
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of styrene maleic anhydride copolymer grafted graphene and its dispersion in aqueous solution
Youqun Wang ,
Mingsheng He*,
JiaJia Cheng and
Songlei Zhang
,
Mingsheng He*,
JiaJia Cheng and
Songlei Zhang
College of Water and Architectural Engineering, Shihezi University, Shihezi, Xinjiang, P. R. China. E-mail: hms1971@163.com
First published on 12th December 2018
Abstract
In this study, three simple methods were described to synthesize covalently functionalized graphene nanoplatelets (GNPs) with poly(styrene-co-maleic anhydride) (SMA) via ester linkages. Furthermore, the dispersibility of modified GNPs in aqueous solution was characterized by several techniques. Anchoring of SMA long-chains on GNPs via chemical bonds was confirmed by Fourier transform infrared spectroscopy. Furthermore, field emission scanning electron microscopy analysis indicated that the SMA unevenly covered the surface and edges of the GNPs. Solubility measurements showed that the modified GNPs prepared by the designed method had excellent dispersibility in aqueous solution, and the grafting rate of modified GNPs showed an obvious positive correlation with the concentration in aqueous solution as revealed by ultraviolet absorbency and thermogravimetric analysis results. Markedly, the modified GNPs during ultrasonic treatment exhibited better dispersibility and grafting rate compared to others. Moreover, the experimental results confirmed that GNP concentration reached up to 0.32 mg mL−1 in aqueous solution and the grafting rate was 18.02%. The content of SMA had little effect on grafting rate; however, the ultrasonic duration significantly affected the process.
1. Introduction
Much attention from scholars for the excellent mechanical,1,2 electrical3,4 and thermal5 properties has been directed towards graphene since it was discovered in 2004.4 In recent years, considerable progress has been generated in the preparation and application of graphene and its composites.6–8 Graphene composites have been extensively used in many industrial applications, such as supercapacitors,9 biosensors,10 fuel cells,11 film materials,12 LCD materials,13 etc. Nevertheless, many industrial processes involving the dispersion of graphene are restricted due to the poor dispersibility of graphene in liquids because of its insolubility in the matrix, van der Waals forces,14 and π–π stacking between the graphene lamellae.15 Therefore, comprehensive understanding of the graphene dispersion methods and its dispersion mechanisms is highly desirable in order to improve its properties and expand its application areas by surface modification.Although the microstructure of graphene is composed of stable hexatomic rings, its edge and defective sites have a higher activity,16 which provides a possibility for the covalent functionalization of graphene. In recent years, the use of covalent modification to obtain graphene dispersions has attracted tremendous attention from scholars. The fundamental thinking is to chemically modify its edges and incomplete parts to obtain stable graphene dispersions. Moreover, notably, carbon nanotubes (CNTs), previously discovered as one-dimensional carbon materials, have similar properties to graphene in many aspects. Numerous studies related to the preparation and functionalization of CNTs have been providing considerable inspiration. Hordy17 et al. adopted a simple, inexpensive, and scalable method to vapour deposit CNTs directly from stainless steel mesh without any pretreatment or the use of an external catalyst. This method is an effective approach with practical application value and has the potential to be applied for the preparation of graphene. At the same time, Hordy18 et al. surface functionalized CNTs via an Ar/O2/C2H6 capacitively coupled radio frequency plasma discharge. As a result, the aqueous nanofluids that are produced by removing the CNTs from the substrate were found to remain stable for more than six months. Fortunately, compared to CNTs, graphene can be oxidized to a higher degree because of its sheet structure. It can be oxidized to produce a large number of hydroxyl groups, carboxyl groups, and other active groups on its surface,19 which provides more active points for covalent modification. A class of effective method of covalent modification to improve graphene's dispersion in the earlier studies was focused on combining small organic molecules with the oxygen-containing functional groups on the surface of graphene oxides via covalent interaction. Stankovich16 et al. modified graphene oxide (GO) with isocyanate groups. The carboxyl and hydroxyl groups on GO surface were converted into amide groups and carbamates, which improved the dispersion stability of GO in DMF, NMP, and DMSO. Besides isocyanate, 1-ethyl-3-(3-(dimethylamino)-1-propylamine)-carbodiimide,20 thionyl chloride,21 and N,N′-dicyclohexylcarbodiimide22 were also adopted to increase the dispersibility of graphene, which also aided in achievement of an ideal effect.
Previous studies on using polymers to improve the dispersibility of graphene were more extensive than those using small molecule organics. The advantage is that polymer-functionalized graphene can be given other useful functions and features by utilizing such as monomer type, topological structure, and molecular weight. Hou23 et al. used the hydroxyl group on the surface of GO to react with silane coupling agent, and then reduced it to obtain the functionalized water-soluble graphene derivative (EDTA-rGO). Wan24 et al. enhanced the dispersibility of graphene in epoxy resin via covalent modification between GO and 3-(trimethoxysilyl)propyl methacrylate. Li25 et al. covalently grafted poly(t-butyl acrylate) onto the surface of GO via the surface-induced atom transfer radical polymerization and obtained the toluene dispersion, where the concentration was up to 1 mg mL−1. Sun26 et al. utilized polymer PS-PAM to obtain the amphiphilic graphene dispersion. The molecular structure of these polymers-modified graphene was relatively complicated, resulting in complicated synthesis and high cost, thus these methods are not conducive to use widely.
