
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSnS/C nanocomposites for high-performance sodium ion battery anodes†
Seung-Ho Yu‡
 ab,
Aihua Jin‡cd,
Xin Huange,
Yao Yanga,
Rong Huangb,
Joel D. Brockbe,
Yung-Eun Sung*cd and
Héctor D. Abruña*a
ab,
Aihua Jin‡cd,
Xin Huange,
Yao Yanga,
Rong Huangb,
Joel D. Brockbe,
Yung-Eun Sung*cd and
Héctor D. Abruña*a
aDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA. E-mail: hda1@cornell.edu
bCornell High Energy Synchrotron Source, Cornell University, Ithaca, NY 14853, USA
cCenter for Nanoparticle Research, Institute for Basic Science (IBS), Seoul 151-742, Republic of Korea
dSchool of Chemical and Biological Engineering, Seoul National University, Seoul 151-742, Republic of Korea. E-mail: ysung@snu.ac.kr
eSchool of Applied and Engineering Physics, Cornell University, Ithaca, New York 14853, USA
First published on 29th June 2018
Abstract
Sodium-ion batteries have been considered as one of the most promising types of batteries, beyond lithium-ion batteries, for large-scale energy storage applications. However, their deployment hinges on the development of new anode materials, since it has been shown that many important anode materials employed in lithium ion batteries, such as graphite and silicon, are inadequate for sodium-ion batteries. We have simply prepared novel SnS/C nanocomposites through a top-down approach as anode materials for sodium-ion batteries. Their electrochemical performance has been significantly improved when compared to bare SnS, especially in terms of cycling stability and rate capabilities. SnS/C nanocomposites exhibit excellent capacity retention, at various current rates, and deliver capacities as high as 400 mA h g−1 even at the high current density of 800 mA g−1 (2C). Ex situ transmission electron microscopy, X-ray diffraction and operando X-ray absorption near edge structure studies have been performed in order to unravel the reaction mechanism of the SnS/C nanocomposites.
Introduction
In recent years, sodium-ion batteries (SIBs) have emerged as attractive alternatives to replace lithium-ion batteries (LIBs) for large-scale energy storage applications because sodium is much more abundant and inexpensive than lithium.1–7 Since both lithium and sodium are alkali metals with one electron in their valence shells, there was the expectation that materials developed for LIBs could be directly applied to SIBs. However, it is much more challenging to identify promising anode materials for SIBs, compared to LIBs, because sodium has a higher equilibrium potential and a larger ionic size.8–10 For example, while graphite, used as anode, played a key role in commercializing LIBs, it has been reported that intercalation of sodium ions into graphite is hindered, so that graphite delivers a much lower capacity when applied to SIB anodes.11–13Recently, there has been a great deal of effort focused on the search for suitable anode materials for SIBs. Although silicon has been intensively studied for LIB anodes, due to its exceptionally high theoretical capacity, the redox potential of silicon is too low for application in SIBs.8,14 Instead, other alloy-type materials such as Sn-, Sb-, and Ge-based materials have been examined in detail as high-capacity anodes for SIBs.15–25 In addition, it has been reported that various transition metal oxides, sulfides and selenides can store sodium ions reversibly through intercalation and/or conversion reactions.26–37 Furthermore, several approaches have been introduced to facilitate intercalation of sodium ions into graphite by using expanded graphite, and ether-based electrolytes.11,38–40 However, these materials still have limitations such as low capacity, poor rate capability, rapid capacity fade and complex preparation process.
Herein, we report a simple preparation method for SnS/C nanocomposites, and their application to sodium-ion battery anodes. The grain size of SnS could reach about 10 nm, and SnS particles were well surrounded by carbon particles thereby increasing conductivity. The nanocomposites exhibited significantly enhanced electrochemical performance when compared to those of bare SnS electrodes. The nanocomposites delivered a reversible capacity of 480.0 mA h g−1 with negligible capacity fade during 50 cycles at a current density of 100 mA g−1. The composites also showed outstanding rate capabilities, delivering ∼93% of their capacity when the current density was increased by a factor of ten (from 50 to 500 mA g−1). Furthermore, the reaction mechanism of SnS/C nanocomposites was studied using ex situ transmission electron microscopy (TEM), X-ray diffraction (XRD) and operando X-ray absorption near edge structure (XANES).
Experimental
Preparation of SnS/C nanocomposites
Tin(II) sulfide (Aldrich) and super P carbon were mixed in a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, and the mixture was transferred to a ball-milling container in an argon-filled glove box. Mechanical ball-milling was conducted with a PULVERISETTE7 (Fritsch) at a speed of 500 rpm for 10 h.
