
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrediction of 1H NMR chemical shifts for ionic liquids: strategy and application of a relative reference standard†
Juanfang
Wang
,
Ying
Liu
 *,
Wen
Li
and
Guanjun
Gao
*,
Wen
Li
and
Guanjun
Gao
College of Chemistry and Chemical Engineering, Inner Mongolia University, Hohhot 010021, China. E-mail: celiuy@imu.edu.cn
First published on 10th August 2018
Abstract
The computational predictions of 1H NMR chemical shifts for ionic liquids were investigated. To calculate the chemical shifts more accurately, the approach of relative reference standard (RRS) was proposed. This straightforward computational technique uses organic compounds and ionic liquids that are similar to the studied ionic liquids as standards. The calculated chemical shifts of single ion pairs were strongly influenced by the anion type and the local environment. Using the RRS methodology, the calculated results agreed well with the experimental chemical shifts due to the cancellation of errors caused by the anion. Ionic clusters consisting of 4 ion pairs were also employed to model the ionic liquids with strongly coordinating anions. Large clusters slightly improve the accuracy by reducing systematic errors. Although the experimental 1H NMR data of the reference ionic liquid should be used, the RRS methodology has been shown to predict 1H NMR chemical shifts efficiently at different levels of theory.
Introduction
In the last decade, ionic liquids (ILs) were widely used in chemical reactions, extraction, and catalysis processes.1 As a type of molten salt, ionic liquids have many favourable properties, including good chemical and physical stability, low vapour pressure, non-flammability, and tuneable properties.2–4 A typical ionic liquid usually consists of organic-type cations and inorganic-type anions. However, it is believed that the ionic liquid is a complicated system.5 Nuclear magnetic resonance (NMR) is one of the most powerful tools to investigate the ionic liquid. The interaction between anion and cation can be detected by means of 1H NMR chemical shifts.6 For example, the proton chemical shift of H2 in the [C4mim]+-based IL is very sensitive to the change of the local environment. A downfield H2 chemical shift can be attributed to hydrogen bond formation,7 and the position of the NMR peaks corresponding to the H2 proton heavily depended on the anion type.8 Therefore, the 1H NMR chemical shifts can help us understand the effects of the local environment and the transport properties of ILs. However, the chemical factors that influence proton chemical shifts of the ILs are still not very clear due to the complication of interionic interactions.9,10Recently, several studies have shown that quantum chemical methods are successful in the prediction of NMR chemical shifts. The 1H and 13C NMR chemical shifts can be predicted reasonably well with quantum chemical calculations for various organic compounds.8 However, in the cases of ILs, large deviations were observed for the computing proton NMR chemical shifts when compared with the experimental values. For imidazolium-based ILs, the absolute errors between the predicted and experimental shift of H2 were usually greater than 1 ppm. The backbone H4 and H5 protons of the imidazolium ring also gave large deviations in the cases of [C4mim]Cl and [C4mim][OAc].9 It is believed that the predicted 1H NMR chemical shifts strongly depended on the geometry of the IL. In addition, hydrogen bond and the type of anion significantly influence the accuracy of the predicted values.11,12
Many efforts have been made to improve the accuracy of the predicted 1H NMR chemical shifts. Using empirical scaling corrections to remove systematic errors can obtain well predicted chemical shifts.13 This method requires a large number of experimental data to obtain scaling factors. However, the experimental values of ionic liquids seem not enough to support this procedure so far. A multi-standard approach (MSTD) for the calculation of 1H NMR chemical shifts of organic molecules also can give relatively accurate results.14 Instead of employing the most common standard tetramethysilane (TMS), the MSTD method uses methanol and benzene as the corresponding reference compounds for sp3 and sp2–sp hybridized carbon atoms, respectively.15 Although this simple modification could obtain much better calculation results for many organic molecules, large deviations exist in the calculations for ionic liquids. Chen et al. suggested that large cluster calculations can improve the accuracy of the prediction of proton chemical shifts. When an IL cluster consisting of 8 ion pairs (IP), it can predict proton chemical shifts with relatively high accuracy. If the computational cost were not a problem, a large IL cluster might give good predicted results: mean absolute errors of 1H chemical shifts in the range of 0.2 to 0.8 ppm.16,17
Ionic liquid is composed of cation and anion pairs, which is very different from the ordinary organic compounds. For computing the 1H NMR chemical shifts of ILs, more effort should be made to obtain much better accuracy at lower computational cost. The following strategy maybe helps us to further improve the accuracy of calculations. First, besides the sp3 and sp2–sp hybridized carbon atoms, the structure of cation also markedly affects the calculation of the proton chemical shifts. If the structure of a reference compound were similar to that of the IL cation, the quantum chemical methods might produce little error with respect to experiment. Therefore, apart from TMS, benzene, and methanol, other organic compounds that similar to the cations of ILs (e.g., 1-methyl-imidazole) should be used as the reference compounds to calculate the proton chemical shifts. Secondly, the high quality prediction of 1H NMR chemical shifts should take into account the impact of anions, and then the IL that similar to the structure of the studied IL should also be employed as a reference standard. Because this reference IL is analogous to the studied IL, the effect of anion on both ILs should be very similar. Using this approach, the calculations for the particular chemical shifts of those hydrogens contacted with oxygen or nitrogen atom (–OH or –NH group) might give more confident results than those computed with TMS standard. In consequence, an approach of relative reference standard (RRS) is proposed to achieve accurate predictions of 1H NMR parameters at any level of theory. The only apparent trade-off is the slight increase in complexity when processing data utilizing relative reference compounds. Thirdly, some studies have revealed that a single ion pair was not considered a reliable model for studying particular chemical shifts.5,18 If more ion pairs were given consideration in the calculation with RRS approach, it might produce good agreement with experiment.
In this paper, we did a systematic study on the prediction of 1H NMR chemical shifts of a number of ILs. We have calculated chemical shifts by the RRS methodology with the above-mentioned strategy: (1) using single ion pairs as far as possible to calculate the chemical shifts of ILs. (2) 1H NMR chemical shifts are calculated by using the organic compound that is similar to the cation of the studied IL as a reference standard. (3) When the studied IL containing stronger coordinating anions such as chloride and acetate, the IL that similar to the studied IL is employed as the reference compound. (4) An ionic cluster approach is also adopted to study changes in the chemical shifts. Finally, the results of the performance of RRS at different levels of theory have been compared with those of TMS standard.
Theoretical methods
All calculations were performed with the Gaussian 09 package.19 Recently, Zahn20 and Li18 have performed an assessment of density functional theory and Møller–Plesset perturbation theory for ILs. Li et al. recommended that the density functional of the Minnesota family of the M0X type with a diffusion function basis set (aug-cc-pvdz or 6-31++g**) would give reliable results for IL calculations. In addition, Grimme et al. noted that dispersion-corrected density functionals, such as B3LYP/6-31++G**-gd3bj (DFT-D3), could also lead to reliable results.21 Therefore, the m06-2x/aug-cc-pvdz (or B3LYP/6-31++G**-gd3bj) level of theory was used to optimize all ionic liquids under study. This level is known to afford good geometry at low computational cost, particularly appropriate to be used for the calculation of weak interaction (e.g., hydrogen bonding).22 The 1H absolute magnetic shielding values were computed using the following levels of theory: (1) B3LYP with DFT-D3 dispersion correction,23 (2) mPW1PW91,24 GGA-based functional which was found to provide best results within the MSTD approach for 1H NMR, (3) WP04,25 similar to B3LYP but parameterized to reproduce NMR chemical shifts in chloroform. (4) B972,26 functional augmented with the empirical dispersion correction, (5) m06-2x,27 a meta-GGA functional, (6) Hartree–Fock (HF), and (7) a second-order Mǿller–Plesset (MP2) theory was also considered, which might obtain reasonable results.In order to investigate the effect of basis sets on the accuracy of the predicted 1H chemical shifts, both Pople and Dunning-type double- and triple-ζ basis sets including 6-31++G(d,p), 6-311++G(d,p), 6-311+(3df,2p), cc-PVTZ, and aug-cc-PVDZ were investigated. Due to the “gauge including atomic orbitals approach” (GIAO) success in accurate calculations of NMR chemical shifts,28 the magnetic shielding constants were computed using this method. The NMR calculations were carried out in solution, using chloroform, acetonitrile, or DMSO-d6 as the solvent. The effect of solvent plays an important role on the geometry optimization of ionic liquids. The SMD solvation model that proposed by Truhlar et al. has been used for the IL calculation very well.29,30 Therefore, the density functional theory (m06-2x and B3LYP with dispersion-corrected) combined with the SMD model was employed to investigate the IL optimization and their proton NMR chemical shifts.
When the 1H NMR chemical shifts of [C4mim]Cl are under studied, the RRS approach should be carried out as the following steps:
(1) The organic molecule similar to the cation of the studied IL was used as the reference compound. In this case, 1-methlyimidozle was employed as a corresponding reference compound.
(2) The 1H absolute magnetic shielding values of H2′, H3′, and H4′ protons in 1-methylimidazole ring were calculated at the certain level of theory.
(3) The shielding constants of [C4mim]Cl with a single ion pair were calculated at the same level as the 1-methylimidazole calculations.
(4) Once the shielding constants were computed, the 1H NMR chemical shifts can be calculated in accordance with the following equation:
| δxcalc = σref,x − σx + δref,x |
(5) Where σref,x and σx are the NMR isotropic magnetic shielding values. Generally, the x hydrogen atoms for the given molecule and for the reference compound have similar structure. And δref,x is the certain proton chemical shift of the reference compound in deuterated chloroform, which was taken from the Spectral Data Base for Organic Compounds (SDBS).31 For instance, the δH2calc of [C4mim]Cl would include the calculated values of σ1-methylimidazole,H2′ and σ[C4mim]Cl,H2, and the experimental data of δ1-methylimidazole,H2′.
(6) H3 and H4 calculations of [C4mim]Cl have the similar procedure of H2, but x in the equation should be replaced by H3 and H4, respectively. The –CH2– or –CH3 hydrogens (H5–H8) in the [C4mim]+ cation were calculated by using TMS (δ = 0.00 ppm) as the reference compound.
In addition, clusters consisting of 4 ion pairs (4 IP) were also modelled for ILs. The clusters of [C4mim]OH, ethanolamine acetate and [C4mim][BF4] were constructed by combining energetically preferable ion pair configurations, while ensuring the resulting cluster occupied the minimal volume. All ionic clusters studied were confirmed as minima on the potential energy surface by the absence of imaginary vibrational frequencies. We used the mean absolute errors (MAE, defined as Σ|δcalc,x − δexp,x|/n) to analyse the results of the calculations, and compare the performances of the methods under study. Root mean square deviations (RMS) were also used to investigate the differences between values predicted by a model and the values actually observed. The RMS being defined as follows:
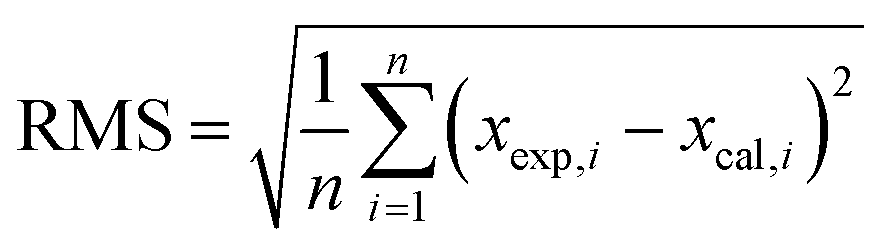
Results and discussion
RRS performance for single ion pairs
To carry out this study, we have worked with a test set of 16 ionic liquids shown in Fig. 1 and S1.† Single ion pair (1 IP) of the studied IL were adopted to investigate the accuracy of theory levels at reasonable computational cost. Table 1 lists the results obtained after carrying out the 1H NMR calculations using TMS and RRS methodology. In the cases of [C4mim]Cl and [C4mim][BF4], it is found that RMSs and MAEs of the calculating results are great even using the “expensive” method mp2/aug-cc-pvdz to optimize and to predict (entries 1 and 2). But a better result could be obtained when using m06-2x/aug-cc-pvdz level of theory for the IL [C4mim][BF4] (entry 3). When an IL with weekly coordinating anions (e.g., [BF4]−) is under the NMR calculation, it is believed that the accuracy of chemical shifts displays a high dependence on the level of theory employed.11,17 Recent studies also show that the calculated 1H NMR chemical shifts for IL systems using either the global minima or all significant configurations might be very similar.8,14 We analysed the performance of TMS and RRS methods for [C4mim][BF4] and [C4mim][NTf2], which could adopt more preferred configurations. However, it is found that no improvement on the RMSs was obtained when using Boltzmann weighted values (entries 4 and 9, respectively). So only the global minimum was considered in all cases of this work. More details of Boltzmann distribution please see the ESI† (Fig. 2 and 3).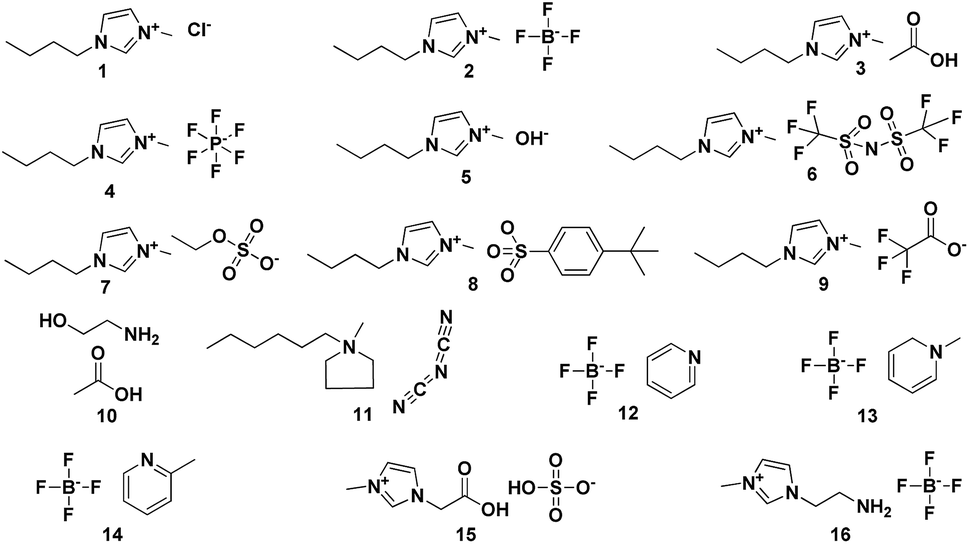 | ||
| Fig. 1 Test set. | ||
| Entry | Ionic liquid | Reference compound | Solvent | Level of theory | MAE | RMS | ||
|---|---|---|---|---|---|---|---|---|
| RRS | TMS | RRS | TMS | |||||
| 1 | [C4mim]Cl | 1-Methyl-imidazole | Chloroform | mp2/aug-cc-pvdz | 0.764 | 0.747 | 1.197 | 1.417 |
| 2 | [C4mim][BF4]9,32 | Chloroform | mp2/aug-cc-pvdz | 0.354 | 0.441 | 0.371 | 0.558 | |
| 3 | [C4mim][BF4] global minimum | Chloroform | m06-2x/aug-cc-pvdz | 0.369 | 0.114 | 0.450 | 0.155 | |
| 4 | [C4mim][BF4] Boltzmann average | Chloroform | m06-2x/aug-cc-pvdz | 0.393 | 0.140 | 0.467 | 0.180 | |
| 5 | [C4mim][OAc]33 | Chloroform | m06-2x/aug-cc-pvdz | 0.750 | 0.649 | 1.221 | 1.318 | |
| 6 | [C4mim][PF6]9 | Chloroform | m06-2x/aug-cc-pvdz | 0.372 | 0.240 | 0.425 | 0.285 | |
| 7 | [C4mim]OH34 | Chloroform | m06-2x/aug-cc-pvdz | 0.459 | 0.289 | 0.573 | 0.511 | |
| 8 | [C4mim][Tf2N]9,35 global minimum | Chloroform | m06-2x/aug-cc-pvdz | 0.612 | 0.632 | 0.977 | 1.134 | |
| 9 | [C4mim][Tf2N] Boltzmann average | Chloroform | m06-2x/aug-cc-pvdz | 0.525 | 0.633 | 0.952 | 1.135 | |
| 10 | [C4mim][MeSO4]36 | Chloroform | m06-2x/aug-cc-pvdz | 0.479 | 0.411 | 0.529 | 0.559 | |
| 11 | [C4mim][OTS]32 | Water | b3lyp/6-31++g(d,p)-d3 | 0.555 | 0.385 | 0.748 | 0.424 | |
| 12 | [C4mim][TFA]32 | Water | b3lyp/6-31++g(d,p)-d3 | 0.817 | 0.610 | 1.291 | 1.370 | |
| 13 | [P16][DCA]37 | Pyrrole | DMSO-d6 | m06-2x/aug-cc-pvdz | 0.360 | 0.330 | 0.387 | 0.350 |
| 14 | [Py][BF4]38 | Pyridine | Acetonitrile | hf-6-31g(d) | 0.854 | 1.159 | 1.295 | 1.439 |
| 15 | [1-me-Py][BF4]38 | Pyridine | Acetonitrile | b3lyp/6-31++g(d,p)-d3 | 0.328 | 0.105 | 0.365 | 0.122 |
 | ||
| Fig. 2 Configuration of [C4mim]Cl optimized by mp2/aug-cc-pvdz. | ||
 | ||
| Fig. 3 Configuration of [C4mim][BF4] optimized by mp2/aug-cc-pvdz. | ||
On the other hands, it is also found that almost all RMSs of those ILs containing strongly coordinating anions (e.g., Cl−, OH−, OAc−) are relatively large. No matter which level of theory was employed for calculating the proton chemical shifts of [C4mim][OAc], the values of MAE are always greater than 1.0 ppm, as Fig. 4 illustrated. These results should be attributed to the influence of the anion to the cation. Ionic liquids are different from the ordinary organic compounds. Due to the complicated relationships of ions, the anion would significantly affect the 1H NMR chemical shifts. When the ionic liquids have strongly coordinating anions, the interionic interactions of these ILs are difficult to model. In the cases of Cnmim-based ILs, the predicted proton NMR chemical shifts of H2 were found to be sensitive to the effect of the anion. If we did not take into consideration the effects of the anions, it is not surprising that the theoretical calculation of a Cnmim-based IL would give a poor result. Particularly, the level of theory is insensitive to the prediction of the IL with strongly coordinating anion and therefore, those methods with high computational cost have no contribution to the improvement of the predicted chemical shifts (Scheme 1).
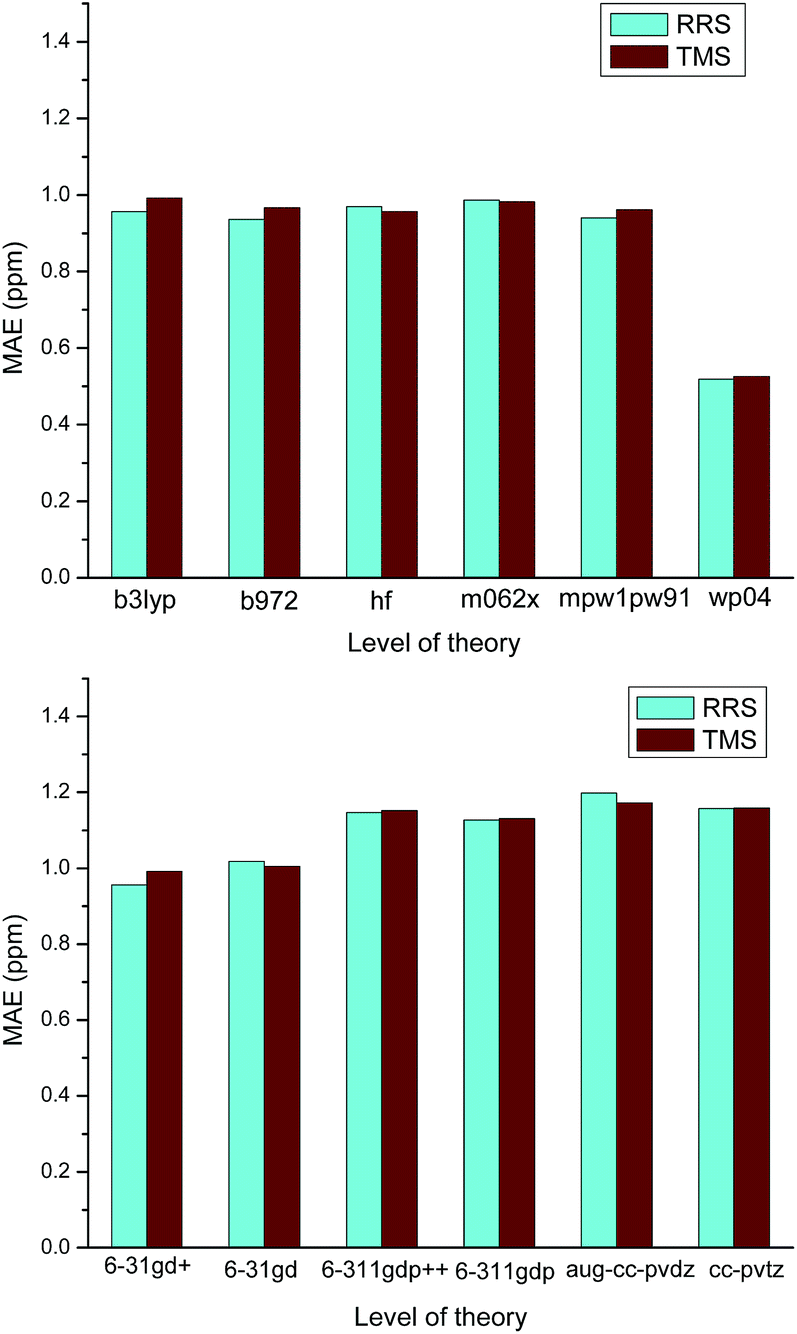 | ||
| Fig. 4 Effect of the method and the basis set used in the calculation of [C4mim][OAC] 1H NMR chemical shifts. | ||
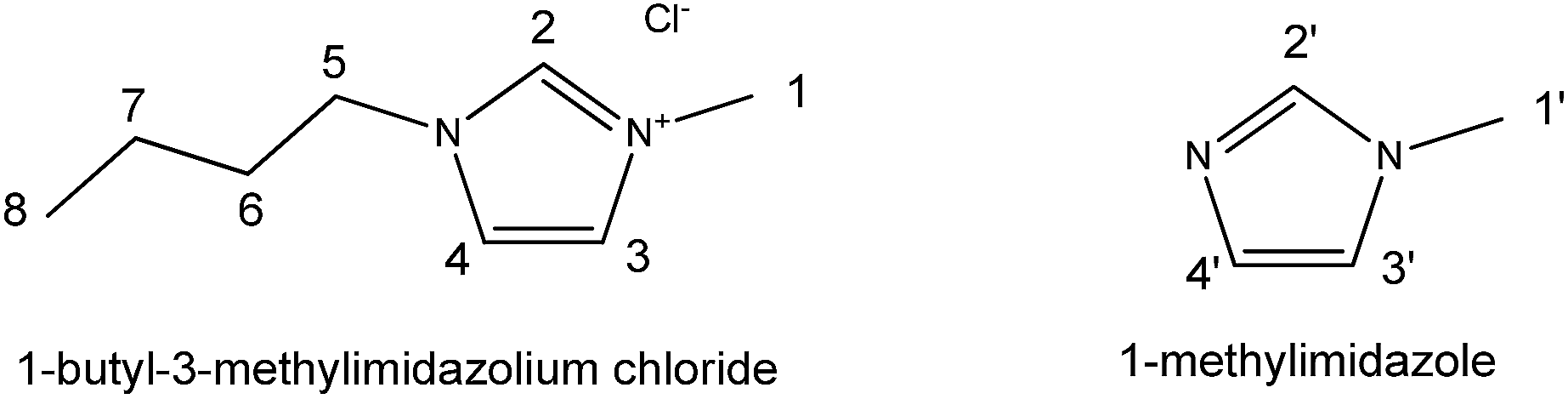 | ||
| Scheme 1 Structure of 1-butyl-3-methylimidazolium chloride and 1-methylimidazole with hydrogen atoms numbered. | ||
Modification of RRS approach
Using the reference compound that similar to the cation of the studied IL, the effects of anion are not completely reflected in the RRS calculation. It is necessary to employ an IL that similar to the studied IL as a reference standard to predict the chemical shifts. This modification should be considered as an upgrade of the RRS approach. The data collected in Table 2 show that the modification of RRS can give more accurate and precise 1H NMR chemical shifts than those obtained with the ordinary RRS standard. Whether pyridinium-based or imidazolium-based ILs, most RMSs are less than 0.3 ppm. The detailed calculations for the cases of [Cnmim]+-based ILs are listed in Table 3. The main improvement in those imidazolium-based ILs came for the H2 proton, whose chemical shift deviation from experiment reduced to 0.2 ppm on average. These results might be explained in terms of error cancellation in isotropic magnetic shielding constants when using relative reference standards that more resemble different types of hydrogens within the studied IL. The RRS approach affords better results than those obtained using TMS, implying that RRS decrease the predicted errors caused by the interaction of anions.| Ionic liquid | Reference compound | Level of theory | MAE | RMS | ||
|---|---|---|---|---|---|---|
| RRS | TMS | RRS | TMS | |||
| [1-me-Py][BF4] | [Py][BF4] | b3lyp/6-31++g(d,p)-d3 | 0.226 | 0.105 | 0.254 | 0.122 |
| wp04/6-31+g(d) | 0.113 | 0.229 | 0.124 | 0.241 | ||
| [2-me-Py][BF4]38 | [Py][BF4] | b3lyp/6-31++g(d,p)-d3 | 0.103 | 0.370 | 0.125 | 0.536 |
| wp04/6-31+g(d) | 0.073 | 0.493 | 0.105 | 0.739 | ||
| P16-DCA | P12-DCA37 | b3lyp/6-31++g(d,p)-d3 | 0.081 | 0.151 | 0.107 | 0.192 |
| [CMI][HSO4]32 | [Emim][HSO4]32 | b3lyp/6-31++g(d,p)-d3 | 0.295 | 0.470 | 0.352 | 0.500 |
| m06-2x/aug-cc-pvdz | 0.265 | 0.539 | 0.296 | 0.579 | ||
| wp04/6-31+g(d) | 0.193 | 0.346 | 0.215 | 0.368 | ||
| [C4mim][MeSO4] | [C2mim][EtSO4]36 | b3lyp/6-31++g(d,p)-d3 | 0.118 | 0.471 | 0.162 | 0.790 |
| m06-2x/aug-cc-pvdz | 0.200 | 0.383 | 0.261 | 0.534 | ||
| wp04/6-31+g(d) | 0.222 | 0.423 | 0.236 | 0.567 | ||
| [NH2Emim][BF4]39 | [C2mim][BF4]17 | m06-2x/aug-cc-pvdz | 0.225 | 0.457 | 0.274 | 0.495 |
| [C4mim][BF4] | [C2mim][BF4] | b3lyp/6-31++g(d,p)-d3 | 0.170 | 0.290 | 0.192 | 0.402 |
| m06-2x/aug-cc-pvdz | 0.134 | 0.204 | 0.167 | 0.214 | ||
| wp04/6-31+g(d) | 0.338 | 0.446 | 0.377 | 0.712 | ||
| [C4mim][PF6] | [C2mim][PF6]17 | b3lyp/6-31++g(d,p)-d3 | 0.190 | 0.192 | 0.252 | 0.247 |
| m06-2x/aug-cc-pvdz | 0.270 | 0.240 | 0.351 | 0.285 | ||
| wp04/6-31+g(d) | 0.262 | 0.301 | 0.266 | 0.356 | ||
| [C4mim][OAc] | [C2mim][OAc]33 | b3lyp/6-31++g(d,p)-d3 | 0.142 | 1.098 | 0.171 | 1.761 |
| m06-2x/aug-cc-pvdz | 0.088 | 0.649 | 0.113 | 1.318 | ||
| [C4mim][OTS] | [C2mim][OTS]32 | b3lyp/6-31++g(d,p)-d3 | 0.117 | 0.385 | 0.131 | 0.424 |
| [C4mim][Tf2N] | [C2mim][Tf2N]35 | b3lyp/6-31++g(d,p)-d3 | 0.099 | 0.354 | 0.157 | 0.707 |
| m06-2x/aug-cc-pvdz | 0.188 | 0.330 | 0.224 | 0.483 | ||
| wp04/6-31+g(d) | 0.222 | 0.423 | 0.236 | 0.567 | ||
| [C4mim][TFA] | [C2mim][TFA]32 | b3lyp/6-31++g(d,p)-d3 | 0.103 | 0.610 | 0.122 | 1.370 |
| Ionic liquid | δ cal | Chemical shifts (ppm) | MAE | RMS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | H3 | H4 | H1 | H5 | H6 | H7 | H8 | ||||
| a The detailed calculation data of Table 2. b Using b3lyp/6-31++g(d,p) with gd3bj correction for optimization and calculation. c Using m06-2x/aug-cc-pvdz for optimization and calculation. | |||||||||||
| [C4mim][PF6] | RRSb | 9.10 | 7.37 | 7.22 | 4.30 | 4.00 | 1.83 | 1.21 | 1.10 | 0.190 | 0.252 |
| TMSb | 9.05 | 7.52 | 7.53 | 4.02 | 4.20 | 1.76 | 1.13 | 1.03 | 0.192 | 0.247 | |
| RRSc | 7.88 | 7.47 | 7.16 | 4.49 | 4.16 | 1.83 | 0.80 | 0.97 | 0.270 | 0.351 | |
| TMSc | 9.54 | 7.88 | 7.82 | 4.12 | 4.36 | 1.95 | 0.92 | 1.09 | 0.240 | 0.285 | |
| δ exp | 8.52 | 7.49 | 7.52 | 4.25 | 3.97 | 1.93 | 1.38 | 0.94 | |||
| [C4mim][MeSO4] | RRSb | 9.15 | 7.59 | 7.57 | 4.25 | 3.60 | 1.60 | 1.37 | 0.84 | 0.118 | 0.162 |
| TMSb | 10.04 | 7.40 | 7.50 | 3.87 | 5.45 | 1.76 | 1.53 | 1.00 | 0.471 | 0.790 | |
| RRSc | 9.15 | 7.53 | 7.54 | 4.35 | 3.62 | 1.29 | 1.16 | 0.87 | 0.200 | 0.261 | |
| TMSc | 10.51 | 7.67 | 7.82 | 4.02 | 4.27 | 1.51 | 1.39 | 1.09 | 0.383 | 0.534 | |
| δ exp | 9.42 | 7.63 | 7.58 | 4.26 | 3.37 | 1.88 | 1.35 | 0.94 | |||
| RRSb | 8.54 | 7.38 | 7.43 | 4.14 | 4.04 | 1.81 | 1.46 | 0.97 | 0.170 | 0.192 | |
| [C4mim][BF4] | TMSb | 9.56 | 7.44 | 7.51 | 3.89 | 5.06 | 1.84 | 1.50 | 1.01 | 0.290 | 0.402 |
| RRSc | 8.89 | 7.42 | 7.68 | 3.95 | 4.06 | 1.64 | 1.39 | 0.86 | 0.134 | 0.167 | |
| TMSc | 9.03 | 7.76 | 7.89 | 3.84 | 3.98 | 1.78 | 1.53 | 1.14 | 0.204 | 0.214 | |
| δ exp | 8.77 | 7.63 | 7.69 | 4.04 | 4.32 | 1.93 | 1.38 | 0.94 | |||
| [C4mim][OAc] | RRSb | 9.90 | 7.91 | 7.85 | 4.22 | 3.83 | 1.76 | 1.47 | 1.05 | 0.142 | 0.171 |
| TMSb | 14.16 | 7.36 | 7.43 | 3.88 | 6.75 | 1.74 | 1.44 | 1.03 | 1.098 | 1.761 | |
| RRSc | 9.95 | 7.85 | 7.82 | 4.22 | 3.82 | 1.50 | 1.21 | 0.96 | 0.088 | 0.113 | |
| TMSc | 13.82 | 7.74 | 7.85 | 4.26 | 4.60 | 1.67 | 1.38 | 1.13 | 0.649 | 1.318 | |
| δ exp | 10.18 | 7.88 | 7.96 | 4.2 | 3.89 | 1.6 | 1.21 | 0.84 | |||
| [C4mim][Tf2N] | RRSb | 9.02 | 7.73 | 7.42 | 4.22 | 3.87 | 1.87 | 1.41 | 0.98 | 0.099 | 0.157 |
| TMSb | 10.59 | 7.26 | 7.27 | 4.07 | 3.85 | 1.92 | 1.46 | 1.02 | 0.354 | 0.707 | |
| RRSc | 8.93 | 7.80 | 7.76 | 4.22 | 3.87 | 1.69 | 1.10 | 0.97 | 0.188 | 0.224 | |
| TMSc | 9.79 | 7.81 | 7.95 | 4.17 | 4.09 | 1.91 | 1.32 | 1.40 | 0.330 | 0.483 | |
| δ exp | 8.63 | 7.54 | 7.46 | 4.24 | 3.96 | 1.92 | 1.4 | 0.97 | |||
| [C4mim][OTS] | RRSb | 8.52 | 7.74 | 7.37 | 4.23 | 3.65 | 1.88 | 1.37 | 0.97 | 0.117 | 0.131 |
| TMSb | 7.89 | 7.28 | 7.53 | 3.33 | 3.91 | 2.08 | 1.56 | 1.16 | 0.385 | 0.424 | |
| δ exp | 8.53 | 7.66 | 7.29 | 4.02 | 3.78 | 1.71 | 1.22 | 0.87 | |||
| [C4mim][TFA] | RRSb | 8.87 | 7.56 | 7.49 | 4.17 | 3.73 | 1.60 | 1.29 | 0.89 | 0.103 | 0.122 |
| TMSb | 12.51 | 7.30 | 7.40 | 3.71 | 4.00 | 1.70 | 1.39 | 0.96 | 0.610 | 1.370 | |
| δ exp | 8.67 | 7.43 | 7.39 | 4.14 | 3.85 | 1.79 | 1.26 | 0.86 | |||
The modified RRS approach can provide a high quality prediction of 1H NMR chemical shifts regardless the level of theory employed, as can be observed in Fig. 5. Further observations can be drawn from the data collected in Table S23 of the ESI.† In general, m06-2x and B3LYP with DFT-D3 corrections were found to provide good results for the RRS approach. Although the results would be better when triple-ζ basis sets such as aug-cc-pvdz and 6-31++G(d,p) were used, the low cost basis sets such as wp04/6-31+G(d) can give good results as well (Fig. 6).
