TGA data correlate well with FT-IR patterns acquired for uncalcined and calcined nanofibers (Fig. 1b). The differential thermal analysis (DTA) curve, shown in Fig. 1a, indicates two prominent transformations that correspond with TGA curve. The exothermic peak at 500 °C represents the formation of anatase TiO2 phase. The FT-IR spectrum of uncalcined as-prepared nanofibers shows several vibrational bands, which are originating from PVP polymer. The band at 1654 cm−1 can be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–C stretching frequencies while bands at 1531 and 1420 cm−1 are ascribed to CH deformation of cyclic CH2 groups. In addition, the peak that appears at 1289 cm−1corresponds to amide C–N vibration.40 On the other hand, the FT-IR spectrum of the calcined-nanofiber photocatalyst is featureless and agrees with TGA data.
O and N–C stretching frequencies while bands at 1531 and 1420 cm−1 are ascribed to CH deformation of cyclic CH2 groups. In addition, the peak that appears at 1289 cm−1corresponds to amide C–N vibration.40 On the other hand, the FT-IR spectrum of the calcined-nanofiber photocatalyst is featureless and agrees with TGA data.
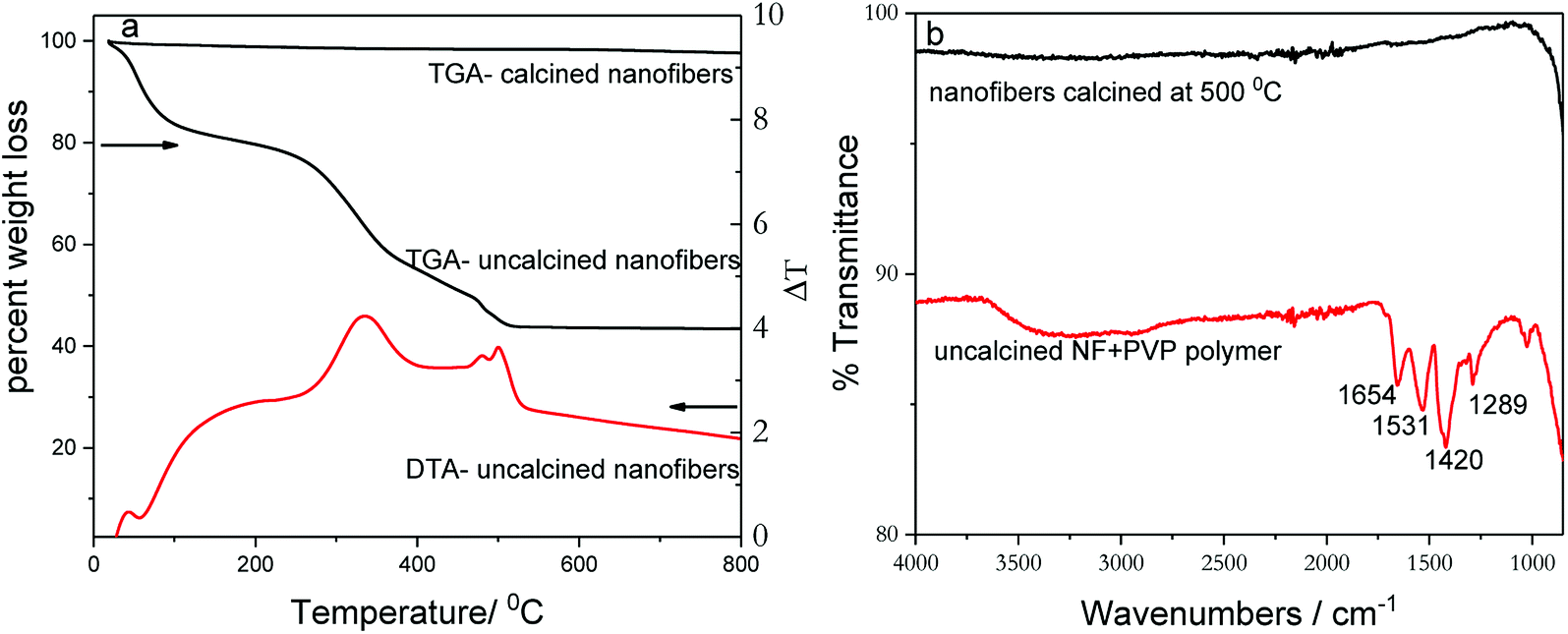 | ||
| Fig. 1 TGA and DTA plots of (a) calcined and uncalcined TiO2 nanofibers acquired by heating in air. (b) FTIR spectra of calcined and uncalcined TiO2 nanofibers. | ||
The powder X-ray diffraction (PXRD) technique was used to investigate the crystal structure of nanofibers and to elucidate alloying. TiO2 nanofiber photocatalysts deposited with 2% metal co-catalysts show no diffraction peaks originating from metal or metal alloy co-catalysts due to the presence of very small percentage of respective co-catalyst (Fig. S1†). In order to solely elucidate the alloying effect, photocatalysts were deposited with 10% metal or metal alloy co-catalysts under similar reaction conditions using polyol reduction method, and the XRD patterns of those photocatalysts are shown in Fig. 2a. Presence of sharp peaks in XRD pattern indicate that the calcination in air at 500 °C turns as-synthesized nanofibers into a polycrystalline phase of anatase-TiO2 along with some residual rutile-phase (Fig. 2a). The presence of rutile-TiO2 may have an added benefit because its band gap is 0.2 eV lower than that of anatase-TiO2 (3.0 vs. 3.2 eV), thus the electron excitation from valence band (VB) to conduction band (CB) is easier in the rutile phase. Due to the fact that anatase has a slightly higher CB, the conduction band electrons can be effectively transferred to CB of rutile TiO2. This process tends to increase the charge separation and decrease recombination.16 Fig. 2b shows magnified XRD patterns of 2%PtCu/TiO2, 2%Pt/TiO2, and 2%Pt2Pd/TiO2 nanofiber photocatalysts. The XRD pattern of 2%PtCu/TiO2 shows the diffraction peaks corresponding to (111) and (200) facets of a typical face-centered cubic (fcc) crystal structure of Pt–Cu alloy. All the resulting diffraction peaks of the Pt–Cu alloy of PtCu/TiO2 nanofiber photocatalyst are shifted to higher 2θ angles in comparison to diffraction peaks of pure Pt (JCPDS standard 65-2868) and are located between the diffraction peaks originated from pure Pt and Cu. This observation indicates the formation of Pt–Cu alloy. The peak shifting can be ascribed to lattice parameter contraction in Pt–Cu alloy, which originates from partial substitution of bigger Pt atoms (atomic radius 1.77 Å) with smaller Cu atoms (atomic radius 1.45 Å).41 Similar to the Pt–Cu alloy, the XRD pattern of the 2%Pt2Pd/TiO2 nanofiber photocatalyst shows a positive shift towards higher 2θ angles with respect to the peak positions in 2%Pt/TiO2 nanofiber catalyst (Fig. 2b). Again, the shift of Pt–Pd diffraction peaks is indicative of alloy formation between Pt and Pd, which is caused by the lattice contraction as a result of partial incorporation of smaller Pd atoms (atomic radius 1.69 Å) with bigger Pt atoms.42 The degree of peak shift in Pt–Pd alloy is smaller than that of Pt–Cu alloy due to the fact that the difference of atomic radii of Pt and Pd is quite small compared to the atomic radii difference between Pt and Cu.
