
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of SiO2 amount on heterogeneous base catalysis of SiO2@Mg–Al layered double hydroxide†
Mahiro Shirotoria,
Shun Nishimura *ab and
Kohki Ebitani*ab
*ab and
Kohki Ebitani*ab
aSchool of Materials Science, Japan Advances Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: s_nishim@jaist.ac.jp; Fax: +81-761-51-1149; Tel: +81-761-51-1613
bGraduate School of Advanced Science and Technology, Japan Advances Institute of Science and Technology, Japan
First published on 6th August 2018
Abstract
The effects of SiO2 amount on the base catalysis of highly active finely crystallized Mg–Al type layered double hydroxides prepared by the co-precipitation method with coexistence of SiO2 spheres, denoted as SiO2@LDHs, were investigated. With the Si/(Mg + Al) atomic ratios of 0–0.50, the highest activity for the Knoevenagel condensation was observed in the case of Si/(Mg + Al) = 0.17, as the reaction rate of 171.1 mmol g(cat)−1 h−1. The base activity increased concomitantly with decreasing LDH crystallite size up to Si/(Mg + Al) atomic ratio of 0.17. However, above the Si/(Mg + Al) atomic ratio of 0.17, the reaction rate and TOFbase were decreased although the total base amount was increased. Results of TEM-EDS and 29Si CP-MAS NMR suggest that the co-existing SiO2 causes advantages for dispersion and reduction of the LDH crystallite to improve the base catalysis of SiO2@Mg–Al LDH, whereas the excess SiO2 species unfortunately poisons the highly active sites on the finely crystallized LDH crystals above a Si/(Mg + Al) atomic ratio of 0.17. According to these results, we inferred that the amount of spherical SiO2 seeds in the co-precipitation method is an important factor to increase the base catalysis of SiO2@LDHs; i.e. the control of Si/(Mg + Al) atomic ratio is necessary to avoid the poisoning of highly active base sites on the LDH crystal.
Introduction
Heterogeneous solid catalysts have been widely used in various chemical reactions in the chemical industry, exhaust gas purification, and environmentally friendly reactions. They are more suitable for reactions at high temperatures than homogeneous catalysts, and are removed easily from the reactor. In particular, solid acid catalysts are used in many important processes related to petroleum refining and petrochemical production. Consequently, numerous studies have specifically examined solid acid catalysts. On the other hand, fewer efforts have been devoted to heterogeneous base catalysts.1 Because general base sites on a solid base catalyst are readily poisoned by moisture and carbon dioxide in the atmosphere,2 the development of a promotion strategy for solid base catalysts is more difficult than for solid acid catalysts.Layered double hydroxide, generally called LDH, is a well-known layered clay mineral that is known to act as a unique solid base catalyst. Actually, LDH comprises brucite-like positively charged two-dimensional sheets denoted as [M1−x2+Mx3+(OH)2]x+ and interlayer parts denoted as Ax/nn−·mH2O, where An− corresponds to interlayer anions such as carbonate and hydroxide. The positively charged sheets and the interlayers are alternately laminated to compensate for the charge between sheets.3–5 Base sites on LDH are mainly regarded as identical Brønsted basic OH− and HCO3− anions, which are adsorbed onto the LDH surface. These unique basic sites can act even in an air atmosphere and can exhibit basic characteristics without pretreatment.4 As a solid base catalyst, LDH is well-known to catalyze various organic transformations such as aldol condensation,6–9 Knoevenagel condensation,10–13 epoxidation,14–16 and transesterification.17–19 Recent studies have indicated that LDHs can promote advanced environmentally friendly reactions such as biomass-derived saccharide conversion13,20–25 and photocatalytic conversion of CO2 in an aqueous solution.26–28 Therefore, the development of highly active LDH catalysts is eagerly sought.
The main base sites on the LDH surface are generally regarded as adsorbed anions located at the corner and edge of a crystal.29 However, the anions in the interlayer space cannot participate in the chemical reactions because of the high charge density of the LDH layers and the high contents of anionic species and water molecules, resulting in strong interlayer electrostatic interactions between the sheets.4,30 Therefore, the delamination of LDH nanosheets30–36 and the fine crystallization of LDH on appropriate carriers37–42 have been conducted to increase the number of exposed active base sites. As-prepared LDH materials also have been evaluated carefully to assess their characteristics and utility as photocatalysts and electrocatalysts,43,44 high active base catalysts for Knoevenagel condensation45 and epoxidation,46 magnetic separation of proteins,37 pseudocapacitance,38 flame retardancy of epoxy resins,40 and as adjuvants.41 Nevertheless, no report describes a study of improvement of base catalysis of LDH itself by fine crystallization followed by in situ growth method of SiO2@LDH nanoparticles.
An earlier study revealed a co-precipitation method for preparation of small-crystallized LDH catalysts on SiO2 nanospheres and explored the superior base catalyses for the Knoevenagel condensation of benzaldehyde compared with conventional LDHs.47 This method is applicable for the preparation of SiO2@LDH nanoparticles with various compositions and element combinations: i.e. SiO2@M2+–M3+LDH (M2+: Mg2+ or Ni2+, M3+: Al3+ or Ga3+, and M2+/M3+: 1 or 3). Various characterizations of SiO2@LDH nanoparticles using XRD, TEM-EDS, and 29Si CP-MAS NMR techniques revealed that the co-existence of small SiO2 sphere (ca. 40 nm diameter) surface generated the starting points of LDH growth via Si–O–M covalent bond formation, leading to the formation of fine-crystallized LDH and enhancement of base catalysis for the Knoevenagel condensation of benzaldehyde with ethyl cyanoacetate. However, the roles of Si–O–M covalent bonds in the fine-crystallization of LDH and base catalysis have not been explored well. Therefore, in this paper, we investigated the base properties and structural parameters of as-prepared SiO2@Mg–Al LDH materials with Mg2+/Al3+ = 3, and discussed the base catalysis with different Si to (Mg + Al) ratios to reveal the mechanism of our strategy.
Experimental
Materials and synthesis of catalysts
Tetraethyl orthosilicate (TEOS), triethanolamine (TEA), and benzaldehyde were purchased from Sigma-Aldrich Corp. Sodium carbonate (Na2CO3), sodium hydroxide (NaOH) and toluene were supplied by Kanto Chemical Co. Inc. Cetyltrimethylammonium bromide (CTAB), magnesium nitrate hexahydrate (Mg(NO3)2·6H2O), aluminum nitrate enneahydrate (Al(NO3)3·9H2O), benzaldehyde and benzoic acid were obtained from Wako Pure Chemical Industries Ltd. Benzaldehyde was purified by distillation under 0.4 Pa pressure. Ethyl cyanoacetate was purchased from Tokyo Chemical Industry Co. Ltd. and was used without further purification.Spherical SiO2 (40 nm) was prepared according to descriptions in earlier reports.41,46 First, 96 mmol of TEA and 2 mL of TEOS were combined in a 200 mL eggplant flask. The two-phase mixture was heated in an oil bath at 363 K for 20 min without stirring. When the mixture was removed from the oil bath, 26.0 mL of an aqueous solution (2.8 wt%) of CTAB pre-heated at 333 K was added immediately as a structure-directing agent in a condensation process. Then, it was stirred continuously for 24 h at room temperature. Thereafter, the resulting mixture was added to 50 mL of ethanol to obtain colloidal aqueous suspension. The obtained precipitate was centrifuged for 5 min at 4000 rpm. After decantation, the sediment was re-dispersed through vigorous stirring in 50 mL of an ethanolic solution of ammonium nitrate (20 g L−1), and then refluxed for 1 h. This procedure was repeated three times. The same operation was performed with a solution of concentrated hydrochloric acid in ethanol (5 g L−1) to replace the ammonium ions. The final sediment was washed with ethanol, and then dried in vacuo. The obtained spherical SiO2 powder was calcined at 823 K under 1 L min−1 of air flow for 6 h.
The SiO2@(Z)LDH catalysts (Z: desired Si/(Mg + Al) atomic ratio) were prepared via an in situ co-precipitation method according to a previous report.42 Spherical SiO2 (40 nm) was dispersed in 20 mL of water using ultrasound treatment. After 30 min, 0.96 mmol of Na2CO3 was added to the solution. Then, after an additional 5 min of sonication was conducted, 19.2 mL of metal nitrate aqueous solution ([Mg] + [Al] = 0.075 M) was slowly dropped into the spherical SiO2 dispersed solution, followed by stirring at room temperature. The pH was maintained at 10.0 by an aqueous NaOH solution (1 M) during titration. The obtained suspension was stirred for an additional 1 h. After the resulting paste was filtered, it was washed with 1 L of water and ethanol. Then, it was dried at 383 K overnight. The Si/(Mg2+ + Al3+) atomic ratios were varied from 0 to 0.50 whereas the Mg/Al atomic ratio was adjusted to 3.
Reaction
Knoevenagel condensation of benzaldehyde with ethyl cyanoacetate was proceeded in a 20 mL Schlenk tube under an N2 flow (30 mL min−1). The reaction was performed using 1.0 mmol of benzaldehyde, 1.2 mmol of ethyl cyanoacetate, 10 mg of catalysts and 3 mL of toluene at 313 K. The obtained products were analyzed using a GC-FID (GC-2014, Shimadzu Corp.) equipped with a polar column (DB-FFAP, Agilent Technologies Inc.).Characterizations
X-ray diffraction patterns (XRD) were collected using a SmartLab (Rigaku Corp.) with a Cu Kα X-ray source (40 kV, 30 mA). The LDH (003) and (110) crystallite sizes were calculated using the Scherrer equation: Dhkl = Kλ/(β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) (K: Scherrer number (0.9), λ: incident ray wavelength (0.1542 nm), β: peak width at half height (rad), θ; Bragg angle). 29Si cross polarization magic angle spinning nuclear magnetic resonance (29Si CP-MAS NMR) measurements were obtained by an Avance III 500 (Bruker Analytik GmbH) in a 4 mm ZrO2 rotor. The spinning rate was 8 kHz. The 29Si chemical shifts are referenced to hexamethylcyclotrisiloxane (taken to be at δ = −9.6875 ppm). Transmission electron microscope – energy dispersive X-ray spectroscopy (TEM-EDS) elemental mapping analytical techniques were done with a JEM-ARM200F (JEOL) at 200 kV. Inductively coupled plasma – atomic emission spectrometry (ICP-AES) was operated by an iCAP 6300 Duo (Thermo Fisher Scientific Inc.) to estimate the actual amount of precipitated M(OH)x and SiO2 in as-prepared SiO2@Mg–Al LDHs with various Si/(Mg + Al) atomic ratio.
cosθ) (K: Scherrer number (0.9), λ: incident ray wavelength (0.1542 nm), β: peak width at half height (rad), θ; Bragg angle). 29Si cross polarization magic angle spinning nuclear magnetic resonance (29Si CP-MAS NMR) measurements were obtained by an Avance III 500 (Bruker Analytik GmbH) in a 4 mm ZrO2 rotor. The spinning rate was 8 kHz. The 29Si chemical shifts are referenced to hexamethylcyclotrisiloxane (taken to be at δ = −9.6875 ppm). Transmission electron microscope – energy dispersive X-ray spectroscopy (TEM-EDS) elemental mapping analytical techniques were done with a JEM-ARM200F (JEOL) at 200 kV. Inductively coupled plasma – atomic emission spectrometry (ICP-AES) was operated by an iCAP 6300 Duo (Thermo Fisher Scientific Inc.) to estimate the actual amount of precipitated M(OH)x and SiO2 in as-prepared SiO2@Mg–Al LDHs with various Si/(Mg + Al) atomic ratio.
Results and discussion
Optimization of Si/(Mg + Al) ratio in SiO2@LDH
We prepared SiO2@LDHs with various loading amounts of SiO2 to ascertain the optimized SiO2@LDH structure for high catalytic reactivity. The sphere morphology and the diameter of SiO2 were confirmed from SEM and TEM observations, as presented in an earlier report.47 The correlation between Si/(Mg + Al) atomic ratio, base catalysis, and structural properties of SiO2@LDHs were investigated in the range of 0–0.50 on Si/(Mg + Al) atomic ratio. The prepared catalysts are designated as SiO2@(Z)LDH, where Z is a Si/(Mg + Al) atomic ratio. The actual ratios of Si/(M2+ + M3+) and M2+/M3+ in the obtained materials are presented in Table 1.| Sample | Si/(M2+ + M3+) ratio | M2+/M3+ ratio | ||
|---|---|---|---|---|
| Precursor | Obtained material | Precursor | Obtained material | |
| LDH(CP) | 3.0 | 2.8 | ||
| SiO2@(0.13)LDH | 0.13 | 0.14 | 3.0 | 2.5 |
| SiO2@(0.17)LDH | 0.17 | 0.18 | 3.0 | 2.6 |
| SiO2@(0.25)LDH | 0.25 | 0.28 | 3.0 | 2.5 |
| SiO2@(0.50)LDH | 0.50 | 0.53 | 3.0 | 2.2 |
Fig. 1 presents catalytic activity for the Knoevenagel condensation over SiO2@(Z)LDHs as (A) time-based reaction progression on benzaldehyde conversion and (B) reaction rate. The detailed results are listed in Table S1 (see ESI†). Among various Si/(Mg + Al) atomic ratio from 0 to 0.50, actually, the 0.17 was found to be the best catalyst with a reaction rate of 171.1 mmol g(cat)−1 h−1. This reaction rate is 2.2 times higher than conventional LDH prepared with the same co-precipitation method without SiO2 seeds (Si/(Mg + Al) = 0).
 | ||
| Fig. 1 Activities for Knoevenagel condensation of benzaldehyde with ethyl cyanoacetate over as-prepared SiO2@LDHs with various Si/(Mg + Al) atomic ratio; (A) time-based reaction progression on benzaldehyde conversion and (B) reaction rate. Reaction conditions: benzaldehyde (1.0 mmol), ethyl cyanoacetate (1.2 mmol), catalyst (10 mg), toluene (3 mL), 313 K, N2 flow (30 mL min−1). Number in (A) denotes Si/(Mg + Al) atomic ratio. | ||
The XRD patterns and crystal properties of SiO2@(Z)LDHs are portrayed respectively in Fig. 2 and Table 2. All prepared catalysts showed an LDH-originated diffraction pattern. The intensity of LDH originated peaks decreased in accordance with Si loading amount, whereas that of amorphous SiO2 increased slightly. Lattice parameters a and c, respectively calculated from LDH (003) and (110) diffraction peaks, are almost identical among Si/(Mg + Al) atomic ratios of 0–0.50. This result indicates clearly that these SiO2@(Z)LDHs have the same LDH crystal unit. However, the crystallite size of LDH is unquestionably affected by Si/(Mg + Al) atomic ratio. The crystallite size of D(003) is reduced in accordance with the Si loading amount. The crystallite size of D(110) is almost identical in the region among Si/(Mg + Al) ratio of 0–0.13, but it is reduced from ca. 13 nm to 8 nm when a Si/(Mg + Al) ratio increased. In our earlier research, it was revealed that the co-precipitation method with co-existence of spherical SiO2 caused dispersion of starting points of LDH crystal growth on the SiO2 surface through the Si–O–Al and Si–O–Mg covalent bonds to lead generation of fine-crystallized LDH nanocrystal.47 Fig. 3(A) shows that the spherical SiO2 (40 nm) showed three peaks at −91, −100 and −109 ppm, which respectively correspond to Q2, Q3, and Q4 species48–50 where Qn designated the Si-centered tetrahedral structural species; Q refers to silicon atom and n denotes the number of bridging oxygens. Furthermore, SiO2@(Z)LDHs showed broad resonance between −70 to −115 ppm, which include some peaks attributed to Q0 and/or Q1 (−60 to −83 ppm)49 and a Si-centered tetrahedral structure that possesses Si–O–Al and Si–O–Mg bonds (−73 to −105 ppm).42
 | ||
| Fig. 2 XRD patterns of as-prepared SiO2@LDHs with various Si/(Mg + Al) atomic ratio. | ||
| Si/(Mg + Al) atomic ratio | Lattice parameter c/nm | Crystallite size (003)a/nm | Lattice parameter a/nm | Crystallite size (110)a/nm |
|---|---|---|---|---|
| a The crystallite sizes of LDHs were calculated by Scherrer equation: Dhkl = Kλ/(βcosθ) (K: Scherrer number (0.9), λ: incident ray wavelength (0.1542 nm), β: peak width at half height (rad)). |
||||
| 0 | 2.33 | 7.6 | 0.31 | 13.4 |
| 0.13 | 2.32 | 5.2 | 0.31 | 13.7 |
| 0.17 | 2.36 | 4.5 | 0.31 | 10.3 |
| 0.25 | 2.38 | 3.7 | 0.31 | 8.0 |
| 0.50 | 2.43 | 2.8 | 0.31 | 8.3 |
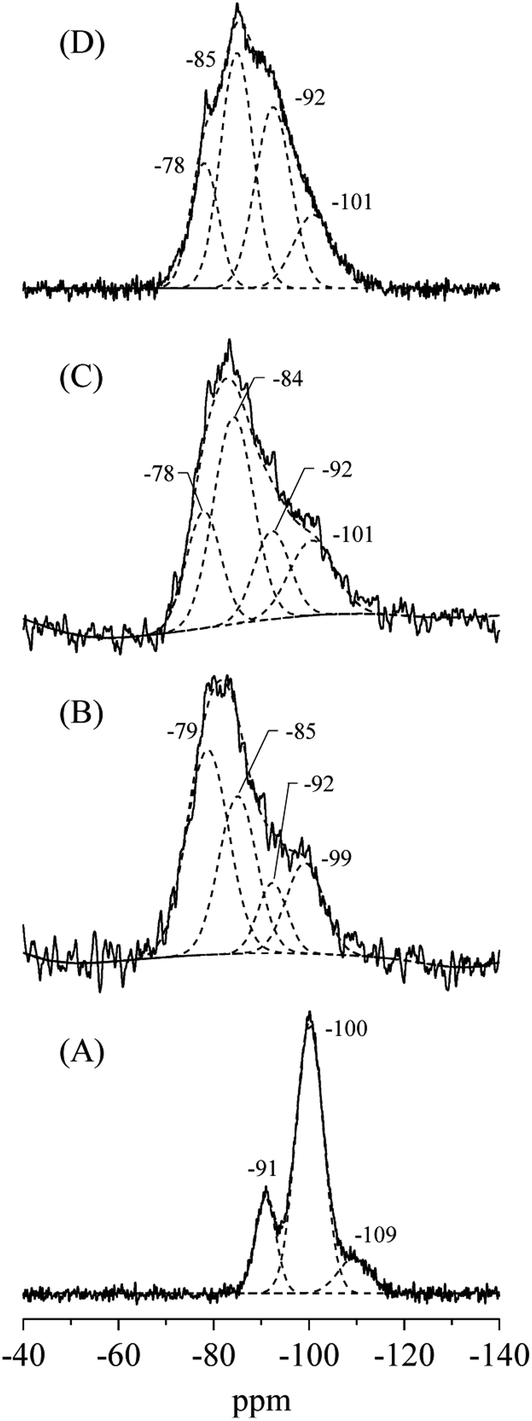 | ||
| Fig. 3 29Si CP-MAS NMR spectra of (A) spherical SiO2 (40 nm), (B) SiO2@(0.13)LDH, (C) SiO2@(0.17)LDH and (D) SiO2@(0.50)LHD. | ||
These results suggest that, in the case of lower Si/(Mg + Al) atomic ratio (<0.13), the LDH crystal is immobilized onto the SiO2 surface through the Si–O–Al and Si–O–Mg bonds to inhibit ab-face stacking without reducing the plane crystallite size. This result seems to be attributable to the lower number of starting points of LDH crystal growth: the amount of metal constituting one crystal did not change compared with conventional LDH prepared without SiO2 seeds (Si/(Mg + Al) = 0). Actually, the proportion for the 29Si CP-MAS NMR peaks attributed to Si–O–Mg and Si–O–Al bonds on the SiO2@(0.13)LDH is only ≤40%, whereas that of SiO2@(0.17)LDH is ≤61%, as shown in Fig. 3 and Table 3. Therefore, we infer that the number of Si–O–Mg and Si–O–Al bonds on the SiO2 surface deeply affected reduction of the crystallite size not only in the stacking direction but also in the plane direction when below Si/(Mg + Al) < 0.17–0.25.
| Sample | Assignment | δ/ppm | Percentage/% |
|---|---|---|---|
| SiO2 (40 nm) sphere | Q4 | −109 | 11 |
| Q3 | −100 | 70 | |
| Q2 | −91 | 19 | |
| SiO2@(0.13)LDH | Q3 | −99 | 17 |
| Q2 | −92 | 10 | |
| Q4(3Al), Q3(1Mg) | |||
| Q4(4Al), Q3(1Al), Q2(1Mg) | −85 | 30 | |
| Q1 and/or Q0 | −79 | 43 | |
| SiO2@(0.17)LDH | Q3 | −100 | 18 |
| Q2 | −92 | 17 | |
| Q4(3Al), Q3(1Mg) | |||
| Q4(4Al), Q3(1Al), Q2(1Mg) | −85 | 44 | |
| Q1 and/or Q0 | −78 | 21 | |
| SiO2@(0.50)LDH | Q3 | −101 | 14 |
| Q2 | −92 | 31 | |
| Q4(3Al), Q3(1Mg) | |||
| Q4(4Al), Q3(1Al), Q2(1Mg) | −85 | 37 | |
| Q1 and/or Q0 | −78 | 17 |
Correlation between the base amount and catalytic activity for Knoevenagel condensation over SiO2@(Z)LDHs is presented in Table 4. Although the base amount of SiO2@(0.13)LDH was lower than that of conventional LDH, the reaction rate and the apparent TOF per base site (TOFbase) for SiO2@(0.13)LDH are higher than those of LDH. The D(003) of SiO2@(0.13)LDH was smaller than LDH, whereas D(110) of SiO2@(0.13)LDH and LDH are almost identical, as shown in Table 2. Therefore, these indicated that the immobilization of LDH crystal onto SiO2 with inhibition of the ab-face stacking led to increase in the number of highly active base sites located on the surface LDH layer. Above the Si/(Mg + Al) atomic ratio of 0.13, a base amount increased in accordance with Si/(Mg + Al) ratio from 0.32 to 0.49 mmol g(cat)−1. Furthermore, the activity was maximized at Si/(Mg + Al) ratio of 0.17 with the reaction rate of 171.1 mmol g(cat)−1 h−1. It is particularly interesting that the reaction rate per obtained LDH phase and TOFbase were also maximized at Si/(Mg + Al) ratio of 0.17 with the reaction rate of 193.6 mmol g(LDH)−1 h−1 and TOFbase of 450 h−1. Above the Si/(Mg + Al) ratio of 0.17, both the reaction rate per LDH phase and TOFbase were decreased respectively to 158.4 mmol g(LDH)−1 h−1 and 238 h−1 at Si/(Mg + Al) ratio of 0.50. These results strongly suggest that the Si/(Mg + Al) atomic ratio affects not only the LDH crystallite size and base amount but also the type of base sites and these fractions.51
| Sample | Amount of LDHa/wt% | Reaction rateb | Base amountc/mmol g(cat)−1 | TOFbase/h−1 | |
|---|---|---|---|---|---|
| mmol g(cat)−1 h−1 | mmol g(LDH)−1 h−1 | ||||
| a The amount of LDH in the SiO2@(Z)LDHs was calculated by ICP-AES with an assumption: all SiO2@(Z)LDHs are composed of mixture of LDH and SiO2.b Reaction rate for the Knoevenagel condensation of benzaldehyde with ethyl cyanoacetate.c Base amount calculated from poisoning test by benzoic acid titration. | |||||
| LDH(CP) | 100 | 76.9 | 76.9 | 0.42 | 183 |
| SiO2@(0.13)LDH | 90.8 | 124.5 | 137.0 | 0.32 | 389 |
| SiO2@(0.17)LDH | 88.3 | 171.1 | 193.6 | 0.38 | 450 |
| SiO2@(0.25)LDH | 82.9 | 158.4 | 191.0 | 0.40 | 396 |
| SiO2@(0.50)LDH | 73.6 | 116.7 | 158.4 | 0.49 | 238 |
The LDH crystallite size of SiO2@(0.50)LDH is at least smaller than that of SiO2@(0.17)LDH. Therefore, the base catalysis of SiO2@(0.50)LDH is expected to be better than that of SiO2@(0.17)LDH if the base catalysis is only influenced by the crystallite size. 29Si CP-MAS NMR spectra showed that the proportion of terminal Si–OH species assigned as Q0 and/or Q1 decreased in accordance with Si/(Mg + Al) atomic ratio, as shown in Table 3, indicating first that a surface Si–O–Si bond is cleaved to generate terminal Si–OH species and then that these act as cross-link point with Mg and Al ions. Consequently, when there are the excess free terminal Si–OH species in the solution after the generation of SiO2@LDH, these excess Si–OH species cover the LDH crystal to produce Si–O–Mg and Si–O–Al covalent bonds. Although the base amount is increased even the region from SiO2@(0.13)LDH to SiO2@(0.50)LDH, this phenomenon might take place only with difficulty on the inferior base sites located at a flat plane of LDH. However, the decrease of TOFbase strongly suggests that the high active base sites are poisoned by Si species in the case of a higher Si/(Mg + Al) atomic ratio.
Dark-field TEM images and results of EDS elemental mapping of SiO2@(Z)LDHs are presented in Fig. 4. In the case of lower Si/(Mg + Al) atomic ratio such as 0.13 and 0.17, the LDH crystal is generated with covering the SiO2 phase to form a SiO2 core – LDH shell-like structure, as shown in Fig. 4(A)–(J). Furthermore, results show that the boundary between SiO2 phase and LDH phase becomes ambiguous in accordance with the increase of Si/(Mg + Al) atomic ratio (Fig. 4(K)–(T)). These indicate that first the LDH crystal grows up from the SiO2 surface to generate the immobilized SiO2 core – LDH shell structure. If there are excess dissolved SiO2 species possessing a free terminal Si–OH group, then these produced Si–O–Mg and Si–O–Al covalent bonds with the LDH crystal to cover the LDH shell.
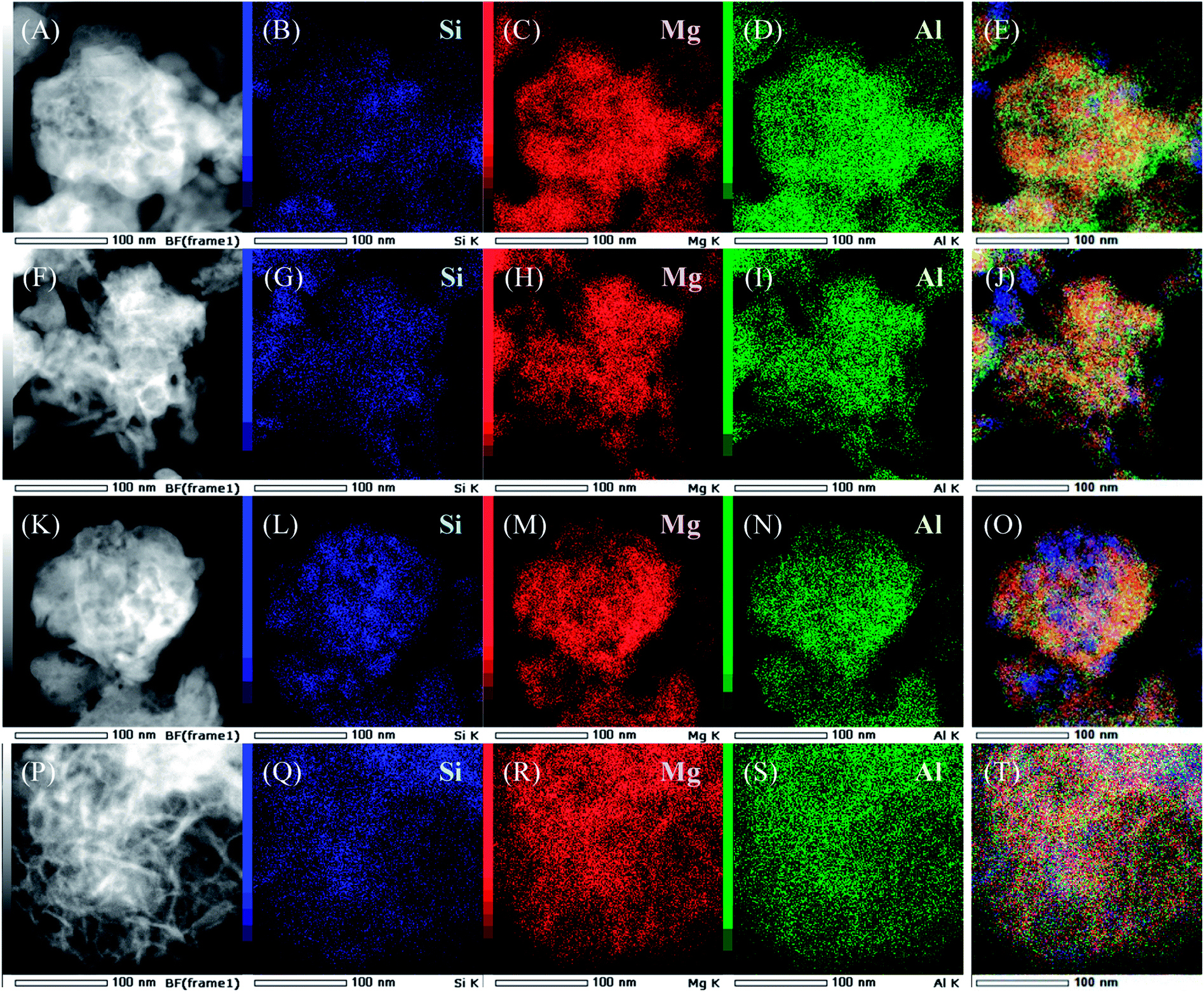 | ||
| Fig. 4 (A, F, K and P) Dark-field TEM images of as-prepared SiO2@LDHs with various Si/(Mg + Al) atomic ratio: (A) 0.13, (F) 0.17, (K) 0.25 and (P) 0.50. Also shown are (B–E), (G–J), (L–O), (Q–T) EDS mapping results of as-prepared SiO2@LDH with various Si/(Mg + Al) atomic ratio. | ||
Accordingly, we infer the correlation between Si/(Mg + Al) atomic ratio and base catalysis of prepared SiO2@(Z)LDHs as follows: (i) below Si/(Mg + Al) atomic ratio of 0.13, the LDH crystal is just immobilized onto the SiO2 surface with inhibition of the ab-face stacking. The exposed corner and edge located at the surface layer act as highly active base sites. (ii) Fine crystallization occurs not only in the stacking direction, but also in the plane direction to increase the amount of base sites, especially base sites with high activity up to Si/(Mg + Al) atomic ratio of 0.17. (iii) Above a Si/(Mg + Al) atomic ratio of 0.17, excess terminal Si–OH species covered the highly active base sites to produce Si–O–Mg and Si–O–Al covalent bonds, thereby lowering the activity.
Conclusions
The effects of SiO2 loading amounts on the crystallite sizes, basicity, and catalytic activity of SiO2@LDH catalysts were investigated. SiO2@LDHs were prepared using co-precipitation with the coexistence of various amounts of spherical SiO2 with particle sizes of ca. 40 nm. The XRD results suggest that the LDH crystallite size of D(003) is simply reduced in accordance with the Si loading amount. Furthermore, the crystallite size of D(110) is almost identical below Si/(Mg + Al) of 0.13, although it is reduced above 0.13. Base catalysis of SiO2@LDHs was evaluated using Knoevenagel condensation of benzaldehyde and ethyl cyanoacetate. Both the reaction rate and the apparent TOF per base site were increased in the region between Si/(Mg + Al) atomic ratio of 0–0.17, whereas the base amount is increased linearly from the Si/(Mg + Al) atomic ratio of 0.13 to 0.50 with reduction of the LDH crystallite size. The results of 29Si CP-MAS NMR and STEM-EDS suggest that a surface Si–O–Si bond is cleaved, generating terminal Si–OH species that act as a cross-link point with Mg and Al ions to form an immobilized SiO2 core – LDH shell structure. However, when the amount of Si becomes excessive with respect to Mg and Al ions, the excess Si–OH group forms Si–O–Mg and Si–O–Al covalent bonds with LDH crystal to cover the LDH shell. From these results, we inferred the effect of SiO2 amount on heterogeneous base catalysis of SiO2@Mg–Al LDH as follows: (i) below Si/(Mg + Al) atomic ratio of 0.13, the highly active base sites located at the corner and edge of surface layer are exposed by immobilization of LDH crystals onto the SiO2 surface, (ii) up to Si/(Mg + Al) atomic ratio of 0.17, the number of exposed highly active base sites is increased in accordance with reduction of LDH crystallite, and (iii) above Si/(Mg + Al) atomic ratio of 0.17. Excess terminal Si–OH species covered the highly active base sites through the Si–O–Mg and Si–O–Al covalent bonds to decrease the activity. This study elucidated the correlation between SiO2 amount, crystal properties, and the basicity of fine-crystallized SiO2@LDH catalysts to present a new technique to improve the base catalysis of the widely used LDH material. After optimization of the Si/(Mg + Al) atomic ratio in SiO2@LDHs, the Si/(Mg + Al) atomic ratio of 0.17 was found to be the best catalyst, with the reaction rate of 171.1 mmol g(cat)−1 h−1, which is a 2.2 times higher value than that of the conventional LDH prepared with same protocol in absence of SiO2 seed agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate financial support from a Grant-in-Aid from the Japan Society for the Promotion of Science (JSPS) for Fellows and Young Scientists (A): No. 15J10050 and 17H04966. Dr Akio Miyasato (Center for Nano Materials and Technology, JAIST) and Dr Koichi Higashimine (Center for Nano Materials and Technology, JAIST) respectively supported on 29Si CP-MAS NMR and TEM-EDS experiments. The ICP-AES analysis was performed by Yamato Environmental Analysis Co. Ltd. (Ishikawa, Japan).References
- Y. Ono and H. Hattori, Solid Base Catalysis, Tokyo Institute of Technology Press, Tokyo, Japan, 2011 Search PubMed.
- H. Hattori, Appl. Catal., A, 2001, 222, 247–259 CrossRef.
- S. Miyata, Clays Clay Miner., 1980, 28, 50–56 CrossRef.
- S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2013, 15, 2026–2042 RSC.
- P. J. Sideris, U. G. Nielsen, Z. Gan and C. P. Grey, Science, 2008, 321, 113–117 CrossRef PubMed.
- Z. An, W. Zhang, H. Shi and J. He, J. Catal., 2006, 241, 319–327 CrossRef.
- H. C. Greenwell, P. J. Holliman, W. Jones and B. V. Velasco, Catal. Today, 2006, 114, 397–402 CrossRef.
- L. Hora, V. Kelbichová, O. Kikhtyanin, O. Bortnovskiy and D. Kubička, Catal. Today, 2014, 223, 138–147 CrossRef.
- D. G. Crivoi, R. A. Miranda, E. Finocchio, J. Llorca, G. Ramis, J. E. Sueiras, A. M. Segarra and F. Medina, Appl. Catal., A, 2016, 519, 116–129 CrossRef.
- M. L. Kantam, B. M. Choudary, C. V. Reddy, K. K. Rao, M. L. Kantam, B. M. Choudary, K. K. Rao and F. Figueras, Chem. Commun., 1998, 1033–1034 RSC.
- M. J. Climent, S. Iborra, K. Epping and A. Velty, J. Catal., 2004, 225, 316–326 CrossRef.
- E. Angelescu, O. D. Pavel, R. Bîrjega, R. Zăvoianu, G. Costentin and M. Che, Appl. Catal., A, 2006, 308, 13–18 CrossRef.
- M. Shirotori, S. Nishimura and K. Ebitani, Catal. Sci. Technol., 2014, 4, 971–978 RSC.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Org. Chem., 2000, 65, 6897–6903 CrossRef PubMed.
- T. Honma, M. Nakajo, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2002, 43, 6229–6232 CrossRef.
- O. D. Pavel, B. Cojocaru, E. Angelescu and V. I. Pârvulescu, Appl. Catal., A, 2011, 403, 83–90 CrossRef.
- M. Fuming, P. Zhi and L. Guangxing, Org. Process Res. Dev., 2004, 8, 372–375 CrossRef.
- E. Li, Z. P. Xu and V. Rudolph, Appl. Catal., B, 2009, 88, 42–49 CrossRef.
- J. Nowicki, J. Lach, M. Organek and E. Sabura, Appl. Catal., A, 2016, 524, 17–24 CrossRef.
- M. Ohara, A. Takagaki, S. Nishimura and K. Ebitani, Appl. Catal., A, 2010, 383, 149–155 CrossRef.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Lett., 2010, 39, 838–840 CrossRef.
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562–1565 CrossRef.
- J. Tuteja, S. Nishimura and K. Ebitani, Bull. Chem. Soc. Jpn., 2012, 85, 275–281 CrossRef.
- M. Shirotori, S. Nishimura and K. Ebitani, Chem. Lett., 2016, 45, 194–196 CrossRef.
- M. Shirotori, S. Nishimura and K. Ebitani, Catal. Sci. Technol., 2016, 6, 8200–8211 RSC.
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 8008–8011 CrossRef PubMed.
- S. Iguchi, K. Teramura, S. Hosokawa and T. Tanaka, Catal. Today, 2015, 251, 140–144 CrossRef.
- S. Iguchi, K. Teramura, S. Hosokawa and T. Tanaka, Phys. Chem. Chem. Phys., 2016, 18, 13811–13819 RSC.
- M. B. Roeffaers, B. F. Sels, I. H. Uji, F. C. de Schryver, P. A. Jacobs, D. E. de Vos and J. Hofkens, Nature, 2006, 439, 572–575 CrossRef PubMed.
- M. Adachi-Pagano, C. Forano and J. P. Besse, Chem. Commun., 2000, 91–92 RSC.
- E. Gardner, K. M. Huntoon and T. J. Pinnavaia, Adv. Mater., 2001, 13, 1263–1266 CrossRef.
- S. O'Leary, D. O'Hare and G. Seeley, Chem. Commun., 2002, 1506–1507 RSC.
- T. Hibino, Chem. Mater., 2004, 16, 5482–5488 CrossRef.
- W. Chen, L. Feng and B. Qu, Chem. Mater., 2004, 16, 368–370 CrossRef.
- Z. Liu, R. Ma, M. Osada, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2006, 128, 4872–4880 CrossRef PubMed.
- H. Kang, Y. Shu, Z. Li, B. Guan, S. Peng, Y. Huang and R. Liu, Carbohydr. Polym., 2014, 100, 158–165 CrossRef PubMed.
- M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2012, 134, 1071–1077 CrossRef PubMed.
- M. Shao, F. Ning, Y. Zhao, J. Zhao, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2012, 24, 1192–1197 CrossRef.
- C. Chen, P. Wang, T. T. Lim, L. Liu, S. Liu and R. Xu, J. Mater. Chem. A, 2013, 1, 3877–3880 RSC.
- S. D. Jiang, Z. M. Bai, G. Tang, L. Song, A. A. Stec, T. R. Hull, Y. Hu and W. Z. Hu, ACS Appl. Mater. Interfaces, 2014, 6, 14076–14086 CrossRef PubMed.
- J. Wang, R. Zhu, B. Gao, B. Wu, K. Li, X. Sun, H. Liu and S. Wang, Biomaterials, 2014, 35, 466–478 CrossRef PubMed.
- C. Chen, R. Felton, J. C. Buffet and D. O'Hare, Chem. Commun., 2015, 51, 3462–3465 RSC.
- J. L. Gunjakar, T. W. Kim, H. N. Kim, I. Y. Kim and S. J. Hwang, J. Am. Chem. Soc., 2011, 133, 14998–15007 CrossRef PubMed.
- C. Zhang, J. Zhao, L. Zhou, Z. Li, M. Shao and M. Wei, J. Mater. Chem. A, 2016, 4, 11516–11523 RSC.
- T. Hara, J. Kurihara, N. Ichikuni and S. Shimazu, Chem. Lett., 2010, 39, 304–305 CrossRef.
- J. Kobler, K. Möller and T. Bein, ACS Nano, 2008, 2, 791–799 CrossRef PubMed.
- M. Shirotori, S. Nishimura and K. Ebitani, J. Mater. Chem. A, 2017, 5, 6947–6957 RSC.
- N. Gunawidjaja, M. A. Holland, G. Mountjoy, D. M. Pickup, R. J. Newport and M. E. Smith, Solid State Nucl. Magn. Reson., 2003, 23, 88–106 CrossRef PubMed.
- A. M. B. Silva, C. M. Queiroz, S. Agathopoulos, R. N. Correia, M. H. V. Fernandes and J. M. Oliveira, J. Mol. Struct., 2011, 986, 16–21 CrossRef.
- M. Lewandowski, G. S. Babu, M. Vezzoli, M. D. Jones, R. E. Owen, D. Mattia, P. Plucinski, E. Mikolajska, A. Ochenduszko and D. C. Apperley, Catal. Commun., 2014, 49, 25–28 CrossRef.
- Discussion on the surface area values of LDHs itself in as-prepared SiO2@(Z)LDHs were hardly estimated because the obtained surface area in experiment included not only LDH shell but also SiO2 core contributions. Note that a tentative discussion had been attempted in our previous paper of ref. 47.
Footnote |
| † Electronic supplementary information (ESI) available: Reactivity of SiO2@Mg–Al LDH. See DOI: 10.1039/c8ra04925d |
| This journal is © The Royal Society of Chemistry 2018 |