
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
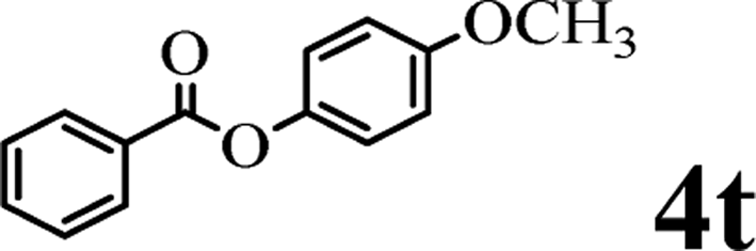
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransesterification of (hetero)aryl esters with phenols by an Earth-abundant metal catalyst†
Jianxia Chen,
E. Namila,
Chaolumen Bai,
Menghe Baiyin,
Bao Agula and
Yong-Sheng Bao *
*
College of Chemistry and Environmental Science, Inner Mongolia Key Laboratory of Green Catalysis, Inner Mongolia Normal University, Hohhot, 010022, China. E-mail: sbbys197812@163.com; Tel: +86-471-4392442
First published on 13th July 2018
Abstract
Readily available and inexpensive Earth-abundant alkali metal species are used as efficient catalysts for the transesterification of aryl or heteroaryl esters with phenols which is a challenging and underdeveloped transformation. The simple conditions and the use of heterogeneous alkali metal catalyst make this protocol very environmentally friendly and practical. This reaction fills in the missing part in transesterification reaction of phenols and provides an efficient approach to aryl esters, which are widely used in the synthetic and pharmaceutical industry.
Introduction
Esters are important classes of chemicals that are widely found in, polymers, agrochemicals, natural products, and biological systems and they have found wide application as versatile building blocks in organic synthesis.1 Accordingly, the development of new methodologies to access these valuable molecules continues to be of great importance in synthetic organic chemistry. Transesterifications as classic organic reactions are widely applied to the preparation of esters in organic synthesis and chemical industry.2 Transesterification is a process where an ester is transformed into another through interchange of the alkoxy moiety under the catalysis of acids or bases (see Scheme 1a). However, lower reactivity of the ester carbonyl functionality and the reversible nature of the reaction equilibrium resulting in partial conversion are two major limitations associated with this class of reaction.3 In recent years, the main efforts have been devoted to developing Lewis acids,4 organic and inorganic bases5 and N-heterocyclic carbene (NHC)6 as efficient catalysts for improvement of the transesterifications of esters with alcohols. Compared with alcohols, the transesterifications of esters with phenols are more challenging due to the weaker nucleophilicity of phenols.7 Phenols are particularly attractive starting materials because most of them are readily available from fuel and biomass and the global annual production of phenols has grown to millions of tons.8 Phenols and their salt derivatives have been successfully employed as substitutes for aryl halides in many transition-metal-catalyzed transformations,9 but there are only a few reports of ester based acylation of phenols. Furthermore, the substrate scopes of these methods were limited to vinyl acetates3 or specific lactones.7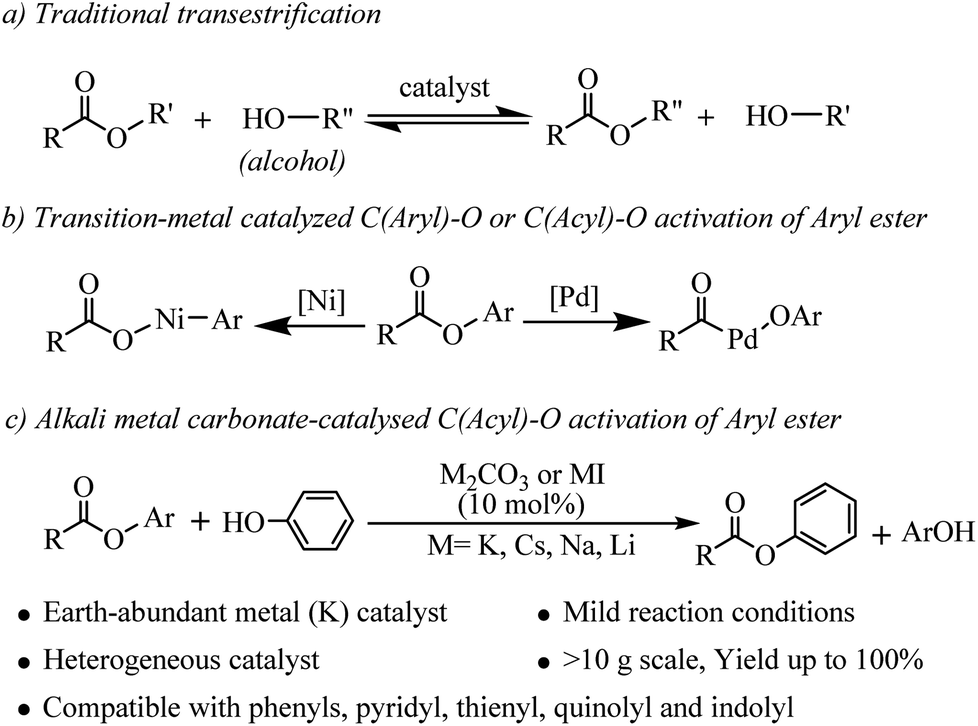 | ||
| Scheme 1 C–O bond activation of esters. | ||