:3, and the mixture was transferred to a ball-milling container in an argon-filled glove box. Mechanical ball-milling was conducted with a PULVERISETTE7 (Fritsch) at a speed of 500 rpm for 10 h.
Characterizations
Field-emission scanning electron microscopy (FE-SEM) analysis was conducted with a SUPRA 55VP (Carl Zeiss). X-ray diffraction patterns were obtained using a Bruker D-5005 with Cu Kα radiation. Operando XANES experiments were conducted at the F-3 station of the Cornell High Energy Synchrotron Source (CHESS). High-resolution transmission electron microscopy (HRTEM) images, and corresponding selected area electron diffraction (SAED), were obtained using an FEI field-emission-gun (FEG) Tecnai F-20 operated at 200 kV.Electrochemical characterization
A slurry, composed of 70 wt% of active material, 15 wt% of super P carbon and 15 wt% of poly(acrylic acid) in N-methyl-2-pyrrolidinone solvent, was coated on the current collector. The electrodes were subsequently vacuum dried in an oven overnight at 120 °C. The loading mass of active material on electrode is about 1.4 mg cm−2. The coin cells (CR-2032) were assembled in an argon-filled glove box with sodium metal as both counter and reference electrodes. A glass fiber filter (GF/C, Whatman) was used as separator. 1.0 M NaClO4 in ethylene carbonate (EC)/propylene carbonate (PC) (1:1 by volume) with 5 wt% fluoroethylene carbonate (FEC) as additive was used as the electrolyte. Coin cells were tested on BT-2000 (Arbin Instruments) and WBCS3000 cyclers (WanA Tech) over a voltage range from 0.01 V to 3.0 V (vs. Na+/Na).
Results and discussion
Characterization of prepared SnS/C nanocomposites
Fig. 1 presents X-ray diffraction (XRD) patterns of bare commercial SnS and SnS/C nanocomposites. The XRD peaks of bare SnS powder are well matched to values of the SnS reference card (JCPDS no. 14-0620, orthorhombic structure, a = 4.33, b = 11.19 and c = 3.98). The (040) peak of bare SnS is overwhelmingly stronger when compared to other peaks, indicating that the SnS layers are preferentially stacked in the [010] direction (see crystal structure in inset of Fig. 1). The XRD peaks of the SnS/C nanocomposites are much broader than those of bare SnS, indicating that the grain size is much smaller. The calculated grain size of SnS/C nanocomposites, by the Scherrer equation is about 10 nm. It is worthwhile to note that the intensities of the (040) and (080) peaks decreased more significantly than other peaks after ball-milling. This suggests that the layers of SnS are more easily broken in the [010] direction than others during ball-milling (see Fig. 2a and b). It is worthwhile to note that obtaining nanosized material (<100 nm) using ball-milling is difficult in general, but intrinsically bare SnS has two-dimensional character with preferential direction, and thickness in the other direction is small. Therefore, small-sized SnS particles can be obtained simply using ball-milling, even size can reach to about 10 nm. XANES spectrum of SnS/C nanocomposites is overlapped with that of bare SnS, indicating that no phase transition (reduction/oxidation) was occurred during ball-milling process (see Fig. S1 in ESI†). | ||
| Fig. 1 Schematic illustration of morphology of (a) bare SnS and (b) SnS/C nanocomposites. (c) SEM image and (e) TEM images of bare SnS. Inset of (e) shows a fast Fourier transform (FFT) image. (d) SEM image and (f) TEM images of SnS/C nanocomposites. | ||
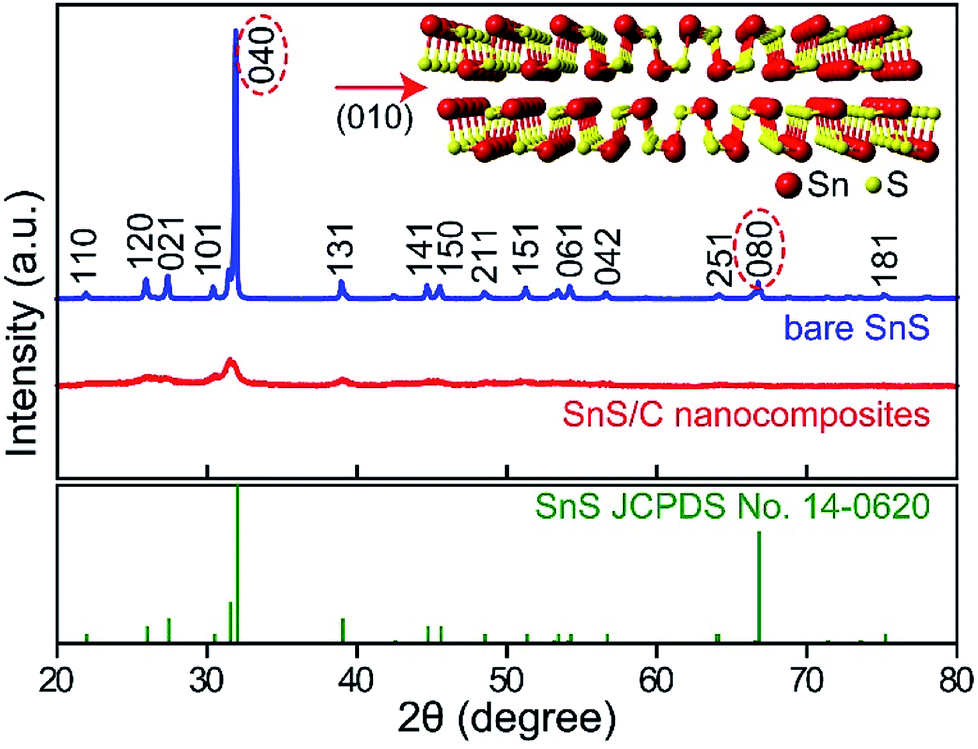 | ||
| Fig. 2 XRD patterns of bare commercial SnS and SnS/C nanocomposites (top panel) and SnS from JCPDS no. 14-0620 (bottom panel). Inset shows crystal structure of SnS. | ||
In scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of bare SnS (Fig. 2c and e), several thin (∼50 nm) micron-sized SnS layers are observed. Based on the FFT (inset to Fig. 2e), the most prominently observed interplanar distance is 0.29 nm for the (101) planes, supporting our earlier statement that the layers are preferentially oriented along the [010] direction. In the SnS/C nanocomposites, the SnS particles have a nonhomogeneous size distribution, and they are well covered by carbon particles (Fig. 2d and f). In the HRTEM and STEM images of SnS/C nanocomposites (Fig. 2f, S2 and Fig S3†), it can be observed that they still have 2D characteristic, and the typical grain size is ∼10 nm, which is in good agreement with value calculated from the XRD results presented above.
Electrochemical properties of SnS/C nanocomposites electrochemical characterization
The electrochemical properties of the bare SnS and SnS/C nanocomposites were evaluated as anode materials for SIBs. The cells were galvanostatically discharged and charged at a current density of 100 mA g−1 over the voltage range of 0.01–3.0 V (vs. Na+/Na) (Fig. 3). For the bare SnS, the initial sodiation and desodiation capacities were 740.2 and 592.5 mA h g−1, respectively (Fig. 3a). During the initial sodiation process a large plateau was observed at about 0.85 V. However, the plateau moved to higher voltages in subsequent cycles. Similarly, in the cyclic voltammetric responses, an intense peak was observed in the first reduction cycle. However, in subsequent cycles, this peak disappeared and was replaced by two separate peaks at higher potentials (0.97 and 0.67 V) (Fig. 3b). Such activation processes during the initial discharge are typically observed in conversion reaction-based materials in LIBs and SIBs and often result in a dramatic decrease in the domain size.41–43 Fig. 3c and d present discharge/charge profiles and cyclic voltammograms (CVs) for SnS/C nanocomposites, respectively. While in Fig. 3c, the first and second sodiation profiles are still different, the differences are much smaller when compared to the first two profiles for bare SnS shown in Fig. 3a. The