
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ insights into the nanoscale deposition of 5,6-dihydroxyindole-based coatings and the implications on the underwater adhesion mechanism of polydopamine coatings†
Qinghua Lyua,
Hongyan Songb,
Nikolai L. Yakovlevb,
Wui Siew Tan*b and
Christina L. L. Chai *a
*a
aDepartment of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore 117543. E-mail: phacllc@nus.edu.sg
bInstitute of Materials Research and Engineering, A*STAR, 2 Fusionopolis Way, Singapore 138634. E-mail: wuisiew@gmail.com
First published on 3rd August 2018
Abstract
The biomimetic coating polydopamine (PDA) has emerged as a promising coating material for various applications. However, the mechanism of PDA deposition onto surfaces is not fully understood, and the coating components of PDA and its relation to the putative intermediate 5,6-dihydroxyindole (DHI) are still controversial. This investigation discloses the deposition mechanisms of dopamine (DA)-based coatings and DHI-based coatings onto silicon surfaces by monitoring the nanoscale deposition of both coatings in situ using high-precision ellipsometry. We posit that the rapid and instantaneous nano-deposition of PDA coatings onto silicon surface in the initial stages critically involves the oxidation of DHI and/or its related oligomers. Our studies also show that the slow conversion of DA to DHI in PDA solution and the coupling between DA and DHI-derived precursors could be crucial for subsequent PDA coating growth. These findings elucidate the critical role of DHI, acting as an ‘initiator’ and a ‘cross linker’, in the PDA coating formation. Overall, our study provides important information on the early stage nano-deposition behavior in the construction of PDA coatings and DHI-based coatings.
Introduction
Underwater adhesion is a remarkably difficult process to effect due to the presence of a thin hydration layer that prevents the contact between a polymer and the surface. Yet in nature, there are many examples of marine adhesives that are able to glue surfaces underwater with high strength and durability. One example is the marine mussel adhesive proteins (MAPs) that enable mussels to adhere to surfaces (e.g. intertidal rocks) with great tenacity.1–3 In the context of MAPs, studies have identified that the presence of 3,4-dihydroxyphenylalanine (DOPA) and lysine residues are critical for the adhesive properties.4–9 Inspired by this, a number of biomimetic catechol(amine)-based materials have been developed in the past decade.10–15 Among these, polydopamine (PDA) holds the greatest promise due to the ease of formation, almost universal substrate scope, versatility and ease of further functionalization; these properties have garnered significant interest in applications such as anti-bacterial and anti-fouling coatings, nanotechnology and biomedicine.16–20Despite the promising applications of PDA films in various fields, several important issues remain unresolved.21 One of these issues pertains to the deposition mechanisms of PDA coatings, which remains unclear. Currently, limited techniques are available to investigate the complex underwater self-assembly of PDA coatings onto surfaces. In addition, despite the numerous reports on the proposed structures of PDA, the structure and the constituent building block(s) of PDA are still controversial. Different models, including the ‘open-chain polycatechol quinone’ model; 5,6-dihydroxyindoline/its quinone supramolecular structure; covalent polymers derived from dopamine (DA) and its oxidized cyclization product dihydroxyindole (DHI); and single DHI-based eumelanin model etc., have been proposed.22–28 While a number of intermediates have been proposed to be involved in PDA coating formation, the common understanding is that DHI is the key precursor involved in the subsequent oxidative polymerization to form coatings, in a manner that is similar to the biosynthesis of melanin.29,30 Hong et al. reported the isolation of a physical DHI/(DA)2 non-covalent complex from DA–phosphate buffer solution,23 while Vecchia and Liebscher et al. demonstrated the presence of DHI-based units in PDA oligomers using a chemical degradation approach and high resolution mass spectrometry respectively.25,26 However, the most recent report by Alfieri et al. found that the use of DHI under classical conditions i.e. Tris buffer pH 8.5 did not give rise to any coatings.31 Due to the complicated aggregation/organization of PDA, a definitive relationship between PDA coating and its putative intermediate DHI has not been demonstrated unequivocally.
In this study, we aimed to explore the deposition mechanism of PDA coatings onto silicon wafers by the in situ monitoring of the nano-coating process via high-precision ellipsometry (HPEL). This purpose-built equipment with a 0.1 nm sensitivity has been successfully utilised in the study of adsorption and desorption of single molecules.32 DA and the putative intermediate DHI were used in this study to gain a better understanding of the structural features needed for adhesion onto surfaces. To the best of our knowledge, this is the first reported in situ study on the nanoscale deposition of PDA- and DHI-based coatings using HPEL. Using a combination of techniques including Atomic Force Microscope (AFM), Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR) and Nuclear Magnetic Resonance (NMR), we were able to elucidate the deposition mechanisms of both PDA and DHI-derived coatings. Our studies revealed new insights into the relationship between PDA coating and its putative intermediate DHI.
Results and discussion
The setup for in situ HPEL is shown in Fig. S1.† The general experimental protocol can be briefly described as follows: 1 mL sample of DA or DHI was prepared and injected into the HPEL cuvette. This was allowed to incubate for a predetermined time to allow for deposition onto the silicon substrates. Then de-ionized water was flowed into the system in order to test the robustness of the film/coating formed underwater. The initial deposition kinetics of DA on silicon wafers in different pH Tris-buffers under static conditions were first examined. As displayed in Fig. 1, DA samples at pH 5 and 7 only gave rise to coatings with a thickness of 0.4 ± 0.1 nm after a 20 min incubation. The thickness of these deposited coatings was close to the reported self-assembly monolayer.33 However, these layers were readily washed away during the subsequent flow of de-ionized water, indicating that the adsorption of DA at these pH values is physical and reversible. At pH 8.5, significant deposition was observed and the deposited coatings were robust (i.e. adhered to the surface) despite treatment with water flow (for the effects of different concentrations and buffers on deposition kinetics, see Fig. S2†). It was also observed that the deposition rate dropped rapidly with time under Tris conditions: the coatings were deposited linearly in the first 8 min with a deposition rate of 0.15 ± 0.01 nm min−1, while the deposition rate decreased to half i.e. 0.07 ± 0.02 nm min−1 in the following 8 min. After 20 min, the deposition rate was much slower than the initial stage (Fig. S2†), implying that the coating growth had reached steady state. These results were in line with the findings of Bernsmann et al. in which the growth of PDA coating on silicon oxide was examined using quartz crystal microbalance: a 2.46 nm-thick coating was formed in 15 min and a drop in deposition rate in Tris buffer was also noted in their study.34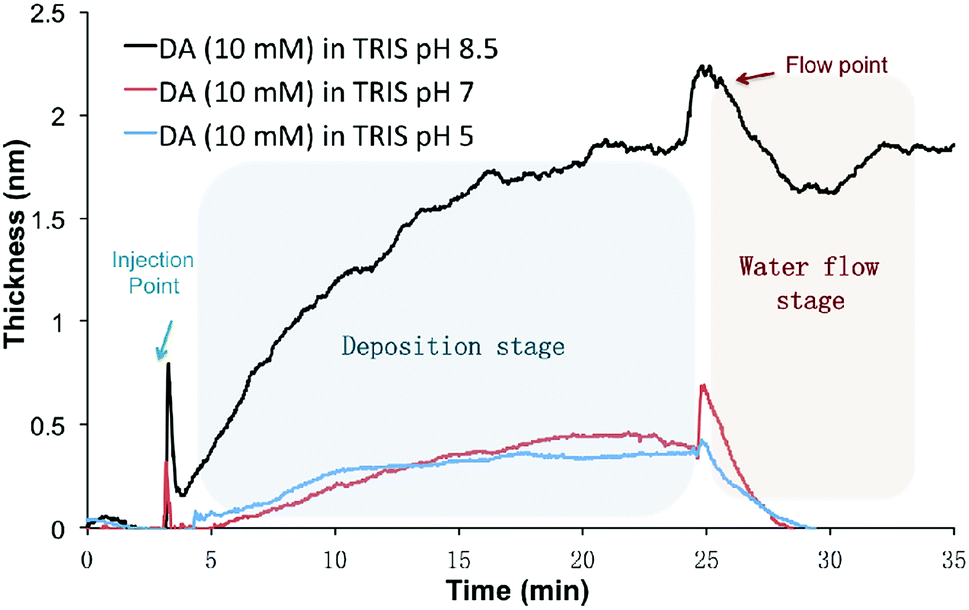 | ||
| Fig. 1 The nano-deposition kinetics of DA-based coatings on Si wafer at pH 5, 7 and 8.5 in the first 30 min (10 mM DA, 10 mM Tris buffer). | ||
The fast deposition of films in alkaline buffer is consistent with previous reports that the formation of PDA coatings requires an elevated pH (mildly alkaline) to initiate the oxidative polymerization of the DA monomer.35,36 It should be noted that this rapid and instantaneous deposition differs from the slow self-assembly of DA at lower pH conditions (Fig. 1). Although the presence of the catechol moiety is one important contributor to the process of underwater adhesion,37–42 some other interactions must also be responsible for this rapid deposition onto the silicon surface, e.g. electrostatic or π–π stacking.43,44 Others have hypothesized that the deposition is initiated by radical species at the early stages, followed by the polymerization of these species to coat the surfaces.34,45–47 In the next section, we examined if the putative intermediate DHI can initiate a fast and spontaneous deposition on surfaces.
A quantitative study of DHI formation in PDA solution (10 mM DA, 10 mM Tris buffer 8.5) was carried out for the first time. The authentic, air-sensitive DHI compound was freshly prepared via the reported method.48 In our experiment that mimics the coating process (minus the silicon substrate), a 100 mL DA–Tris solution was exposed to air for a predetermined time following which the organics were extracted using ethyl acetate and the concentrated extracts were analysed. From the 1H-NMR spectra (Fig. 2), DHI was shown to be the main intermediate present, along with the presence of unreacted DA monomer. Interestingly, these 1H-NMR signals matched the spectra of the mixture of DHI/DA, but were not in good agreement with the spectrum of pure DHI (Fig. S3†), suggesting the presence of