Poly(styrene-co-maleic anhydride) (SMA) is a type of polymer material with excellent performance and cost effectiveness. It is commonly used in pigment dispersants,27,28 emulgator,29,30 epoxy resin curing agents,31 and other fields. The maleic anhydride segments at the SMA after hydrolysis have high reactivity, thus the experimental conditions for functionalization of graphene using SMA may be very mild compared to other covalent modification cases. Moreover, SMA is inexpensive, thus it has the potential for large-scale production and offers the possibility of using graphene for low-value industrial products such as cement-based materials. In this study, a simple and reliable method was introduced for grafting the long-chains of SMA onto GNPs to achieve the purpose of preparing highly dispersed aqueous solution of graphene.
2. Experimental
2.1. Materials and methods
Liquid phase exfoliated graphene nano platelets (GNPs) with around 10 layers were supplied by Nanjing XFNANO Materials Tech Co., Ltd. (China). Poly(styrene-co-maleic anhydride) (SMA-1000) with an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio was obtained from Cray Valley (France), and its physical and chemical properties are listed in Table 1. Sulfuric acid (95–98%), nitric acid (65%), and N,N-dimethylformamide (DMF) were purchased from Sinopharm Chemical Reagent Co., Ltd.
:1 mole ratio was obtained from Cray Valley (France), and its physical and chemical properties are listed in Table 1. Sulfuric acid (95–98%), nitric acid (65%), and N,N-dimethylformamide (DMF) were purchased from Sinopharm Chemical Reagent Co., Ltd.
| Acid Number, mg KOH/g | 480 |
| Number-average molecular weight, Mn | 2000 |
| Weight-average molecular weight, MW | 5500 |
| Solubility, g/100 mL | |
| Solvent at 25 °C | |
| n-Hexane | <1 |
| Toluene | <1 |
| Ethanol-190 proof | <1 |
| Acetone | >50 |
| MEK | >50 |
| THF | >50 |
| Ethyl acetate | >50 |
| DMF | >50 |
The synthesized samples were prepared separately for analysis and testing by various methods. Fourier transform infrared (FTIR, Nicolet iS10, Thermo Fisher Scientific, USA) spectroscopy was used to measure the functional groups of the modified GNPs and tested in the range of 500 to 4000 cm−1. Thermogravimetric analysis (TGA) was carried out using an STA449F3 analyzer (Netzsch, Germany) from room temperature to 800 °C at a heating rate of 10 °C min−1 under the nitrogen atmosphere. The microstructure of GNPs was characterized by field emission scanning electron microscopy (FESEM, SU8010, HITACHI, Japan). The absorbance of the dispersion was characterized using a DR6000 ultraviolet-visible (UV-vis) spectrophotometer (Hach, USA) with a selected wavelength of 270 nm.
2.2. Synthesis
In order to minimize the destruction of the surface structure of graphene and introduce active groups at its edges, the mixed acid was first utilized to treat the GNPs. The specific approach was as follows: sulfuric acid (30 mL) and nitric acid (10 mL) were mixed quantitatively with GNPs in an ice-water bath. Then the mixture of reactants was dispersed under ultrasonication at 30 °C for 6 h. Next, the mixture was filtered after being diluted ten times. Finally, the reactant was rinsed until the pH of the filter liquor reached 7, and the powdery product was achieved after lyophilization.The activity of hydroxyl group linked to graphene has not been reported in previous literature. Therefore, the reaction conditions for modifying graphene should be considered primarily. Herein, three reaction methods were designed as follows:
(a) It is assumed that the hydroxyl group on the surface of the modified graphene is equivalent to that of the primary alcohol on account of the fact that it is generally highly reactive and reacts easily with maleic anhydride. At the same time, in order to assure that the graphene after modification still remains connected with the carboxylic group, SMA was adopted in this study to directly modify graphene as follows: pretreated GNPs (200 mg) were added to the quantitative DMF containing excess SMA, and the mixture reactant was dispersed under ultrasonication for 30 min. Next, the mixture was left to stir at 60 °C for 2 h. Then, the reactant was filtered and rinsed with plenty of pure water to remove the solvent and unreacted reactants. Following these procedures, the powdery product was achieved after lyophilization.
(b) Modification of GNPs under high temperature conditions: pretreated GNPs (200 mg) were added to the DMF containing excess SMA, and the mixture reactant was dispersed under ultrasonication for 30 min. Then the intermediate was left to stir under reflux at 150 °C for 4 h. The solvent and unconverted reactants were removed in the manner similar to that mentioned above to obtain the black powder.
(c) Modification of GNPs under ultrasonic conditions: mixed pretreated GNPs (200 mg) were added to the DMF containing excess SMA, and the mixture reactant was oscillated under ultrasonic condition at 60 °C for 4 h. The solvent and unconverted reactants were removed in the same manner as mentioned above to obtain the black powder. The schematic illustration of synthesis is shown in Fig. 1.