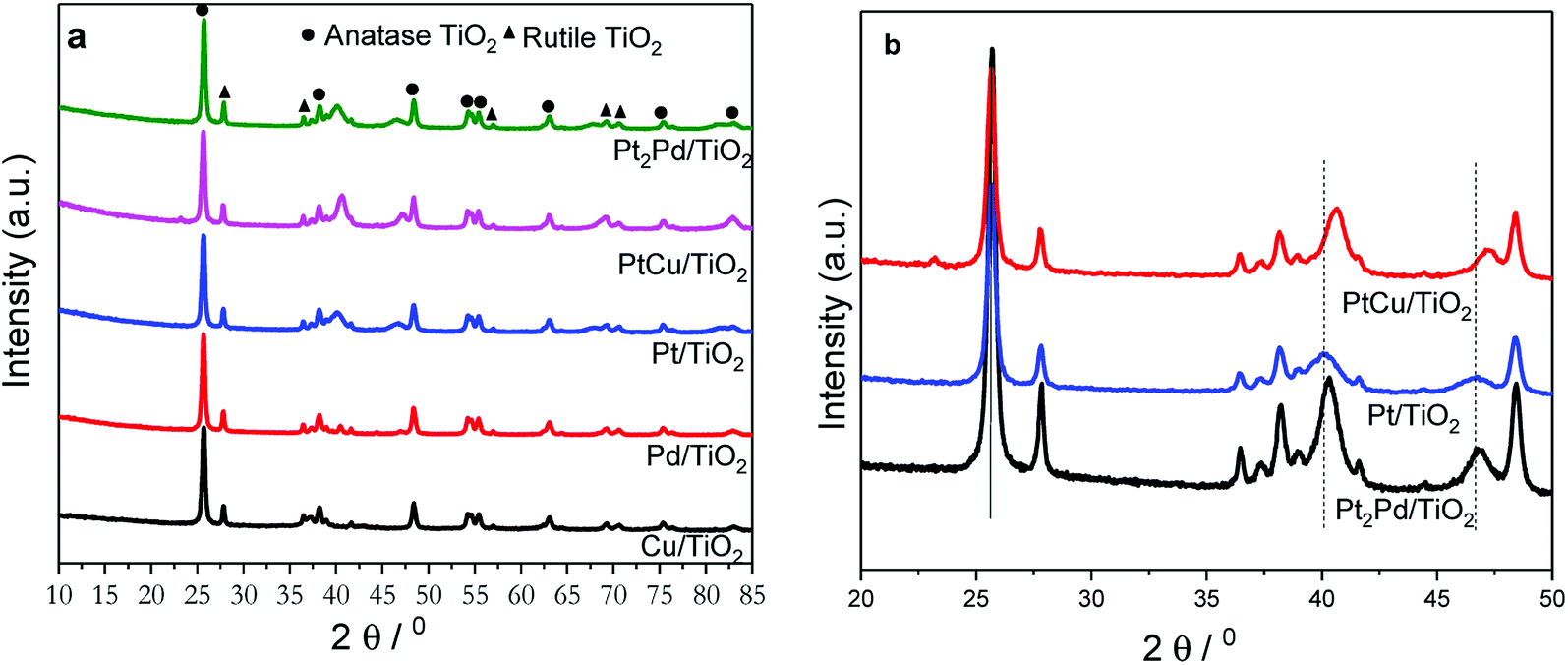 | ||
| Fig. 2 PXRD patterns of TiO2 nanofiber photocatalysts (a) deposited with various co-catalysts. (b) PXRD patterns of 2%Pt/TiO2, 2%Pt2Pd/TiO2, and 2%PtCu/TiO2 nanofiber photocatalysts in the region 20° ≤ 2θ ≤ 50°. | ||
Morphology and microstructure of photocatalysts were investigated using scanning electron and transmission electron microscopy (SEM and TEM). The SEM image of TiO2 nanofiber photocatalyst, shown in Fig. 3, is displaying the morphology of the calcined fibers. The TiO2 nanofibers are uniform in the long axial direction with an average diameter of approximately 60.0 ± 5.0 nm calculated by analyzing at least 150 individual fibers. The electrospinning method facilitates the formation of longer nanofibers, perhaps in centimeter lengths, if proper synthesis conditions are maintained to control the continuity of spinning process. The low magnification SEM image of TiO2 nanofibers produced by calcination in air at 500 °C indicates that the fibers can be grown into millimeter lengths along the fiber axial direction (Fig. 3a). Literature reports suggest that the fibers produced from electrospinning process are more flexible,37,43 and the Young's modulus of fibers is reported to be improved even though the tensile strength evidently decreases with calcinations.44 However, it is possible for calcined nanofibers to break into smaller segments of fibers during applied stress such as sonication. In this study, the calcined TiO2 fibers were sonicated in ethylene glycol for about one hour prior to co-catalyst deposition. We have observed that sonication has an impact on fiber length and it breaks fibers into smaller fibers (Fig. 3b). However, even after this fragmentation, fiber lengths are still in the micron range.
 | ||
| Fig. 3 SEM micrographs of (a) TiO2 nanofiber photocatalyst produced by calcination at 500 °C and (b) TiO2 nanofiber photocatalyst calcined and sonicated; TEM micrographs of (c) Pt co-catalyst deposited calcined TiO2 nanofiber photocatalyst; and (d) high magnification image, which shows TiO2 nanoparticle stacking. | ||
High resolution SEM and TEM images (Fig. 3b inset and Fig. 3d, respectively) clearly show that the electrospun nanofibers are made from agglomerated individual TiO2 nanoparticles, which are tightly packed and stacked along the axial direction of the nanofibers. Therefore, TiO2 nanofibers are expected, as suggested by literature,16 to exhibit similar XRD patterns and XPS spectra as of TiO2 nanoparticles. The inset in Fig. 3b shows a high magnification SEM image of TiO2 nanofibers, which exhibits a porous nature in calcined fibers. The decomposition of the PVP polymer and residual organic solvents and their removal as carbon dioxide during calcinations are responsible for the porosity of TiO2 nanofibers. However, surface area of the photocatalysts collected by employing N2 adsorption–desorption isotherms are quite low compared to literature-reported surface area values16,45 and are in the range of 15–25 m2 g−1. As noted by previous publications, the surface area of TiO2 decreases with increased annealing temperature. In the current study, TiO2 nanofibers were annealed at 500 °C. In comparison to previous reports, the average diameter of the TiO2 nanofibers was larger, which explains the decreased surface area per gram. Presence of co-catalyst (e.g. Pt) is evident from dark spots observed in TEM micrograph (Fig. 3c).