strong inter-molecular interactions between DA and DHI. Hong et al. also noted a similar non-covalent interaction between DA and DHI in PDA solution and isolated a self-assembled physical complex of DHI/(DA)2 from DA-phosphate buffer.23 We carefully quantified the DHI formed in solution by 1H-NMR spectroscopy using dimethylsulfoxide (DMSO) as an internal standard and found that the real-time concentration of DHI was ca. 4.3 μM as measured at the 30 min time-point. Further experiments showed that the concentration of DHI at 1, 4 and 8 h were also at micromolar levels, ranging from 3.6 to 0.9 μM. These data demonstrated that DHI is one main oxidized intermediate formed in the PDA solution.
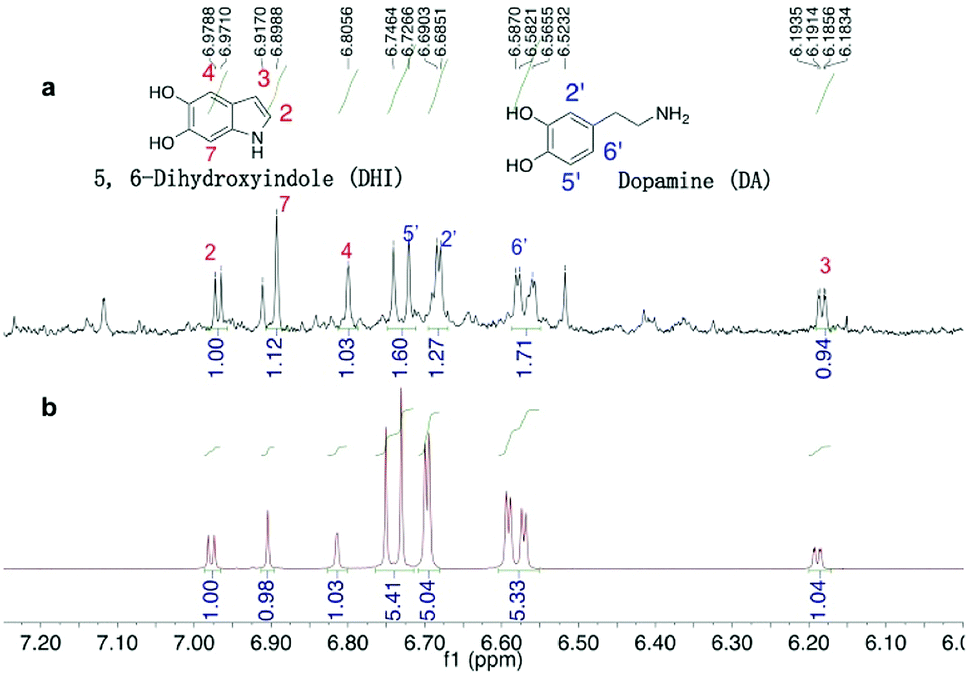 | ||
| Fig. 2 1H NMR spectra of (a) organic extracts of PDA solution and (b) authentic DA/DHI mixture. | ||
The deposition of DHI on silicon surfaces was then studied to examine whether this intermediate can form coatings on its own. DHI solutions were prepared under conditions similar to that used for PDA coatings (10 mM precursor, Tris buffer 8.5). Once DHI sample was injected, a fast and instantaneous deposition was noted; after a few minutes, however, the beam intensity dropped significantly as the solution darkened with time, and subsequent deposition kinetics could not be monitored (Fig. 3, indicated as asterisk). When fresh DHI–Tris samples (10 mM) at pH 5 and 7 were used, rapid depositions onto surfaces were observed at both the pH values. The thickness of coatings formed at pH 7 and pH 5 after 30 min were 7.3 ± 0.2 nm and 5.7 ± 0.2 nm respectively. These coatings were robust enough to endure the treatment with water-flow. The successful depositions of these coatings onto the silicon substrate were verified by further AFM analyses (discussed further below). In addition, rapid drop in deposition rates with time at both pH 5 and 7 were observed. The decrease of deposition rates may be explained by the saturation of binding sites on the silicon surfaces and/or competing self-aggregation pathway of DHI in solutions. The latter was supported by the observation that aged DHI solution (10 mM, pH 7, exposed to air for 10 min) indeed led to a slower deposition of coatings on a clean silicon substrate (Fig. 3, indicated as red dashed line) than when a fresh DHI solution was used. This indicates that DHI and/or its low molecular weight oligomers are capable of coating silicon substrates rapidly, while high molecular weight oligomers or aggregates of DHI may possess weak adhesion property, possibly due to their inability to stack efficiently.49,50
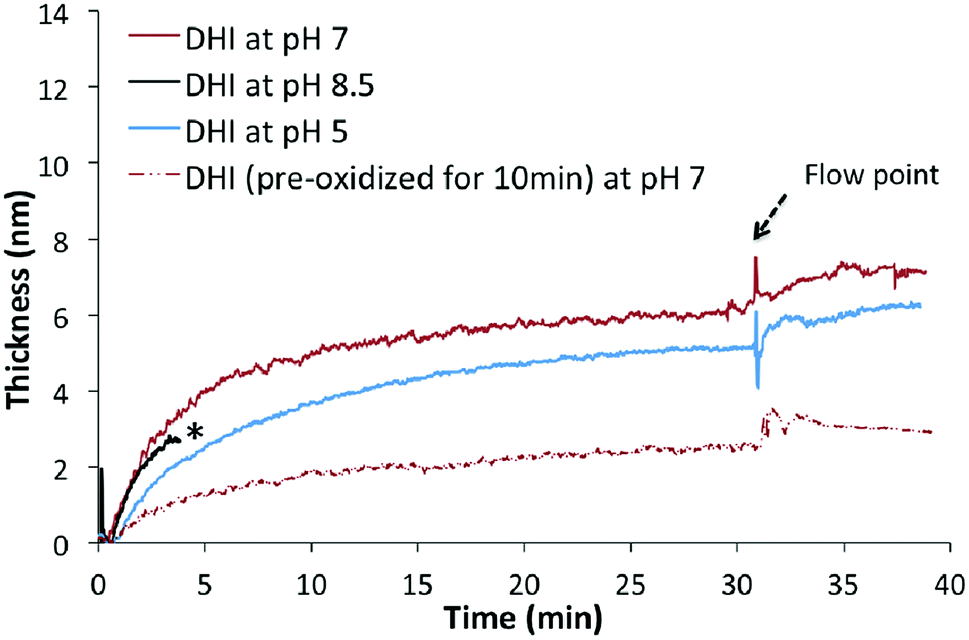 | ||
| Fig. 3 The nano-deposition kinetics of DHI-based coatings at different pHs in the first 30 min. | ||
We further investigated the role of oxidation in the initial stages of DHI coating deposition under neutral conditions (pH 7). Pre-treatment of DHI–Tris solution (pH 7) with N2 degassing resulted in a relative lower deposition rate. Similarly, the addition of reducing agent Na2S2O4 into the DHI–Tris solution significantly slowed down the deposition process. In this case, only a 0.5 ± 0.1 nm-thick coating was formed after 30 min of incubation (Fig. S4†). The above observation suggests that the oxidation of DHI is critical in initiating rapid coatings onto the silicon surface. This is in accordance with previous studies on the electrochemical self-assembly of DHI-based films.51,52 Based on these findings, the deposition mechanism of DHI can be interpreted as the initial attachment of small oxidized species, followed by the polymerization of these deposited species. To further support this notion, the durability of DHI coatings in the initial stage (1–10 min in this context) was further examined. The initial deposits (1.3 ± 0.1 nm) formed at 1 min were robust under water flow conditions, while treatment with aqueous Na2S2O4 showed an instantaneous decrease in coating thickness to ca. 0.5 ± 0.2 nm (Fig. 4a). Similar reducing treatment to the coating obtained after above 4 min deposition time did not show any significant effect on the stability of the coatings (Fig. 4b), suggesting that these deposited species may have already formed irreversible linkages e.g. through covalent bonding and the coating is not compromised by the treatment with reducing agents.
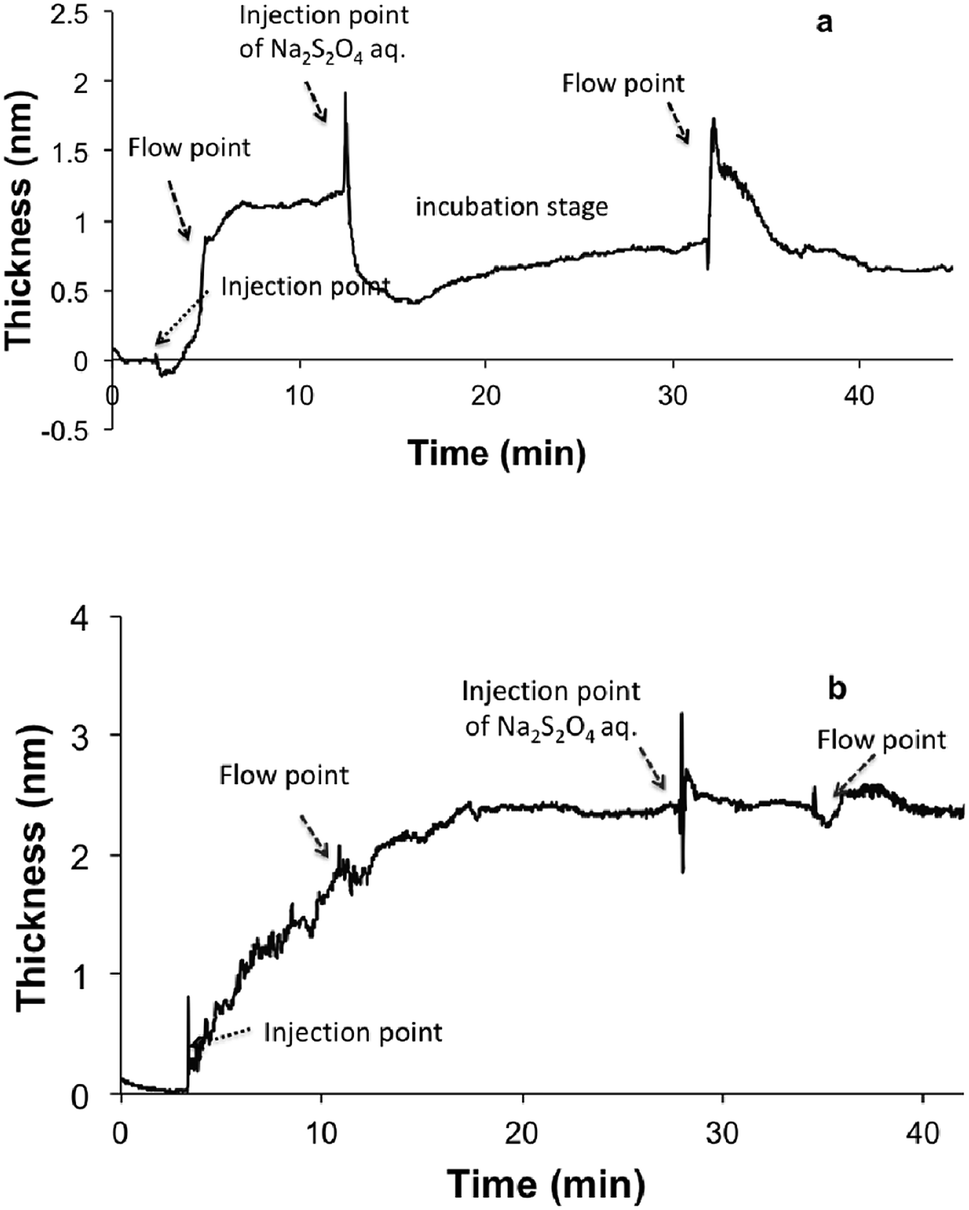 | ||
| Fig. 4 The stability of DHI-based nano-coatings in the initial (a) and later (b) stages following treatment with reducing agent. | ||
Further AFM studies verified that coatings were formed on the silicon substrates in the deposition study (Fig. 3, 10 mM DHI, Tris buffer pH 5, 7 or 8.5, 30 min). However, a smaller amount of DHI-coating was formed at pH 8.5 and the surface was not fully coated (Fig. 5a), while at low pH values the substrates were well coated and uniform disk-shaped topographies were observed (Fig. 5b and S5†) which were consistent with the observations in recent DHI-based coating studies using spin-coating,53,54 supporting the notion of stacking interactions (presumably π–π) leading to larger aggregates.55–58 We tentatively rationalized that the poor DHI-coating obtained at alkaline pH maybe the result of rapid oxidation and self-aggregation effects in solutions.
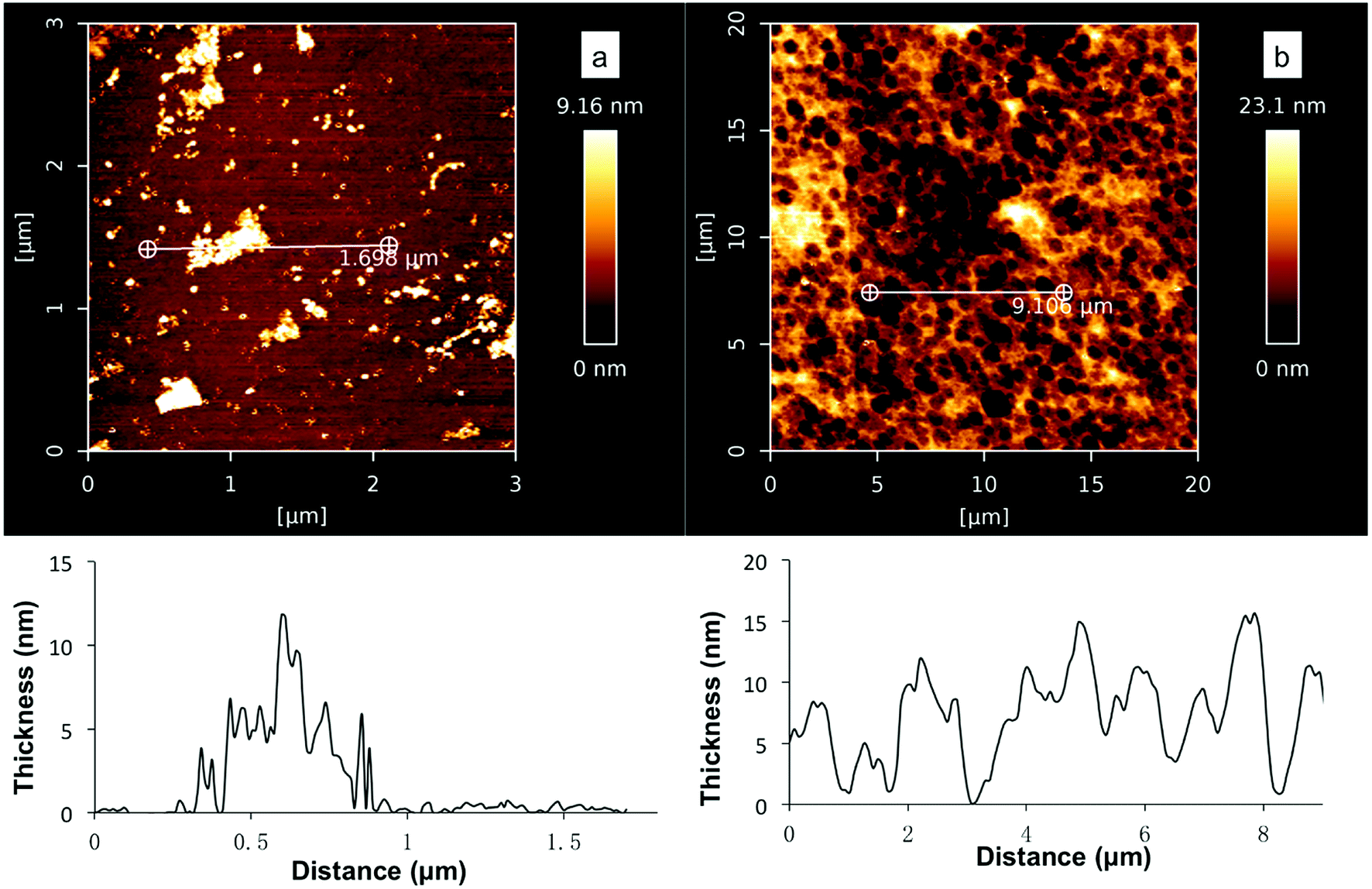 | ||
| Fig. 5 AFM images of DHI-coatings after 30 min at (a) pH 8.5; (b) pH 7. | ||
It was also found that at pH 8.5 and 7, no significant increase in the amount of coating was observed after 1 h and 4 h respectively. At pH 8.5, slightly more amorphous granules with 20–40 nm thickness were noted but the surfaces were not well coated (Fig. S6†), while at pH 7 the maximal thickness of DHI coatings at 15 ± 2 nm was observed after a long deposition time (4 h). It should be noted that the thickness of DHI-based coatings obtained at pH 7 (10 mM DHI) was still less than that of PDA coatings prepared under the same concentrations (ca. 40 nm, 10 mM DA, Tris pH 8.5). These results show that DHI-related species are able to adhere efficiently and robustly to silicon surfaces but competing self-aggregation in solutions can significantly affect the deposition process. This proposed competing process shares similarities with the findings of Arzillo et al. on the buildup of eumelanin that DHI can rapidly adsorb onto the surfaces of DHI-eumelanin particles.57
Our observations strongly suggest that the deposition process of DHI-based coatings is dominated by monomeric species of DHI, low molecular weight oligomers and/or small nano-aggregates, while larger aggregates and particles cannot deposit onto silicon surfaces but competitively suppress the deposition process via the sequestration of these species, leading to a self-limiting thickness of coatings. As the competing self-aggregation of DHI/its oligomers in solution is unavoidable, it can be speculated that neither the use of small nor large amounts of DHI can yield substantially thicker coatings. Small quantities of DHI cannot provide enough DHI-related species as feeds for sustaining coating growth, while the use of larger amounts of DHI may lead to rapid coating growth in the early stages, but the simultaneous rapid formation of large aggregates/particles will affect the subsequent coating growth. Therefore, an ideal coating protocol for DHI-based components would be a continual supply of fresh DHI in small quantities as feed, lest a significant amount of aggregates/particles are formed in solution. Interestingly, the formation of PDA coatings meets this prerequisite as the DHI generated in situ is used as feed at low concentrations, as verified in our quantitative studies. The slow conversion of DA to DHI in PDA solutions and the strong non-covalent interactions between DA and DHI could be important factors in retarding the rapid self-polymerization/aggregation of DHI at alkaline pHs.59 These notions can explain the observed thickness difference between PDA coatings and DHI coatings and also the observed self-limiting thickness of PDA coatings reported in our studies (Fig. S2†) and other studies.34,60,61
A question that arises here is whether PDA coating formation is mainly based on DHI-derived components. To characterise DHI-based coatings and PDA coatings by ATR-FTIR, dip-coating studies with DA and DHI precursors, respectively, were carried out on quartz substrates. Consistent with our deposition studies on silicon substrate, no significant DHI-based coating was formed at pH 8.5, while at pH 7 a rough DHI-coating film with a thickness 18–23 nm was obtained after 24 h (Fig. S7†). AFM analyses on the DHI coating and PDA coating revealed that the morphology of DHI coatings was different to that from PDA films, as shown in Fig. 6a and b. Further characterization of the DHI-based film and PDA film based on ATR-FTIR analyses showed that the fingerprint region from 1880–780 cm−1 of PDA films was different to that of DHI-based film (Fig. 6c). The latter has higher intensity bands around 1180 cm−1 that can be assigned to the stretching of C–N, C–C, C–H bending vibrations and indole ring vibrations and also a higher intensity band centered at 856 cm−1 which is possibly due to the bending N–H vibrations, suggesting that the constituents of PDA coatings are not identical to coatings derived from DHI. Collectively, this study and reported studies by Alfieri et al.,31 did not support the notion that the primary building blocks of PDA coatings are DHI-only based oligomers, e.g. trimer complex in Ding's study27 or porphyrin-type DHI-tetramer in Chen's modeling study.62 As the classical PDA solution comprises a large amount of DA precursor, micromolar-quantities of DHI and also other possible intermediates present at low quantities (not detectable by 1H-NMR spectroscopy), it is possible that the highly reactive DHI/its oligomers can couple with DA to form the main adhesive/cohesive units of the PDA coating.23,25,63
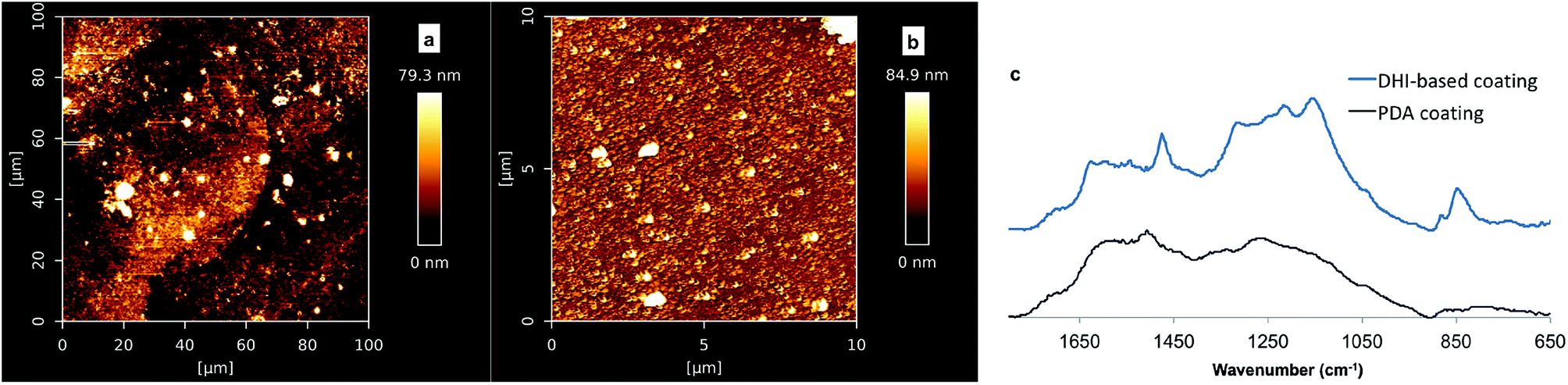 | ||
| Fig. 6 AFM images of (a) DHI-coating (pH 7, 24 h, ca. 18–23 nm) and (b) PDA coatings (pH 8.5, 24 h, ca. 40 nm) on quartz substrates; (c) ATR-FTIR spectra of DHI-based coating film and PDA coating film on quartz slides. | ||
To support the view of conjugation between DA and DHI/its oligomers, ESI- and APCI-Mass analyses was carried out on the following samples: (i) mixture of DA/DHI sample (10 mM DA, 1 mM DHI, Tris pH 8.5), (ii) DA–Tris sample (10 mM, Tris pH 8.5) and (iii) DHI–Tris sample (1 mM DHI, Tris pH 8.5). The main peak patterns observed from the DA/DHI sample (t = 30 min) from mass spectral analysis were consistent with that of DA sample (4 h) in our study and other reported PDA studies (Fig. S8†).25,26 Higher intensity signals of common peaks e.g. 390, 402, 420, 432 and 450 m/z, were found in the spectra of DA/DHI sample (30 min) as compared to that of the DA sample. In contrast, these peaks were not observed to be significant in the sample derived from DHI only. Assuming that the components in solution mirror that of the coatings, these observations suggest that DA and DHI/its oligomers can form covalent adducts,23 which may rationalize the observed difference between PDA coating and DHI-based coating. As many studies have reported the necessity of a free amine structural element in the formation of catecholic-based coating materials,64–67 the presence of conjugation adducts derived from both DA and DHI is likely to be the major determinants of the adhesion/cohesion properties of PDA films. The precise structures of DA/DHI conjugates remain to be further identified.
Conclusion
In summary, this investigation provides in situ insights into the nanoscale deposition of DHI-based coatings using HPEL and elucidates the critical roles of DHI in the formation of PDA coatings. To our knowledge, this is the first report of the deposition mechanism of DHI-based coatings. It is noted that the deposition of DHI-based nano-coating is oxidation-dependent and pH-dependent. Our studies also show that competing self-aggregation of DHI oligomers can significantly inhibit the coating process, especially at alkaline pHs. It can be rationalized that DHI is able to play a dual role to initiate the coating process as well as to act as a ‘cross-linker’ in the formation of PDA coating films. Specifically, we propose that fast nano-deposition of PDA coatings onto the silicon substrate in the initial stages of the coating process critically involves the oxidation of DHI and/or its related oligomers. This is followed by the coupling of DHI/its oligomers with DA-related species to provide the adhesive/cohesive units for PDA film formation. Our data also supports the notion that the DA monomer provides a critical structural element i.e. the free primary amine.68 However we acknowledge that PDA coatings may possess heterogeneity and our studies do not preclude other parallel pathways to PDA coatings. The results of our research provide critical information on the deposition and formation of PDA coatings, which could be used to improve the properties of PDA and DHI-based coatings, as well as provide new insights for the development of catecholic coating materials.Experimental
In situ ellipsometry study on deposition kinetics of DA and DHI precursors
Ellipsometry is a technique which measures the polarization of light reflected off the sample surface (Fig. S1†). It derives sensitivity from measuring the phase shift Δ between normal (p) and in-plane (s) components of polarization vector. For layers much thinner than wavelength, d ≪ λ, Δ is proportional to the thickness. Precision ellipsometry (PREL) uses modulation of polarization, making it much more sensitive. In the present setup, the light from laser pointer, λ = 650 nm, passes through linear polarizer onto the substrate. Adsorbed molecules or ultrathin layers change reflected polarization to elliptic; the retarder converts ellipticity Δ into rotation γ, which is measured using polarization modulator. Thus, γ is proportional to Δ, hence proportional to the thickness. For organic molecules on silicon, 1 nm corresponds to 2 milliradians of polarization rotation. This coefficient was used to obtain thickness scale on all figures. With compact modulator made in A*STAR's Institute of Materials Research and Engineering, this system measures polarization rotation down to microradians. This translates to sensitivity of 0.1 nm of attached layer thickness.General procedure for deposition studies
(i) A clean silicon water (0.5 cm × 1.5 cm) was set inside the cuvette of ellipsometry; when opening the pump and flow control valve, deionized-water (DI-H2O) was flowed through the ellipsometry system; (ii) after the reading output was stabilized at a certain value, the flow control valve was closed and 1 mL fresh DA or DHI samples at the specified buffers were injected to drain the water out; (iv) the pump was then closed and the injected samples were incubated for a predetermined time (generally 20–30 min) to allow the coatings to deposit onto silicon surfaces; (v) then both of the flow control valve and pump system were opened and DI-H2O water was flowed through the cuvette for 15–20 min to wash off the injected sample solutions and to test the underwater adhesion. In separate experiments, Na2S2O4 (20 mM) was then injected into the cuvette and incubated for a predetermined time (5–6 min), followed by flowing DI-H2O to test the durability of coatings deposited on silicon. For each experiment, at least three samples were tested.Substrates used for coating studies and general dip and coat procedure
Silicon (Si) wafers were obtained from Mitsubishi Silicon America, USA. Quartz microscope slides (fused, 76.2 mm × 25.4 mm × 1.0 mm) were purchased from Alfa Aesar. Si wafers were pre-cleaned in a fresh H2O/NH4OH/H2O2 (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) solution at 150 °C for 10 min, and then rinsed in deionized (DI) water and ethanol for 10 min each. Quartz slides substrates were first cleaned in an ultrasound water bath for 30 min, rinsed with ethanol, and then blown dry under nitrogen gas. The substrates were then immersed in 20 mL of Tris buffers as indicated with specified precursors. After coating for a specified time, the substrates were rinsed with water and ethanol, and dried with a stream of nitrogen gas. These samples were subsequently characterized as described below.
:1:1) solution at 150 °C for 10 min, and then rinsed in deionized (DI) water and ethanol for 10 min each. Quartz slides substrates were first cleaned in an ultrasound water bath for 30 min, rinsed with ethanol, and then blown dry under nitrogen gas. The substrates were then immersed in 20 mL of Tris buffers as indicated with specified precursors. After coating for a specified time, the substrates were rinsed with water and ethanol, and dried with a stream of nitrogen gas. These samples were subsequently characterized as described below.
Characterization of coatings
AFM characterization was carried out using a Nanowizard III instrument (JPK Instruments AG, Berlin, Germany) equipped with NanoWizard head and controller. The triangular shaped silicon nitride cantilevers (Nano World, PNP-TR) were used throughout the scanning and the spring constant was calibrated using the thermal noise method, in the range of 0.07–0.09 N m−1. The experiments were performed in air, letting the system equilibrate for 30–60 min. To carry out the measurement, the coated samples were fixed on glass slides before each measurement. The quantitative imaging mode (QITM) was performed for imaging. The QITM is a force spectroscopy based imaging mode that enables the user to have the full control over the tip-sample force at each pixel of the image so that the lateral forces can be greatly minimized, making nondestructive imaging straightforward. All images were processed using the JPK data processing software (Version 6.1.22). Attenuated total reflection (ATR)-Fourier transform infrared spectroscopy (FTIR) analysis was carried out on a Bruker Alpha spectrometer equipped with (RT) DLaTGS detector (Bruker, USA) and a platinum ATR accessory with a 2 mm diameter single reflection diamond crystal. A clean silicon wafer and a quartz slide were used as references respectively.Synthesis of 5,6-dihydroxyindole (DHI)
All reagents and solvents were purchased from Sigma Aldrich or Alfa Aesar and were used without further purification unless otherwise specified. NMR spectra were recorded on a Bruker Avance III 400 MHz spectrometer at 400 MHz for 1H and at 100 MHz for 13C with methanol-d4 as solvent. The chemical shifts are given in ppm, using the proton solvent residue signal (CD3OD: δ 3.31) as a reference in the 1H NMR spectrum. The deuterium coupled signal of the solvent was used as a reference in 13C-NMR (CD3OD: δ 49.00). The following abbreviations were used to describe the signals: s = singlet, d = doublet, t = triplet, m = multiplet, q = quartet. DHI was prepared under nitrogen atmosphere using the reported method.48 A mixture of K3[Fe(CN)6] (6.6 g, 20 mmol) and NaHCO3 (2.5 g, 30 mmol) in H2O (60 mL) was added dropwise over 5 min to a stirred solution of L-DOPA (0.99 g, 5 mmol) in 500 mL H2O. The resulting solution was stirred at room temperature under nitrogen atmosphere for 3 h following which 600 mg Na2S2O4 was added. The solution was then adjusted to pH 4 with 3 M HCl aq. and extracted with ethyl acetate (250 mL × 3). The combined organic phases were washed with saturated brine (100 mL × 3) and were dried over Na2SO4. Evaporation of ethyl acetate to 5 mL followed by the addition of hexane (50 mL) yielded a pale brown solid; the solids were then re-dissolved in ethyl acetate (5 mL) followed by recrystallization from hexane (50 mL) to give 287 mg (27% yield) of DHI as an off-white solid. 1H-NMR (CD3OD): δ (ppm) 6.209 (1H, dd, J = 0.92, 3.08 Hz), 6.84 (1H, d, J = 0.92 Hz), 6.946 (1H, s), 6.972 (1H, d, J = 3.08 Hz); 13C-NMR (CD3OD): δ (ppm) 98.0, 101.4, 101.5, 122.4, 123.7, 132.1, 141.2, 143.5.Quantitative study of DHI formed in classic DA–Tris solution
2 g DA sample was dissolved in 1 L TRIS buffer (10 mM, pH 8.5) and the solution was stirred at room temperature to ensure the efficient supply of oxygen. At predetermined time points (0.5, 4, 12 and 24 h), 100 mL DA–Tris solution was extracted twice with 80 mL ethyl acetate (80 mL × 2). The organic layers were combined, washed with 1 M HCl and then concentrated. The residue was dissolved in 0.5 mL CD3OD containing 6 uM DMSO as an internal standard. 1H-NMR analyses of the samples were carried out on a Bruker Avance III spectrometer at 400 MHz. For each time point, three samples were prepared.1H-NMR study on the consumption kinetics of DA in different buffers
The consumption kinetics of DA in different buffers were monitored using NMR spectroscopy. DA (10 mM) was dissolved in Tris-d11 buffer (10 mM, pH 8.5) or phosphate–D2O buffer (10 mM, pH 8.5) containing 1.75 mM DMSO as an internal standard. The solution was incubated at room temperature. At predetermined time points (0, 0.5, 2, 4, 8, 16 and 24 h), 1H-NMR analysis of the samples was carried out using a Bruker Avance III spectrometer at 400 MHz.Mass analyses on the major intermediates in DA, DA/DHI and DHI solution
DA (10 mM), DA (10 mM)/DHI (1 mM) and DHI (1 mM) were dissolved in 20 mL Tris buffer (10 mM, pH 8.5). After a predetermined time (0.5, 4, 12 and 24 h), 1 mL aliquots of these samples were acidified with 1 M HCl to pH 2–4 and filtered using 0.2 μm syringe filter (Millex®-FG, Millipore Corporation, USA). Then 20 μL of the samples prepared was injected for mass analyses. Mass analyses were performed on LC/MS-2020, SHIMADZU Scientific instrument using ESI-MS and APCI-MS respectively. At each condition and time-point, three to four samples were prepared and analyzed. Blank Tris buffer samples were subjected to the same treatment of acidification and filtration and used for comparison.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by Ministry of Education Academic Research Fund Tier 1 Grant (No. R148-000-252-114) Singapore and supported by the Institute of Materials Research and Engineering, Agency of Science, Technology and Research (A*STAR), under project fund IMRE/17-1P1207.Notes and references
- J. H. Waite and M. L. Tanzer, Science, 1981, 212, 1038–1040 CrossRef PubMed.
- D. J. Crisp, G. Walker, G. A. Young and A. B. Yule, J. Colloid Interface Sci., 1985, 104, 40–50 CrossRef.
- J. H. Waite, Integr. Comp. Biol., 2002, 42, 1172–1180 CrossRef PubMed.
- J. H. Waite, Ann. N. Y. Acad. Sci., 1999, 875, 301–309 CrossRef PubMed.
- M. Yu, J. Hwang and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 5825–5826 CrossRef.
- V. V. Papov, T. V. Diamond, K. Biemann and J. H. Waite, J. Biol. Chem., 1995, 270, 20183–20192 CrossRef PubMed.
- J. H. Waite and X. Qin, Biochemistry, 2001, 40, 2887–2893 CrossRef PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U.S.A, 2006, 103, 12999–13003 CrossRef PubMed.
- W. Zhang, H. Yang, F. Liu, T. Chen, G. Hu, D. Guo, Q. Hou, X. Wu, Y. Su and J. Wang, RSC Adv., 2017, 7, 32518–32527 RSC.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef PubMed.
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- J. Sedo, J. Saiz-Poseu, F. Busque and D. Ruiz-Molina, Adv. Mater., 2013, 25, 653–701 CrossRef PubMed.
- E. Faure, C. Falentin-Daudre, C. Jerome, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236–270 CrossRef.
- P. Kord Forooshani and B. P. Lee, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 9–33 CrossRef PubMed.
- N. Patil, C. Jérôme and C. Detrembleur, Prog. Polym. Sci., 2018, 82, 34–91 CrossRef.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef PubMed.
- M. E. Lynge, P. Schattling and B. Staedler, Nanomedicine, 2015, 10, 2725–2742 CrossRef PubMed.
- Y. H. Ding, M. Floren and W. Tan, Biosurface and Biotribology, 2016, 2, 121–136 CrossRef PubMed.
- K. S. Schanze, H. Lee and P. B. Messersmith, ACS Appl. Mater. Interfaces, 2018, 10, 7521–7522 CrossRef PubMed.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef.
- T. Shalev, A. Gopin, M. Bauer, R. W. Stark and S. Rahimipour, J. Mater. Chem., 2012, 22, 2026–2032 RSC.
- N. F. Della Vecchia, R. Avolio, M. Alfe, M. E. Errico, A. Napolitano and M. d'Ischia, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef.
- J. Liebscher, R. Mrowczynski, H. A. Scheidt, C. Filip, N. D. Hadade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef PubMed.
- Y. Ding, L. T. Weng, M. Yang, Z. Yang, X. Lu, N. Huang and Y. Leng, Langmuir, 2014, 30, 12258–12269 CrossRef PubMed.
- D. Schaubroeck, Y. Vercammen, L. Van Vaeck, E. Vanderleyden, P. Dubruel and J. Vanfleteren, Appl. Surf. Sci., 2014, 303, 465–472 CrossRef.
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and T. Sarna, Angew. Chem., Int. Ed., 2009, 48, 3914–3921 CrossRef PubMed.
- M. d'Ischia, A. Napolitano, V. Ball, C. T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef PubMed.
- M. L. Alfieri, R. Micillo, L. Panzella, O. Crescenzi, S. L. Oscurato, P. Maddalena, A. Napolitano, V. Ball and M. d'Ischia, ACS Appl. Mater. Interfaces, 2018, 10, 7670–7680 CrossRef PubMed.
- N. L. Yakovlev, S. D. Ow and T. Tandiono, Singapore Patent, WO 2015183201, 2015.
- M. Rodenstein, S. Zürcher, S. G. P. Tosatti and N. D. Spencer, Langmuir, 2010, 26, 16211–16220 CrossRef PubMed.
- F. Bernsmann, A. Ponche, C. Ringwald, J. Hemmerle, J. Raya, B. Bechinger, J. C. Voegel, P. Schaaf and V. Ball, J. Phys. Chem. C, 2009, 113, 8234–8242 CrossRef.
- M. Müller and B. Keßler, Langmuir, 2011, 27, 12499–12505 CrossRef PubMed.
- V. Ball, D. D. Frari, V. Toniazzo and D. Ruch, J. Colloid Interface Sci., 2012, 386, 366–372 CrossRef PubMed.
- S. A. Mian, L. C. Saha, J. Jang, L. Wang, X. Gao and S. Nagase, J. Phys. Chem. C, 2010, 114, 20793–20800 CrossRef.
- S. A. Mian, X. Gao, S. Nagase and J. Jang, Theor. Chem. Acc., 2011, 130, 333–339 Search PubMed.
- S. A. Mian, L. M. Yang, L. C. Saha, E. Ahmed, M. Ajmal and E. Ganz, Langmuir, 2014, 30, 6906–6914 CrossRef PubMed.
- M. Guardingo, E. Bellido, R. Miralles-Llumà, J. Faraudo, J. Sedó, S. Tatay, A. Verdaguer, F. Busqué and D. Ruiz-Molina, Small, 2014, 10, 1594–1602 CrossRef PubMed.
- P. Das and M. Reches, Nanoscale, 2016, 8, 15309–15316 RSC.
- T. Utzig, P. Stock and M. Valtiner, Angew. Chem., Int. Ed., 2016, 55, 9524–9528 CrossRef PubMed.
- C. Lim, J. Huang, S. Kim, H. Lee, H. Zeng and D. S. Hwang, Angew. Chem., Int. Ed., 2016, 55, 3342–3346 CrossRef PubMed.
- L. Klosterman, J. K. Riley and C. J. Bettinger, Langmuir, 2015, 31, 3451–3458 CrossRef PubMed.
- J. Jiang, L. Zhu, L. Zhu, B. Zhu and Y. Xu, Langmuir, 2011, 27, 14180–14187 CrossRef PubMed.
- V. Ball, D. Del Frari, M. Michel, M. J. Buehler, V. Toniazzo, M. K. Singh, J. Gracio and D. Ruch, J. Bionanosci., 2012, 2, 16–34 CrossRef.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef PubMed.
- L. K. Charkoudian and K. J. Franz, Inorg. Chem., 2006, 45, 3657–3664 CrossRef PubMed.
- S. Lin, C. T. Chen, I. Bdikin, V. Ball, J. Gracio and M. J. Buehler, Soft Matter, 2014, 10, 457–464 RSC.
- D. Chai, Z. Xie, Y. Wang, L. Liu and Y. J. Yum, ACS Appl. Mater. Interfaces, 2014, 6, 17974–17984 CrossRef PubMed.
- V. Horak and G. Weeks, Bioorg. Chem., 1993, 21, 24–33 CrossRef.
- I. G. Kim, H. J. Nam, H. J. Ahn and D. Y. Jung, Electrochim. Acta, 2011, 56, 2954–2959 CrossRef.
- A. Pezzella, M. Barra, A. Musto, A. Navarra, M. Alfe, P. Manini, S. Parisi, A. Cassinese, V. Criscuolo and M. d'Ischia, Mater. Horiz., 2015, 2, 212–220 RSC.
- M. Salomäki, M. Tupala, T. Parviainen, J. Leiro, M. Karonen and J. Lukkari, Langmuir, 2016, 32, 4103–4112 CrossRef PubMed.
- E. Kaxiras, A. Tsolakidis, G. Zonios and S. Meng, Phys. Rev. Lett., 2006, 97, 218102 CrossRef PubMed.
- G. S. Lorite, V. R. Coluci, M. I. N. da Silva, S. N. Dezidério, C. F. O. Graeff, D. S. Galvão and M. A. Cotta, J. Appl. Phys., 2006, 99, 113511 CrossRef.
- M. Arzillo, G. Mangiapia, A. Pezzella, R. K. Heenan, A. Radulescu, L. Paduano and M. d'Ischia, Biomacromolecules, 2012, 13, 2379–2390 CrossRef PubMed.
- C. T. Chen, V. Ball, J. J. de Almeida Gracio, M. K. Singh, V. Toniazzo, D. Ruch and M. J. Buehler, ACS Nano, 2013, 7, 1524–1532 CrossRef PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Chem. Sci., 2013, 4, 3796–3802 RSC.
- F. Bernsmann, O. Ersen, J. C. Voegel, E. Jan, N. A. Kotov and V. Ball, ChemPhysChem, 2010, 11, 3299–3305 CrossRef PubMed.
- F. Bernsmann, V. Ball, F. Addiego, A. Ponche, M. Michel, J. J. d. A. Gracio, V. Toniazzo and D. Ruch, Langmuir, 2011, 27, 2819–2825 CrossRef PubMed.
- C. T. Chen, F. J. Martin-Martinez, G. S. Jung and M. J. Buehler, Chem. Sci., 2017, 8, 1631–1641 RSC.
- F. Ponzio and V. Ball, Colloids Surf., A, 2014, 443, 540–543 CrossRef.
- H. Hu, J. C. Dyke, B. A. Bowman, C. C. Ko and W. You, Langmuir, 2016, 32, 9873–9882 CrossRef PubMed.
- Y. Li, T. Wang, L. Xia, L. Wang, M. Qin, Y. Li, W. Wang and Y. Cao, J. Mater. Chem. B, 2017, 5, 4416–4420 RSC.
- M. A. Gebbie, W. Wei, A. M. Schrader, T. R. Cristiani, H. A. Dobbs, M. Idso, B. F. Chmelka, J. H. Waite and J. N. Israelachvili, Nat. Chem., 2017, 9, 473–479 CrossRef PubMed.
- M. V. Rapp, G. P. Maier, H. A. Dobbs, N. J. Higdon, J. H. Waite, A. Butler and J. N. Israelachvili, J. Am. Chem. Soc., 2016, 138, 9013–9016 CrossRef PubMed.
- L. Klosterman, Z. Ahmad, V. Viswanathan and C. J. Bettinger, Adv. Mater. Interfaces, 2017, 4, 1700041 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04472d |
| This journal is © The Royal Society of Chemistry 2018 |