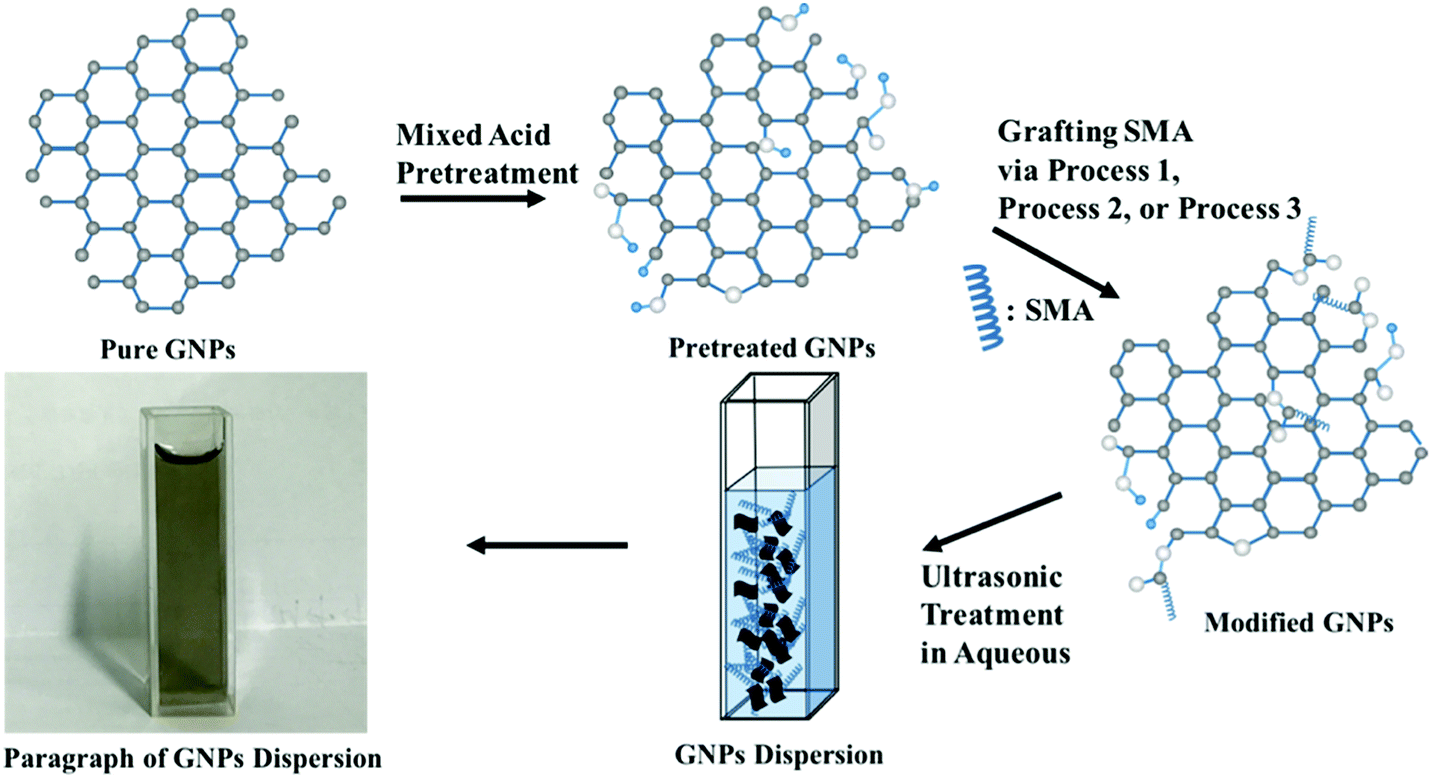 | ||
| Fig. 1 The schematic illustration of synthesis of modified GNPs. | ||
3. Results and discussion
3.1. Characterization and mechanism analysis of modified GNPs
The enhanced interfacial interactions between GNPs and SMA were confirmed by FTIR spectroscopy. Fig. 2 shows the FTIR spectra of the modified GNPs under different reaction conditions. Notably, the FTIR spectrum of GNPs under mixed acid process (Fig. 2a) illustrates bands with distinct intensities at 3439, 1626, and 1052 cm−1, which correspond to O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and C–O–C stretching vibrations, respectively. This indicates that under the mixed acid process a number of oxygen-containing functional groups were introduced onto graphene.32 The spectra of GNPs under the method (a) (Fig. 2c) and (c) (Fig. 2d) illustrate that an absorption peak for the carbonyl stretching vibration of anhydrides is observed at approximately 1710–1790 cm−1, and the peaks at 1377 and 1383 cm−1 are attributed to the antisymmetric extension peak of cyclic anhydride, which represents the reaction product of maleic anhydride. At the same time, the intensity of the hydroxyl stretching vibration absorption peak at 3444 cm−1 is significantly reduced, which might have resulted from the interaction of the residual oxygen functionalities on graphene with the polar anhydride groups of SMA33 However, the high-temperature reaction method (method (b), Fig. 2b) does not show many changes compared to the GNPs modified under the mixed acid treatment.
O, and C–O–C stretching vibrations, respectively. This indicates that under the mixed acid process a number of oxygen-containing functional groups were introduced onto graphene.32 The spectra of GNPs under the method (a) (Fig. 2c) and (c) (Fig. 2d) illustrate that an absorption peak for the carbonyl stretching vibration of anhydrides is observed at approximately 1710–1790 cm−1, and the peaks at 1377 and 1383 cm−1 are attributed to the antisymmetric extension peak of cyclic anhydride, which represents the reaction product of maleic anhydride. At the same time, the intensity of the hydroxyl stretching vibration absorption peak at 3444 cm−1 is significantly reduced, which might have resulted from the interaction of the residual oxygen functionalities on graphene with the polar anhydride groups of SMA33 However, the high-temperature reaction method (method (b), Fig. 2b) does not show many changes compared to the GNPs modified under the mixed acid treatment.