grain size of the initial SnS/C nanocomposites is much smaller (∼10 nm) than that of the bare SnS, so the differences, before and after the first cycle in SnS/C nanocomposites are less significant. In Fig. 3d, many peaks are observed during the reduction and oxidation processes in SnS/C nanocomposites. In an effort to match reduction and anodic peaks, we performed cyclic voltammetry with different cut-off voltages after full sodiation (Fig. 3e). Based on these results, we feel confident in correcting the various cathodic peaks at 1.08, 0.65, 0.30, and 0.01 V and anodic peaks at 1.70, 1.07, 0.74, and 0.28, as representing redox pairs, respectively. It is also worth making some additional observations regarding the voltammetric behavior. First, the CVs of SnS/C nanocomposites well overlapped (Fig. 3d), indicating that stability of SnS/C nanocomposites was enhanced. Secondly, the peaks in the SnS/C nanocomposites are much better defined and with a lower overpotential than those of bare SnS, indicating improved reaction kinetics in the SnS/C composites. Fig. 3f presents the cycling performance of bare SnS and SnS/C nanocomposites. The initial desodiation capacity of SnS/C nanocomposites was 480.0 mA h g−1, which translates to 685.7 mA h g−1 when calculated based only on the weight of SnS. While the capacity of the bare SnS electrode decreased continuously during the first 50 cycles, the capacity retention of SnS/C nanocomposites was dramatically higher, reaching 96.3% after 50 cycles.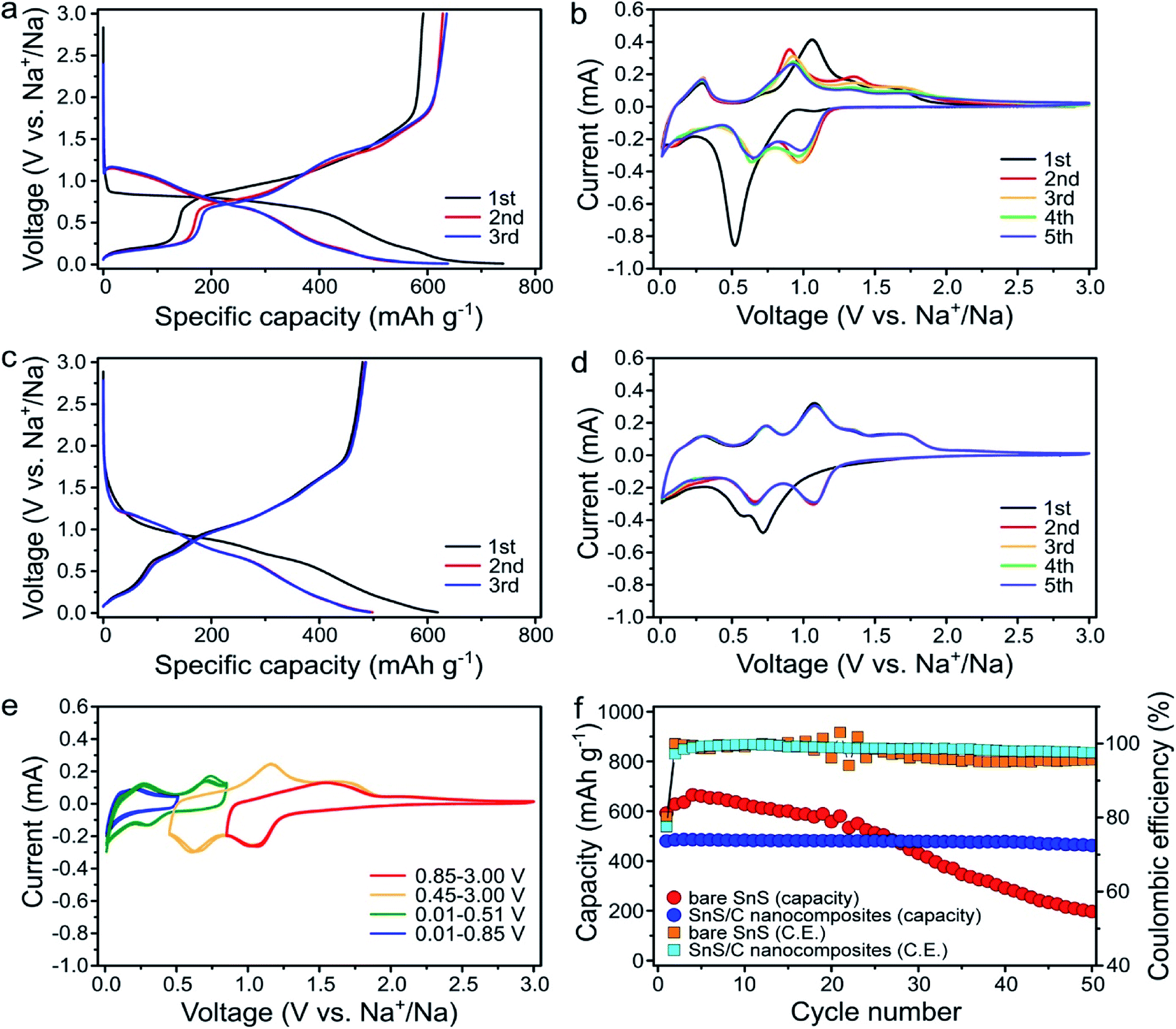 | ||
| Fig. 3 (a) First three voltage profiles of bare SnS at a current density of 100 mA g−1. (b) Cyclic voltammograms of bare SnS at a scan rate of 0.1 mV s−1. (c) First three voltage profiles of SnS/C nanocomposites at a current density of 100 mA g−1. (d) Cyclic voltammograms of SnS/C nanocomposites at a scan rate of 0.1 mV s−1 between 0.01 and 3.0 V (e) cyclic voltammograms of SnS/C nanocomposites at a scan rate of 0.1 mV s−1 with different cut off voltage ranges. (f) Cycle performance of bare SnS and SnS/C nanocomposites at a current density of 100 mA g−1. | ||
Rate properties of bare SnS and SnS/C nanocomposites were evaluated by varying the current density stepwise from 50 to 800 mA g−1 (Fig. 4a). Upon increasing the current density, the capacity of bare SnS decreased significantly. At the high current density of 800 mA g−1, bare SnS delivered a low capacity (<200 mA h g−1), while SnS/C nanocomposites delivered a much higher capacity of about 400 mA h g−1. In addition, SnS/C composites exhibited stable cycling even at a current density of 800 mA g−1. This is especially important when one considers that this current density enables two full sodiation or desodiation processess in one hour. When the current density was changed back to 50 mA g−1, after charging/discharging at 800 mA g−1, the capacity of SnS/C nanocomposites recovered their initial value, indicating that there was no structural damage to the material during high rate cycling. The voltage profiles for the fifth cycle of each current step are plotted in Fig. 4b and c for bare SnS and SnS/C nanocomposites, respectively. For bare SnS (Fig. 4b), the voltage profiles exhibited large overpotentials and the reaction plateaus were ill-defined at high current densities. Contrary to that, the SnS/C composites exhibited much smaller overpotentials in the voltage profiles, and the reaction plateaus were also clearly defined even at a current density of 800 mA g−1. Fig. 4d shows the cycle performance of SnS/C nanocomposites at various current densities (see Fig. S4† for cycle performance of ball-milled SnS without carbon at 500 mA g−1). The first reversible capacities at current densities of 50 and 500 mA g−1 were 488.2 and 441.2 mA h g−1, indicating a capacity retention of over 90% even after a tenfold increase in current density. In addition, SnS/C nanocomposites exhibited excellent cycling stability during 50 cycles at all current densities from 50 to 500 mA g−1. In particular, the capacity retention was 98.7% at 500 mA g−1; a value that is much higher than those of other reported anode materials.44–47 We also prepared SnS/C nanocomposites using mesoporous carbon (CMK), instead of super P carbon, and these exhibited much improved cycling stability and rate performance (Fig. S5†). This confirms that SnS can exhibit excellent electrochemical properties when it is ball-milled with carbon, with virtually no dependence on the type of carbon employed, so that SnS/C nanocomposites are very attractive not only in terms of their battery performance but also in practical terms.
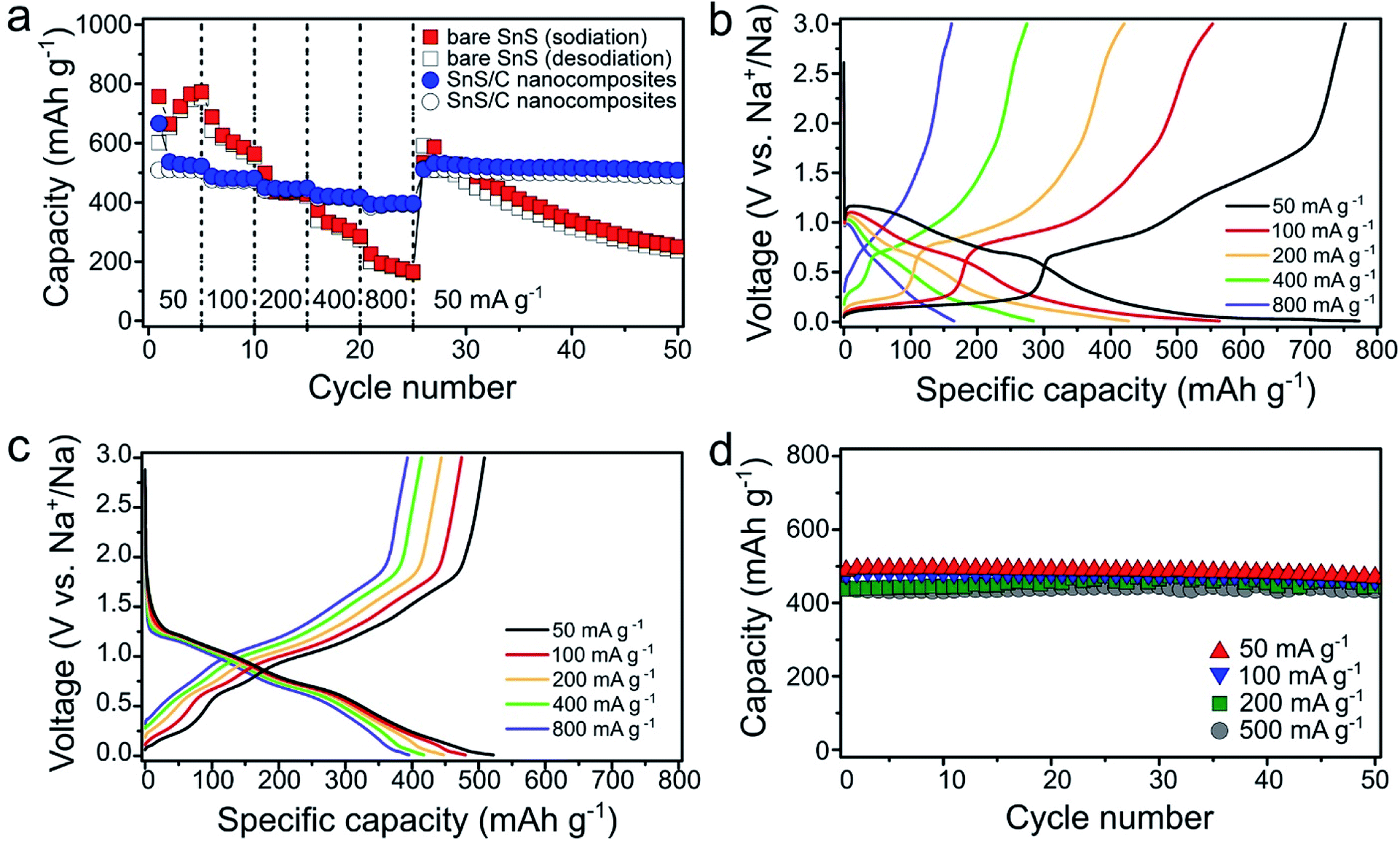 | ||
| Fig. 4 (a) Rate properties of bare SnS and SnS/C nanocomposites. Voltage profiles of (b) bare SnS and (c) SnS/C nanocomposites at various current densities. (d) Cycle performance of SnS/C nanocomposites at various current densities. | ||
Reaction mechanism of SnS/C nanocomposites
In order to better understand the reaction mechanism of the SnS/C nanocomposites, ex situ XRD and operando X-ray absorption near edge structure (XANES) analyses were performed (Fig. 5). Both Al and Cu can be used as current collectors for SIB anodes, because Al does not react with Na ions. Al is less expensive than Cu, so Al is more frequently used for SIB anodes. However, since the main XRD peaks of SnS is overlapped with Al XRD peaks, Cu foil was used as a current collector for XRD experiment. The broad XRD peak from SnS started to weaken during the initial sodiation, and disappeared at 0.67 V. At the same time, the peaks ascribed to Sn metal appeared, indicating that the conversion reaction of SnS occurred to form Sn metal clusters and amorphous sodium sulfide. At the fully sodiated state (0.01 V), Sn metal peaks can be seen in the XRD patterns. Additionally, weak electron diffraction patterns, corresponding to Sn metal and the Na15Sn4 phase were observed in the SAED pattern at the fully sodiated state (0.01 V) (Fig. S6b†). This suggests that some of the Sn metal clusters reacted with sodium to form Na15Sn4 through alloy reactions, but it was difficult to detect diffraction patterns of Na–Sn alloy phases likely because of their small particle size and low crystallinity.48,49 During desodiation, XRD peaks from Sn totally disappeared, and no other peaks nor patterns, from active material, were observed at the fully desodiated state (3.0 V). Similarly, only carbon electron diffraction patterns were observed at the fully desodiated state (Fig. S6d†). In the Sn XANES spectra, the peak height continuously decreased during sodiation (Fig. 5b), reached an intensity comparable to that of the Sn reference metal at 0.48 V, and further decreased until fully discharged. This indicates that the conversion reaction of SnS to Sn metal was complete at around 0.5 V and that further sodiation processes into Sn metal occurred at lower potentials. During desodiation (Fig. 5b), the peak height continuously increased, reached an intensity comparable to that of the Sn reference metal at about 1.00 V, and kept increasing further, indicating that the Na–Sn alloy oxidized to Sn metal, and continuously changed to SnS above 1.0 V. It is worth noting that the XANES spectra of the SnS/C nanocomposites before testing, and after the first and second cycles, at the charged state, totally overlapped, indicating that the reactions of SnS/C nanocomposites are fully reversible although the reactions go through multiple steps involving conversion and alloying reactions. When compared to tin oxide-based materials, which also go through both conversion and alloying reactions similarly to tin sulfide, their conversion reactions are known to be irreversible.50,51 | ||
| Fig. 5 (a) Ex situ XRD patterns and (b) operando XANES spectra of SnS/C nanocomposites at the Sn K edge during the first sodiation and desodiation process. (c) XANES spectra of SnS/C nanocomposites at the Sn K edge before battery testing, and after the first and second sodiation and desodiation processes. | ||
Conclusions
SnS/C nanocomposites were obtained through a practical and economical top-down approach. The unique structural properties of SnS enable to reach small nanodomain of about 10 nm using ball-milling process. The SnS/C nanocomposites exhibited superior electrochemical performance, compared to bare SnS, for sodium ion batteries. The SnS/C nanocomposites delivered a high desodiation capacity of about 480 mA h g−1 (about 686 mA h g−1 when calculated based on the weight of SnS alone) at a current density of 100 mA h g−1 through multiple reaction steps including conversion and alloying reactions. They also showed excellent cycling stability at various current densities, with more than 90% of its capacity delivered when the current increased from 50 to 500 mA g−1. The sodiation/desodiation reaction mechanism of SnS/C composites was studied by ex situ TEM, XRD and operando XANES analyses. The reactions of SnS/C nanocomposites were remarkably reversible, as established by cyclic voltammetry and operando XANES analysis. We believe that the approach that we have used in this study, for the preparation of SnS/C nanocomposites, will provide valuable insights into developing practical and promising anode materials for sodium-ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS) which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under NSF award DMR-1332208. S.-H. Yu acknowledges support from CHESS and the Energy Materials Center at Cornell (emc2). Y.-E. Sung acknowledges the financial support by IBS-R006-A2.Notes and references
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947 CrossRef.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 RSC.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636 CrossRef PubMed.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710 CrossRef.
- Y. Wang, X. Yu, S. Xu, J. Bai, R. Xiao, Y.-S. Hu, H. Li, X.-Q. Yang, L. Chen and X. Huang, Nat. Commun., 2013, 4(2365), 6 Search PubMed.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529 RSC.
- K. Kubota and S. Komaba, J. Electrochem. Soc., 2015, 162, A2538 CrossRef.
- Y. Kim, K.-H. Ha, S. M. Oh and K. T. Lee, Chem.–Eur. J., 2014, 20, 11980 CrossRef PubMed.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 125, 4731 CrossRef.
- M. Dahbi, N. Yabuuchi, K. Kubota, K. Tokiwa and S. Komaba, Phys. Chem. Chem. Phys., 2014, 16, 15007 RSC.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033 CrossRef PubMed.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859 CrossRef.
- E. Irisarri, A. Ponrouch and M. Palacin, J. Electrochem. Soc., 2015, 162, A2476 CrossRef.
- V. Chevrier and G. Ceder, J. Electrochem. Soc., 2011, 158, A1011 CrossRef.
- L. D. Ellis, T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2012, 159, A1801 CrossRef.
- Y. Liu, N. Zhang, L. Jiao, Z. Tao and J. Chen, Adv. Funct. Mater., 2015, 25, 214 CrossRef.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657 CrossRef PubMed.
- B. Zhang, G. Rousse, D. Foix, R. Dugas, D. A. D. Corte and J.-M. Tarascon, Adv. Mater., 2016, 28, 9824 CrossRef PubMed.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805 CrossRef PubMed.
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255 CrossRef PubMed.
- L. Hu, X. Zhu, Y. Du, Y. Li, X. Zhou and J. Bao, Chem. Mater., 2015, 27, 8138 CrossRef.
- L. Baggetto, J. K. Keum, J. F. Browning and G. M. Veith, Electrochem. Commun., 2013, 34, 41 CrossRef.
- Z. Li, X. Tan, P. Li, P. Kalisvaart, M. T. Janish, W. M. Mook, E. J. Luber, K. L. Jungjohann, C. B. Carter and D. Mitlin, Nano Lett., 2015, 15, 6339 CrossRef PubMed.
- S. H. Choi and Y. C. Kang, Nano Res., 2015, 8, 1595 CrossRef.
- L. Wu, H. Lu, L. Xiao, J. Qian, X. Ai, H. Yang and Y. Cao, J. Mater. Chem. A, 2014, 2, 16424 RSC.
- Y. Jiang, M. Hu, D. Zhang, T. Yuan, W. Sun, B. Xu and M. Yan, Nano Energy, 2014, 5, 60 CrossRef.
- Y. Liu, N. Zhang, C. Yu, L. Jiao and J. Chen, Nano Lett., 2016, 16, 3321 CrossRef PubMed.
- K. He, F. Lin, Y. Zhu, X. Yu, J. Li, R. Lin, D. Nordlund, T.-C. Weng, R. M. Richards, X.-Q. Yang, M. M. Doeff, E. A. Stach, Y. Mo, H. L. Xin and D. Su, Nano Lett., 2015, 15, 5755 CrossRef PubMed.
- Y. Denis, P. V. Prikhodchenko, C. W. Mason, S. K. Batabyal, J. Gun, S. Sladkevich, A. G. Medvedev and O. Lev, Nat. Commun., 2013, 4, 2922 CrossRef PubMed.
- T. Zhou, W. K. Pang, C. Zhang, J. Yang, Z. Chen, H. K. Liu and Z. Guo, ACS Nano, 2014, 8, 8323 CrossRef PubMed.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759 CrossRef PubMed.
- Y. L. Ding, P. Kopold, K. Hahn, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2016, 28, 7774 CrossRef PubMed.
- Y. Xiao, S. H. Lee and Y.-K. Sun, Adv. Energy Mater., 2017, 7, 1601329 CrossRef.
- Z. Hu, Q. Liu, S.-L. Chou and S.-X. Dou, Adv. Mater., 2017, 29, 1700606 CrossRef PubMed.
- P. Ge, C. Zhang, H. Hou, B. Wu, L. Zhou, S. Li, T. Wu, J. Hu, L. Mai and X. Ji, Nano Energy, 2018, 48, 617 CrossRef.
- M. Wan, R. Zeng, K. Chen, G. Liu, W. Chen, L. Wang, N. Zhang, L. Xue, W. Zhang and Y. Huang, Energy Storage Materials, 2018, 10, 114 CrossRef.
- P. Ge, H. Hou, X. Ji, Z. Huang, S. Li and L. Huang, Mater. Chem. Phys., 2018, 203, 185 CrossRef.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169 CrossRef PubMed.
- A. P. Cohn, K. Share, R. Carter, L. Oakes and C. L. Pint, Nano Lett., 2016, 16, 543 CrossRef PubMed.
- H. Kim, J. Hong, G. Yoon, H. Kim, K.-Y. Park, M.-S. Park, W.-S. Yoon and K. Kang, Energy Environ. Sci., 2015, 8, 2963 RSC.
- S.-H. Yu, S. H. Lee, D. J. Lee, Y.-E. Sung and T. Hyeon, Small, 2016, 16, 2146 CrossRef PubMed.
- M. A. Lowe, J. Gao and H. D. Abruña, J. Mater. Chem., 2013, 1, 2094 RSC.
- F. Klein, B. Jache, A. Bhide and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 15876 RSC.
- Y.-M. Lin, P. R. Abel, A. Gupta, J. B. Goodenough, A. Heller and C. B. Mullins, ACS Appl. Mater. Interfaces, 2013, 5, 8273 CrossRef PubMed.
- P. K. Dutta, U. K. Sen and S. Mitra, RSC Adv., 2014, 4, 43155 RSC.
- Y. Zhu, X. Han, Y. Xu, Y. Liu, S. Zheng, K. Xu, L. Hu and C. Wang, ACS Nano, 2013, 7, 6378 CrossRef PubMed.
- Z. Jian, B. Zhao, P. Liu, F. Li, M. Zheng, M. Chen, Y. Shi and H. Zhou, Chem. Commun., 2014, 50, 1215 RSC.
- J. W. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett., 2012, 12, 5897 CrossRef PubMed.
- C. Ma, J. Xu, J. Alvarado, B. Qu, J. Somerville, J. Y. Lee and Y. S. Meng, Chem. Mater., 2015, 27, 5633 CrossRef.
- I. A. Courtney and J. Dahn, J. Electrochem. Soc., 1997, 144, 2045 CrossRef.
- M. Gu, A. Kushima, Y. Shao, J.-G. Zhang, J. Liu, N. D. Browning, J. Li and C. Wang, Nano Lett., 2013, 13, 5203 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04421j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |