
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of DNA-guided silver nanoparticles on a graphene oxide surface: enhancing the antibacterial effect and the wound healing activity†
Chunyi Tong‡
 a,
Wei Zou‡b,
Weimin Ning‡a,
Jialong Fana,
Li Lia,
Bin Liu*a and
Xuanming Liu*a
a,
Wei Zou‡b,
Weimin Ning‡a,
Jialong Fana,
Li Lia,
Bin Liu*a and
Xuanming Liu*a
aCollege of Biology, Hunan Province Key Laboratory of Plant Functional Genomics and Developmental Regulation, Hunan University, Changsha, 410082, PR China. E-mail: binliu2001@hotmail.com; xml05@hnu.edu.cn; Fax: +86 731 89720939; Tel: +86 731 89720939
bKey Laboratory of Hunan Provincial TCM Administration for TCM in Obstetrics & Gynecology, Hunan Provincial Maternal and Child Health Care Hospital, Changsha 410008, PR China
First published on 7th August 2018
Abstract
The occurrence of antibiotic resistance against pathogens is rapidly increasing and endangering the efficacy of antibiotics. Thus, finding a way to address this problem has become a major challenge due to the inability of conventional antibiotics to kill these multidrug-resistant bacteria. In order to further enhance the antibacterial ability and reduce the possibility of antibiotic resistance, we developed a simple two-step approach and synthesized a new nanocomposite by directly loading single-stranded DNA (ssDNA)-guided silver nanoparticles (AgNPs) on graphene oxide (ssDNA-AgNPs@GO). Through systematically evaluating the bactericidal activity and wound healing capability, we found that ssDNA-AgNPs@GO exhibited synergistic antibacterial activity against Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus and Bacillus subtilis with low minimum inhibitory concentrations (6.8 μg mL−1, 6.8 μg mL−1, 11.9 μg mL−1 and 10.2 μg mL−1, respectively) and large-diameter inhibition zones (12.83 ± 0.63 mm, 13.14 ± 0.37 mm, 8.6 ± 0.9 mm and 8.93 ± 0.47 mm, respectively). Furthermore, the wound healing experiment indicated that it has a striking ability to remedy wound infection caused by Staphylococcus aureus bacteria. In conclusion, the properties of ssDNA-AgNPs@GO with enhanced antibacterial and wound healing capability will give it broad applications in the future.
1 Introduction
Over recent decades, bacterial infections, which cause millions of infection-related morbidities and mortalities annually, have seriously threatened human health and become global public health issues.1–3 Furthermore, the increasing occurrence of antibiotic resistance against pathogens, especially “superbugs” with multidrug resistance such as Staphylococcus aureus (SA)4–6, has become the most common clinical drug-resistant infection in burn wounds, with the SA wound infection rate accounting for more than 60%. Moreover, the number of different SA strains isolated sharply increased. Thus, the control of wound infection has become a global health problem. Although widely used, antibiotics, which are efficient bactericidal agents, resolved this public health crisis to some extent7–9 – however, abuse or overdose of them resulted in ineffective infection treatment, adverse drug reactions, rapid increase in drug-resistant pathogens, and even food and drinking water contamination. In addition, the currently used inorganic antimicrobial or organic agents commonly exhibit some cytotoxicity and instability.10–13 Thus, it is still urgent to develop new antibiotics. Recently, advancements in nanotechnology have provided an alternative approach to resolve the challenges by developing new antibacterial materials with ultra-high treatment efficiencies and low side effects.14,15 Among these metallic bactericides, silver nanoparticles (AgNPs) have been recognized as one of the most excellent antibacterial agents due to their broad spectrum of antibacterial activity, limited bacterial resistance and relatively low toxicity towards mammalian cells.16–18 Until now, they have been widely used in many fields19,20 (e.g. surgical wound dressing, medical device coatings and preventing pathogen contamination) and daily consumer products21,22 (e.g. cotton fabrics, air filter, water disinfection, food preservation and cosmetics). However, the antibacterial capacity of AgNPs is often affected by their size and surface potential. In addition, the aggregation-prone nature of bare AgNPs also decreases their stability, which in turn greatly weakens their antibacterial capability.23,24 Thus, finding a way to improve the homogeneous dispersion and stability of AgNPs is an important issue for tailoring their performance. In general, polymeric and chelating agents, which can efficiently regulate the particle size and shape of AgNPs, were used as supporting substances to retain their physicochemical characteristics.25,26 However, these agents often deactivate their functions through unavoidable organic wrapping on the particle surface.27,28 Recently, many studies have demonstrated that the immobilization of AgNPs on other nanomaterials is very useful for improving antimicrobial efficacy and biological safety.29Graphene oxide (GO) is the oxidized form of graphene, composed of a single layer of sp2-bonded carbon atoms. The surface functional groups, including epoxy, carboxylic acid and hydroxyl groups, offers active sites for further functionalization through non-covalent interactions. Moreover, highly hydrophilic GO can be easily produced on a large scale without a high cost. Due to these excellent properties, coupled with a good carrying capacity and low biological toxicity, GO has been used to prepare AgNP-loaded nanohybrids (GO–Ag) and the obtained GO-based nanocomposites showed extraordinary antibacterial properties.30–32 However, unfortunately, the process for AgNP synthesis is time-consuming and sophisticated.
In this study, we report a simple two-step method for the synthesis of ssDNA-guided AgNP-loaded GO (ssDNA-AgNPs@GO) nanocomposites through the reduction of Ag+ by sodium borohydride (NaBH4) in aqueous solution. These stable ssDNA-AgNPs@GO nanocomposites with uniform size exhibited excellent antibacterial efficiency against both Gram-positive and Gram-negative bacteria. In addition, the antibacterial mechanism and wound healing capability of the ssDNA-AgNPs@GO nanocomposites were thoroughly explored.
2 Materials and methods
2.1 Chemicals
Single-layer GO with a 0.5–3 μm size range (2 g L−1) was purchased from Xianfeng Nano-Tech (Nanjing, China). AgNO3 (99%), NaBH4 (98%) and 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) were obtained from Sigma-Aldrich. Sodium nitrate (NaNO3) was obtained from Alfa-Aesar (USA). Single-stranded DNA with a sequence of 5′-CCCTTAATCCCC-3′ was purchased from Takara (Dalian, China).2.2 Synthesis of ssDNA-AgNPs@GO nanocomposites
The synthesis of ssDNA-AgNPs@GO was accomplished through the reduction of AgNO3 with NaBH4. GO (final concentration of 20 μg mL−1) was firstly mixed with DNA solution (final concentration of 1 μM) in 10 mL HEPES buffer. After 30 min of incubation, the mixture was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min. The precipitated ssDNA@GO was redissolved in 1 mL HEPES buffer, followed by the addition of AgNO3 with a final concentration of 100 μM with vigorous stirring. After 10 min of incubation, an icy freshly prepared solution of NaBH4 (final concentration of 500 μM) was injected dropwise into the mixture and stirred vigorously for 30 min. The solution was centrifuged at 12000 rpm for 10 min until the supernatant became clear. The precipitate was redissolved and stored in the HEPES buffer for further characterization. The control ssDNA-AgNPs were synthesized according to similar steps.
000 rpm for 20 min. The precipitated ssDNA@GO was redissolved in 1 mL HEPES buffer, followed by the addition of AgNO3 with a final concentration of 100 μM with vigorous stirring. After 10 min of incubation, an icy freshly prepared solution of NaBH4 (final concentration of 500 μM) was injected dropwise into the mixture and stirred vigorously for 30 min. The solution was centrifuged at 12000 rpm for 10 min until the supernatant became clear. The precipitate was redissolved and stored in the HEPES buffer for further characterization. The control ssDNA-AgNPs were synthesized according to similar steps.
2.3 Characterization
UV-vis adsorption spectra were recorded on a UV-1800 model spectrophotometer (Shimadzu, Japan). The hydrodynamic diameter and zeta potential were measured using a Zetasizer Nano ZS (Malvern, United Kingdom) instrument. Transmission electron microscope (TEM) and high-resolution TEM images were obtained using a JEM-2100F (JEOL, Japan) instrument operated at an accelerating voltage of 200 kV. Samples were prepared by placing a drop of AgNP dispersion on the carbon-coated copper grid and drying at room temperature. AFM images were recorded using a Bioscope System atomic force microscope (Brucker, USA).2.4 The minimal inhibitory concentration (MIC) test
Bacteria including Escherichia coli (E. coli) and Pseudomonas aeruginosa (P. aeruginosa) (Gram-negative), Staphylococcus aureus (S. aureus) and Bacillus subtilis (B. subtilis) (Gram-positive) were stored in 50% glycerol at −80 °C. All of them were firstly cultivated on a Luria-Brentani (LB) solid medium at 37 °C for 12 h. Then, the pure colonies were further plated on the LB plates and incubated as described above. Bacterial colonies from the LB plates were cultured in LB medium at 37 °C, 250 rpm and 80% humidity. When bacteria were grown to an OD600 of 0.6, which corresponds to the exponential phase, the bacteria were serially diluted to a concentration of 5 × 105 CFU mL−1. Then, different materials (GO, ssDNA-AgNPs and ssDNA-AgNPs@GO) were separately added into wells containing bacterial cultures and shaken for 24 h at 37 °C, 250 rpm and 80% humidity. After incubation for 12 h, the well with the lowest concentration which completely inhibited bacteria growth was determined as the sample of MIC. Each sample was carried out 3 times in parallel.2.5 The growth curve of bacteria
The susceptibility of E. coli, P. aeruginosa, S. aureus and B. subtilis to the nanomaterials was determined using the growth curve method. Mixtures of bacteria (OD600 = 0.1) and the different materials were cultured in the LB liquid medium for 12 h. The concentration of ssDNA-AgNPs@GO in a final volume of 500 μL was used to determine the MIC and IC50 values. GO and ssDNA-AgNPs with the same concentration were used as controls. Growth curves were determined by measuring the OD600 value every hour. Each sample was carried out 3 times in parallel.2.6 Agar diffusion test
The agar diffusion method was used to determine the antimicrobial effect of different nanomaterials. Under sterile conditions, bacterial suspensions (109 CFU mL−1) of E. coli, P. aeruginosa, S. aureus and B. subtilis were spread onto solid nutrient agar plates to form a uniform carpet. After drying in air, 0.2 mL of HEPES buffer containing GO, ssDNA-AgNPs or ssDNA-AgNPs@GO with concentrations equal to the IC50 were carefully dropped onto the Oxford Cups which were placed on the surface of the agar plate (four Oxford Cups in each plate, one was used for control with HEPES buffer). The plates were incubated overnight at 37 °C and the diameters of the growth inhibition zones were measured and expressed in mm. Each treatment was carried out three times in parallel.2.7 Confocal fluorescence microscopy
The membrane damage was investigated using the DNA-binding fluorescent dye staining method. E. coli, P. aeruginosa, S. aureus and B. subtilis with a final concentration of 108 CFU mL−1 were inoculated with GO, ssDNA-AgNPs or ssDNA-AgNPs@GO for 12 h at 37 °C, followed by centrifugation at 3500 rpm at 4 °C. The precipitate was resuspended in a fresh LB liquid medium containing calcein-AM and PI and incubated for 15 min at 37 °C. All samples were detected using a multidimensional living cell imaging system (OLYMPUS FV1200, Japan). Each sample was carried out 3 times in parallel.2.8 Determination of cellular ROS
The reactive oxygen species (ROS) level of bacteria induced by ssDNA-AgNPs@GO was detected using a DCFH-DA assay. E. coli, P. aeruginosa, S. aureus and B. subtilis with a final concentration of 108 CFU mL−1 were inoculated with same concentration of GO, ssDNA-AgNPs or ssDNA-AgNPs@GO for 12 h. Then, 10 μM of DCFH-DA probe was added and incubated for another 30 min. After treatment, the bacteria suspension was centrifuged at 1000 rpm for 10 min, washed 3 times, and resuspended in 1 mL ultrapure water. The samples were detected using a fluorescence detector and a multidimensional living cell imaging system.2.9 ATP assay
The ATP level of bacteria was investigated using an enhanced ATP assay kit. E. coli, P. aeruginosa, S. aureus and B. subtilis in the fresh medium were treated with same concentration of GO, ssDNA-AgNPs or ssDNA-AgNPs@GO for 12 h. The final concentration of the bacteria cells was 108 CFU mL−1. The bare medium was used as a control. All samples were centrifuged at 12000 rpm for 15 min at 4 °C and resuspended in 100 μL of lysis buffer after treatment. Then, the samples were transferred into an ultrasonic cell disruption instrument and treated with 30% power (3 s on then 5 s off) for 5 min in an ice-bath. The collected supernatant was detected on a multi-mode detection platform.
2.10 Cytotoxicity assay
The cytotoxicity of different nanomaterials to the murine NIH-3T3 cell line was evaluated using a MTT assay, which is commonly used to investigate the possible harmful effects of nanomaterials on cells. Murine NIH-3T3 seeded in the 96-well plate (1 × 104 cells per well) was cultured in the DMEM medium containing 10% FBS under a 5% CO2 incubator for 24 h. Then, 100 μL of GO, ssDNA-AgNPs and ssDNA-AgNPs@GO with different concentrations were added in triplicate. After incubation for 24 h, the 96-well plate was washed 3 times with PBS buffer. Then, the medium in the well was replaced by a mixture of 90 μL fresh medium and 10 μL MTT (5 mg mL−1) reagent followed by incubation for another 4 h. 100 μL of DMSO solution was added per well followed by reaction for 15 min in the shaker. Finally, the optical density was determined on a multi-mode detection platform in absorbance mode at 490 nm.2.11 Hemolysis assay of the AgNPs
A hemolysis assay was performed using fresh human blood samples provided by the Third Xiangya Hospital of Central South University. The erythrocytes were collected via centrifugation at 3000 rpm for 5 min at 4 °C. The stock dispersion was prepared by mixing 50 μL of erythrocytes into 950 μL of PBS. After adding different amounts of nanomaterial into the stock dispersion, the mixture was incubated for 3 h at 37 °C. The percentage of hemolysis of the supernatant was measured at 540 nm absorbance after centrifugation at 3000 rpm for 5 min. PBS was used as the negative control and pure water was used as the positive control. The percentage of hemolysis was calculated with the following formula:| hemolysis (%) = (Atest − Aneg)/(Apos − Aneg) × 100% |
2.12 In vivo infected wound healing and histological analysis
All experimental procedures and postoperative animal care were conducted in accordance with the National Institute of Health’s Guidelines for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee of Hunan University (Hunan, China). Blab/c mice (20 ± 2 g) purchased from Hunan SJA Laboratory Animal Co., Ltd (Changsha, China) were maintained on a 12 h light/dark cycle in a room at 22–25 °C and allowed free access to food and water at all times. The surgical area was shaved using shaving machine and disinfected with 75% alcohol. Full-thickness cutaneous wounds of approximately 1.5 mm were generated on the dorsal side by removing epidermal and dermal layers with sterile surgical scalpel, and then a 200 μL liquid of S. aureus suspension (108 CFU mL−1) was added on the wound. Then, the wounds were covered with HEPES buffer and GO, ssDNA-AgNPs or ssDNA-AgNPs@GO, then bandaged with 3 gauze bandages. The area of the wound was determined every day to evaluate the healing rate. The mouse serum at day 14 was subjected to liver function analysis with analysis kits (Beijing Leadmanbio Institute, China). Meanwhile, parts of the liver tissue and healed skin tissue were dissolved in 69% HNO3. The mixture of 200 μL sample solution with 9.8 mL H2O were used to assess the body’s absorption of AgNPs by ICP-MS. Wound closure (WC, %) area was calculated using the following formula:where Ao is the original wound area and At is the open area of the wound at the time of sacrifice. Wound tissues were gathered at postoperative day 14 for histological analysis. All tissues were placed in 4% paraformaldehyde solution. Isometric continuum cut sections were obtained using a microtome in vertical planes of each fixed tissue, stained with hematoxylin and eosin (H&E), and imaged under an optical microscope (TE-2000, Nikon, Japan).
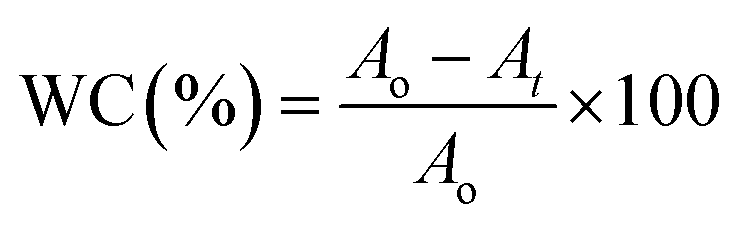
3 Results and discussion
3.1 Preparation and characterization of ssDNA-AgNPs@GO
The strategy of ssDNA-AgNPs@GO synthesis using a two-step method is shown in Scheme 1. Firstly, ssDNA@GO was directly prepared through π–π stacking between GO and ssDNA. In this study, the ssDNA containing 7 cytosine bases possesses high affinity with Ag0. Then, Ag0 deposited on the surface of ssDNA@GO was reduced to Ag0 by NaBH4 so as to form ssDNA-AgNPs@GO. This kind of nanomaterial with a dark yellow color showed two strong emission peaks at 260 nm and 400 nm under UV illumination (Fig. 1A), which resulted from the surface plasmon resonance of ssDNA and the AgNPs. Compared to the ssDNA-AgNPs, the color of ssDNA-AgNPs@GO became darker (inset Fig. 1A). This phenomenon also suggested that the ssDNA-AgNPs loaded on the surface of GO maintained their small size instead of aggregating into large-sized particles, which was evidenced by the similar emission peaks between ssDNA-AgNPs@GO and the bare DNA-AgNPs. Meanwhile, a minor red shift of the emission wavelengths was observed compared to those of the DNA-AgNPs. This can be explained by the interaction of the ssDNA-AgNPs with GO. The AFM and TEM images provided more direct evidence for the successful formation of hybrids. Although the thickness of ssDNA-AgNPs@GO increased by about 0.5 nm due to the coverage of the ssDNA-AgNPs on the basal plane of GO, it still presented a lateral size of 50–200 nm, similar to that of GO (Fig. 1B and S1C†). The TEM image in Fig. 1D shows that the AgNP-loaded GO exhibited a transparent and stable sheet structure and the spherical-like AgNPs uniformly covered the surface of the GO sheet. This result suggested that the attachment of the AgNPs to the GO surface is mainly through the interaction between Ag+ and the oxygen-containing groups of ssDNA and the GO sheet, which provides nucleation sites for the anchoring and growth of AgNPs.33 Finally, we investigated the difference in zeta potential between ssDNA-AgNPs and ssDNA-AgNPs@GO. As GO is a nanomaterial with negative potential (Fig. S1B†), the zeta potential of the ssDNA-AgNPs thus decreased from −8.65 mV to −27.5 mV after loading on the GO surface (Fig. 1C). The zeta potential change directly resulted in the stability enhancement of ssDNA-AgNPs@GO by increasing the repulsion force between charged molecules34 (Fig. S3†).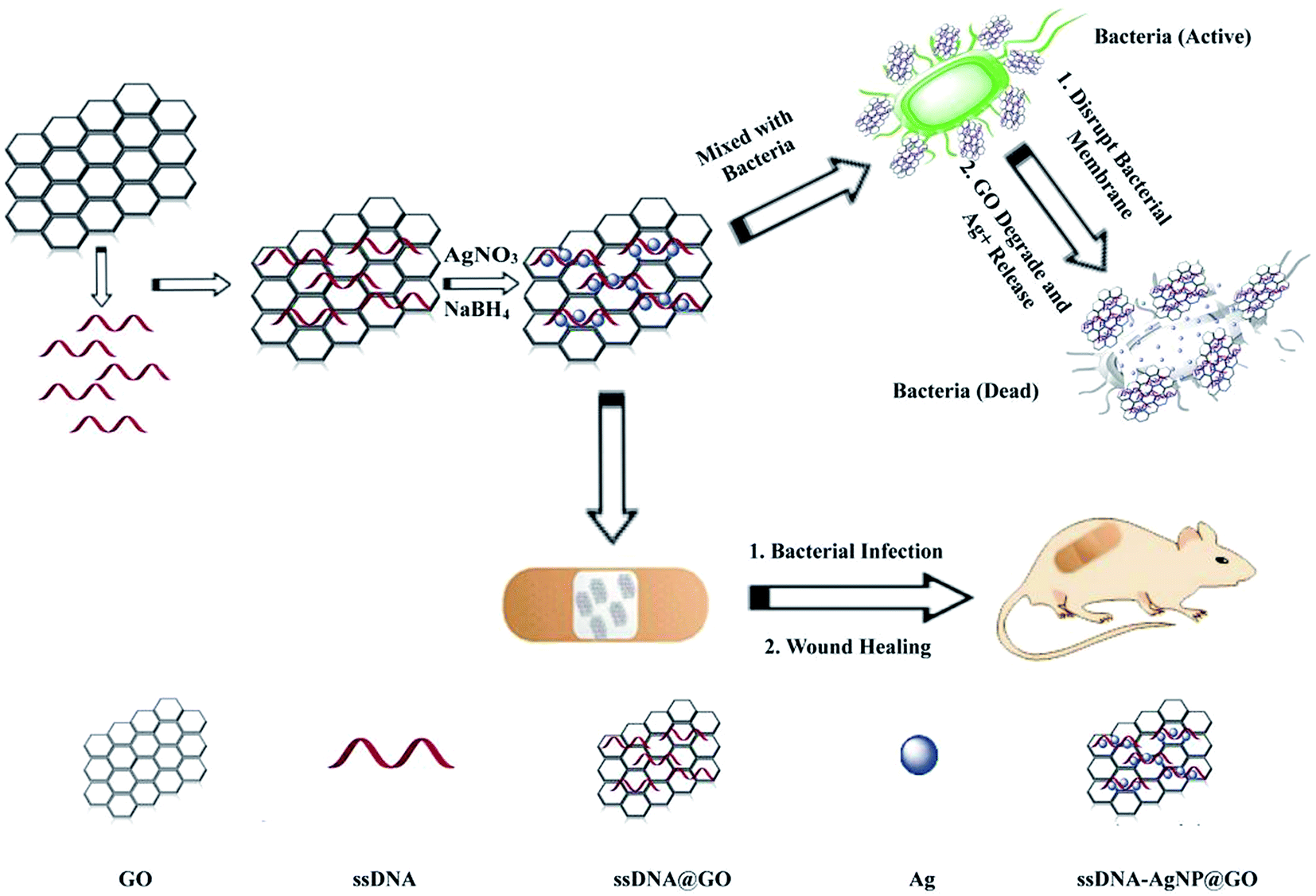 | ||
| Scheme 1 Illustration of the synthesis of antibacterial ssDNA-AgNPs@GO and the application to wound healing. | ||
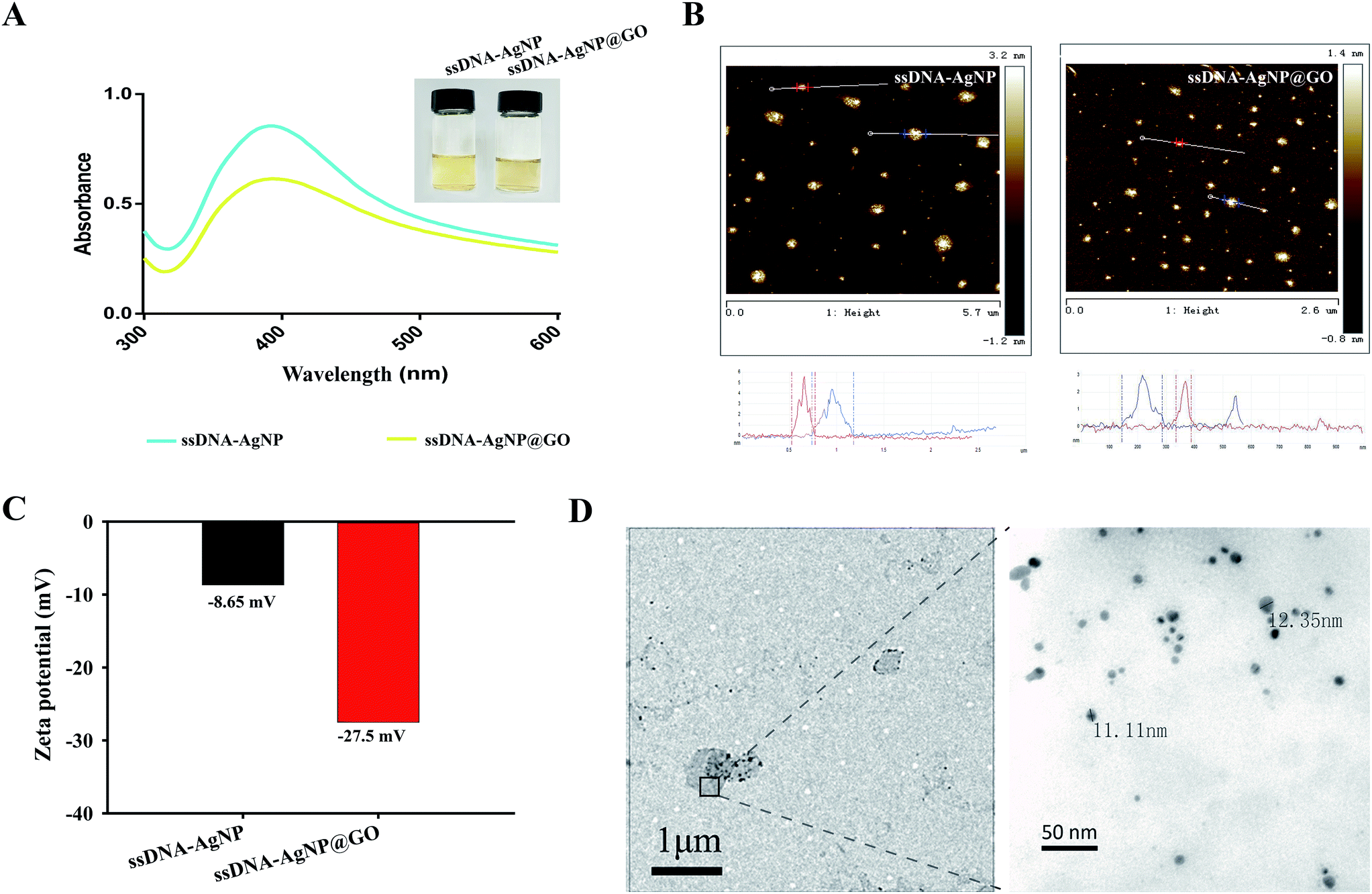 | ||
| Fig. 1 (A) UV-vis absorption spectra of ssDNA-AgNPs and ssDNA-AgNPs@GO. Inset: photographs of ssDNA-AgNPs (left) and ssDNA-AgNPs@GO (right) solutions under natural light. (B) AFM images of ssDNA-AgNPs and ssDNA-AgNPs@GO. (C) Zeta potentials of ssDNA-AgNPs and ssDNA-AgNPs@GO. (D) TEM image of ssDNA-AgNPs@GO. | ||
3.2 Antibacterial activity of ssDNA-AgNPs@GO
Owing to the high local concentration of Ag+ assembled on the GO surface, ssDNA-AgNPs@GO showed superior antibacterial activity compared to ssDNA-AgNPs. 4 different opportunistic bacteria, including two types of Gram-negative bacteria (E. coli and P. aeruginosa), as well as Gram-positive S. aureus and B. subtilis, were selected as bacterial models to study the antimicrobial properties of ssDNA-AgNPs@GO. By investigating the MIC of ssDNA-AgNPs@GO for these bacteria, we found that the MIC values of the ssDNA-AgNPs were 10.2 μg mL−1, 10.2 μg mL−1, 13.6 μg mL−1 and 11.9 μg mL−1 against E. coli, P. aeruginosa, S. aureus and B. subtilis, respectively. However, the corresponding MIC values of ssDNA-AgNPs@GO for these bacteria reduced to 6.8 μg mL−1, 6.8 μg mL−1, 11.2 μg mL−1 and 10.9 μg mL−1, respectively. The decrease in MIC values indicated that ssDNA-AgNPs@GO possessed higher antibacterial activity than ssDNA-AgNPs. In should be noted that the Gram-positive bacteria were more susceptible to the nanomaterial than Gram-negative bacteria. The reason for this can be explained by the difference in cell wall structure and peptidoglycan content of these bacterias.35 In addition, the half MIC of ssDNA-AgNPs and GO was 5.1 μg mL−1 and 9 μg mL−1, respectively. However, the MIC of ssDNA-AgNPs@GO was only 6.8 μg mL−1 (Table S1†). According to the equation for synergistic effect evaluation, we can conclude that the hybrids of ssDNA-AgNPs with GO have a significant synergistic effect on the antibacterial properties. In addition, a medium turbidity assay also evidenced the strong antimicrobial ability. A mixture of the 4 kinds of bacteria in a LB liquid medium became turbid after culturing for 12 h and 24 h. However, the mixture became clear in the presence of ssDNA-AgNPs@GO due to its effective antibacterial activity (Fig. S4†). These data suggested that the existence of ssDNA increased the adhesive force between ssDNA-AgNPs@GO and the bacterial cell membrane. Meanwhile, the uniform distribution of AgNPs on GO is helpful for nonspecifically attaching and wrapping bacteria as well. As a result, the interaction between ssDNA-AgNPs@GO and bacteria greatly improved the antibacterial properties.The dynamics of bacteria showed that the E. coli and P. aeruginosa bacteria began to grow after incubation for 2 h, without adding any material or with only GO (Fig. 2A and B). In contrast, the growth of the two bacteria was significantly inhibited at the first 6 h in the presence of 6.8 μg mL−1 ssDNA-AgNPs and 3.4 μg mL−1 ssDNA-AgNPs@GO, or even wholly inhibited in the presence of 6.8 μg mL−1 (ssDNA-AgNPs@GO). Thus, the concentration of 6.8 μg mL−1 was the MIC of ssDNA-AgNPs@GO and the concentration of 3.4 μg mL−1 was half of the MIC. Meanwhile, a similar phenomenon was found for Gram-positive bacteria (S. aureus and B. subtilis) (Fig. 2C and D). These results indicated that the half concentration of ssDNA-AgNPs@GO possessed similar antibacterial capability as the full concentration of ssDNA-AgNPs in all four kinds of bacteria. These results further confirmed the synergistic antibacterial properties of ssDNA-AgNPs@GO.
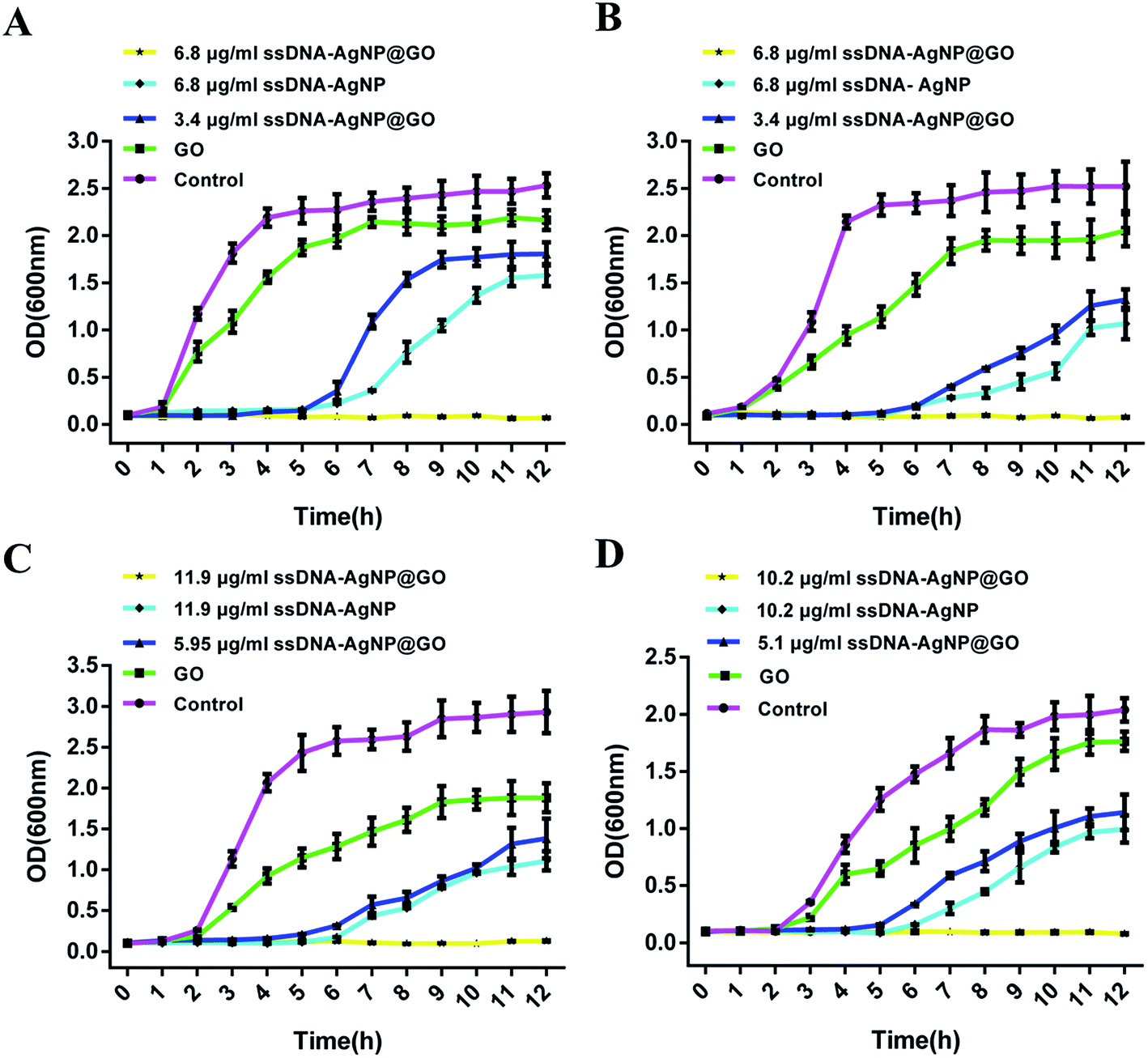 | ||
| Fig. 2 The growth curves of (A) E. coli, (B) P. aeruginosa, (C) S. aureus and (D) B. subtilis in the presence of GO, ssDNA-AgNPs and ssDNA-AgNPs@GO. Error bars represent the standard error of 3 parallel experiments. | ||
The result of the inhibition zone assay also reflected the improved antimicrobial ability of ssDNA-AgNPs@GO. Fig. 3 shows that the diameters of the inhibition zones for ssDNA-AgNPs@GO against E. coli, P. aeruginosa, S. aureus and B. subtilis were 12.83 ± 0.63 mm, 13.14 ± 0.37 mm, 8.6 ± 0.9 mm and 8.93 ± 0.47 mm, respectively. In comparison, the diameters for ssDNA-AgNPs against these bacteria were 7.04 ± 0.5 mm, 6.86 ± 0.97 mm, 5.13 ± 4.2 mm and 4.5 ± 1.03 mm, respectively. These data clearly demonstrated the higher antibacterial activity of ssDNA-AgNPs@GO compared to that of ssDNA-AgNPs. Meanwhile, we found that the activity of ssDNA-AgNPs@GO against E. coli and P. aeruginosa was higher than that against S. aureus and B. subtilis. By combining the results of no antibacterial activity observed for GO at the investigated concentration, we concluded that the synergistic effect for antibacterial activity appeared when the ssDNA-AgNPs were loaded on the GO.
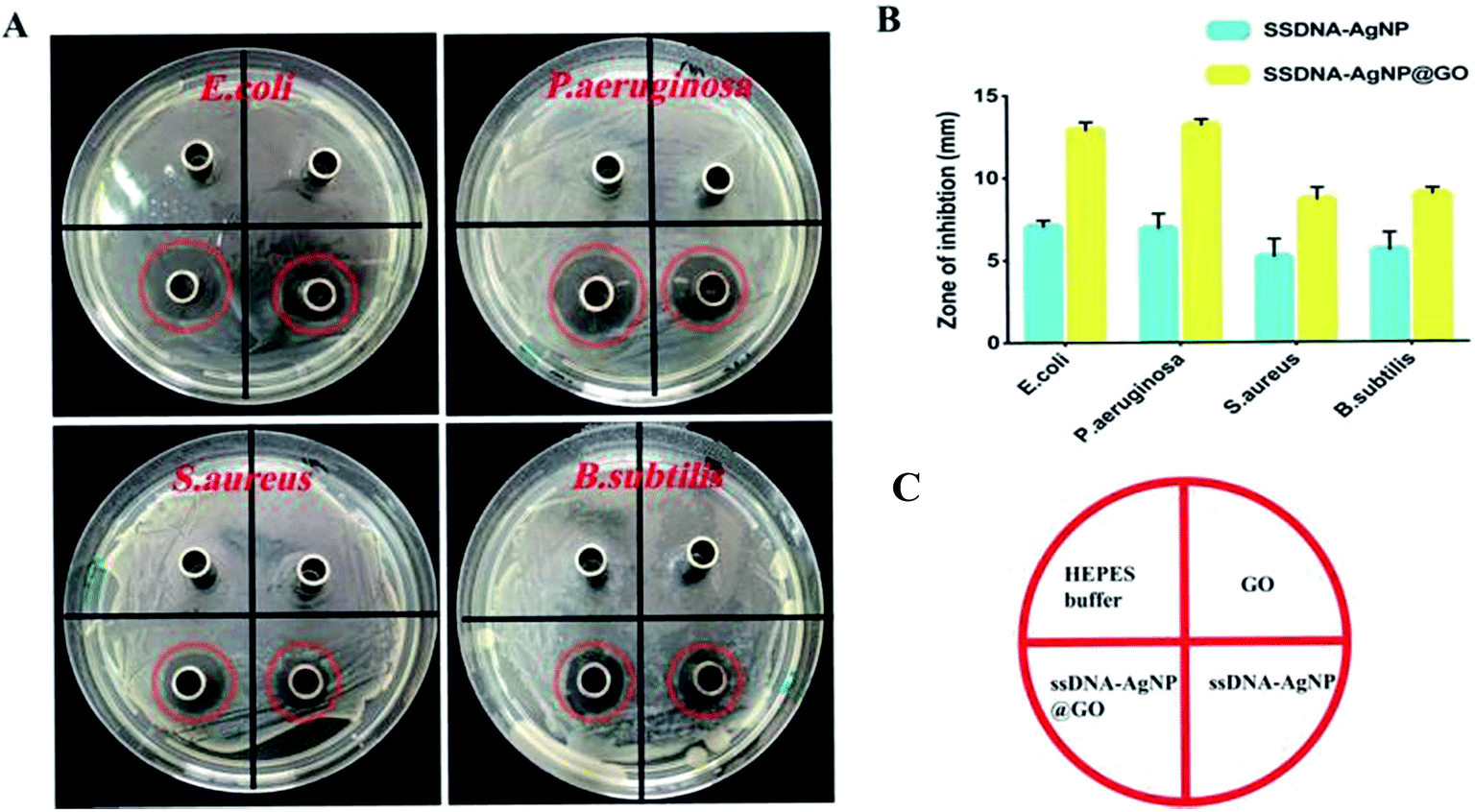 | ||
| Fig. 3 (A) Digital images showing the inhibition halos caused by HEPES, GO, ssDNA-AgNPs and ssDNA-AgNPs@GO against E. coli, P. aeruginosa, S. aureus and B. subtilis. The diameter of the Oxford cup is 7 mm. (B) Statistical column graph showing the antibacterial effect of the materials on the bacteria. Error bars represent the standard error of 3 parallel experiments. (C) Schematic diagram of the different sections in (A). | ||
3.3 Antibacterial mechanism of ssDNA-AgNPs@GO
In order to explore the mechanism of ssDNA-AgNPs@GO against bacteria, the double staining method of PI and calcein-AM was used to selectively investigate the degree of the membrane-damaging effects of E. coli and S. aureus. Fig. 4A and B show that bacteria with PBS treatment only emitted green fluorescence, and half of them emitted green and half emitted red in the GO or ssDNA-AgNPs treatment groups. However, only red fluorescence was observed in the ssDNA-AgNPs@GO treatment group. As the red fluorescence intensity of PI directly reflected the extent of membrane damage, the high red fluorescence intensity that appeared in the ssDNA-AgNPs@GO treatment group indicated that this nanomaterial killed most of the bacteria by destroying the cell membranes.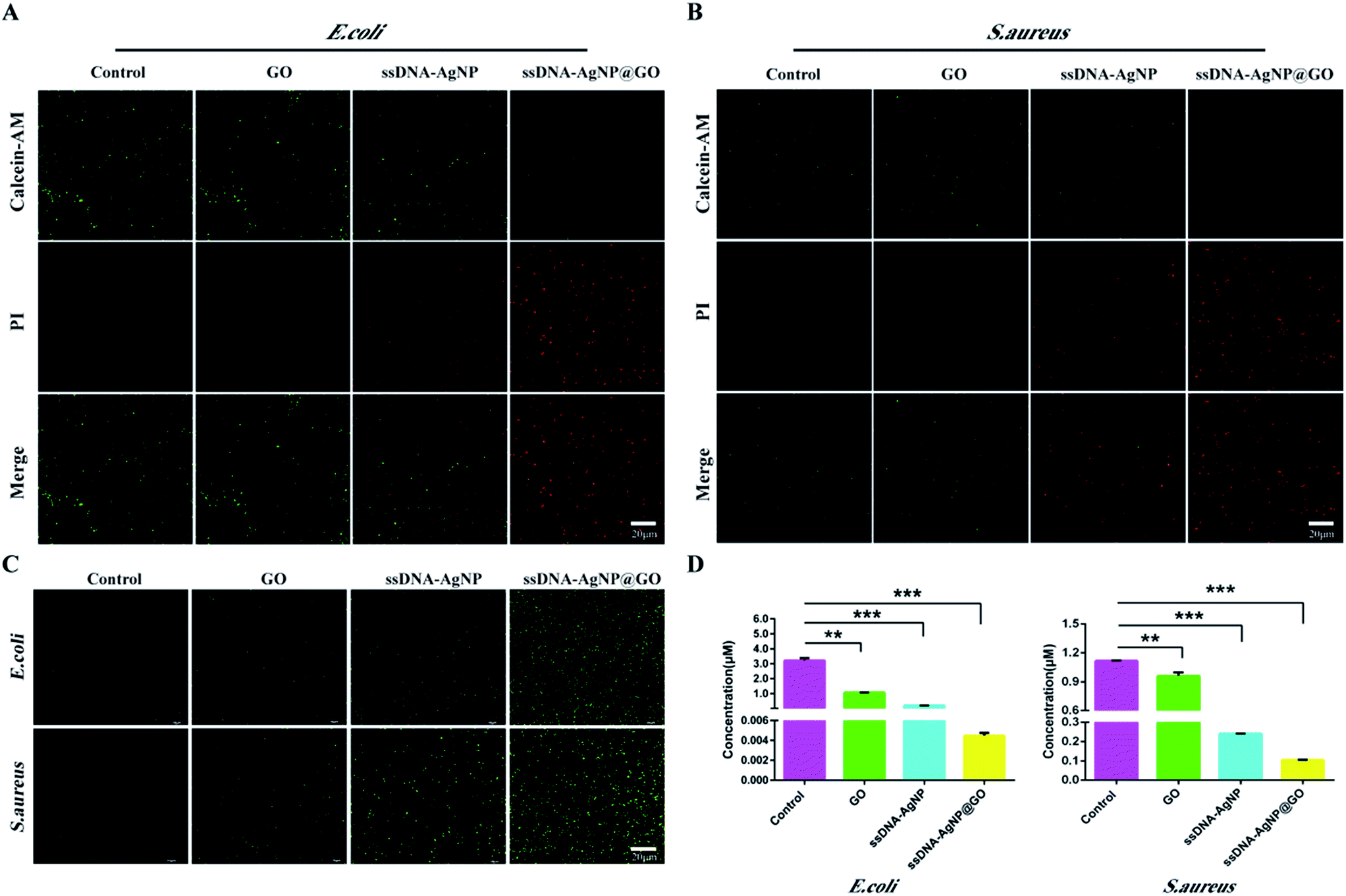 | ||
| Fig. 4 Antibacterial mechanisms of ssDNA-AgNPs@GO. (A) and (B) live–dead fluorescence images of E. coli and S. aureus treated with GO, ssDNA-AgNPs and ssDNA-AgNPs@GO for 12 h. Green fluorescence represents live bacteria stained with calcein-AM, while red fluorescence represents dead bacteria stained with PI. (C) Total ROS of E. coli and S. aureus treated with GO, ssDNA-AgNPs and ssDNA-AgNPs@GO, detected using a multidimensional living cell imaging system and a fluorescence spectrometer. Green fluorescence represents the ROS. (D) The level of ATP for E. coli and S. aureus with different nanomaterial treatments. Error bars represent the standard error of 3 parallel experiments. | ||
It has been reported that the AgNP-triggered antibacterial effect is related to the generation of free radicals, which can be monitored using DCFH-DA.36 By using the DCFH-DA staining method, we found that treatment with GO, ssDNA-AgNPs or ssDNA-AgNPs@GO differentially induced the production of ROS. As expected, the level of ROS was the highest among these groups (Fig. 4C). This data suggested that ssDNA-AgNPs@GO caused severe damage to the cell membrane, which allowed more antimicrobial hybrids to enter into the cell and produce more intracellular ROS.
In addition, ATP, an energy storage and transportation molecule, which plays an important role in various physiological and pathological processes of bacteria, often declines under apoptosis, necrosis or toxic conditions.37 In this study, we found that the ATP level of E. coli and S. aureus significantly decreased in the ssDNA-AgNPs@GO treatment group. However, GO or ssDNA-AgNPs only weakly decreased the ATP level of bacteria compared to the PBS treatment group (Fig. 4D). According to these data, we suspected that ssDNA-AgNPs@GO could be easily adsorbed on the surface of the bacteria, destroying the membrane with the help of GO, which was confirmed by the ROS production and ATP decrease.38 Moreover, the maintenance of the small size of the AgNPs by GO conferred a higher and more synergistic antibacterial effect compared to that of the ssDNA-AgNPs.
3.4 Biological safety investigation of ssDNA-AgNPs@GO
MTT and hemolysis methods were used to evaluate the biosafety of ssDNA-AgNPs@GO. According to GB/T 16886.5-2003 (ISO 10993-5:1999), samples with a cell viability higher than 75% can be considered as non-cytotoxic. Fig. 5A shows that the cell viabilities of NIH-3T3 cells treated with 3.4, 6.8, 10.2 and 13.6 μg mL−1 of ssDNA-AgNPs were 98.15% ± 1.11%, 89.63% ± 3.51%, 96.67% ± 2.36% and 85.68% ± 5.53%, respectively, while the cell viabilities were 88.84% ± 4.71%, 85.46% ± 2.12%, 91.09% ± 4.21% and 80.79% ± 3.82%, respectively, after treatment with same concentration of ssDNA-AgNPs@GO. Although the viability of cells in the ssDNA-AgNPs@GO treatment group slightly decreased, we can still conclude that both ssDNA-AgNPs and ssDNA-AgNPs@GO are safe for wound healing in vivo. In addition, hemocytolysis ratios caused by different nanomaterials were also investigated. According to the American Society for Testing and Materials (ASTM F 756-00, 2000), materials can be classified in three different categories: hemolytic (hemolysis over 5%), slightly hemolytic (between 5% and 2%) and non-hemolytic (below 2%). Fig. 5B shows that the hemolysis rate was only 2.1 ± 0.26%, even when the concentration of ssDNA-AgNPs@GO was at 15.3 μg mL−1. Fig. S5† also indicates that GO and ssDNA-AgNPs did not cause hemocytolysis of erythrocytes, even at high concentration. The above data strongly indicates the high biocompatibility of these nanomaterials.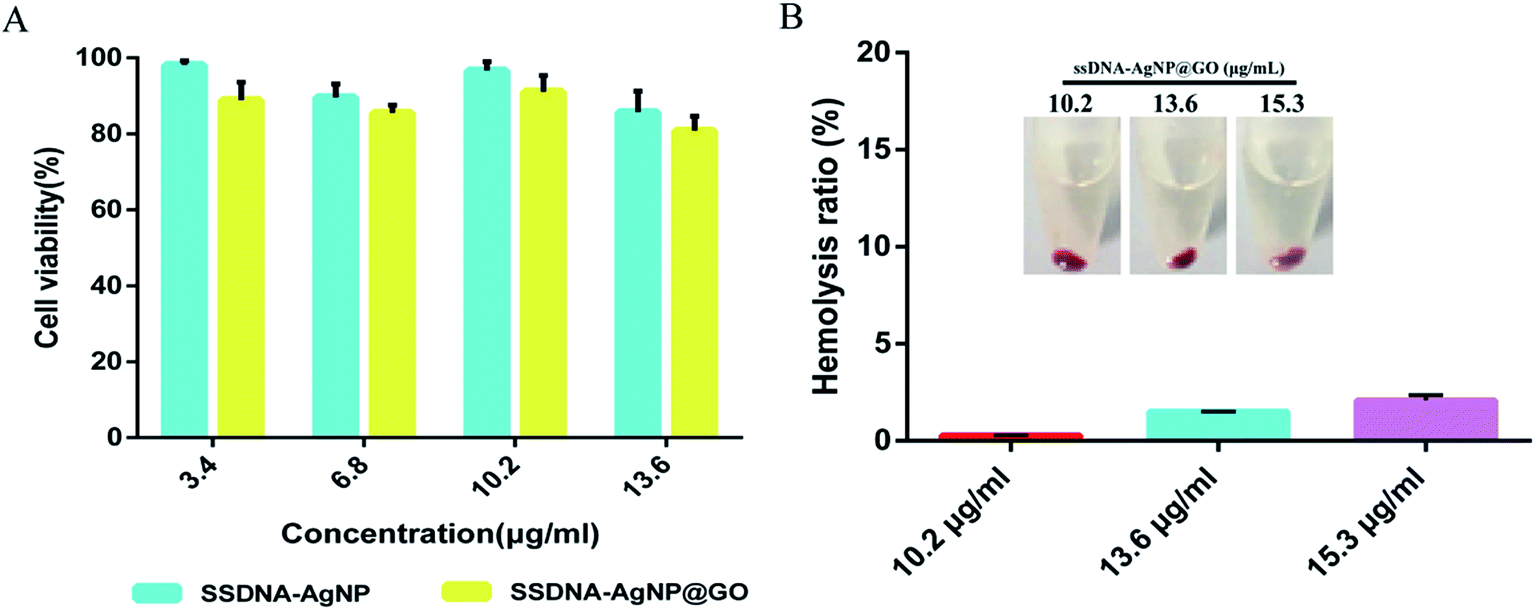 | ||
| Fig. 5 (A) Viability of NIH-3T3 cells treated with ssDNA-AgNPs and ssDNA-AgNPs@GO for 24 h. (B) Hemolysis ratio of human blood incubated with different nanomaterials. The inner figure shows photographs of hemocytolysis. | ||
3.5 In vivo evaluation of the treatment of ssDNA-AgNPs@GO in a wound healing model
Finally, animal experiments were carried out to evaluate the wound healing capability of ssDNA-AgNPs@GO in vivo. Fig. S6† shows the excellent healing effect in ssDNA-AgNPs@GO-treated mice after 7 days. Moreover, the regenerated skin with GO, ssDNA-AgNP or ssDNA-AgNPs@GO treatment is smooth and similar to normal skin without scar formation, indicating their good healing ability. By further quantitating the extent of wound closure at different times, we found that the wound treated with ssDNA-AgNPs@GO achieved a closure of 32.71 ± 2.95%, compared to the GO, ssDNA-AgNPs and PBS-treated groups, which only showed 23.28 ± 3.36%, 23.87 ± 2.81% and 23.64 ± 1.13% closure, respectively, after treatment for 3 days. After 11 days, the wound closure in the ssDNA-AgNPs@GO-treated group significantly enhanced to 91 ± 1.71%, which was about 20% higher than that of the control group. Complete wound closure was observed 13 days later for the ssDNA-AgNPs@GO treatment group, while the closure rates for the PBS, GO and ssDNA-AgNPs treatment groups were 92.69 ± 1.09%, 95.39 ± 1.57% and 95.75 ± 0.39% at this time point (Fig. 6A). In addition, we investigated the morphological difference of wound healing tissues in mice with different treatments. It has been reported that the major steps in the process of wound healing are re-epithelialization and the formation of granulation tissues. The signals of re-epithelization are migration and the proliferation of fibroblasts and epidermal cells, because elongated fibroblasts are helpful for the formation of mature fibrous granulation tissue. In contrast, the presence of lymphocytes and neutrophils indicates an inflammatory response.39 From Fig. 6E, we found that a cuticular layer emerged, with elongated fibroblasts and epithelial cells in all wound dressing groups at day 14. Among them, most of the elongated fibroblasts appeared in the ssDNA-AgNPs@GO treatment group. On the contrary, many lymphocyte and neutrophil cells were observed in the GO or ssDNA-AgNPs treatment groups, which reflected the inflammatory response of the two groups. These results demonstrate that ssDNA-AgNPs@GO can provide effective support for re-epithelialization and granulation tissue formation to accelerate wound healing.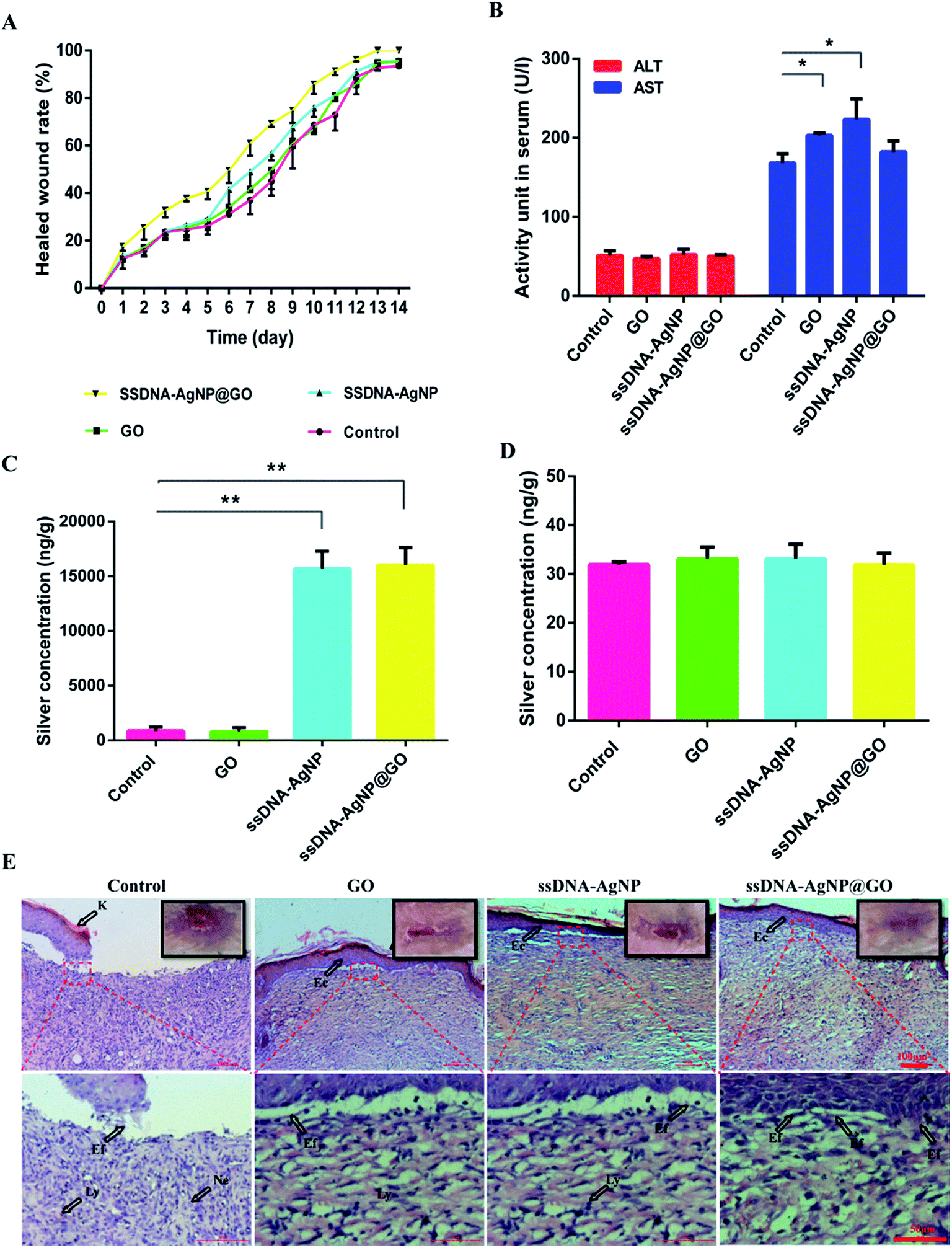 | ||
| Fig. 6 The effects of GO, ssDNA-AgNPs and ssDNA-AgNPs@GO on wound infection and healing of MRSA in mice. (A) Quantitation of the topical wound healing rate. (B) AST and ALT level assay of mouse blood at the 14th day. (C) and (D) ICP-MS analysis for silver content in healed skin and liver tissues at the 14th day. Error bars represent the standard error of 3 parallel experiments. (E) Histology of the wound healing process in various groups on 14 days with H&E staining at magnifications of ×10 and ×40. Lines indicate wound healing events. K = keratin, Ly = lymphocyte, Ne = neutrophil, Ec = epithelial cells and Ef = elongated fibroblasts. The presence of Ly and Ne indicate an inflammatory response. Ec and Ef were the signals of re-epithelization, which is beneficial for the formation of matured fibrous granulation tissue. | ||
In the following experiment, the levels of ALT and AST in mice were investigated. Fig. 6B shows that the ALT levels in the PBS, GO, ssDNA-AgNPs and ssDNA-AgNPs@GO treatment groups were 51.00 ± 6.00, 47.00 ± 3.00, 52.00 ± 6.00 and 50.00 ± 2.00 U L−1, respectively. These data demonstrated that treatment with these nanomaterials did not cause liver injury. Meanwhile, the AST levels in the corresponding groups were 168.00 ± 12.00, 203.00 ± 3.00, 223.00 ± 26.00 and 182.00 ± 14.00 U L−1, respectively. Compared to the PBS group, the AST level in mice increased only by about 7% in the ssDNA-AgNPs@GO treatment group, which is much lower than that for the GO treatment group (21%) or the ssDNA-AgNPs group (32%). According to our results, the increase in AST level perhaps resulted from dermatomycosis caused by the nanomaterials. In order to further assess the body’s absorption of AgNPs, liver and healed skin tissues of mice with administration for 14 days were used for ICP-MS analysis. Fig. 6C indicates that the silver contents in the healed skin tissues treated with ssDNA-AgNPs and ssDNA-AgNPs@GO were 15692.03 ± 1592.03 and 16020.12 ± 1597.41 ng g−1, respectively, while the contents in the control and GO groups were 860.83 ± 364.54 and 800.32 ± 367.08 ng g−1, which were significantly lower than those for the ssDNA-AgNPs and ssDNA-AgNPs@GO groups. In contrast, the silver content in mouse liver treated with HEPES buffer, GO, ssDNA-AgNPs and ssDNA-AgNPs@GO was 31.93 ± 0.59, 33.11 ± 2.39, 33.09 ± 2.99 and 31.88 ± 2.40 ng g−1, respectively (Fig. 6D). These data indicated that ssDNA-AgNPs@GO reduced accumulation in the liver compared to GO and ssDNA-AgNPs. As expected, the major organs (heart, liver, spleen, lung and kidney) of mice did not show abnormal effects or damage after treatment with the 3 kinds of nanomaterial (Fig. S7†). In summary, these results demonstrated the good biocompatibility of ssDNA-AgNPs@GO.
4 Conclusions
In summary, we developed a facile method to synthesize ssDNA-AgNPs@GO nanocomposites with high stability and good biocompatibility. The new nanocomposites exhibited enhanced antibacterial activity to Gram-negative (E. coli and P. aeruginosa) and Gram-positive bacteria (S. aureus and B. subtilis) compared to GO and ssDNA-AgNPs. The antibacterial mechanisms of ssDNA-AgNPs@GO include damage to the cell integrity, production of ROS, decrease of ATP and bacterial metabolism. In vivo wound healing evaluation in mice showed that ssDNA-AgNPs@GO possessed improved wound healing capability compared to that of ssDNA-AgNPs due to the excellent re-epithelialization and dense collagen deposition properties.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was partially supported by the Natural Science Foundation of China (81374062, 81673579 and 31672457) and the National Standardization Project of Traditional Chinese Medicine (ZYBZH-Y-HUN-23). The authors would like to thank Prof. Dr Xiangxian Meng for the help of bacteria culture.References
- P. S. Stewart and J. W. Costerton, Lancet, 2001, 358, 135–138 CrossRef.
- G. Taubes, Science, 2008, 321, 356–361 CrossRef PubMed.
- H. Ji, H. Sun and X. Qu, Adv. Drug Delivery Rev., 2016, 105, 176–189 CrossRef PubMed.
- M. Yu, Z. Wang, M. Lv, R. Hao, R. Zhao, L. Qi, S. Liu, C. Yu, B. Zhang and C. Fan, ACS Appl. Mater. Interfaces, 2016, 8, 19866–19871 CrossRef PubMed.
- K. Hiramatsu, L. Cui, M. Kuroda and T. Ito, Trends Microbiol., 2001, 9, 486–493 CrossRef PubMed.
- L. J. Zhang, C. F. Guerrero-Juarez, T. Hata, S. P. Bapat, R. Ramos, M. V. Plikus and R. L. Gallo, Science, 2015, 347, 67–71 CrossRef PubMed.
- B. J. Hong, O. C. Compton, Z. An, I. Eryazici and S. B. T. Nguyen, ACS Nano, 2012, 6, 63–73 CrossRef PubMed.
- M. Li, S. A. Nyantakyi, P. Gopal, D. B. Aziz, T. Dick and M. L. Go, ACS Med. Chem. Lett., 2017, 8, 1165–1170 CrossRef PubMed.
- M. Vasakova, F. Morell, S. Walsh, K. Leslie and G. Raghu, Am. J. Respir. Crit. Care Med., 2017, 196, 680–689 CrossRef PubMed.
- J. W. Beaber, B. Hochhut and M. K. Waldor, Nature, 2004, 427, 72–74 CrossRef PubMed.
- P. V. Asharani, K. M. G. Low, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef PubMed.
- N. J. Croucher, S. R. Harris, C. Fraser, M. A. Quail, J. Burton, M. V. D. Linden, L. Mcgee, A. V. Gottberg, J. H. Song and K. S. Ko, Science, 2011, 331, 430–434 CrossRef PubMed.
- V. Stojanoski, B. Sankaran, B. V. V. Prasad, L. Poirel, P. Nordmann and T. Palzkill, BMC Biol., 2016, 14, 81 CrossRef PubMed.
- A. Hebeish, A. Elshafei, S. Sharaf and S. Zaghloul, Carbohydr. Polym., 2014, 113, 455–462 CrossRef PubMed.
- Y. M. Sahin, M. Yetmez, F. N. Oktar, O. Gunduz, S. Agathopoulos, E. Andronescu, D. Ficai, M. Sonmez and A. Ficai, Curr. Med. Chem., 2014, 21, 3391–3404 CrossRef PubMed.
- F. Cao, E. Ju, Y. Zhang, Z. Wang, C. Liu, W. Li, Y. Huang, K. Dong, J. Ren and X. Qu, ACS Nano, 2017, 11, 4651–4659 CrossRef PubMed.
- C. Mao, Y. Xiang, X. Liu, Z. Cui, X. Yang, Y. Kwk, H. Pan, X. Wang, P. K. Chu and S. Wu, ACS Nano, 2017, 11, 9010–9021 CrossRef PubMed.
- A. P. Richter, J. S. Brown, B. Bharti, A. Wang, S. Gangwal, K. Houck, E. A. Cohen Hubal, V. N. Paunov, S. D. Stoyanov and O. D. Velev, Nat. Nanotechnol., 2015, 10, 817–823 CrossRef PubMed.
- H. Qin, H. Cao, Y. Zhao, G. Jin, M. Cheng, J. Wang, Y. Jiang, Z. An, X. Zhang and X. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 10785–10794 CrossRef PubMed.
- R. H. Dong, Y. X. Jia, C. C. Qin, L. Zhan, X. Yan, L. Cui, Y. Zhou, X. Jiang and Y. Z. Long, Nanoscale, 2016, 8, 3482–3488 RSC.
- Y. S. Ko, H. J. Yun, M. Seo, K. Lim, J. Hwang and K. Woo, J. Mater. Chem. B, 2014, 2, 6714–6722 RSC.
- X. Zeng, D. T. Mccarthy, A. Deletic and X. Zhang, Adv. Funct. Mater., 2015, 25, 4344–4351 CrossRef.
- J. Liu, X. Chen, X. Huang, C. Zheng, Y. Liu and T. Xu, J. Mater. Chem. B, 2015, 3, 7020–7029 RSC.
- S. Kellici, J. Acord, A. Vaughn, N. P. Power, D. J. Morgan, T. Heil, S. P. Facq and G. I. Lampronti, ACS Appl. Mater. Interfaces, 2016, 8, 19038–19046 CrossRef PubMed.
- A. Dawn, A. Pratap Mukherjee and A. K. Nandi, Langmuir, 2007, 23, 5231–5237 CrossRef PubMed.
- A. Pyatenko, M. Yamaguchi and M. Suzuki, J. Phys. Chem. B, 2005, 109, 21608–21611 CrossRef PubMed.
- K. Chaloupka, Y. Malam and A. M. Seifalian, Trends Biotechnol., 2010, 28, 580–588 CrossRef PubMed.
- L. Rizzello and P. P. Pompa, Chem. Soc. Rev., 2014, 43, 1501–1518 RSC.
- K. K. Wong and X. Liu, MedChemComm, 2010, 1, 125–131 RSC.
- H. M. Hegab, A. Elmekawy, L. Zou, D. Mulcahy, C. P. Saint and M. Ginic-Markovic, Carbon, 2016, 105, 362–376 CrossRef.
- S. Wei, X. Liu, H. Min, G. Dong, Q. Feng and S. Zuo, ACS Appl. Mater. Interfaces, 2015, 7, 6966–6973 CrossRef PubMed.
- J. Chen, H. Peng, X. Wang, F. Shao, Z. Yuan and H. Han, Nanoscale, 2014, 6, 1879–1889 RSC.
- Q. Li, C. Yong, W. Cao, X. Wang, L. Wang, J. Zhou and X. Xing, J. Colloid Interface Sci., 2018, 511, 285–295 CrossRef PubMed.
- R. Baber, L. Mazzei, N. Thanh and A. Gavriilidis, Nanoscale, 2017, 9, 14149–14161 RSC.
- M. Guzman, J. Dille and S. Godet, Nanomedicine, 2012, 8, 37–45 CrossRef PubMed.
- T. C. Dakal, A. Kumar, R. S. Majumdar and V. Yadav, Front. Microbiol., 2016, 7, 1831 Search PubMed.
- S. Wang, R. Chen, Z. Zhong, Z. Shi, M. Chen and Y. Wang, Am. J. Chin. Med., 2014, 42, 1279–1300 CrossRef PubMed.
- Y. G. Yuan, Q. L. Peng and S. Gurunathan, Int. J. Mol. Sci., 2017, 18, 569 CrossRef PubMed.
- J. C. Provenzano, H. S. Antunes, F. R. F. Alves, I. N. Rôças, W. S. Alves, M. R. S. Silva and J. F. Siqueira Jr, J. Endod., 2016, 42, 880–885 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04933e |
| ‡ These authors contributed equal and were co-first authors. |
| This journal is © The Royal Society of Chemistry 2018 |