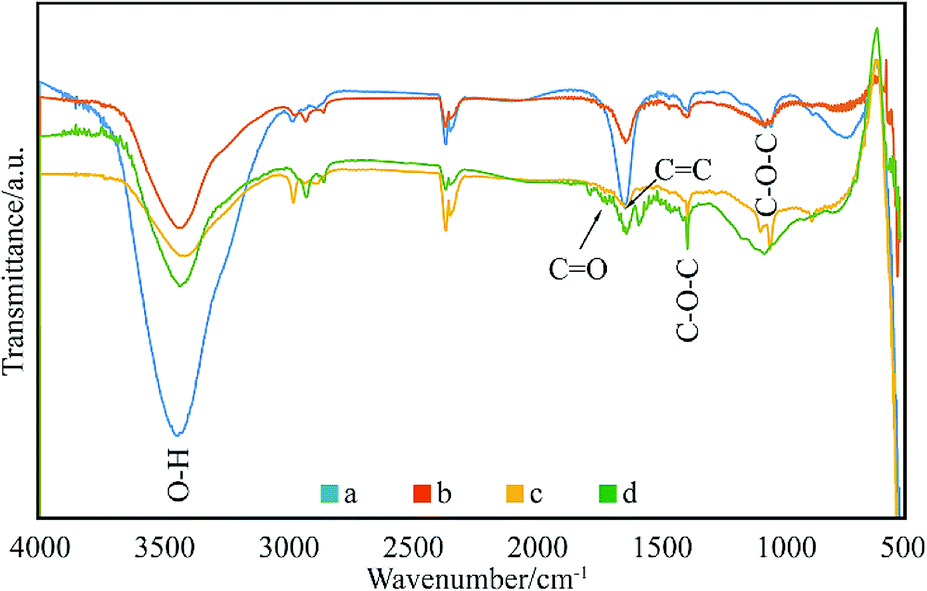 | ||
| Fig. 2 FTIR spectra of: (a) GNPs during mixed acid treatment, (b) modified GNPs reacted at 150 °C, (c) modified GNPs reacted at 60 °C, and (d) modified GNPs reacted at 60 °C under sonication. | ||
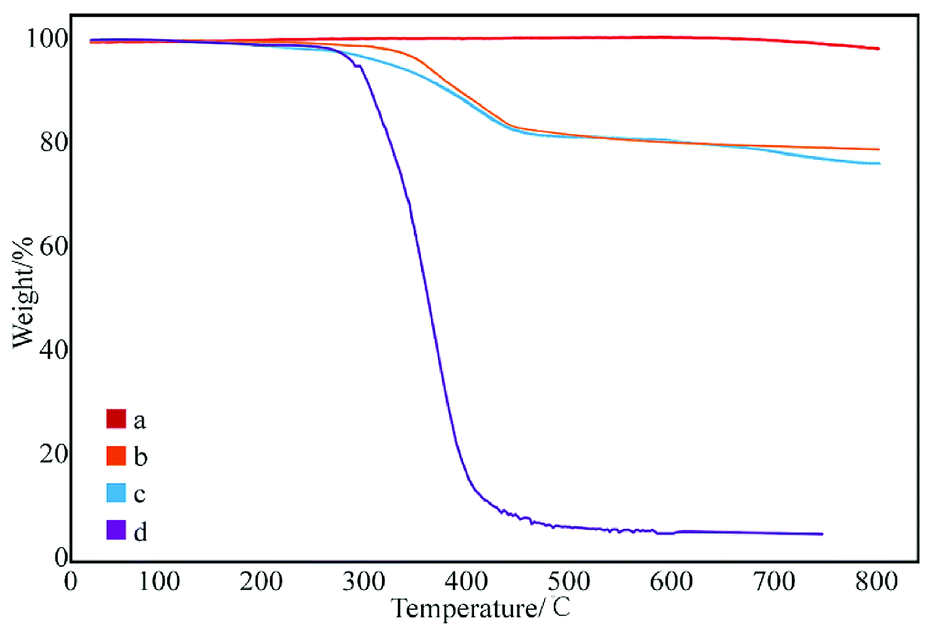
Fig. 3 shows the thermal degradation curve of the modified GNPs under different reaction modes. Fig. 3e demonstrates that the decomposition temperature of SMA is about 300 °C and the maleic anhydride-modified GNPs show a significant agravic stair in the process of weight loss at 300–450 °C, which is caused by the decomposition of the grafted polymer on the graphene surface. Moreover, it further demonstrates that the long-chain molecules of SMA are not a simple physical package for graphene, but with a chemical effect. Quantitatively, the weight degradation rate of the unmodified GNPs (Fig. 3a) is merely 2% at 800 °C, and nearly all the SMA is decomposed at the same temperature. Furthermore, the weight degradation rates of GNPs under methods (a) (Fig. 3b), (b) (Fig. 3c), and (c) (Fig. 3d) in the temperature range from room temperature to 800 °C are 5.13, 9.04, 18.02%, respectively. When SMA was linked onto the surface of the GNPs, its degradation rate was significantly slower than that of the pure SMA, which indicated strong interactions of the SMA with the GNPs.
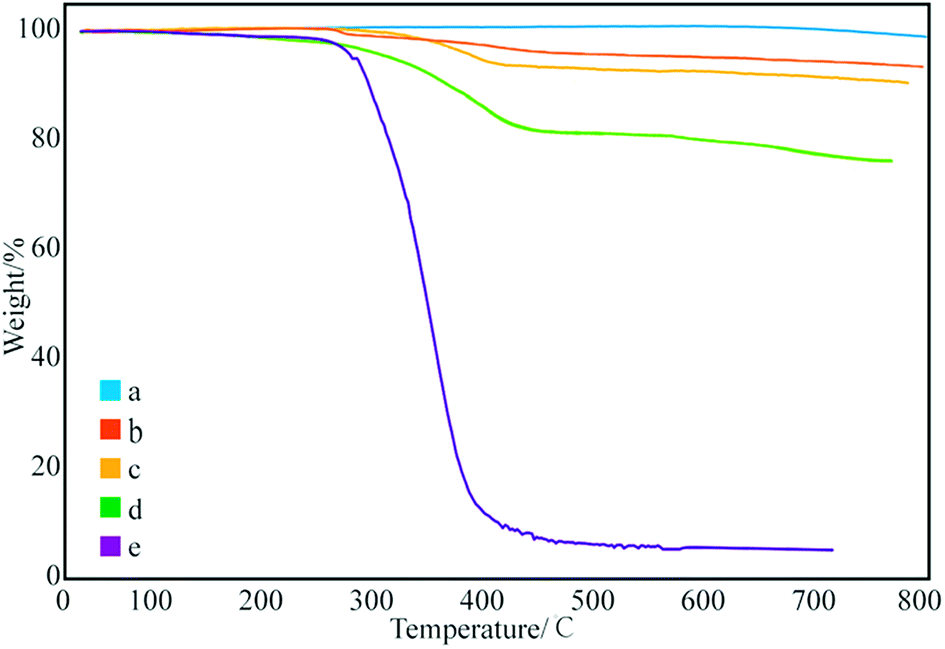 | ||
| Fig. 3 Thermogravimetric curves of: (a) pure GNPs, (b) modified GNPs reacted at 150 °C, (c) modified GNPs reacted at 60 °C, (d) modified GNPs reacted at 60 °C under sonication, and (e) SMA. | ||
FESEM was used to observe the morphology of the graphene dispersions. Fig. 4 shows the FESEM image, exhibiting the modified GNPs under ultrasonic treatment (method (c)) and corresponding graphene dispersion (Fig. 4d). The analysis of image of graphene dispersions indicates that most of the graphene sheets are submicron sized and multi-layered. Fig. 4a shows that the smooth surface of pure GNPs may be due to the regular structure of graphene. In contrast, the image shows the existence of clusters of uneven tumour-shape polymer-coating on the surface (Fig. 4b) and edge (Fig. 4c) of the modified GNPs, which was caused by the different reactivity of the graphene surface. Noteworthy, This further proves the occurrence of chemical grafting reaction between SMA and GNPs. Because if it is physical coating, the distribution of SMA on the surface of GNPs should be uniform. With GNPs bonded with long chain molecule of SMA, the vast long chain molecules drive the GNPs suspending in the aqueous medium.
 | ||
| Fig. 4 FESEM micrographs of: (a) pure GNPs, (b) surface of the modified GNPs, (c) edge of the modified GNPs, and (d) the modified GNPs in aqueous solution. | ||
3.2. Effect of reaction conditions on the modification efficiency of GNPs
After confirming the type of interacting mechanism between SMA and GNPs, the influence of reaction conditions on the modification effect of GNPs was subsequently investigated. First, the relationship between the ultrasonic duration and the grafting rate of modified GNPs was evaluated. Fig. 5 demonstrates that pure SMA basically decomposes at 450 °C, and its effect on the grafting rate can be ignored. Therefore, the difference-value in the weight degradation rate between the treated GNPs and the pure GNPs at 450 °C was adopted in order to evaluate the grafting rate between GNPs and SMA.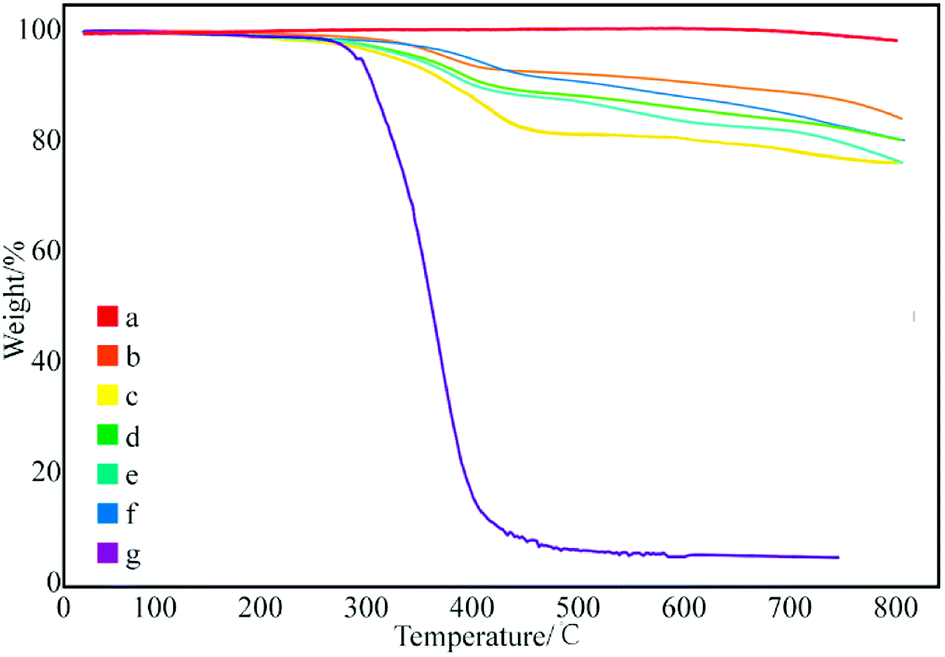 | ||
| Fig. 5 Thermogravimetric curves of modified GNPs at ultrasonication time: (a) 0 h, (b) 2 h, (c) 4 h, (d) 8 h, (e) 12 h, (f) 24 h, and (g) SMA. | ||
Subsequently, with the increase in the ultrasonic duration, the grafting rate gradually decreases. The grafting rate of the samples obtained by ultrasonication for 4 h was the largest, which reached to 18.02% (Fig. 5c). As the time of ultrasonic expression continues to increase, the grafting rate begins to decrease. This can be confirmed based on the fact that the ultrasonic treatment can assist the dispersion of GNPs and increases the efficiency of chemical bonding between SMA and the surface functional groups of GNPs; however, ultrasonic treatment can break chemical bonds under prolonged ultrasonication. The long-chain molecules of SMA that have already been grafted may be broken again if the reaction duration is too long, which reduces the grafting rate. Therefore, under the specific condition, the optimal ultrasonic assisted duration for GNPs grafted SMA is 4 h.
Fig. 6 shows the effect of concentration of SMA in DMF on the grafting rate. The grafting rate modified with 1 and 5% SMA in DMF solution was about 18%, which proves that increasing concentration of SMA has little effect on the grafting rate. This may be ascribed to the fact that the surface active points of the GNPs used in different samples are the same. The grafting reaction of GNPs is already completed, while the concentration of SMA is 1%. Therefore, the concentration of SMA does not help to increase the grafting rate, which can be explained that the degree of grafting is related to the content of active points on the surface of GNPs.
 | ||
| Fig. 6 Thermogravimetric curves of GNPs modified with different content of SMA: (a) pure GNPs, (b) GNPs modified with 1% SMA, (c) GNPs modified with 5% SMA, and (d) SMA. | ||
3.3. Study on the dispersibility of modified GNPs
According to the Lambert Beer's law:where A is UV absorbency, T is light transmittance, E is absorption coefficient, c is the solution concentration, and l is wavelength. This relationship indicates that the absorbance of the solution is proportional to the product of the concentration of the solution and the wavelength. Thus, at a constant wavelength, the concentration of the solution can be expressed indirectly by the absorbance of the solution.34,35 In this experiment, UV spectrophotometry was used to test the maximum solubility of aqueous dispersion of graphene prepared by different treatments. The experimental procedure is as follows: the weighed modified-GNPs were prepared under different treatments (concentrations ranging from 0.025 to 0.125 mg mL−1 in aqueous solution) were added into the aqueous solution for preparing modified GNPs suspensions. These suspensions were oscillated in ultrasonic homogenizer for 30 min and then scanned for their respective spectrum using UV-vis spectrophotometer. The maximum absorption peak at 270 nm was selected to obtain its calibration curve of the variation of modified GNPs concentration with respect to UV absorbency (Fig. 7). Then, the aqueous dispersions containing excess GNPs prepared by different treatments were oscillated in ultrasonic homogenizer for 30 min, and subsequently centrifuged in the high-speed centrifuge. The obtained supernatant could be regarded as a saturated solution of modified GNPs. Next, the supernatant was diluted for 5 times and scanned for obtaining the UV-vis spectrum (for the low concentration of pure GNPs, the absorbance of the supernatant after dilution was not within the range of 0.2–0.8, which did not correspond with the preconditions of the Lambert Beer's law. Thus, the supernatant of pure graphene dispersion after centrifugation did not require dilution), and the concentration of saturated liquid was calculated. The results are presented in Table 2.
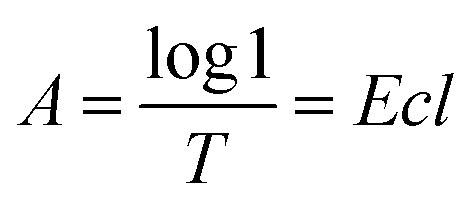
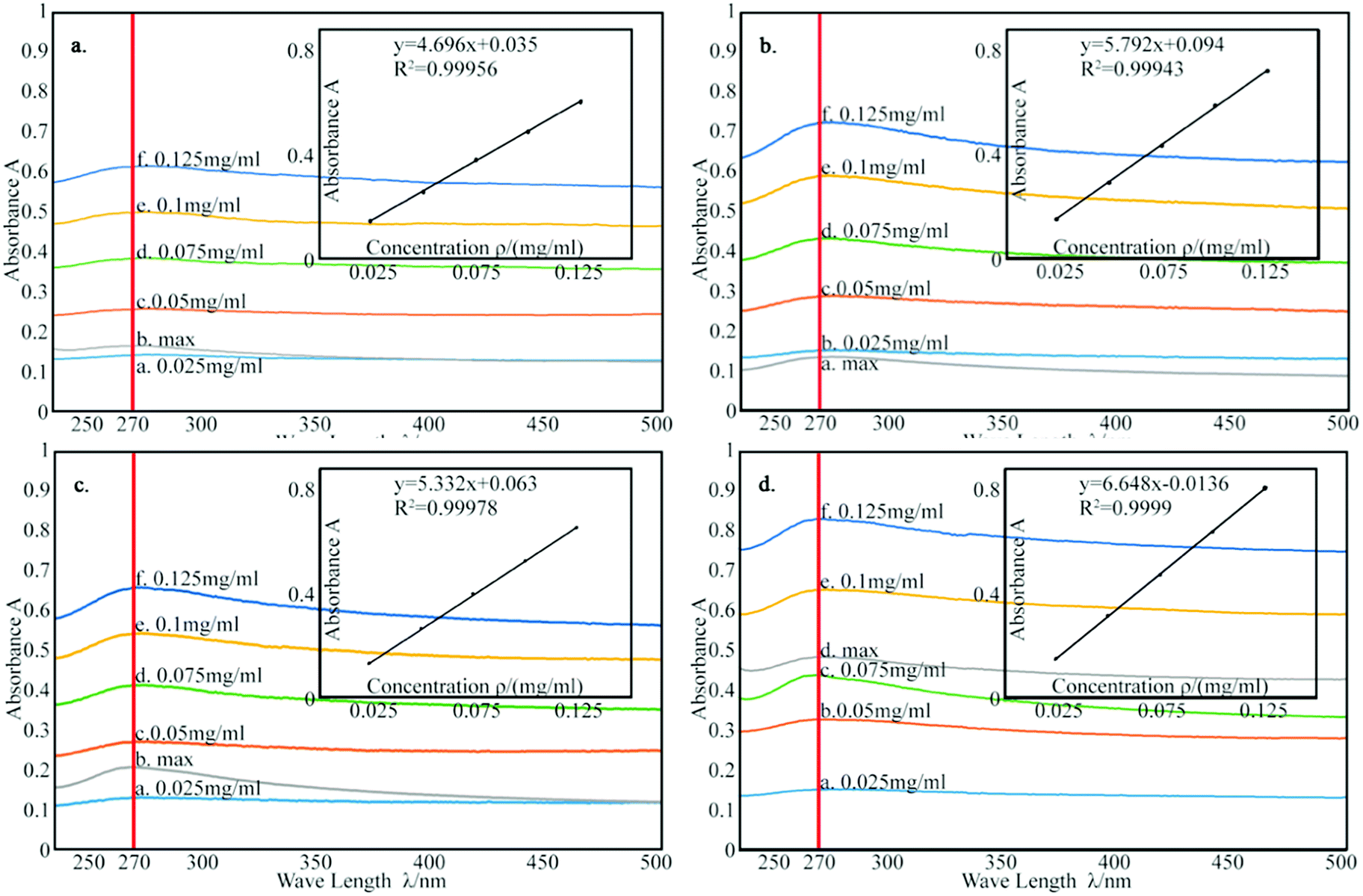 | ||
| Fig. 7 The calibration curve of: (a) pure GNPs, (b) modified GNPs reacted at 150 °C, (c) modified GNPs reacted at 60 °C, and (d) modified GNPs reacted at 60 °C under sonication. | ||
| Name | Curve fitting equation | Correlation coefficient | Maximum absorbance | Maximum solubility/(mg mL−1) |
|---|---|---|---|---|
| Pure GNPs | Y = 4.696x + 0.035 | 0.99956 | 0.172 | 0.029 |
| GNPs-SMA reacted at 150 °C | Y = 5.792x + 0.0094 | 0.99943 | 0.139 | 0.117 |
| GNPs-SMA reacted at 60 °C | Y = 5.332x + 0.063 | 0.99978 | 0.211 | 0.192 |
| GNPs-SMA reacted at 60 °C under sonication | Y = 6.648x − 0.0136 | 0.434 | 0.434 | 0.32 |
Table 2 summarizes that the concentration of the modified graphene dispersion is higher than that of the pure graphene dispersion, and the concentration of the graphene dispersion obtained by ultrasonic process (method (c)) is the highest, reaching 0.32 mg mL−1. Combined with the results of thermal degradation curve, it expresses that the grafting rate of SMA and GNPs is positively related to the dispersion of GNPs. Fig. 8 shows the aqueous dispersion of modified GNPs treated by sonication (method (c)) and aqueous dispersion of pure graphene were standed for one month. Clearly, the modified GNPs have good dispersibility in the aqueous phase, which demonstrates the successful introduction of SMA into GNPs. This result can be explained as follows: SMA has good solubility in aqueous phase, and the long-chain molecules of SMA are stably grafted on the surface of the GNPs and formed steric hindrance in the solution. Then the vast long-chain molecules of SMA led to the steady suspension of the GNPs into these solvents. In contrast, pure modified GNPs are almost non-dispersible in the aqueous phase.
 | ||
| Fig. 8 The photographs of the GNPs dispersion which were allowed to stand for one month: (A) modified GNPs and (B) pure GNPs. | ||
4. Conclusion
SMA was covalently grafted onto the GNPs by the facile technique, and stable graphene dispersion was produced in this study. The main findings are summarized as follows:1. FTIR spectra and FESEM images show that SMA was successfully grafted onto the GNPs, and the SMA long-chain molecules existed unevenly on the edge and surface of GNPs. The grafting rate of modified graphene obtained by ultrasonic processing (method (c)) was the highest according to the thermal degradation curve. Therefore, the ultrasonic procession is an effective method for obtaining SMA-grafted graphene.
2. UV-vis spectrum shows that modified graphene was prepared by method (c) and the concentration of its dispersion could reach 0.32 mg mL−1, which was about 11 times higher than that of pure graphene. Moreover, no deposition occurred for even a month when the dispersion was allowed to stand. Therefore, formation of SMA-grafted graphene is a good method to disperse graphene.
3. Moreover, concentration of the modified graphene under saturated dispersion is positively correlated with the grafting rate based on the combined analysis of thermogravimetric curve and UV-vis spectra. At the same time, the duration of ultrasonic treatment profoundly impacted the grafting rate, and the grafting rate of modified graphene prepared under ultrasonication for 4 h could reach up to 18.02%. Noteworthy, the modified graphene with different dispersibility could be prepared by controlling the time of the ultrasonic wave treatment. On the other hand, SMA concentration in DMF had little effect on grafting yield because possibly the surface active points of GNP used in discrete samples are the same.
The modified GNPs can achieve a stable dispersion without any surfactant, and the conditions required under ultrasonic processing in this study are mild with low cost, thus exhibiting the potential and characteristics of large-scale preparation. The modified graphene has a broad application prospect that can be applied to low-value industrial products such as concrete, geogrid, and sewage adsorption materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express appreciation for the financial support from the National Natural Science Foundation of China (51468057).Notes and references
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, F. Rui, Z. Dai, A. N. Marchenkov, E. H. Conrad and P. N. First, J. Phys. Chem., 2004, 108, 19912–19916 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, science, 2004, 306, 666–669 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336–9344 CrossRef CAS.
- Q. Fang, Y. Shen and B. Chen, Chem. Eng. J., 2015, 264, 753–771 CrossRef CAS.
- S. Gadipelli and X. G. Zheng, Prog. Mater. Sci., 2015, 69, 1–60 CrossRef CAS.
- J. E. Kim, T. H. Han, S. H. Lee, J. Y. Kim, C. W. Ahn, J. M. Yun and S. O. Kim, Angew. Chem., 2011, 123, 3099–3103 CrossRef.
- H. Shen, L. Zhang, M. Liu and Z. Zhang, Theranostics, 2012, 2, 283 CrossRef CAS.
- C. Vallés, C. Drummond, H. Saadaoui, C. A. Furtado, M. He, O. Roubeau, L. Ortolani, M. Monthioux and A. Pénicaud, J. Am. Chem. Soc., 2008, 130, 15802–15804 CrossRef PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. B. T. Nguyen and R. S. Ruoff, Nature, 2016, 448, 457–460 CrossRef.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson and E. W. Hill, Nano Lett., 2008, 8, 1704–1708 CrossRef PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z. F. Ma and D. Li, Angew. Chem., 2011, 50, 7325–7328 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- N. Hordy, N.-Y. Mendoza-Gonzalez, S. Coulombe and J.-L. Meunier, Carbon, 2013, 63, 348–357 CrossRef CAS.
- N. Hordy, S. Coulombe and J. L. Meunier, Plasma Processes Polym., 2013, 10, 110–118 CrossRef CAS.
- C. Shan, L. Wang, D. Han, F. Li, Q. Zhang, X. Zhang and L. Niu, Thin Solid Films, 2013, 534, 572–576 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- X. Zhang, Y. Huang, Y. Wang, Y. Ma, Z. Liu and Y. Chen, Carbon, 2009, 47, 334–337 CrossRef CAS.
- L. M. Veca, F. Lu, M. J. Meziani, L. Cao, P. Zhang, G. Qi, L. Qu, M. Shrestha and Y. P. Sun, Chem. Commun., 2009, 2565–2567 RSC.
- S. Hou, S. Su, M. L. Kasner, P. Shah, K. Patel and C. J. Madarang, Chem. Phys. Lett., 2010, 501, 68–74 CrossRef CAS.
- Y. J. Wan, L. X. Gong, L. C. Tang, L. B. Wu and J. X. Jiang, Composites, Part A, 2014, 64, 79–89 CrossRef CAS.
- G. L. Li, G. Liu, M. Li, D. Wan, K. G. Neoh and E. T. Kang, J. Phys. Chem. C, 2010, 114, 12742–12748 CrossRef CAS.
- H. L. Sun, D. R. Dreyer, J. An, A. Velamakanni, R. D. Piner, S. Park, Y. Zhu, O. K. Sang, C. W. Bielawski and R. S. Ruoff, Macromol. Rapid Commun., 2010, 31, 281–288 CrossRef.
- S. Kumari, D. Nigam and I. Nigam, Int. J. Plast. Technol., 2011, 15, 112–132 CrossRef CAS.
- A. Shakhnovich, J. Carroll and D. Williams, Polymeric Dispersants with Specific Affinity to Pigments for Ink Jet Applications, NIP & Digital Fabrication Conference, Society for Imaging Science and Technology, 2009, vol. 2009, no. 1 Search PubMed.
- F. Yu, Z.-H. Chen and X.-R. Zeng, Colloid Polym. Sci., 2009, 287, 549–560 CrossRef CAS.
- J. Sun, C. Yi, W. Wei, D. Zhao, Q. Hu and X. Liu, Langmuir, 2014, 30, 14757–14764 CrossRef CAS PubMed.
- R. A. Vora, H. C. Trivedi, C. P. Patel, J. T. Guthrie, A. Kazlauciunas and M. C. Trivedi, High Perform. Polym., 1996, 8, 281–293 CrossRef CAS.
- H. Hu, X. Wang, J. Wang, F. Liu, M. Zhang and C. Xu, Appl. Surf. Sci., 2011, 257, 2637–2642 CrossRef CAS.
- Z. He, B. Zhang, H. B. Zhang, X. Zhi, Q. Hu, C. X. Gui and Z. Z. Yu, Compos. Sci. Technol., 2014, 102, 176–182 CrossRef CAS.
- W. Bao-Min, Z. Yuan, G. U. O. Zhi-Qiang, H. A. N. Yu and M. A. Hai-Nan, Nano, 2012, 7, 2175–2382 Search PubMed.
- L. Jiang, G. Lian and S. Jing, J. Colloid Interface Sci., 2003, 260, 89–94 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |