
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA MOF-based carrier for in situ dopamine delivery†
Alessandra Pinna a,
Raffaele Ricco'b,
Rossana Mighelic,
Gaia Rocchittac,
Pier Andrea Serrac,
Paolo Falcarob,
Luca Malfattid and
Plinio Innocenzi*d
a,
Raffaele Ricco'b,
Rossana Mighelic,
Gaia Rocchittac,
Pier Andrea Serrac,
Paolo Falcarob,
Luca Malfattid and
Plinio Innocenzi*d
aDepartment of Materials, Imperial College London, South Kensington Campus, London, SW72AZ, UK
bGraz University of Technology, Institute of Physical and Theoretical Chemistry, Stremayrgasse 9, 8010 Graz, Austria
cDipartimento di Medicina Clinica e Sperimentale, Università di Sassari, Viale S. Pietro 43 B, 07100 Sassari, Italy
dLaboratorio di Scienza dei Materiali e Nanotecnologie, CR-INSTM, Università di Sassari, Dipartimento di Chimica e Farmacia, Via Vienna 2, 07100 Sassari, Italy. E-mail: plinio@uniss.it
First published on 18th July 2018
Abstract
MIL-88A (Fe) MOF crystals were nucleated and grown around a polymer core containing superparamagnetic nanoparticles to assemble a new class of biocompatible particles for magnetophoretic drug delivery of dopamine. The carrier enabled efficient targeted release, dopamine protection from oxidative damage, long-term delivery and improved drug delivery cost-efficiency. After loading, dopamine was stable within the carrier and did not undergo oxidation. Drug release monitoring via spectrofluorimetry revealed a shorter burst effect and higher release efficiency than silica based carriers. The in vitro cytotoxicity at different MOF concentrations and sizes was assessed using PC12 cells as the neuronal cell model. The drug was directly uptaken into the PC12 cells avoiding possible side effects due to oxidation occurring in the extracellular environment.
Introduction
Parkinson's disease is a neurodegenerative disorder of the central nervous system, characterised by a progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta brain region, resulting in a substantial loss of dopamine (DA) in the striatum.1 To date, the most widely used treatment for Parkinson's disease is based on the administration of L-3,4-dihydroxyphenylalanine (L-dopa), a carboxylate precursor of DA able to cross the blood–brain barrier thanks to a specific L-amino acid transporter.2 However, L-dopa can also be metabolised in other bodily fluid compartments, resulting in side effects and patient discomfort. Moreover, long-term treatment with L-dopa has been linked to dopamine dysregulation syndrome.3 A controlled release of DA administration at the target site is therefore clearly needed.Targeting the brain via non-invasive nasal administration of DA could be an alternative pharmacokinetic approach to the current L-dopa therapy in Parkinson's disease.4 To date, a number of studies have focused on the administration of DA via nasal passages using various carrier matrices. Silica particles, liposomes, micelles and polymeric nanoparticles have been investigated to achieve a controlled release of the drug; however, none of these carriers has proved to protect dopamine, which is easily oxidised.5–7 In addition, these systems have provided poor bioavailability due to the complexity of the olfactory region that prevents the direct delivery of particles to the brain once inhaled through the nostrils.8 A promising method recently developed by Xi et al.9 exploited the magnetophoretic forces of superparamagnetic iron oxide particles in an attempt to overcome the geometrical complexity of the olfactory region and nasal clearance and improve the delivery efficiency. Magnetophoresis allows for directing particles towards the brain through the axon of the olfactory nerve instead of blood vessels.8 In this way, the carrier uptake into the cells is not required because the drug can be released into the synaptic cleft and then uptaken into the neuronal cells. The particle size selection is crucial to provide a proper drug delivery and, for this specific type of administration, large particles (in the range of tens of micrometers) are much more efficient than the typical nanocarriers (with an average size between 20–200 nm). Particles larger than 20 μm preferentially deposit in the anterior part of the nasal cavity, thus maximising their contact to the olfactory nerve, whilst particles smaller than 5 μm tend to escape the nasal cavity following the air streamlines, and to deposit in the lungs.10 Optimisation of the carrier particle density and size distribution is, therefore, the key to maximising this drug delivery mechanism, as it avoids lung inhalation and blood–brain barrier (BBB) issues.
The nasal administration strategy can be further improved by using porous engineered microsystems whose high surface area allows a substantial drug loading.
Metal–Organic Frameworks (MOFs) are a class of organic–inorganic hybrid porous materials consisting of metal ions or cluster nodes and organic linkers as bridges. The unique properties of MOFs – such as high specific surface areas, very large porosity and crystalline structure – make them suitable for loading and release of drugs with different physical–chemical properties.11,12 Recently, an increasing number of studies reports the use of MOFs as a drug carrier of therapeutic molecules such as anti-inflammatory,13,14 anticancer,15–18 and antiviral.19,20 Amongst ZIF-8,21,22 UiO-66,23 HKUST-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 and iron(III) carboxylate MOFs, the MIL-88 class stands out as a suitable candidate for biomedical and pharmaceutical applications25 due to its low toxicity, biocompatibility and broad spectrum of drug loading capacities.12 Moreover, in the case of MIL-88, the intrinsic magnetic properties of the metal atoms enable its use as MRI contrast agent26 and the flexibility of the porous framework allows for drug incorporation through pore enlargement.27 Additional properties can be provided by combining MOFs with inorganic nanoparticles28–31 to design a multifunctional composite material.32,33
24 and iron(III) carboxylate MOFs, the MIL-88 class stands out as a suitable candidate for biomedical and pharmaceutical applications25 due to its low toxicity, biocompatibility and broad spectrum of drug loading capacities.12 Moreover, in the case of MIL-88, the intrinsic magnetic properties of the metal atoms enable its use as MRI contrast agent26 and the flexibility of the porous framework allows for drug incorporation through pore enlargement.27 Additional properties can be provided by combining MOFs with inorganic nanoparticles28–31 to design a multifunctional composite material.32,33
MOFs-based magnetic composites, prepared by introducing magnetic particles into MOFs during synthesis, are promising carriers for brain-targeted drug-delivery and controlled-release, because of the high porosity and the possibility to be positioned, moved by using a magnetic field.34–38 Although some publications claim the optimization of a versatile synthesis for biological applications,39 this type of composite has been mainly applied for catalysis,40 water remediation41,42 and concentration of biological species.43,44 The research on magnetic framework composites for drug delivery, therefore, remains in its early developmental stage and a few publications exploiting both magnetic and drug-delivery properties have been reported so far.45
In this work, the porosity, the high surface area of MOF and magnetism of iron oxide particles (Fe3O4) were synergistically used to engineer DA-delivery systems with potential magnetophoretic applications. To the best of our knowledge, this is the first time that DA was incorporated into MOF composites. The structure of the MILL-88A was selected to design this molecule, which is typically prone to oxidation in a biological environment. The composite synthesis makes use of a controlled in situ growth of a MIL-88A (Fe) MOF shell on carboxyl-functionalised particles bearing magnetic properties. A controlled particle size growth allowed tuning the conditions for the dopamine loading and release. The amount of dopamine released by the carriers was measured in vitro, in a phosphate buffer medium, with fluorescence spectroscopy, and in nerve pheochromocytoma cell line (PC12) culture, by HPLC-EC chromatography. This cell line was chosen because it shows several physiological properties characteristic of dopaminergic neurons in Parkinson's disease.46,47
Results and discussion
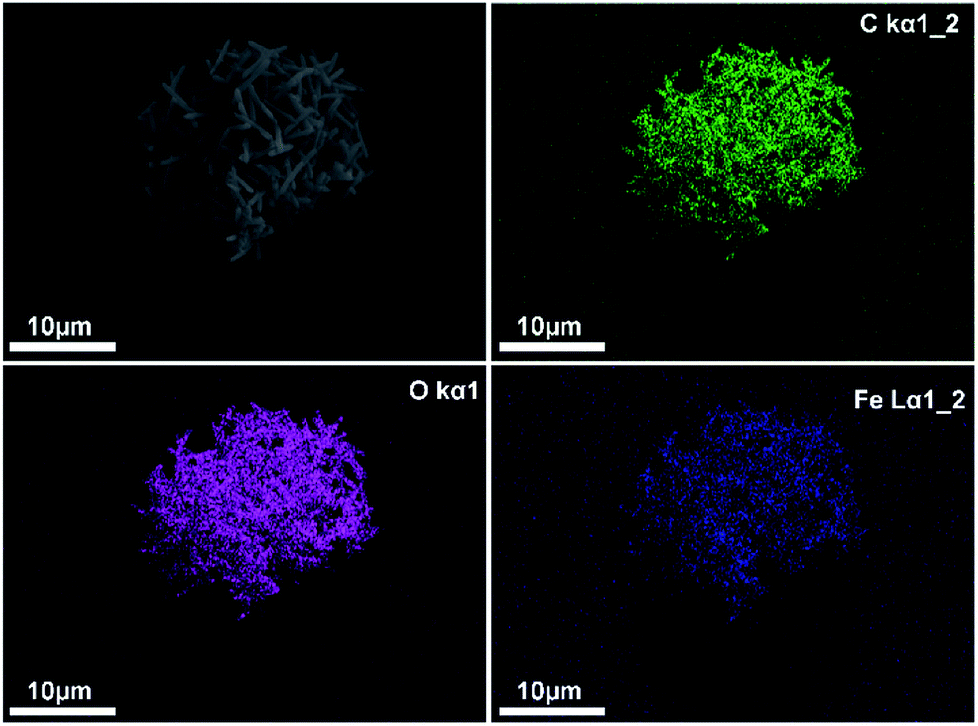
The growth of MIL-88A (Fe) crystals was induced on the surface of carboxyl-functionalized polymeric magnetic particles (PMPs, with a size distribution of 3.0–6.0 μm), to allow the formation of PMP@MIL-88A systems. PMPs were mixed in an aqueous solution with MIL-88A precursors (fumaric acid and iron(III) chloride) to obtain the composite. Dimensional tuning was obtained by repeating the MIL-88A growth on the previous generation of PMP@MIL-88A system.Particles with controlled dimensions, from 8 to 86 μm, as shown by SEM analysis (Fig. 1a–e) were obtained. Fig. 1e shows the changes in the size of the PMP@MIL-88A particles as a function of the number of repeated growth cycles. Since the particles size was not homogeneous (Fig. S1, ESI†), the mean size was calculated by considering the particles as equiaxial and measuring their average diameter. Although the variability of this measure, the PMP@MIL-88A particle mean size increased over the generations: 8 ± 1 μm, 18 ± 4 μm, 49 ± 5 μm, and 86 ± 8 μm for the first, second, third and fourth growth cycles respectively. The elemental distribution in the PMP@MIL-88A particles, as analysed by SEM with EDX mapping (Fig. 2), shows a homogeneous distribution of C, O, and Fe in the shell of the composite particles, corresponding to the MOFs composition. The magnetic nanoparticles inside the composite MOF-based structure cannot be identified because both core and shell of the materials contain a large amount of iron.
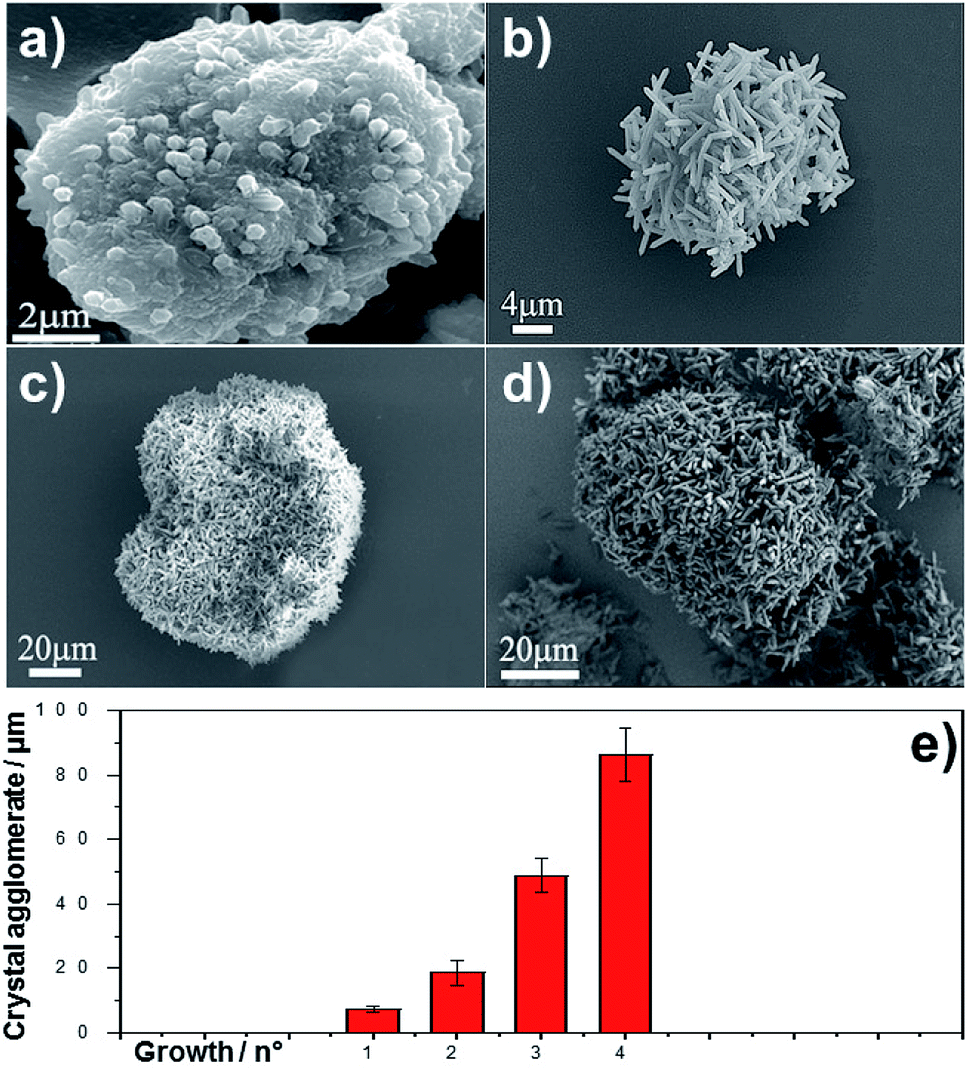 | ||
| Fig. 1 SEM images of MIL-88A MOF at different generations on PMP particles. (a) 1st, (b) 2nd, (c) 3rd and (d) 4th generation. (e) Crystal agglomerate diameter (μm) as a function of the generations. Results are the mean ± SD of ten measurements of PMP@ MIL-88A MOF agglomerate. | ||
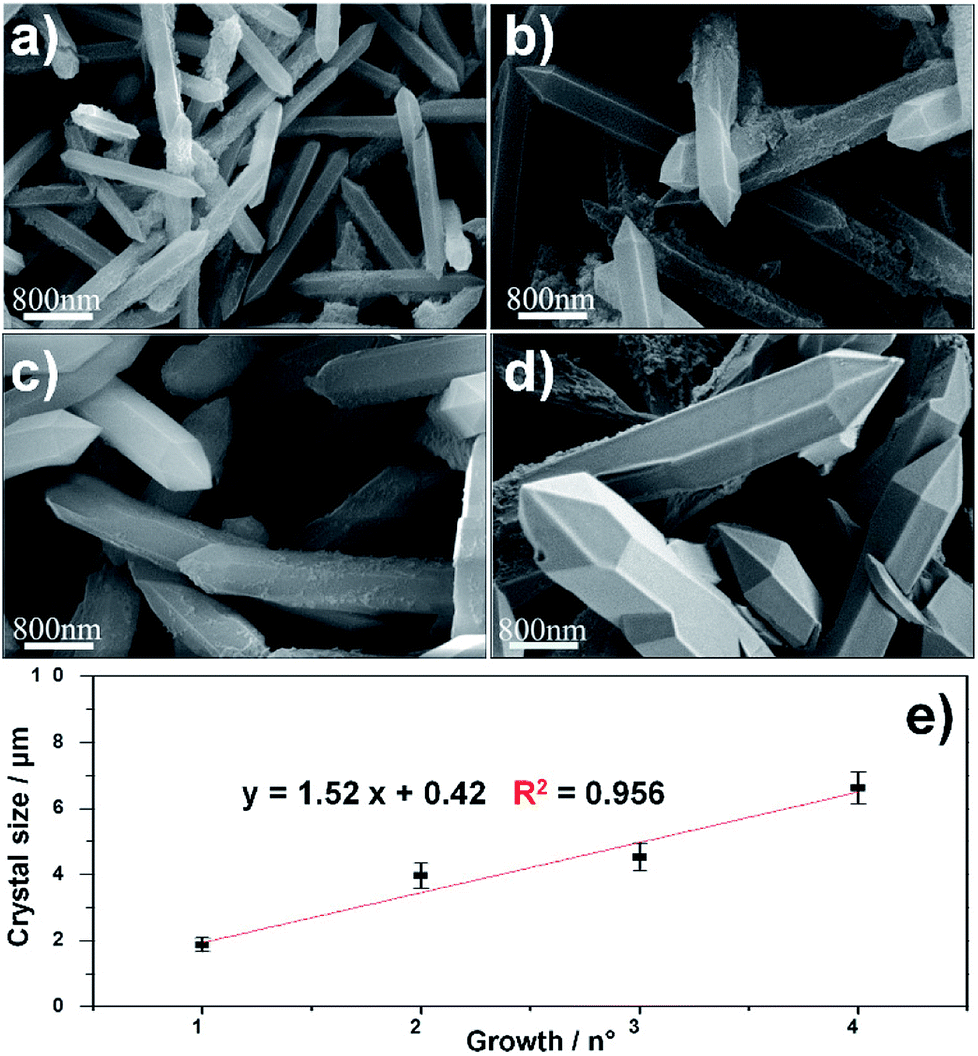
 | ||
| Fig. 2 SEM images of MIL-88A crystals at different generation on PMP particles. (a) 1st (b) 2nd (c) 3rd and (d) 4th. (e) Crystals size (μm) as a function of the growths. Results are the mean ± SD of ten measurements of crystal size. | ||
The SEM characterization (Fig. 3a–d) clearly shows that the repetitive growths not only affect the average size of the composite particles but also the typical size of the MOF crystals surrounding the PMP core. In particular, an increase in the crystal size was observed from the first to the fourth growth cycle. The correlation between MIL-88A growths and crystal size is shown in Fig. 3e. A linear increase in dimension is observed: 1.8 ± 0.2 μm, 3.9 ± 0.3 μm, 4.5 ± 0.4 μm, and 6.6 ± 0.5 μm for first, second, third and fourth growths respectively.
 | ||
| Fig. 3 EDX mapping analysis of elements C (green), O (violet) and Fe (blue) in PMP@MIL-88A (Fe) MOF 2nd generation. | ||
PMP@MIL-88A could be considered as a potential carrier in magnetophoretic drug delivery. The PMP core of the MIL-88A particles allows the manipulation of carrier motion in the nasal passage to reach the olfactory nerve, which is directly connected to the central nervous system.8 Interestingly, the administration through the nose can allow the maximisation of the carrier size without the limitation required in other pharmaceutical administrations.
Particles larger than 7 μm can produce vascular occlusions in the capillary blood vessels in the case of intravenous injection with a reduction of blood flow.48 Conversely, if the administration is done through the nose, particles larger than 10 μm can be employed. This is reflected in a higher drug loading delivered to the brain49 and avoids the side effects related to systemic administration. Previous works reported an optimal particle size of 15 μm for magnetophoretic administration in the olfactory region.8 Based on these previous findings we considered the 2nd growth PMP@MIL-88A a potential candidate for dopamine delivery through magnetophoretic administration. Additionally, 2nd generation composite showed a better stability in the dopamine uptake (Fig. 4). The yellow solution in Fig. 4a is due to the oxidation of DA which occurs by a multistep reaction.50 Initially, the solution containing DA was colourless, but the oxidation process resulted in a shift towards an orange intermediate (dopamine-o-quinone) progressing towards a black final coloration (dopaminochrome). The improved DA uptake of the 2nd generation composite is attributed to a denser and thicker structure of the MOF crystals shell. Thermogravimetric analysis (TGA) was performed on PMP@MIL-88A samples to estimate the number of iron oxide particles encapsulated into the MOF shell. Fig. 5 shows the TG curves of carboxyl-magnetic particles (red curve), MIL-88A (black) and PMP@MIL-88A (blue). In the multistage decomposition of PMP particles, the first degradation step (270 °C) corresponds to decarboxylation, while the second step (450 °C) is related to the combustion of the polystyrene core in the particles. When the temperature rises up to 700 °C, only iron oxide remains, providing a residual mass of 18.5% with respect to the non-treated particles.
 | ||
| Fig. 4 Image of DA-PMP@MIL-88A (black powder), (a) 1st and (b) 2nd generations, after several washing cycles with PBS. The particles are sensitive to the magnetic field and can be collected by a magnet. | ||
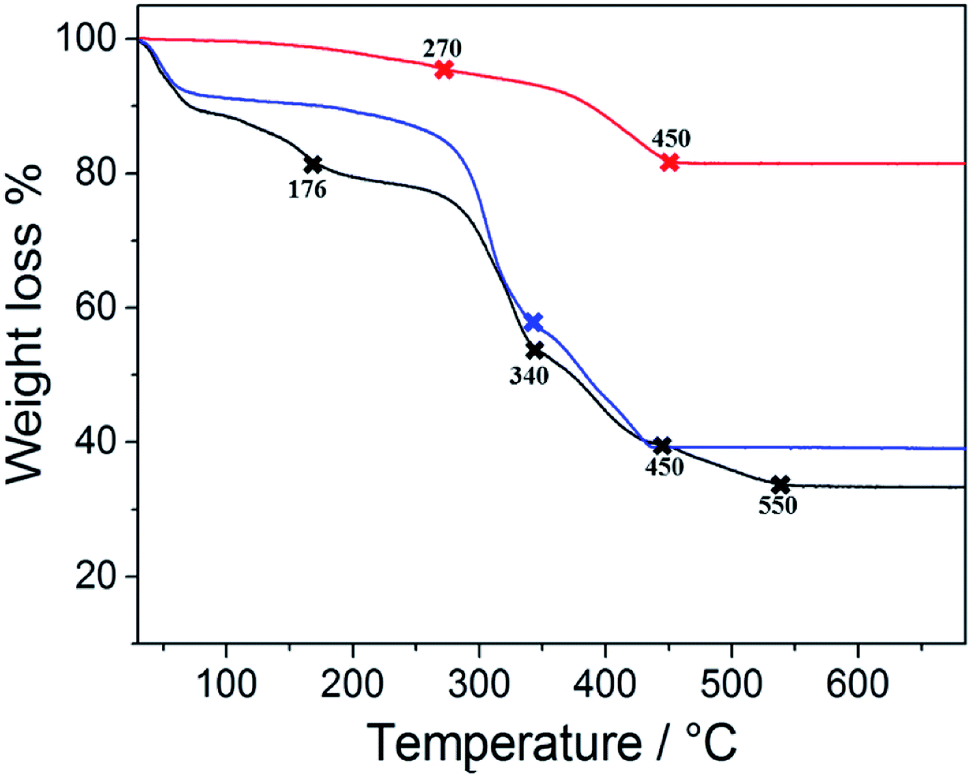 | ||
| Fig. 5 TGA analysis of PMP particles (red), MIL-88A MOF (black) and PMP@MIL-88A (Fe) (blue) in air at a heating rate of 10 °C min−1. | ||
The TGA results give the following PMP particle composition: 81.5% polystyrene and 18.5% magnetite (Fe3O4). The black curve shows the thermogravimetric decomposition of MIL-88A MOF. A first decrease in weight up to 176 °C corresponds to a loss of water and/or other gas physio-absorbed on the MOF.51 The second degradation step (340 °C), accounting for a 50% weight loss, corresponds to fumaric acid decomposition, which was completed at 550 °C.52 As expected, the multistage decomposition of the PMP@MIL-88A composite is a combination of the features shown by the two previous materials, i.e. PMP particles and MOFs (blue curve of Fig. 5). After a faint degradation due to organic combustion of carboxyl linker, the first clear weight loss of ca. 50% (340 °C) is due to the degradation of the fumaric acid in MIL-88A. Then, the second step (450 °C) is attributed to the thermal removal of polystyrene from the particle core. PMPs account for a 59 wt% content in the composite (0.96 mg of particles in 1.62 mg of PMP@MIL-88A) and this explains why the weight loss up to 176 °C is not visible in the PMP@MIL-88A sample.
Assuming that the magnetic particles have a stoichiometric composition and are perfectly spherical with an average size of 4.5 μm (as reported in the materials specification), the number of magnetic PMP particles corresponding to a weight of 0.96 mg is 13. Fig. S2 (ESI†) shows N2 sorption/desorption isotherms of MIL-88A and dopamine-loaded MIL-88A, which are a combination of type II and III sorption isotherms. The surface area was calculated as 27.5 ± 0.4 m2 g−1 and 18.1 ± 0.3 m2 g−1 for MIL-88A alone and dopamine-loaded respectively. Pore sizes of 2.4 ± 0.2 nm and 2.2 ± 0.1 nm were calculated for MIL-88A alone and dopamine-loaded respectively. The 33% surface area reduction is attributed to the dopamine occupancy of the framework pores, resulting in a reduced area available for the N2 gas.
The relatively low surface area exhibited by this type of MOF (SBET < 30 m2 g−1) is consistent with other reports.53–56
The encapsulation of dopamine into PMP@MIL-88A is fundamental to the oxidation prevention of the molecule. The catechol portion of the molecule is sensitive to air, pH, and light,57 incurring in the formation of quinone-based highly reactive by-products, such as dopamine-o-quinone (DQ), aminochrome (AC) and indole-5,6-quinone (IQ).58
The release of dopamine from PMP@MIL-88A was assessed by optical spectroscopy in a PBS buffer 1 mM at pH 7.4. The amount of dopamine in PMP@MIL-88A was set to 0.61 mg mg−1, considering that only 5–10% of the drug contained in the commercial L-dopa tablets for oral administration (Sinemet® 250 mg) is able to cross the blood–brain barrier.59 To calculate the amount of dopamine released from the PMP@MIL-88A composite, a calibration curve was created by measuring the fluorescence emission at 327 nm (λexc: 305 nm) of increasing quantities of dopamine in PBS buffer, from 0 to 2 mg mL−1 (Fig. 6a). In a water-based solution, the dopamine tends to be converted to quinone as a consequence of oxidation (Fig. 6b). The dopamine oxidation to dopamine-o-quinone (DQ) can be monitored in solution by measuring the decrease of fluorescence intensity at 327 nm (λexc = 390 nm) which is the characteristic emissions of DA.60,61
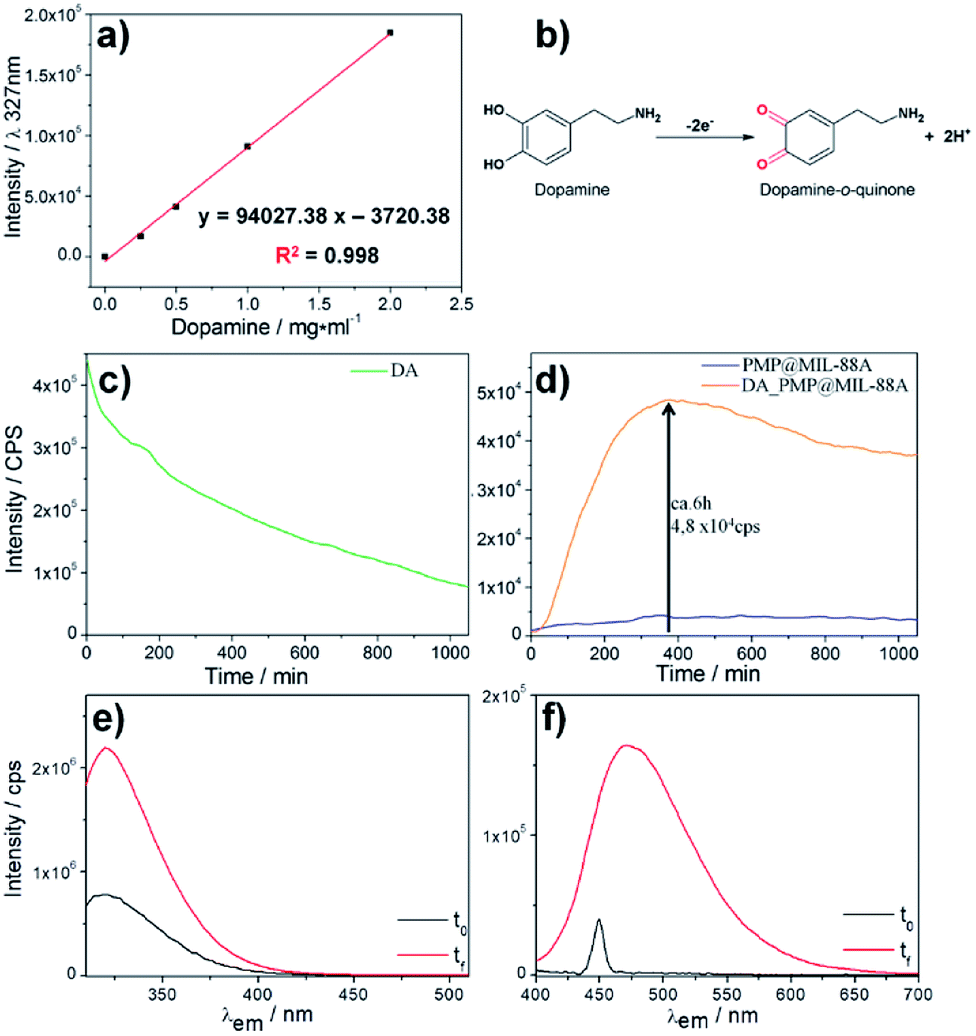 | ||
| Fig. 6 (a) Calibration curve of the dopamine, emission intensity λem: 327 nm vs. concentration in PBS solution. (b) Scheme of dopamine oxidation. (c) Fluorescence spectrum of dopamine in PBS as function of time. (d) Fluorescence spectra of PMP@MIL-88A (blue line) and dopamine release from PMP@MIL-88A (orange line) in PBS solution as function of time. λexc: 305 nm and λem: 327 nm. (e) DA (λexc: 305 nm) and (f) DQ (λexc: 390 nm) before (black line) and after the 10 hours (red line) required for the release test in PBS solution. | ||
Fig. 6c shows the bleaching dopamine emission in PBS as a function of time. The variation of fluorescence intensity was used to monitor the DA release from the PMP@MIL-88A and the subsequent DA oxidation. Fig. 6d shows the kinetic profile of dopamine release from PMP@MIL-88A (orange line) obtained by measuring the emission at 327 nm (λexc 305 nm). The fluorescence intensity of the empty carrier (PMP@MIL-88A) at the same wavelength emission over time (blue line) is also reported for comparison. The carrier reveals negligible variations in the emission which remain stable even after 1000 minutes. On the contrary, there is an increase of fluorescence intensity in the DA-PMP@MIL-88A system with a maximum at ca. 6 h (360 min). This represents a burst release of the drug, followed by a slower release of dopamine. A maximum DA concentration of 0.56 mg mL−1 was measured by using the calibration curve reported in Fig. 6a. After the burst release, a decrease in fluorescence intensity is observed, due to the oxidation of DA into quinone by-product (not emitting at the considered excitation wavelength) in the buffer solution. Further studies are needed to understand the mechanism of DA release. However, a tentative explanation of the release mechanism can be provided by assuming that the premature release of DA is due to the presence of the drug near the surface of the PMP@MIL-88A particles, while the delayed released can be attributed to the gradual drug diffusion from the porous carrier to the external medium and to a partial MIL-88A dissolution. It is also important to note that the PMP@MIL-88A can potentially allow for a stimuli-controlled drug release. In fact, it was shown that in presence of an alternate magnetic field, the magnetic core of the PMP@MIL-88A increases the local temperature of the materials and, therefore, the MOF pore size, facilitating the drug release.62
A similar trend, i.e. a burst release followed by a slower release of dopamine, was also observed in other nanoscale-designed drug carries, such as nanostructured silica.57 However, with respect to these systems, PMP@MIL-88A shows a 75%-reduced burst effect, which ends within 6 h, whilst a 24 h burst is observed in the nanostructured silica carries. This can be considered beneficial for a practical usage, reducing the pharmacological danger, increasing a longer-term delivery, achieving target effect, and improving the cost efficiency.63
The spectrofluorimetry also allows determining the concentration of DA and DQ before (black line) and after 22 h (red line) the dopamine release process. (Fig. 6e–f, λexc/em: 305/327 nm for DA; λexc/em: 390/470 nm for DQ). The amount of dopamine released by 1.14 mg of DA-PMP@MIL-88A systems, was calculated as 3.65 μM. This value exceeds by 3 orders of magnitude the basal level of dopamine, as determined in the brain of rats by using microdialysis technique, which is in the scale of nanomoles per liter.64–66 The DA concentration released by DA-PMP@MIL-88A, therefore, is potentially sufficient to preserve the basal level of dopamine in rat brain. The cytotoxicity of different concentrations (10, 20, 40 μg mL−1) and generations (1st, 2nd, 3rd) of PMP@MIL-88A and DA-PMP@MIL-88A were evaluated in vitro by assessing the viability of PC12 neuronal cell models with the trypan blue assay (Fig. 7).67 The free DA used to treat the PC12 cell can be considered as the positive control and its amount (0.87 μM, 1.75 μM, and 3.5 μM) was designed considering the highest amount 0.56 mg mL−1 (3.5 μM) of DA released in PBS solution. The concentration of DA used in the cells studies was calculated considering the maximum of DA released in the in vitro experiment (Fig. 6d).
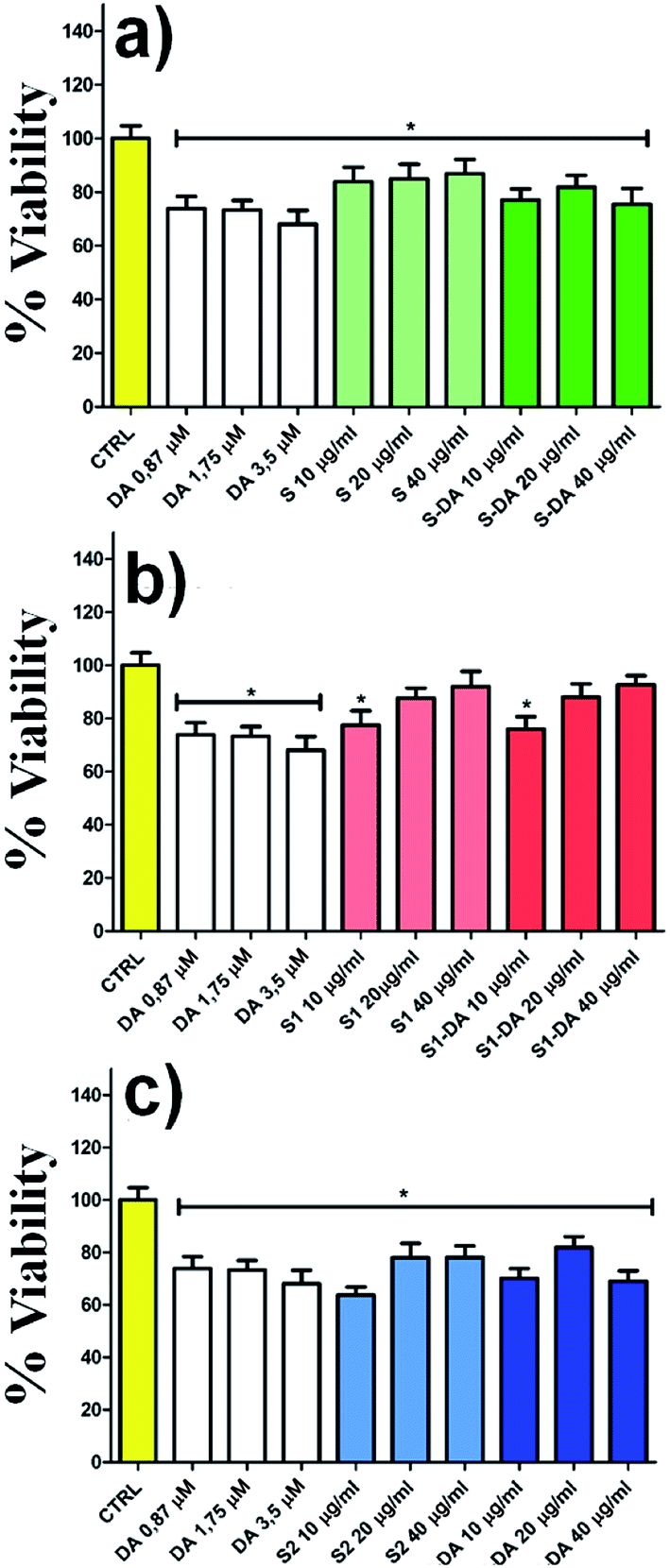 | ||
| Fig. 7 The effect of PMP@MIL-88A and DA-PMP@MIL-88A (10, 20, 40 μg mL−1) at different generations (a) 1st, (b) 2nd, (c) 3rd on viability of PC12 for 24 h evaluated by trypan blue assay. P-value < 0.05 vs. control group. Legend: S= 1st generation, S1 = 2nd generation, S2 = 3rd generation, suffix-DA = loaded with dopamine. | ||
The bar graphs in Fig. 7a–b show that there are no toxic effects on PC12 treated with 1st (green bar) and 2nd (red bar) growth of PMP@MIL-88A respectively, even when loaded with dopamine (DA-PMP@MIL-88A). Conversely, the 3rd growth samples (Fig. 7c, blue bar) of PMP@MIL-88A and DA-PMP@MIL-88A, exhibit a limited toxicity. This is an indication of the good biocompatibility of the PMP@MIL-88A delivery system.
Nearly 100% of cells viability was reached using the 2nd growth of PMP@MIL-88A at a concentration of 40 μg mL−1.
The effect of the dopamine-loaded PMP@MIL-88A particles on cellular dopamine levels was evaluated on PC12 cells by High-Pressure Liquid Chromatography Electrochemical detection (HPLC-EC). This technique can determine the cellular compartment of the released dopamine, by measuring the extracellular and the intracellular DA concentration. The amount of DA was evaluated using the optimized sample, that is the 2nd growth PMP@MIL-88A loaded with 40 μg mL−1 of DA. Fig. 8a reports the extracellular DA measured after 2, 4, 6 and 24 h. In both PMP@MIL-88A and DA-PMP@MIL-88A systems, DA levels are comparable to basal DA concentration in the control. The amount of DA released in the first 24 h in the extracellular environment cannot be detected most likely due to multiple chemical reactions occurring in the cell medium. PC12 cells can methylate DA by catechol O-methyltransferase (COMT) to 3-methoxytyramine (3-MT). Furthermore, monoamine oxidase (MAO) can oxidize cytoplasmic DA to 3, 4-dihydroxyphenylacetate (DOPAC), which COMT may further convert to homovanillate (HVA).68
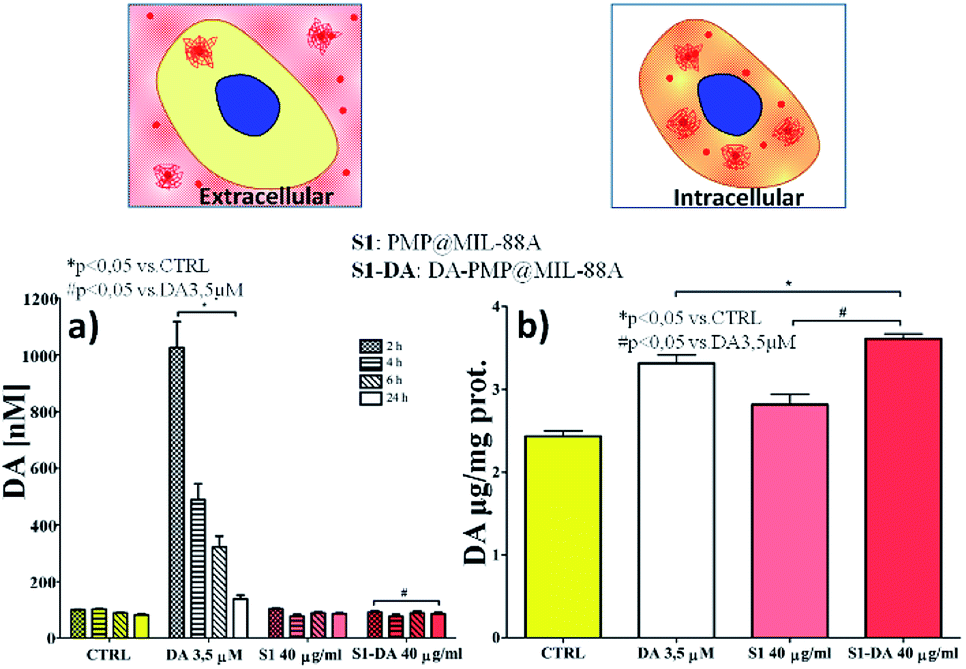 | ||
| Fig. 8 (a) Effect of DA (0.664 μg mL−1), PMP@MIL-88A and DA-PMP@MIL-88A (DA loading solution: 40 μg mL−1) on a extracellular levels of dopamine in PC12 cells after 2, 4, 6, and 24 hours exposure, and (b) intracellular levels of dopamine in PC12 cells after 24 hours exposure. | ||
A complete in situ monitoring of all these chemical compounds would allow a quantitative determination of the DA oxidation pathways; this is however beyond the scope of the present article. On the other hand, as depicted in Fig. 8b, the amount of intracellular dopamine assessed after 24 h of DA-PMP@MIL-88A treatment (red bar) is higher than the basal level in the control group and slightly higher than the group treated with only DA 0.66 μg mL−1 (white bar). Furthermore, the treatment with unloaded PMP@MIL-88A did not cause any change in the extracellular and intracellular amount of DA, confirming the MOF biocompatibility.
In summary, the experimental data indicate an increase of DA inside the cells when the PMP@MIL-88A composites are incubated with PC12 model cells. This confirms that PMP@MIL-88A composites can be used to enable or improve DA delivery. The capability of MOF to protect dopamine from the oxidation process is well supported by the experimental evidence. No dopamine release in the extracellular environment was detected, thus avoiding side effects related to quinone species formation. The intracellular level of dopamine released by PMP@MIL-88A carrier is very similar to the basal level shown in the control.
Experimental
PMP@MIL-88A (Fe) carrier synthesis
The PMP@MIL-88A carriers were obtained by an in situ synthesis of MIL-88A (Fe) metal organic framework on the surface of commercial carboxyl-functionalized polystyrene coated magnetite particles (PMP) (CMX-40-10, 3.0–6.0 μm, 1.58 g mL−1, Spherotech) through repetitive growth cycles. First, 0.30 g (0.0025 mol) of fumaric acid (C4H4O4, ≥99.0%, Sigma Aldrich) were dissolved in 50 mL of deionized water (DI) and left under stirring in water-bath until a homogeneous solution was obtained. 0.68 g (0.0025 mol) of iron(III) chloride hexahydrate (FeCl3·6H2O, 97%, Fluka) were subsequently added under stirring, and soon after 2 mL of PMP particles were added. The solution was left under stirring at room temperature for 72 h by using a rotor stirrer. This 1st generation PMP@MIL-88A particles were washed three times with 50 mL of water using a magnet and dispersed in 2 mL of water.The further growth cycles were obtained as follows; 0.68 g (0.05 mol) of FeCl3·6H2O were dissolved in 50 mL of DI water (solution A). In a separate vial, 0.30 g (0.05 mol) of C4H4O4 was dissolved in 50 mL of DI water (solution B) and left under stirring in a water-bath until a homogeneous solution was obtained. 5 mL of solution A was subsequently mixed with 5 mL of solution B and 0.32 mL of the first generation PMP@MIL-88A particles were then added. The reaction mixture was left in a thermal bath at 70 °C for half an hour. The obtained second generation PMP@MIL-88A particles were washed three times with 50 mL of DI water, separated by a magnet, and eventually dispersed in 2 mL of water. The entire process was repeated four times until the fourth generation of PMP@MIL-88A particles was produced.
The nth-generation PMP@MIL-88A particles were dried at 60 °C under vacuum overnight. The amount and number of PMP particles in the MIL-88A core were evaluated by TGA analysis. The PMP@MIL-88A particles size and the MIL-88A crystal size were measured by scanning electron microscopy (SEM). Elemental distribution of C, O, and Fe in the PMP@MIL-88A particles was analysed by Energy Dispersive X-ray Analysis (EDX).
Dopamine loading/release of nth growth PMP@MIL-88A particles
Prior to loading with dopamine (DA), nth-generation PMP@MIL-88A powder was treated at 150 °C for 16 h in order to completely remove the solvent from the porous structure. 20 mg of dopamine hydrochloride (DA·HCl, Sigma Aldrich) together with 0.033 g PMP@MIL-88A particles of the desired generation were suspended in 1 mL ethanol (EtOH, 99.8% Fluka). The weight ratio between PMP@MIL-88A and DA was 5:3, considering MIL-88A as a flexible metal organic framework (MOF).68 The solution was left under stirring in the dark at 25 °C for 72 h. DA-PMP@MIL-88A particles were washed 3 times with water using a magnet to remove the excess unloaded DA and then dried at 60 °C for 12 hours.
In vitro dopamine release was achieved by soaking 10 mg of DA-PMP@MIL-88A in 0.57 mL of phosphate buffer solution (PBS 1 mM, pH 7.4, Sigma Aldrich) at room temperature. The DA kinetics release was measured continuously for 22 h by fluorescence spectrophotometry.
Fluorescence spectroscopy
Fluorescence spectra and continuous kinetics release were recorded using a “NanoLog” Horiba Jobin Yvon spectrofluorometers: 3D and 2D mapping were recorded with a 450 W xenon lamp as the excitation source. 2D maps were collected by using an excitation wavelength at 305 nm and acquiring the emission in the range of 300−700 nm for DA and exciting at 390 nm and acquiring the emission in the range of 400−700 nm for DQ.Scanning electron microscopy (SEM)
The surface morphologies of samples were observed using a field emission scanning electron microscope (FE-SEM; Merlin; Carl Zeiss Germany) with iridium sputtered coating.Thermogravimetric analysis (TGA)
Thermogravimetric (TG) curves of PMP, MIL-88A (Fe) and PMP@MIL-88A were measured using a thermogravimetric analyzer (Pyris 1 TGA, PerkinElmer) at a heating rate of 10 °C min−1 from 30 to 700 °C in air.Surface area analysis (BET)
Nitrogen sorption isotherms were measured on MIL-88A (Fe) MOF by the volumetric method using a Micromeritics 3Flex instrument. Solid samples with dopamine were prepared using the aforementioned method: 20 mg of DA. HCl was mixed with 33 mg of MOF in 1 mL of ethanol, shaken at 500 rpm in the dark for 24 hours, and dried overnight in vacuum at 60 °C. The samples with and without dopamine were transferred to pre-weighed analysis tubes and evacuated by heating at 100 °C under dynamic vacuum at 0.02 mbar for 16 h. For all isotherms, free space corrections measurements were performed with ultrapure helium. Surface area was calculated using the Brunauer–Emmett–Teller (BET) model. Analysed samples were re-weighed afterwards for the correct mass determination.Cell culture and cell viability assessment
All experiments were performed on PC12 cells derived from rat pheochromocytoma (PC12 ATC CCRL-1721 passage 12-25 PC12). Cells were maintained in an atmosphere of 5% CO2 and 95% air, at 37 °C in Dulbecco's modified Eagle's medium (DMEM/F-12, HEPES, no phenol red, Life Technologies), containing 10% horse serum (Life Technologies) and 5% fetal bovine serum (Life Technologies). Cells were exposed for 24 h, to increasing concentrations of 1st, 2nd, and 3rd generation PMP@MIL-88A (10, 20, 40 μg mL−1) with and without DA.For each experiment, 1 × 105 PC12 cells cm−2 were seeded and treated 24 h later. Experiments were done in triplicate. After treatment, cell viability was evaluated with trypan blue (0.4%, Sigma Aldrich) exclusion assay, based on the capability of viable cells to exclude the dye.
Chromatographic analysis
The determination of DA level was performed by High performance liquid chromatography with electro-chemical detection (HPLC-EC). Chromatographic analysis was done using an Alltech 426 HPLC pump equipped with a Rheodyne injector, column length 15 cm, internal diameter 64.6 mm, Alltech Adsorbosphere C18 5U, electrochemical detector Antec CU-04-AZ and Varian Star Chromatographic Workstation. The mobile phase contained citric acid 0.5 M (99.5%, Sigma Aldrich), sodium acetate 1.0 M (NaAc, 99%, Sigma Aldrich), ethyl-enediaminetetraacetic acid 12.5 mM (EDTA, 98.5%, Sigma Aldrich), methanol 10% (MeOH, 99.93%, Sigma Aldrich) and sodium octyl sulfate 650 mg (pH 3.0, 95%, Sigma Aldrich); the flow rate was 1.3 mL min−1.Statistical analysis
Data derived from all experiments with PC12 cells are represented as mean values with 95% confidence intervals and statistical significance between control and experimental groups; they were evaluated as significant when two-tailed P values were <0.05. Data were evaluated by One-Way ANOVA analysis of variance test using Graph-Pad Prism 5.0 software (GraphPad Software, Inc, San Diego, CA, USA).Conclusions
A MOF composite drug delivery carrier, produced by combining the magnetic properties of polymer-embedded iron oxide particles with the high surface area and biocompatibility of MIL-88A (iron(III) fumarate) MOF, was designed for controlled dopamine release. Up to four generations of composites were produced by repetitive growths of MIL-88A on PMP or PMP@MIL-88A cores. This approach allowed the fabrication of particles with controlled dimension and enhanced drug loading, whose release efficiency is higher than other systems (e.g. silica nanoparticles).The dopamine-releasing capacity of PMP@MIL-88A MOF both in PBS solution and in PC12 cell environment was also evaluated, and the low toxicity of the nanocomposite carrier verified. In addition, dopamine oxidation was not observed when loaded into the nanocomposite carrier. This could help to keep the concentration of neurotransmitter in Parkinson's patient close to that of a healthy person, mitigating disease symptoms. The experimental data indicate that dopamine can be released into the intracellular compartment using the PMP@MIL-88A carrier, thus avoiding side effects linked to its degradation in the extracellular environment. The effective drug release from PMP@MIL-88A is, therefore, expected to reduce the pharmacological danger by enabling increased targeting efficiency, enhanced long-term delivery, and improved cost efficiency.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Paul Jones is gratefully acknowledged for his support.Notes and references
- E. A. Garrett, Dialogues Clin. Neurosci., 2004, 6, 259–280 Search PubMed.
- T. Kageyama, M. Nakamura, A. Matsuo, Y. Yamasaki, Y. Takakura, M. Hashida, Y. Kanai, M. Naito, T. Tsuruo, N. Minato and S. Shimohama, Brain Res., 2000, 8, 79115–79121 Search PubMed.
- D. Merims and N. Giladi, Parkinsonism Relat. Disord., 2008, 14, 273–280 CrossRef PubMed.
- M. A. de Souza Silva, B. Topic, J. P. Huston and C. Mattern, Synapses, 2008, 62, 176–184 CrossRef PubMed.
- S. Di Gioia, A. Trapani, D. Mandracchia, E. De Giglio, S. Cometa, V. Mangini, F. Arnesano, G. Belgiovine, S. Castellani, L. Pace, M. A. Lavecchia, G. Trapani, M. Conese, G. Puglisi and T. Cassano, Eur. J. Pharm. Biopharm., 2015, 9, 4180–4193 Search PubMed.
- M. Dahlin, U. Bergman, B. Jansson, E. Bjork and E. Brittebo, Pharm. Res., 2000, 17, 737–742 CrossRef.
- G. Cuenca, H. Jiang, S. N. Hochwald, M. Delano, W. G. Cance and S. R. Grobmyer, Cancer, 2006, 107, 459–466 CrossRef PubMed.
- X. Si, J. Xi, J. Kim, Y. Zhou and H. Zhong, Respir. Physiol. Neurobiol., 2013, 186, 22–32 CrossRef PubMed.
- J. Xi, Z. Zhang and X. A. Si, Int. J. Nanomed., 2015, 10, 1211–1222 CrossRef PubMed.
- S. B. Yarragudi, R. Richter, H. Lee, G. F. Walker, A. N. Clarkson, H. Kumar and S. B. Rizwan, Carbohydr. Polym., 2017, 163, 216–226 CrossRef PubMed.
- W. Cai, C. C. Chu, G. Liu and Y. J. Wáng, Small, 2015, 11, 4806–4822 CrossRef PubMed.
- S. Wuttke, M. Lismont, A. Escudero, B. Rungtaweevoranit and W. J. Parak, Biomaterials, 2017, 123, 172–183 CrossRef PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, Angew. Chem., Int. Ed. Engl., 2006, 45, 5974–5978 CrossRef PubMed.
- F. Ke, Y. P. Yuan, L. G. Qiu, Y. H. Shen, A. J. Xie, J. F. Zhu, X. Y. Tian and L. D. Zhang, J. Mater. Chem., 2011, 21, 3843–3848 RSC.
- K. M. Taylor-Pashow, J. Della Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef PubMed.
- K. R. Deng, Z. Y. Hou, X. J. Li, C. X. Li, Y. X. Zhang, X. R. Deng, Z. Y. Cheng and J. Lin, Sci. Rep., 2015, 5, 1–5 Search PubMed.
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1–20 Search PubMed.
- T. S. Yarza, M. G. Marqués, R. Mrimi, A. Mielcarek, R. Gref, P. Horcajada, C. Serre and P. Couvreur, Angew. Chem., 2017, 129, 15771–15775 CrossRef.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef PubMed.
- M. Lismont, L. Dreesen and S. Wuttke, Adv. Funct. Mater., 2017, 27, 1–16 Search PubMed.
- J. Zhuang, C. H. Kuo, L. Y. Chou, D. Y. Liu, E. Weerapana and C. K. Tsung, ACS Nano, 2014, 8, 2812–2819 CrossRef PubMed.
- R. R. Salunkhe, C. Young, J. Tang, T. Takei, Y. Ide, . Kobayashia and Y. Yamauchi, Chem. Commun., 2016, 52, 4764–4767 RSC.
- S. Tai, W. Zhang, J. Zhang, G. Luo, Y. Jia, M. Deng and Y. Ling, Microporous Mesoporous Mater., 2016, 220, 148–154 CrossRef.
- F. R. Santos Lucena, L. C. C. de Araújo, M. d. D. Rodrigues, T. G. da Silva, V. R. A. Pereira, G. C. G. Militão, D. A. F. Fontes, P. J. Rolim-Neto, F. F. da Silva and S. C. Nascimento, Biomed. Pharmacother., 2013, 67, 707–713 CrossRef PubMed.
- S. Keskin and S. Kızılel, Ind. Eng. Chem. Res., 2011, 50, 1799–1812 CrossRef.
- A. Zimpel, T. Preiß, R. Röder, H. Engelke, M. Ingrisch, M. Peller, J. O. Rädler, E. Wagner, T. Bein, U. Lächelt and S. Wuttke, Chem. Mater., 2016, 28, 3318–3326 CrossRef.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- W. Chaikittisilp, N. L. Torad, C. Li, M. Imura, N. Suzuki, S. Ishihara, K. Ariga and Y. Yamauchi, Chem.–Eur. J., 2014, 20, 4217–4221 CrossRef PubMed.
- J. Tang and Y. Yamauchi, Nat. Chem., 2016, 8, 638 CrossRef PubMed.
- W. Zhang, X. Jiang, Y. Zhao, A. Carnè-Sànchez, V. Malgras, J. Kim, J. H. Kim, S. Wang, J. Liu, J. S. Jiang, Y. Yamauchi and M. Hu, Chem. Sci., 2017, 8, 3538 RSC.
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, ACS Nano, 2017, 11, 5293–5308 CrossRef PubMed.
- Y. Lei, Y. Sun, H. Zhang, L. Liao, S. T. Lee and W. Y. Wong, Chem. Commun., 2016, 52, 12318–12321 RSC.
- S. Wuttke, A. Zimpel, T. Bein, S. Braig, K. Stoiber, A. Vollmar, D. Müller, K. H. Talini, J. Schaeske, M. Stiesch, G. Zahn, A. Mohmeyer, P. Behrens, O. Eickelberg, D. A. Bölükbas and S. Meiners, Adv. Healthcare Mater., 2017, 6, 1–11 Search PubMed.
- C. M. Doherty, D. Buso, A. J. Hill, S. Furukawa, S. Kitagawa and P. Falcaro, Acc. Chem. Res., 2014, 47, 396–405 CrossRef PubMed.
- R. Ricco, L. Malfatti, M. Takahashi, A. J. Hill and P. Falcaro, J. Mater. Chem. A, 2013, 1, 13033–13045 RSC.
- P. Falcaro, R. Ricco, C. M. Doherty, K. Liang, A. J. Hill and M. J. Styles, Chem. Soc. Rev., 2014, 43, 5513–5560 RSC.
- M. R. Lohe, K. Gedrich, T. Freudenberg, E. Kockrick, T. Dellmannc and S. Kaskel, Chem. Commun., 2011, 47, 3075–3077 RSC.
- F. Ke, L. G. Qiu, Y. P. Yuan, X. Jiang and J. F. Zhu, J. Mater. Chem., 2012, 22, 9497–9500 RSC.
- F. Ke, L. G. Qiu and J. Zhu, Nanoscale, 2014, 6, 1596–1601 RSC.
- X. Zhao, S. Liu, Z. Tang, H. Niu, Y. Cai, W. Meng, F. Wu and J. P. Giesy, Sci. Rep., 2015, 5, 118491–118499 Search PubMed.
- F. Du, Q. Qin, J. Deng, G. Ruan, X. Yang, L. Li and J. Li, J. Sep. Sci., 2016, 39, 2356–2364 CrossRef PubMed.
- Y. Chen, Z. Xiong, L. Peng, Y. Gan, Y. Zhao, J. Shen, J. Qian, L. Zhang and W. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 16338–16347 CrossRef PubMed.
- Z. Xiong, Y. Ji, C. Fang, Q. Zhang, L. Zhang, M. Ye, W. Zhang and H. Zou, Chem.–Eur. J., 2014, 20, 1–8 CrossRef.
- R. Chowdhuri, D. Bhattacharya and S. K. Sahu, Dalton Trans., 2016, 45, 2963–2973 RSC.
- C. Malagelada and L. A. Greene in Parkinson's Disease, ed. R. Nass and S. Przedborski, Elsevier, New York, 1st edn, 2008, ch. 29, pp. 375–387 Search PubMed.
- A. Pinna, L. Malfatti, G. Galleri, R. Manetti, S. Cossu, G. Rocchitta, R. Migheli, P. A. Serra and P. Innocenzi, RSC Adv., 2015, 5, 20432–20439 RSC.
- J. Wong, A. Brugger, A. Khare, M. Chaubal, P. Papadopoulos, B. Rabinow, J. Kipp and J. Ning, Adv. Drug Delivery Rev., 2008, 60, 939–954 CrossRef PubMed.
- L. Pereswetoff-Morath, Adv. Drug Delivery Rev., 1998, 29, 185–194 CrossRef PubMed.
- F. Natalio, R. André, S. A. Pihan, M. Humanes, R. Wever and W. Tremel, J. Mater. Chem., 2011, 21, 11923–11929 RSC.
- K. Y. A. Lin, H. A. Chang and C. J. Hsu, RSC Adv., 2015, 5, 32520–32530 RSC.
- J. Zachary, H. Carpenter and E. E. Carpenter, Dalton Trans., 2014, 43, 12236–12242 RSC.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef PubMed.
- J. Wang, J. Wan, Y. Ma, Y. Wang, M. Puac and Z. Guanac, RSC Adv., 2016, 6, 112502–112511 RSC.
- X. H. Li, W. L. Guo, Z. H. Liu, R. Q. Wang and H. Liu, Appl. Surf. Sci., 2016, 369, 130–136 CrossRef.
- L. Wang, Y. Zhang, X. Li, Y. Xie, J. He, J. Yu and Y. Song, Sci. Rep., 2015, 5, 1–7 CrossRef.
- A. E. Sánchez-Rivera, S. Corona-Avendaño, G. Alarcòn-Angeles, A. Rojas-Hernández, M. T. Ramìrez-Silva and M. A. Romero-Romo, Spectrochim. Acta, Part A, 2003, 59, 3193–3203 CrossRef.
- M. Bisaglia, M. E. Soriano, I. Arduini, S. Mammi and L. Bubacco, Biochim. Biophys. Acta, 2010, 1802, 699–706 CrossRef PubMed.
- S. A. Factor, Neurotherapeutics, 2008, 5, 164–180 CrossRef PubMed.
- S. J. Clarke, C. A. Hollmann, Z. Zhang, D. Suffern, S. E. Bradforth, N. M. Dimitrijevic, W. G. Minarik and J. L. Nadeau, Nat. Mater., 2006, 5, 409–417 CrossRef PubMed.
- H. Y. Wang, Y. Sun and B. Tang, Talanta, 2002, 57, 899–907 CrossRef PubMed.
- E. Guisasola, A. Baeza, M. Talelli, D. Arcos, M. Moros, J. M. de la Fuente and M. Vallet-Regì, Langmuir, 2015, 31, 12777–12782 CrossRef PubMed.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef PubMed.
- G. Bazzu, G. Rocchitta, R. Migheli, M. D. Alvau, M. Zinellu, G. Puggioni, G. Calia, G. Mercanti, P. Giusti, M. S. Desole and P. A. Serra, Brain Res., 2013, 1538, 159–171 CrossRef PubMed.
- G. Bazzu, A. Biosa, D. Farina, Y. Spissu, S. Dedola, G. Calia, G. Puggioni, G. Rocchitta, R. Migheli, M. S. Desole and P. A. Serra, Talanta, 2011, 85, 1933–1940 CrossRef PubMed.
- K. C. Chen, J. Neurochem., 2005, 92, 46–58 CrossRef PubMed.
- W. Strober, Curr Protoc Immunol, 2001, vol. 21, 3B, pp. A.3B.1–A.3B.2 Search PubMed.
- Z. Qi, G. W. Miller and E. O. Voit, PLoS One, 2008, 3, 1–10 Search PubMed.
- D. Cunha, M. B. Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaïm, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767–2776 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04969f |
| This journal is © The Royal Society of Chemistry 2018 |