SEM imaging combined with EDS elemental mapping of the co-catalyst deposited TiO2 nanofiber photocatalysts clearly show the distribution of noble metal co-catalysts on TiO2 nanofiber surface. Fig. 4 shows a SEM image of 2%Pt2Pd/TiO2 nanofiber photocatalyst along with its EDS elemental mapping data. Fig. 4 clearly depicts the presence of Pt and Pd particles and their homogenous distribution over the nanofiber surface. The presence of co-catalysts was evidenced by XRD analysis, but showed small diffraction peaks due to their presence in very minute amounts. Nevertheless, EDS elemental mapping data coupled with XPS data undoubtedly indicate the presence of co-catalysts on the surface of the TiO2 nanofiber photocatalysts. Similar distribution of Pt, Pd, and Cu in Pt–Cu alloy, Pt, Pd, and Cu co-catalysts was observed and respective elemental mapping data are provided in ESI (Fig. S2†).
 | ||
| Fig. 4 SEM image of 2%Pt2Pd/TiO2 nanofiber photocatalyst with related EDS mapping and EDS spectrum of Ti, O, Pd, and Pt. | ||
XPS elucidates the surface chemistry of the electrospun TiO2 nanofiber photocatalysts decorated with various metal and noble metal co-catalysts; Cu, PtCu, Pt2Pd, and Pd (Fig. 5). As evidenced by XPS, the oxidation state of the Ti in the TiO2 nanofiber is Ti4+ for most samples; however, in the TiO2 nanofibers loaded with Cu co-catalyst, the surface of the TiO2 become partially reduced to Ti3+ (Fig. S3†). Introduction of the Ti3+ defect is possibly due to the ethylene glycol reduction method utilized to decorate TiO2 nanofiber surface with metal nanoparticles.46 Shown in Fig. 5a–b are XPS spectra of Pt2Pd co-catalysts. Peaks located at 70.9 and 74.3 eV in Fig. 5a corresponds with Pt 4f7/2 and Pt 4f5/2 binding energies, while peaks appear at 335.0 and 340.3 eV in Fig. 5b originates from Pd 3d5/2 and Pd 3d3/2, respectively. This is further evidence of coexistence of Pt and Pd. Furthermore, most samples show oxidation on the surface of the co-catalyst. XPS also revealed that the surface of the 2%Pt2Pd/TiO2 catalyst contained both oxidized and metallic Pt and Pd species. The two peaks located at 71.93 and 75.83 eV can be attributed to the presence of PtO, while PtO2 presence is evidenced by two peaks appearing at 73.76 and 77.66 eV (Fig. 5a). Similarly, Pd also shows oxidation, which is evidenced by the two peaks at 335.73 and 341.2 eV (Fig. 5b). The surface oxidation of the noble metal-based co-catalysts can be explained on the basis of the utilized chemical synthesis for the photocatalyst fabrication. Microwave-assisted polyol synthesis, which was utilized to decorate TiO2 nanofiber photocatalysts with metal or metal alloy co-catalyst, was conducted in an open container using ethylene glycol as the reducing agent, in a microwave designed for domestic use. It is highly possible for atmospheric surface oxidation to occur under microwave heating because the localized temperature exceeds the boiling point of ethylene glycol. In Fig. 5a, the observed Pt 4f binding energy is lower (by ca. 0.3 eV) than the standard binding energies of bulk Pt (71.2 eV (ref. 47)). This peak shift can be attributed to alloy formation as a consequence of electronic interactions between Pt and Pd atomic orbitals.48
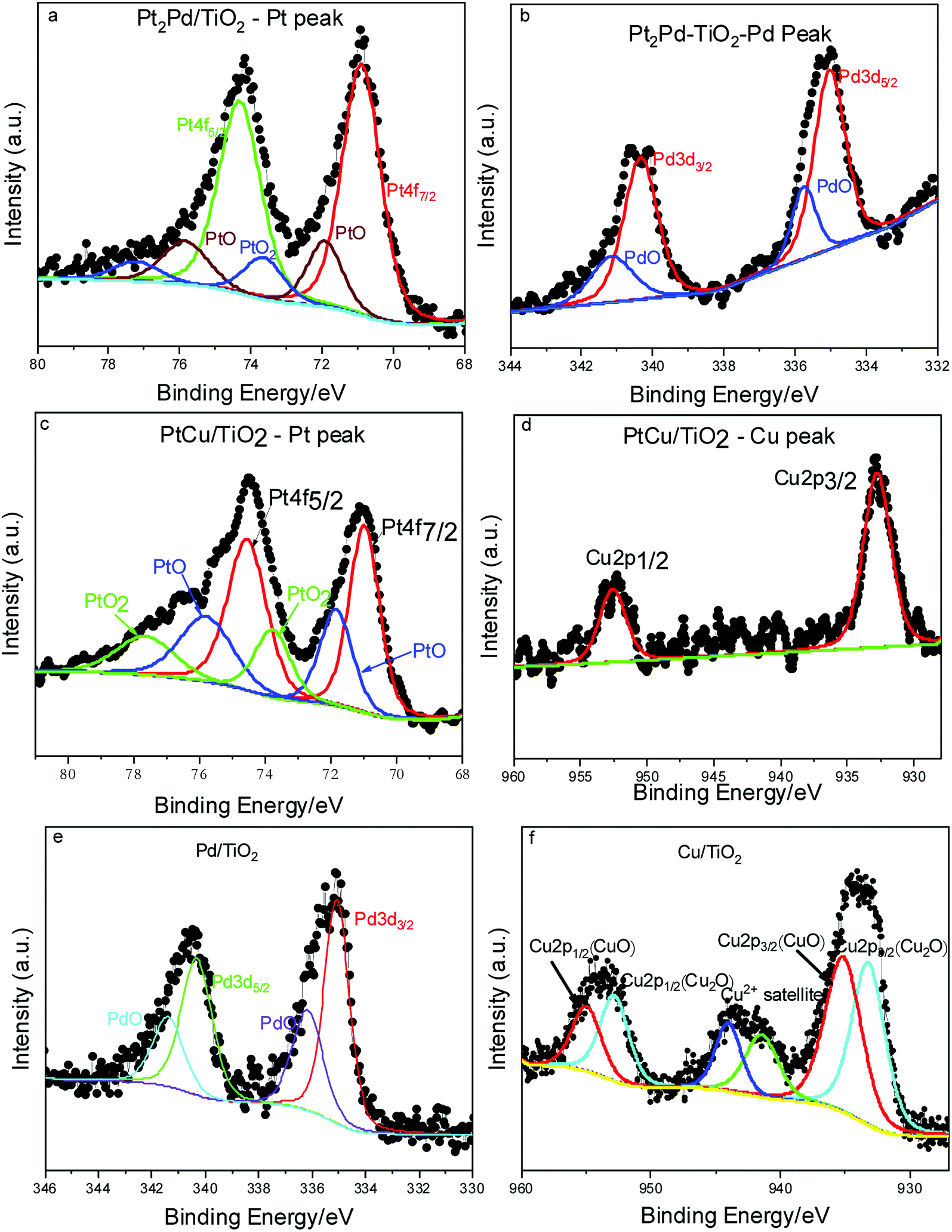 | ||
| Fig. 5 XPS spectra of the co-catalyst deposited TiO2 nanofiber photocatalysts. | ||
In the XPS spectra for the alloyed 2%PtCu/TiO2 catalyst (Fig. 5c and d), Pt 4f7/2 and Pt 4f5/2 peaks appear along with Cu 2p3/2 (932.7 eV) and Cu 2p1/2 (952.6 eV), indicating the coexistence of Pt and Cu. Similar to the Pt2Pd case, PtCu alloy formation can be confirmed due to peak shift of Pt 4f7/2 and 4f5/2 peaks by ca. 0.2 eV towards lower binding energy, as compared to peak positions of bulk Pt. The peak shift is caused by the interaction between Pt and Cu and their varying electro negativities (2.28 and 1.90, respectively).49 The surface oxidation of Pt is apparent, with the presence of both PtO and PtO2 species, while the Cu remains in its reduced state. However, XPS data of 2%Cu/TiO2 nanofiber photocatalyst (Fig. 5f) clearly show that the surface of the Cu co-catalyst is primarily composed of Cu2O (Cu 2p3/2 at 933.2 eV and Cu 2p1/2 at 952.9 eV) and CuO (Cu 2p3/2 at 935.1 and Cu 2p1/2 at 955.0 eV). The two different behaviors of copper can be ascribed to the catalytic reduction of Cu2+ in the presence of pre-formed Pt nuclei. Hence, reduced form of copper is more stable in Pt–Cu alloy than in standalone copper nanoparticles.41 Similarly, the 2%Pd/TiO2 nanofiber photocatalyst contained surface palladium oxide species, as seen in Fig. 5e. Interestingly, after the 2%Pd/TiO2 catalyst was used for photocatalysis, it contained a similar concentration of surface palladium oxide and a similar oxidation state of the TiO2 support, suggesting that the catalyst support is both chemically and photochemically stable under UV irradiation and in the presence of water.
The photocatalytic activity of TiO2 nanofibers decorated with noble metal and metal alloy co-catalysts was evaluated by investigating hydrogen generation under UV-Vis irradiation in a 5:1 (v/v) water-methanol mixture. The photocatalytic experiments were carried out at room temperature, where temperature of the photocatalytic cell was controlled using a water jacket around the cell. A photocatalytic cell containing catalyst dispersion was purged with argon for one hour prior to the experiment. The experiment was conducted in the presence of methanol as sacrificial agent (hole-scavenger). Fig. 6a shows photocatalytic hydrogen generation in millimole H2 per gram of catalyst as a function of time, and Fig. 6b depicts amount of H2 formed in millimole per milligram of noble metal co-catalyst. Pristine TiO2 nanofibers, without co-catalysts show little or no photocatalytic activity in terms of hydrogen generation under UV-Vis irradiation, which is a common behavior shown by most of hydrogen generating or water splitting photocatalysts.24 However, co-catalyst deposited photocatalysts clearly show hydrogen generation and prove co-catalysts play a significant role to generate considerable amount of H2 gas. The amount of H2 generated gradually increases as a function of time for all the catalysts tested. However, it may be possible to observe a plateau due to increase in pressure within the photocatalytic cell as H2 gas accumulates. Among the photocatalysts tested, the nanofiber photocatalyst with 2%Pt2Pd co-catalyst shows the highest H2 generation (4 mmol h−1 gramcatalyst), even higher than that of 2%Pt/TiO2 nanofiber photocatalysts; meanwhile, 2%Cu/TiO2 nanofiber photocatalyst was the least active (0.04 mmol h−1 gramcatalyst). The amount of H2 generated by 2%Pt2Pd/TiO2 nanofiber photocatalyst is approximately 45% higher than the amount of H2 generated by 2%Pt/TiO2 nanofiber photocatalyst. The exact reason for the enhanced catalytic behavior of 2%Pt2Pd/TiO2 nanofiber photocatalyst is not known at this point. The function of Pt cocatalyst is to cause adsorption of H+ from the photolyte, to facilitate the adsorbed protons to combine with photogenerated electron on the surface of the Pt, and to cause molecular H2 to be produced.50–52 Alloying Pt with Pd can fine-tune the co-catalyst's hydrogen adsorption free energy, thus increasing the hydrogen adsorption rate leading into higher photocatalytic activity towards hydrogen generation. Gonzalez et al. has reported that the alloying of Pt with Pd has reportedly decreased the activation energy of Pt by 20% (ref. 42) in Pt–Pd alloy catalyst leading to a higher activity in terms of ethanol oxidation in comparison to Pt only catalyst. Pt–Pd catalyst is not only active in molecular hydrogen generation but also in hydrogenation of organic molecules. As reported by Huang et al., Pt2Pd catalyst has shown the highest turnover frequency (TOF) out of various Pt–Pd catalysts tested for the hydrogenation of nitrobenzene.53 It is true that Pt is one of the most desirable cocatalyst because hydrogen adsorption energy on Pt is optimal. However, alloying of Pt with Pd may further fine-tune hydrogen adsorption energy of Pt–Pd cocatalyst leading to improved photocatalytic hydrogen generation. It is reported that alloying of Pt and Pd with a mol ratio of 2:1 (∼30 mol% Pd) leads to a maximum hydrogen generation and turnover frequency as a result of optimization of hydrogen adsorption energy of Pt by Pd.50 Furthermore, Pt/Pd binary structures are reported to be tolerant to self-poisoning.54 As evident from literature reported photocurrent measurements,50 Pt–Pd cocatalyst exhibits enhanced interfacial electron transfer and decreased electron–hole recombination. Therefore, based on literature reported facts about Pt–Pd cocatalyst and the observed enhanced photocatalytic hydrogen generation data collected for 2%Pt2Pd/TiO2 nanofiber photocatalyst, we can assume that the Pt2Pd cocatalyst effectively reduces photogenerated electron–hole recombination and improves interfacial electron transfer leading to higher photocatalytic activity. Pd is also known to act as a catalytically enhancing agent through modifying the electronic properties of Pt. We assume that a similar phenomenon is taking place with respect to photocatalytic H2 generation by 2%Pt2Pd/TiO2. Even though there is clear positive effect of using Pt2Pd alloy co-catalyst, the alloying of Pt with Cu shows no significant enhancement of hydrogen generation, and observed activity is significantly lower than that of Pt co-catalyst. The activity of functional cocatalysts involves the hydrogen evolution pathway suggested by Zhu and co-workers,55 which can be attributed to three steps; successful surface capture of H atoms through strong H atom adsorption energy, generation of molecular H2 through reduction by photogenerated electrons and the release of molecular H2 from the catalyst surface. It is reported that H atom absorption energy of Cu is relatively small compared to noble metals such as Pt and Pd while molecular H2 adsorption energy is smaller (2.49 eV and 0.001 eV, respectively).56 This in fact suggests that Cu is prone to release molecular H2 quite easily but lacks strong capture of H atoms, which is essential in hydrogen reduction reaction. The observation of relatively low photocatalytic activity by 2%PtCu/TiO2 nanofiber photocatalyst may be attributed to the fact that alloying of Pt with Cu obscures the electronic properties of Pt such as higher H atom adsorption energy.