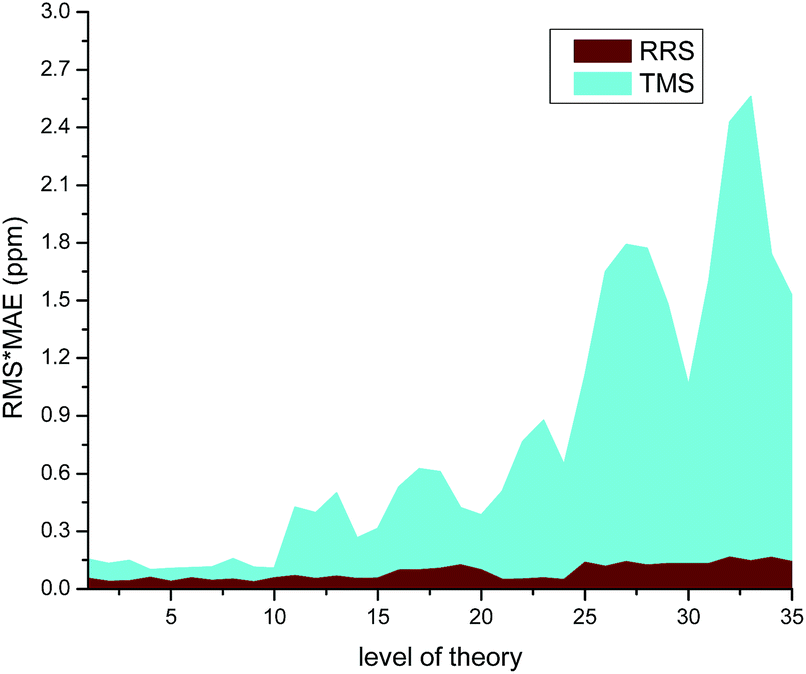 | ||
| Fig. 5 Effect of the level of theory used in the calculation of 1H NMR chemical shifts for various ILs. | ||
 | ||
| Fig. 6 IL cluster of [C4mim][BF4] (4 IP). | ||
However, an apparent limitation of the RRS method is that the experimental 1H NMR shifts of a relatively simple ionic liquid must be used. The purpose of using experimental 1H NMR data is mainly to cancel the errors from the anion influence. On the other hand, for those ionic liquids with long alkyl chains, the alkyl–alkyl chain interactions maybe dominate the bulk. Although it is generally considered that the proton chemical shifts were relatively insensitive to increasing alkyl chain length,40 the effects of long alkyl chain on the prediction of the calculated proton chemical shifts is still ambiguous. Therefore, we set out to calculate 1H NMR chemical shifts of a series of 1-alkyl-3-methylimdazolium ([Cnmim][BF4] with n = 6, 8, and 12) ILs by means of RRS method. The results show that the length of alkyl chains in the imidazolium ring has little impact on the accuracy of calculated chemical shifts. The RMS of [C12mim][BF4] ionic liquid is less than 0.25 ppm. The details see Tables S32–S34 of the ESI.†
IL cluster and RRS for ILs with weakly coordinating anions
Those ILs with strongly coordinating anions, such as 1-butyl-3-methylimidazolium hydroxide and ethanolamine acetate, maybe require a cluster model to improve the accuracy of the prediction. Moreover, some studies have suggested that the cluster should consist of at least 4 ion pairs to do the calculation.17 The predicted chemical shifts of IL clusters together with MAE and RMS were collected in Table 4. However, when TMS was used as the reference standard, the cluster model is clearly not sufficient to predict 1H NMR chemical shifts for the ILs with some particular anions (e.g., OH− and [OAc]−). The 4 ion-paired (4 IP) clusters of [C4mim]OH and ethanolamine acetate did not produce better results when compared to those using single ion pair (1 IP). Their RMSs exceeding 0.6 and even 5.0 ppm on average, respectively. By contrast, good accuracy for these ILs could be obtained when the RRS approach with 1 IP model was used.| Ionic liquid | Size | Level of theory | Reference compound | MAE | RMS |
|---|---|---|---|---|---|
| Ethanolamine acetate | 1 IP | m06-2x/aug-cc-pvdz | Ethanolamine formate (RRS) | 0.111 | 0.126 |
| 1 IP | m06-2x/aug-cc-pvdz | TMS | 4.069 | 5.668 | |
| 4 IP | m06-2x/aug-cc-pvdz | TMS | 3.457 | 5.182 | |
| [C4mim]OH | 1 IP | m06-2x/aug-cc-pvdz | [C2mim]OH (RRS) | 0.086 | 0.125 |
| 1 IP | m06-2x/aug-cc-pvdz | TMS | 0.406 | 0.839 | |
| 4 IP | m06-2x/aug-cc-pvdz | TMS | 0.411 | 0.610 | |
| [C4mim][BF4] | 1 IP | mp2/aug-cc-pvdz | TMS | 0.441 | 0.558 |
| 4 IP | mp2/aug-cc-pvdz | TMS | 0.308 | 0.390 |
In the case of [C4mim][BF4], the cluster with 4 ion pairs worked slightly better than that in the single ion pair. It is worth noting that the isotropic magnetic shielding constants tend to have more equal values when the cluster was used (Tables S25–S29†). Therefore, more ion pairs might decrease the influence of anions or the local arrangement of surrounding ions on the 1H NMR chemical shifts. However, it is found that the cluster model was more likely to influence the prediction accuracy of those ILs with weakly coordinating anions. The calculated chemical shifts are not nearly accurate for the ILs containing strongly coordinating anions, which is obviously due to the fact that the large cluster models could not reduce the effects of anion on the 1H NMR calculations (4 IP [C4mim]OH and ethanolamine acetate).
In addition, a generalised standard might be proposed when the ILs have the weakly coordinating anions. As Table 5 shown, if the IL [C2mim][BF4] were used as a reference standard, the proton NMR chemical shifts of ILs with weakly interacting anions could be well predicted. For example, the RMSs of [C4mim][PF6] and [C2mim][PF6] are 0.242 and 0.284, respectively. Moreover, an order that representing the intensity of anion interactions might also be obtained by means of RMS analysis. That is, larger RMS usually indicates stronger interaction between the cation and the anion. According to the RMS results, the intensity order of the anion interaction is: [C4mim][PF6] ≈ [C2mim][PF6] < [C4mim][NTf2] < [C4mim][OTS] ≈ [C4mim][MeSO4] < [C4mim][TFA] < [C4mim][OAc]. The details see Tables S35–S41 of the ESI.†
| Reference standard | Ionic liquids | RMS |
|---|---|---|
| [C2mim][BF4] | [C4mim][PF6] | 0.242 |
| [C2mim][PF6] | 0.284 | |
| [C4mim][NTf2] | 0.374 | |
| [C4mim][OTS] | 0.477 | |
| [C4mim][MeSO4] | 0.495 | |
| [C4mim][TFA] | 1.064 | |
| [C4mim][OAc] | 1.272 |
Case study
We present a case study from our laboratory to give a further example on the utility of RRS methodology to solve the real life problem.41 As a functional IL, [NH2-Emim][BF4]can be used for SO2 removal.42 However, the 1H NMR spectroscopic data of unsaturated C–H in the imidazolium ring was easy to be confused with the values of N–CH2–CH2–NH2 after SO2 absorption. Consequently, the relationship of [NH2-Emim][BF4] and SO2 was not very clear.43 The quantum chemical calculation gives us an opportunity to investigate the reaction between [NH2-Emim][BF4] and SO2. We have optimized [NH2-Emim][BF4] and SO2–[NH2-Emim]BF4 compounds, and computed the 1H chemical shifts using RRS approach. Fig. 7 presents the experimental values in deuterated chloroform for neat and SO2-absorbed [NH2-Emim][BF4], together with calculated chemical shifts at the m06-2x/aug-cc-pvdz level of theory in the same solution. The calculated results show a good agreement with experimental data. The predicted spectral data reveal that the proton chemical shifts of –NH2 group were significantly changed after the SO2 absorption: from 2.07 to 1.45 and to 3.35 ppm, respectively. The experimental 1H NMR spectrum of the SO2-treated and untreated [NH2-Emim][BF4] IL is represented in Fig. 8. The resonance peaks of H (unsaturated C–H in the imidazole ring, with N–CH2–CH2–NH2 connected to the right) moved downfield from 7.20 to 8.10 ppm due to the impact of [BF4]− and SO2 group, which is basically in line with the predicted chemical shifts. These results suggest that the proposed reaction between [NH2-Emim][BF4] and SO2 had occurred (Fig. 7). For details, please see the ESI.†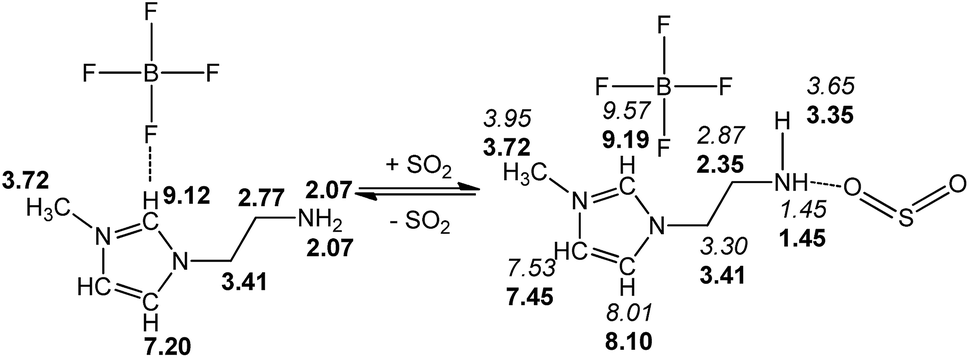 | ||
| Fig. 7 Experimental (in bold) and calculated (in italics) 1H NMR chemical shifts of [NH2-Emim][BF4] before and after SO2 absorption. | ||
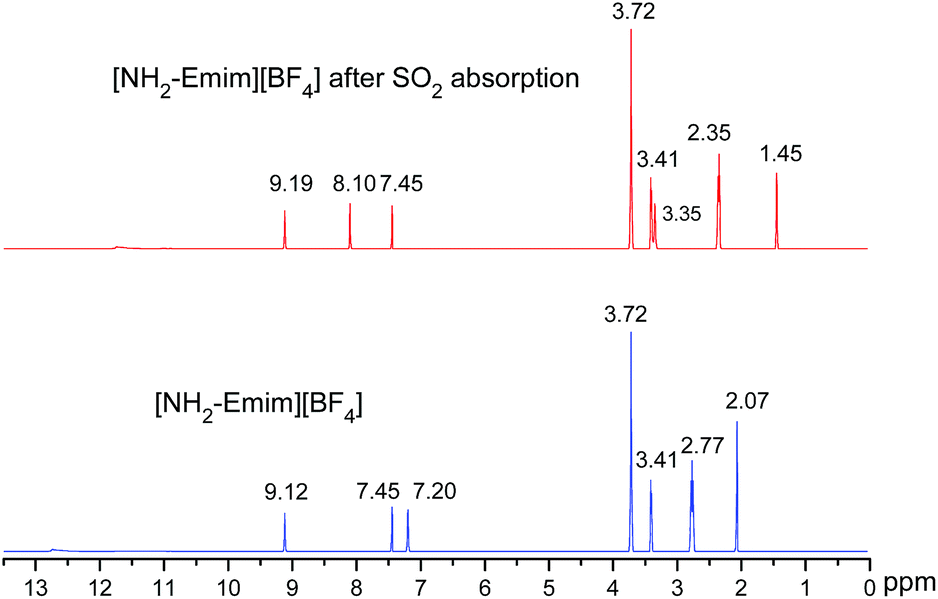 | ||
| Fig. 8 1H NMR spectra of [NH2-Emim][BF4] before and after SO2 absorption. | ||
Conclusions
We have investigated the application of the RRS approach for computing 1H NMR chemical shifts of ILs. The results were compared with those obtained with the use of the typical TMS standard to assess the performance of the RRS methodology. To decrease the errors caused by the anion and the local environment, the ILs that similar to the structure of the studied ILs were used as reference standards in the calculations. m06-2x/aug-cc-pvdz DFT functional can give good prediction results with lower computational cost, which should be as the preferred DFT functional for the prediction. Besides using relative reference standards, ionic clusters consisting of 4 ion pairs were also employed for computing the proton chemical shifts. For the ILs with strongly coordinating anions, the effect of anions was shown to be the main factor contributing to a more reliable prediction of the proton chemical shifts. Single ion pair with the RRS approach is sufficient to produce more accurate and precise 1H NMR chemical shifts for most of the studied ILs. An additional advantage of the RRS method is that the computational results do not depend much on the level of theory used in the calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (21766021 and 21266015).References
- A. S. Amarasekara, Chem. Rev., 2016, 116, 6133–6183 CrossRef PubMed.
- B. Wang, L. Qin, T. Mu, Z. Xue and G. Gao, Chem. Rev., 2017, 117, 7113–7131 CrossRef PubMed.
- Y. Cao and T. Mu, Ind. Eng. Chem. Res., 2014, 53, 8651–8664 CrossRef.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3691 CrossRef PubMed.
- E. I. Izgorodina, Phys. Chem. Chem. Phys., 2011, 13, 4189–4207 RSC.
- D. Bankmann and R. Giernoth, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 63–90 CrossRef.
- S.-T. Lin, M.-F. Ding, C.-W. Chang and S.-S. Lue, Tetrahedron, 2004, 60, 9441–9446 CrossRef.
- E. I. Izgorodina, Z. L. Seeger, D. L. A. Scarborough and S. Y. S. Tan, Chem. Rev., 2017, 117, 6696–6754 CrossRef PubMed.
- A. Bagno, F. D'Amico and G. Saielli, J. Phys. Chem. B, 2006, 110, 23004–23006 CrossRef PubMed.
- A. Dejaegere, R. A. Bryce and D. A. Case, ACS Symposium Series, American Chemical Society, Washington, DC, 1999, pp. 194–206 Search PubMed.
- S. Chen, R. Vijayaraghavan, D. R. MacFarlane and E. I. Izgorodina, J. Phys. Chem. B, 2013, 117, 3186–3197 CrossRef PubMed.
- Y. Chen, Y. Cao, X. Sun and T. Mu, J. Mol. Liq., 2014, 190, 151–158 CrossRef.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef PubMed.
- A. M. Sarotti and S. C. Pellegrinet, J. Org. Chem., 2012, 77, 6059–6065 CrossRef PubMed.
- A. M. Sarotti and S. C. Pellegrinet, J. Org. Chem., 2009, 74, 7254–7260 CrossRef PubMed.
- E. I. Izgorodina, J. Rigby and D. R. MacFarlane, Chem. Commun., 2012, 48, 1493–1495 RSC.
- S. Chen and E. I. Izgorodina, Phys. Chem. Chem. Phys., 2017, 19, 17411–17425 RSC.
- H. P. Li, Y. H. Chang, W. S. Zhu, W. Jiang, M. Zhang, J. X. Xia, S. Yin and H. M. Li, J. Phys. Chem. B, 2015, 119, 5995–6009 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- S. Zahn, D. R. MacFarlane and E. I. Izgorodina, Phys. Chem. Chem. Phys., 2013, 15, 13664–13675 RSC.
- S. Grimme, W. Hujo and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 4875–4883 RSC.
- T. M. Simeon, M. A. Ratner and G. C. Schatz, J. Phys. Chem. A, 2013, 117, 7918–7927 CrossRef PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef.
- K. W. Wiitala, T. R. Hoye and C. J. Cramer, J. Chem. Theory Comput., 2006, 2, 1085–1092 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101–194118 CrossRef PubMed.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed.
- V. S. Bernales, A. V Marenich, R. Contreras, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2012, 116, 9122–9129 CrossRef PubMed.
- Spectral Database for Organic Compounds, https://www.aist.go.jp/RIODB/SDBS/cgi-bin.
- W. Li, S. Dai, D. Li, Q. Zhang, H. Fan, T. Zhang and Z. Zhang, Synthesis, 2016, 49, 1065–1072 CrossRef.
- B. Banerjee, D. Kundu, G. Pugazhenthi and T. Banerjee, RSC Adv., 2015, 5, 85280–85290 RSC.
- B. C. Ranu and S. Banerjee, Org. Lett., 2005, 7, 3049–3052 CrossRef PubMed.
- L. Sun, G. K. Ramesha, P. V. Kamat and J. F. Brennecke, Langmuir, 2014, 30, 6302–6308 CrossRef PubMed.
- J. D. Holbrey, W. M. Reichert, R. P. Swatloski, G. A. Broker, W. R. Pitner, K. R. Seddon and R. D. Rogers, Green Chem., 2002, 4, 407–413 RSC.
- D. R. MacFarlane, S. A. Forsyth, J. Golding and G. B. Deacon, Green Chem., 2002, 4, 444–448 RSC.
- B. D. McCarthy and J. L. Dempsey, Inorg. Chem., 2017, 56, 1225–1231 CrossRef PubMed.
- K. A. Kurnia, M. I. A. Mutalib and B. Ariwahjoedi, J. Chem. Eng. Data, 2011, 56, 2557–2562 CrossRef.
- J. D. Holbrey and K. R. Seddon, J. Chem. Soc., Dalton Trans., 1999, 2133–2139 RSC.
- W. Li, Y. Liu, L. Wang and G. Gao, Energy Fuels, 2017, 31, 1771–1777 CrossRef.
- Z. Lei, J. Yuan and J. Zhu, J. Chem. Eng. Data, 2010, 55, 4190–4194 CrossRef.
- M. Wang, L. Q. Zhang, L. X. Gao, K. W. Pi, J. Y. Zhang and C. G. Zheng, Energy Fuels, 2013, 27, 461–466 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Examples of the RRS approach, Boltzmann averaging and calculation data of ILs. Cartesian coordinates of optimized ionic liquids and details on case study. See DOI: 10.1039/c8ra04822c |
| This journal is © The Royal Society of Chemistry 2018 |