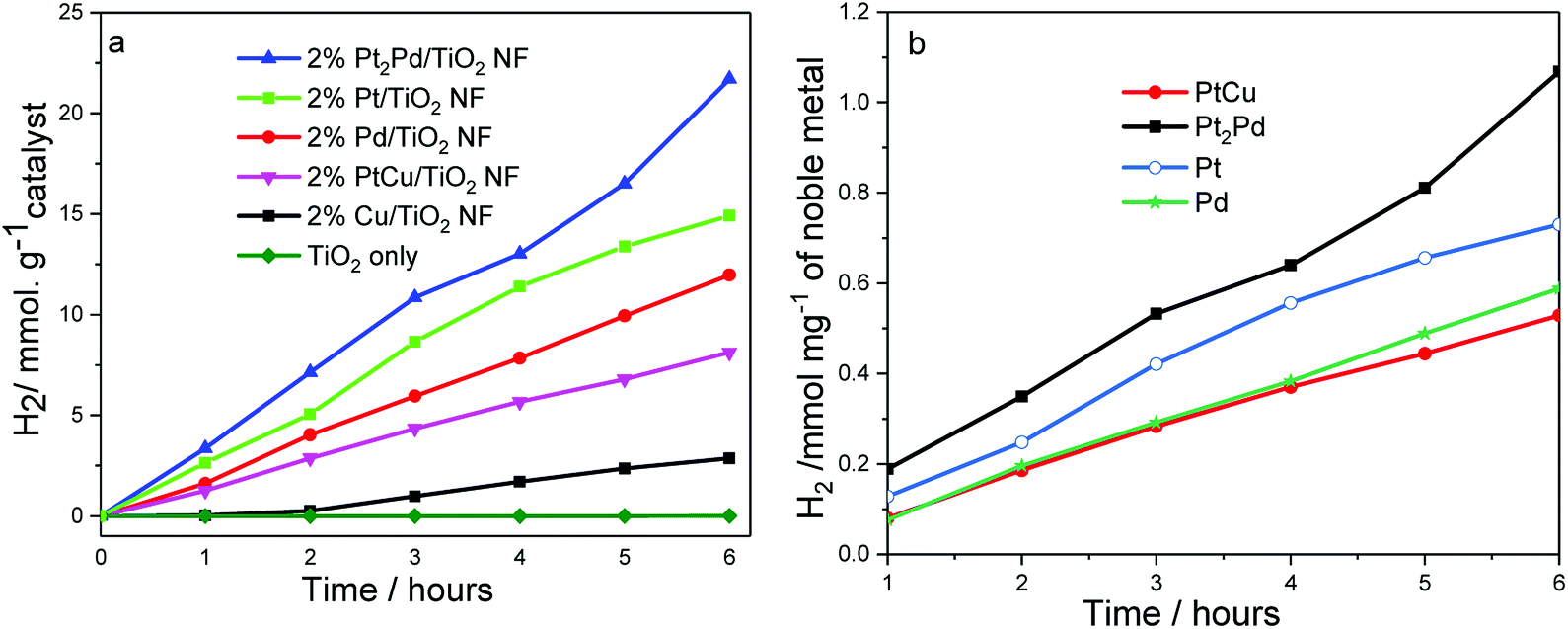 | ||
| Fig. 6 Photocatalytic hydrogen generation by co-catalyst deposited TiO2 nanofiber photocatalysts as a function of time. Reaction conditions: catalyst 0.10 g; distilled water:methanol in 5:1 ratio, 50 mL; 300 W Xe lamp. | ||
The normalized H2 generation per milligram of noble metal co-catalyst (Fig. 6b) of 2%PtCu/TiO2 suggests that the activity is similar to the activity shown by 2%Pd/TiO2 nanofiber photocatalyst. As was indicated in Fig. 6a, a gradual increase in H2 generation was observed throughout the time period in which catalytic activity was tested. The photocatalytic activity that originates from Cu/TiO2 nanofiber photocatalyst is significantly low and copper co-catalyst exists mainly in the form of Cu0, CuO, or Cu2O, as evidenced in XPS data. Surface oxidation of Cu is highly likely due to the experimental conditions of polyol reduction applied to deposit metal co-catalysts. Nonetheless, presence of CuO and Cu2O has little or no negative effect because copper oxides can serve as co-catalysts by rendering the reduction sites for H2 generation.57,58 Besides being a co-catalyst, CuO/TiO2 system may act as a heterojunction, which tend to promote charge separation and extend the lifetime of photogenerated carriers due to the band positions of TiO2 and CuO. Due to the fact that the conduction band (CB) of CuO is more positive than that of TiO2, photoinduced electrons in the CB of TiO2 quickly migrate to CB of CuO and make them readily available for water reduction reaction.59 We speculate that if copper co-catalyst were to present only in elemental form, the photocatalytic activity would be lower than that was observed. However, more data and information are needed to make a concrete conclusion.
The amount of hydrogen produced by various photocatalysts differs due to catalyst properties (surface area, crystallinity, cocatalyst, type of scavengers etc.) as well as the conditions applied in photocatalytic measurements. Photocatalytic activity of Degussa P25 has been used as a baseline to compare photocatalytic data in some literature reports. The photocatalytic hydrogen production values reported for Degussa P25 ranges from 0.004 mmol h−1 g−1 to 5.2 mmol h−1 g−1.16,60,61 A study conducted by Choi and co-workers16 reported that Pt/TiO2 nanofiber photocatalyst to be a better catalyst than Pt deposited Degussa P25 catalyst and have shown a 25% increase in hydrogen production. The increase in catalytic activity is attributed to the higher surface area of Pt/TiO2 nanofiber catalyst as compared to Degussa P25 (96.3 vs. 52 m2 g−1). The photocatalytic hydrogen production by Pt/TiO2 nanofiber catalyst reported by Choi and co-workers are higher than that of measured in this manuscript (6.5 mmol h−1 g−1 vs. 3.2 mmol h−1 g−1) and a comparison of the activity of 2%Pt2Pd/TiO2 nanofiber photocatalyst with literature data is not possible as there are no reports that describe the photocatalytic activity of 2%Pt2Pd/TiO2 nanofiber photocatalyst. The literature reported higher activity of Pt/TiO2 nanofiber photocatalyst may be ascribed to higher catalytic surface area (96.3 vs. 25 m2 g−1).
The higher activity of 2%Pt2Pd/TiO2 catalyst is indicative of higher charge separation of photogenerated electrons and holes. This should be reflected in photocurrent measurements acquired as a function of time. Luo et al. has measured photocurrents of Pt–Pd/CdS and pristine CdS photocatalysts and reported that Pt–Pd/CdS has shown significantly higher photocurrent. This is indicative that Pt–Pd cocatalyst evidently decreases photogenerated electron–hole recombination rate by effectively trapping electrons in the Pt–Pd cocatalyst and transferring from Pt–Pd cocatalyst into hydrogen reduction reaction.50 Additionally, enhanced photocurrent indicates effective interfacial electron transfer from photocatalyst to cocatalyst. Therefore, a higher photocurrent observed for Pt–Pd cocatalyst explains the enhanced photocatalytic activity, in terms of hydrogen production, of Pt–Pd/CdS photocatalyst. In this token, the 2%Pt2Pd/TiO2 nanofiber photocatalyst is expected to behave similarly and show better photocurrents in comparison to pristine TiO2 if measured. Based on the enhanced photocatalytic hydrogen generation data collected for 2%Pt2Pd/TiO2 nanofiber photocatalyst, we can assume that the Pt2Pd cocatalyst effectively reduces photogenerated electron–hole recombination and improves interfacial electron transfer leading to higher photocatalytic activity.
Reusability of catalysts and percent deactivation were investigated by using 2%Pt2Pd/TiO2 nanofiber photocatalyst. The light induced H2 generation was repeated over three successive cycles, and the resulting data is shown in Fig. 7. Evidently, photocatalytic activity slightly decreased after repeated cycles, with an overall decrease of approximately 6% over three consecutive cycles, which indicates that the 2%Pt2Pd/TiO2 nanofiber photocatalyst was reasonably stable under UV-Vis irradiation.
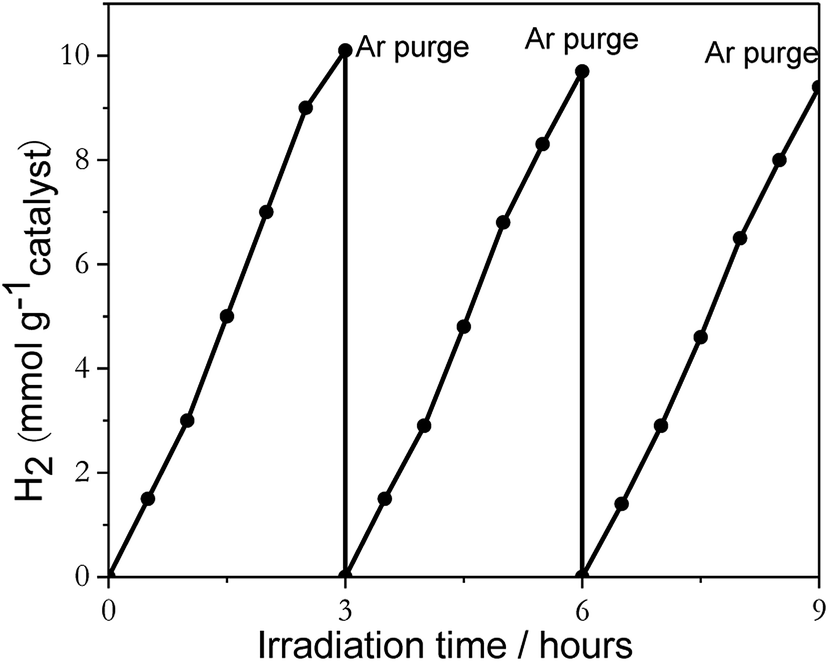 | ||
| Fig. 7 The recyclability data of 2%Pt2Pd/TiO2 nanofiber photocatalysts under UV-Visible light irradiation in the presence of sacrificial reagent methanol. | ||
Due to the fact that photocatalytic activity depends on experimental conditions, apparent quantum yield (AQY) was determined for each catalyst using eqn (4) in order to solely compare activities of nanofiber photocatalysts. The results are summarized in Table 1.
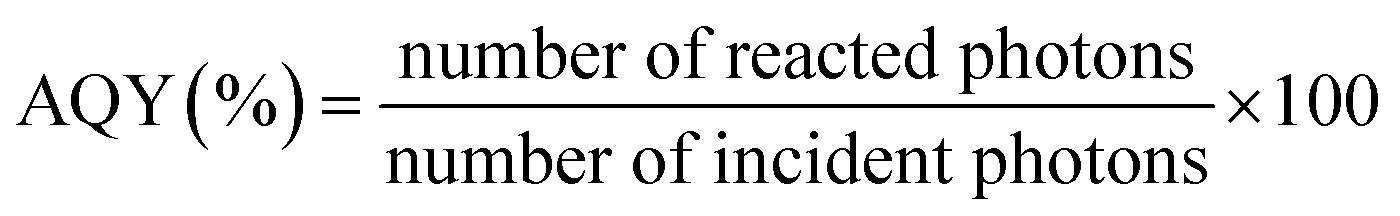 | (4) |
| Photocatalysts | Activity/mmol H2 h−1 g−1 catalyst | AQY (%) |
|---|---|---|
| 2%Pt2Pd/TiO2 | 3.62 | 10 |
| 2%Pt/TiO2 | 2.47 | 6.6 |
| 2%Pd/TiO2 | 1.99 | 5.3 |
| 2%PtCu/TiO2 | 1.37 | 3.6 |
| 2%Cu/TiO2 | 0.47 | 1.3 |
| TiO2 (no co-catalyst) | 1.1 × 10−3 | 2.9 × 10−3 |
The AQY of 2%Pt2Pd/TiO2 nanofiber photocatalyst was calculated to be the highest (10%), with the overall order from greatest to least AQY for each co-catalyst used of Pt2Pd > Pt > Pd > PtCu > Cu. It should be noted that the AQY was calculated based on the intensity of the incident photons at 254 nm. However, in actual experiment, no cutoff filters were used and both UV and Visible irradiance was allowed. Therefore, the actual AQY may be different from what is reported due to an increased number of absorbed and reacted photons. In this report, AQY was primarily used to compare the activities of each photocatalyst.
Conclusions
Metal and metal alloy decorated TiO2 nanofiber photocatalysts were successfully prepared by employing polymer-assisted electrospinning and subsequent microwave-assisted ethylene glycol reduction methods. Simple microwave-assisted co-reduction is sufficient for successfully depositing Pt2Pd and PtCu alloy co-catalysts due to rapid nucleation and high local temperature. One drawback of this method is the surface oxidation of metal co-catalysts, which takes place due the reduction process occurring in ambient air. However, this is not expected to be a detrimental effect on photocatalysis. Co-catalyst deposited TiO2 nanofiber photocatalysts show greater hydrogen generation in comparison with pristine TiO2 nanofiber photocatalyst. The noble metal and metal alloy co-catalyst deposited TiO2 performed exceptionally well, with the catalytic activity change in the order of Pt2Pd > Pt > Pd > PtCu > Cu. Out of all the catalysts tested, 2%Pt2Pd/TiO2 nanofiber photocatalyst showed the highest activity. The photocatalytic activity towards hydrogen generation is reproducible, and the 2%Pt2Pd/TiO2 catalyst loses about 6% of initial activity over 3 consecutive trials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the Faculty Research Awards Program (FRAP) from the School of Graduate Studies and Research-Florida A&M University. The microscopy and XPS analysis were completed at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility. Z. D. H. gratefully acknowledges support from the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1650044 and the Georgia Tech-ORNL Fellowship.References
- M. Yoshida, T. Hirai and K. Maeda, et al., Photoluminescence spectroscopic and computational investigation of the origin of the visible light response of (Ga1-xZnx)(N1-xOx) photocatalyst for overall water splitting, J. Phys. Chem. C, 2010, 114, 15510–15515 CrossRef.
- A. Naldoni, C. L. Bianchi and C. Pirola, et al., Porous TiO2 microspheres with tunable properties for photocatalytic air purification, Ultrason. Sonochem., 2013, 20, 445–451 CrossRef PubMed.
- J. Sato, N. Saito and H. Nishiyama, et al., New photocatalyst group for water decomposition of RuO2-loaded p-block metal (In, Sn, and Sb) oxides with d10 configuration, J. Phys. Chem. B, 2001, 105, 6061–6063 CrossRef.
- C. Massué, X. Huang and A. Tarasov, et al., Microwave-assited synthesis of stable and highly active Ir oxohydroxides for electrochemical oxidation of water, ChemSusChem, 2017, 10, 1958–1968 CrossRef PubMed.
- H. S. Jung, Y. J. Hong and Y. Li, et al., Photocatalysis using GaN nanowires, ACS Nano, 2008, 2, 637–642 CrossRef PubMed.
- G. Wang, A. Pierre and M. G. Kibria, et al., Wafer-level photocatalytic water splitting on GaN nanowire arrays grown by molecular beam epitaxy, Nano Lett., 2011, 11, 2353–2357 CrossRef PubMed.
- A. Wu, B. Liu and W. Yang, et al., Band-gap tailoring and visible-light driven photocatalytic performance of porous (GaN)1-x(ZnO)x solid solution, Dalton Trans., 2017, 46, 2643–2652 RSC.
- A. Kudo and Y. Miseki, Heterogeneous photocatalyst materials for water splitting, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- D. Li and Y. Xia, Electrospinning of nanofibers: Reinventing the wheel?, Adv. Mater., 2004, 16, 1151–1170 CrossRef.
- J. H. Bang and P. V. Kamat, Solar cells by design: photoelectrochemistry of TiO2 nanorod arrays decorated with CdSe, Adv. Funct. Mater., 2010, 20, 1970–1976 CrossRef.
- Y. Liu, H. Wang and Y. Wang, et al., Substrate-free, large-scale, free-standing and two-side oriented single crystal TiO2 nanorod array films with photocatalytic properties, Chem. Commun., 2011, 47, 3790–3792 RSC.
- J. Sun, C. Liu and P. Yang, Surfactant-free, large-scale, solution-liquid-solid growth of gallium phosphide nanowires and their use for visible light driven hydrogen production from water reduction, J. Am. Chem. Soc., 2011, 133, 19306–19309 CrossRef PubMed.
- A. I. Hochbaum and P. Yang, Semiconductor nanowires for energy conversion, Chem. Rev., 2010, 110, 527–546 CrossRef PubMed.
- P. Yang, R. Yan and M. Fardy, Semiconductor nanowire: What's next?, Nano Lett., 2010, 10, 1529–1536 CrossRef PubMed.
- Z. Zhang, C. Shao and X. Li, et al., Hierarchical assembly of ultrathin hexagonal SnS2 nanosheets onto electrospun TiO2 nanofibers: enhanced photocatalytic activity based on photoinduced interfacial charge transfer, Nanoscale, 2013, 5, 606–618 RSC.
- S. K. Choi, S. Kim and S. K. Lim, et al., Photocatalytic comparison of TiO2 nanoparticles and electrospun TiO2 nanofibers: Effects of mesoporosity and interparticle charge transfer, J. Phys. Chem. C, 2010, 114, 16475–16480 CrossRef.
- Y. K. Kim and H. Park, Light-harvesting multi-walled carbon nanotubes and CdS hybrids: Application to photocatalytic hydrogen production from water, Energy Environ. Sci., 2011, 4, 685–694 RSC.
- V. Jovic, Z. H. N. Al-Azria and D. Sun-Waterhouse, et al., Photocatalytic H2 production from bioethanol over Au/TiO2 and Pt/TiO2 photocatalysts under UV irradiation-a comparative study, Top. Catal., 2013, 56, 1139–1151 CrossRef.
- V. Jovic, W.-T. Chen and D. Sun-Waterhouse, et al., Effect of gold loading and TiO2 support composition on the activity of Au/TiO2 photocatalysts for H2 production from ethanol-water mixtures, J. Catal., 2013, 305, 307–317 CrossRef.
- A. Kudo, Development of photocatalyst materials for water splitting, Int. J. Hydrogen Energy, 2006, 31, 197–202 CrossRef.
- K. Maeda, N. Sakamoto and T. Ikeda, et al., Preparation of core-shell-structured nanoparticles (with a noble-metal or metal oxide core and a chromia shell) and their application in water splitting by means of visible light, Chem.–Eur. J., 2010, 16, 7750–7759 CrossRef PubMed.
- S. Ikeda, M. Fubuki and Y. K. Takahara, et al., Photocatalytic activity of hydrothermally synthesized tantalate pyrochlores for overall water splitting, Appl. Catal., 2006, 300, 186–190 CrossRef.
- K. Maeda, K. Teramura and T. Takata, et al., Overall water splitting on (Ga1-xZnx)(N1-xOx) solid solution photocatalyst: Relationship between physical properties and photocatalytic activity, J. Phys. Chem. B, 2005, 109, 20504–20510 CrossRef PubMed.
- T. Histomi, K. Maeda and K. Takanabe, et al., Aspects of the water splliting mechanism on (Ga1-xZnx)N1-xOx) photocatalyst modified with Rh2-yCrO3 o-catalyst, J. Phys. Chem. C, 2009, 113, 21458–21466 CrossRef.
- P. Mani, R. Srivastava and P. Strasser, Dealloyed Pt-Cu core-shell nanoparticle electrocatalysts for use in PEM fuel cell cathodes, J. Phys. Chem. C, 2008, 112, 2770–2778 CrossRef.
- B. C. Beard and P. N. Ross, The structure and reactivity of Pt-Co alloys as oxygen reduction electrocatalysts, J. Electrochem. Soc., 1990, 137, 3368–3374 CrossRef.
- V. R. Stamenkovic, B. Fowler and B. S. Mun, et al., Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability, Science, 2007, 315, 493–497 CrossRef PubMed.
- M. Min, J. Cho and K. Cho, et al., Particle size and alloying effecta of Pt-based alloy catalysts for fuel cell applications, Electrochim. Acta, 2000, 45, 4211–4217 CrossRef.
- M. Ammam and E. B. Easton, Oxygen reduction activity of binary PtMn/C alloy catalysts, J. Power Sources, 2013, 236, 311–320 CrossRef.
- A. K. Shukla, M. Neergat and P. Bera, et al., An XPS study on binary and ternary alloys of transition, etals with platinized carbon and its bearing upon oxygen electroreduction in direct methanol fuel cells, J. Electroanal. Chem., 2001, 504, 111–119 CrossRef.
- H. Colón-Mercado, H. Kim and B. N. Popov, Durability study of Pt3Ni catalysts as cathode in PEM fuel cells, Electrochem. Commun., 2004, 6, 795–799 CrossRef.
- X. Li, S. Park and B. N. Popov, Highly stable Pt and PtPd hybrid catalysts supported on a nitrogen-modified carbon composite for fuel cell applications, J. Power Sources, 2010, 195, 445–452 CrossRef.
- J. Y. Chen, H. C. Chen and J. N. Lin, et al., Effects of polymer media on electrospun mesoporous titania nanofibers, Mater. Chem. Phys., 2008, 107, 480–487 CrossRef.
- K. Senevirathne, R. Hui and S. Campbell, et al., Electrocatalytic activity and durability of Pt/NbO2 and Pt/Ti4O7 nanofibers for PEM fuel cell oxygen reduction reaction, Electrochim. Acta, 2012, 59, 538–547 CrossRef.
- M. Kim, Y.-K. Kim and S.-K. Lim, et al., Efficient visible light-induced H2 production by Au@CdS/TiO2 nanofibers: Synergistic effect of core-shell structured Au@CdS and densely packed TiO2 nanoparticles, Appl. Catal., B, 2015, 166–167, 423–431 CrossRef.
- D. Li and Y. Xia, Fabrication of titania nanofibers by electrospinning, Nano Lett., 2003, 3, 555–560 CrossRef.
- K. Mondal, S. Bhattacharyya and A. Sharma, Photocatalytic degradation of naphthalene by electrospun mesoporous carbon-deoped anatase TiO2 nanfiber mats, Ind. Eng. Chem. Res., 2014, 53, 18900–18909 CrossRef.
- B. Caratão, E. Carneiro and P. Sá, et al., Prpoperties of electrospun TiO2 nanofibers, J. Nanotechnol., 2014, 2014, 1–5 CrossRef.
- Y. Lu, Y. Li and S. Zhang, et al., Parameter study and characterization for polyacrylonitrile nanofibers fabricated via centrifugal spinning process, Eur. Polym. J., 2013, 49, 3834–3845 CrossRef.
- N. P. G. Roeges, A guide to the complete interpretation of infrared spectra of organic structures, John Wiley and Sons, 1994 Search PubMed.
- M. Gong, Z. Yao and F. Lai, et al., Platinum-copper alloy nanocrystals supported on reduced graphene oxide: One-pot synthesis and electrocatalytic applications, Carbon, 2015, 91, 338–345 CrossRef.
- T. Lopes, E. Antolini and E. R. Gonzalez, Carbon supported Pt-Pd alloy as an ethanol tolerant oxygen reduction catalysts for direct ethanol fuel cells, Int. J. Hydrogen Energy, 2008, 33, 5563–5570 CrossRef.
- S. V. Fridrikh, J. H. Yu and M. P. Brenner, et al., Controlling the fiber diameter during electrospinning, Phys. Rev. Lett., 2003, 90, 144502–144508 CrossRef PubMed.
- S.-J. Park, G. G. Chase and K.-U. Jeong, et al., Mechanical properties of titania nanofiber mats fabricated by electrospinning of sol-gel precursor, J. Sol-Gel Sci. Technol., 2010, 54, 188–194 CrossRef.
- S. Chuangchote, J. Jitputti and T. Sagawa, et al., Photocatalytic activity for hydrogen evolution of electrospun TiO2 nanofibers, ACS Appl. Mater. Interfaces, 2009, 1, 1140–1143 CrossRef PubMed.
- Y. Xu, S. Wu and P. Wan, et al., Introducing Ti3+ defects based on lattice distortion for enhanced visible photoreactivity in TiO2 microspheres, RSC Adv., 2017, 7, 32431–32467 Search PubMed.
- K. D. Schierbaum, S. Fischer and M. C. Torquemada, et al., The interaction of Pt with(110) surfaces: a comparative XPS, UPS, ISS, and ESD study, Surf. Sci., 1996, 345, 261–273 CrossRef.
- Y. Xu and X. Lin, Facile fabrication of and electrocatalytic activity of Pt0.9Pd0.1 alloy film catalysts, J. Power Sources, 2007, 170, 13–19 CrossRef.
- M. Gong, Z. Yao and F. Lai, et al., Paltinum-copper alloy nanocrystals supported on reduced graphine oxide: One-pot synthesis and electrocatalytic applications, Carbon, 2015, 91, 338–345 CrossRef.
- M. Luo, P. Lu and W. Yao, et al., Shape and composition effects on photocatalytic hydrogen production for Pt-Pd alloy cocatalysts, ACS Appl. Mater. Interfaces, 2016, 8, 20667–20674 CrossRef PubMed.
- M. Luo, Y. Hong and W. Yao, et al., Facile removal of polyvinylpyrrolidone (PVP) adsorbates from Pt alloy nanoparticles, J. Mater. Chem. A, 2015, 3, 2770–2775 RSC.
- M. Luo, W. Yao and C. Huang, et al., Shape effects of Pt nanoparticles on hydrogen production via Pt/CdS photocatalysts under visible light, J. Mater. Chem. A, 2015, 3, 13884–13891 RSC.
- X. Huang, Y. Li and Y. Li, et al., Synthesis of PtPd bimetal nanocrystals with controllable shape, composition, and their, tunable catalytic properties, Nano Lett., 2012, 12, 4265–4270 CrossRef PubMed.
- H. J. Lee, S. E. Habas and G. A. Somorjai, et al., Localized Pd overgrowth on cubic Pt nanocrystals for enhanced electrocatalytic oxidation of formic acid, J. Am. Chem. Soc., 2008, 130, 5406–5407 CrossRef PubMed.
- H. Zhu, J. Zhang and R. Yanzhang, et al., When cubic cobalt sulfide meets layered molybdenum disulfide: A core–shell system toward synergetic electrocatalytic water splitting, Adv. Mater., 2015, 27, 4752–4759 CrossRef PubMed.
- Z. Lin, J. Li and L. Li, et al., Manipulating the hydrogen evolution pathway on composition-tunable CuNi nanoalloys, J. Mater. Chem. A, 2017, 5, 773–781 RSC.
- J. Yu and J. Ran, Facile preparation and enhanced photocatalytic H2-production activity of Cu(OH)2 cluster modified TiO2, Energy Environ. Sci., 2011, 4, 1364–1371 RSC.
- K. Maeda and K. Domen, A copper and chromium based nanoparticulate oxide as a noble-metal-free cocatalyst for photocatalytic water splitting, Chem. Sci., 2011, 2, 1362–1368 RSC.
- S. S. Lee, H. Bai and Z. Liu, et al., Optimization and an insightful properties-Activity study of electrospun TiO2/CuO composite nanofibers for efficient photocatalytic H2 generation, Appl. Catal., B, 2013, 140–141, 68–81 CrossRef.
- R. Abe, K. Sayama and K. domen, et al., A new type of water-splitting system composed of two different TiO2 photocatalysts (anatase, rutile) and IO3-/I- shuttle redox mediator, Chem. Phys. Lett., 2001, 344, 339–344 CrossRef.
- R. Abe, K. Sayama and H. Sugihara, Development of new photocatalytic water splitting into H2 and O2 using two different semiconductor photocatalysts and a shuttle redox mediator IO3-/I-, J. Phys. Chem. B, 2005, 109, 16052–16061 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04148b |
| This journal is © The Royal Society of Chemistry 2018 |