Recently, aryl esters have gained significant interest as new aryl-coupling partners via nickel catalyzed C(aryl)–O cleavage (see Scheme 1b).10 Actually, the bond dissociation energy (BDE) of the C(acyl)–O bond is more lower than C(aryl)–O bond in aryl esters.10h But, catalyzed by transition-metal, there are challenges associated with C(acyl)–O bond cleavage because of decarbonylation phenomenon.11 In 2001, Yamamoto reported the first example of palladium catalyzed coupling reaction of aryl esters with organoboron compounds via C(acyl)–O cleavage with carbonyl retention. However, the reaction was limited to electronically activated esters, such as perfluoroaliphatic carboxylic esters.12 Chatani reported the palladium catalyzed coupling reaction of 2-pyridyl esters with organoboron compounds.13 More recently, Newman developed an NHC-based Pd catalyst which can catalyzed Suzuki–Miyaura coupling of phenyl esters or cross-coupling between phenyl esters and aniline.14 Our previous work has confirmed that aryl esters can generate an activated acyl intermediate and perform an amidation reaction with tertiary amines or formamides to form amides under Pd or Au catalyzed conditions.15 As concluded: (1) these transformations rely on catalysts derived from rare and expensive precious metals, such as Pd, Ni and Au, which can be a significant limitation, particularly for large-scale syntheses; (2) the pyridine or benzene ring serves as a directing group for C–O cleavage of the ester and that coordination of the nitrogen atom to the catalyst is favorable for the catalyzed reaction; (3) whether in coupling reaction via C(aryl)–O activation or via C(acyl)–O activation, the excess base or alkali metal halides was needed but the effect of these alkali metal salts on C–O bond cleavage is not clear.
More presently, alkali metal salts or bases have been proven to be efficient and competitive catalysts for many reactions, such as C–H bond silylation,16 Heck type coupling,17 intramolecular anionic cyclization,18 hydrogenation of aromatic aldehydes,19 aminobromination,20 arylsulfonylation,21 C–H hydroxylation of carbonyl compounds22 etc. Unlike general base catalyzed traditional reactions, only catalytic amount of alkali metal salt or base was used as catalyst in these reactions. These exciting results prompted us to reconsider the use of Earth-abundant alkali metal bases or salts for the catalytic C(acyl)–O activation of aryl esters.
Herein, we disclose a mild, efficient, and general Earth-abundant alkali metal species catalyzed transesterification of aryl or heteroaryl esters with phenols (relatively non-nucleophilic oxygen species) which is a challenging and under developed transformation (see Scheme 1c). The alkali metal catalyst is compatible with a range of functional groups including pyridyl, thienyl, quinolyl and indolyl, making this novel transesterification method immediately applicable to medicinal chemistry and alkaloid natural product synthesis.
Results and discussion
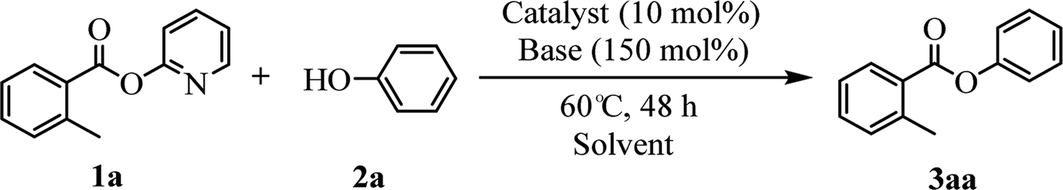
Our evaluation of the reaction system was initiated from the palladium catalyzed transesterification of pyridin-2-yl 2-methylbenzoate 1a with phenol 2a under air in the presence of K2CO3 as base at 60 °C. To our delight, the expected transesterifying product phenyl 2-methylbenzoate 3aa was isolated in 99% yield (Table 1, entry 1). We were surprised to find that the transesterification reaction also took place efficiently with just a catalytic amount (10 mol%) of K2CO3 and the desired product 3aa was obtained quantitatively (entry 2). The Pd, Au, Ni, Cu, Fe, Ru, Ir and Ag content in the K2CO3 and standard reaction solution was less than δ = 0.2 ppm in each case (ICP-MS analysis; see the ESI†), which indicated that this transesterification reaction is promoted by K2CO3 itself rather than catalyzed by trace metal impurities. All potassium salts and Cs2CO3 could catalyze the transesterification reaction smoothly (entries 2–6). As with similar many reported alkali-metal catalyzed reaction,16–22 it is becoming increasingly apparent that the identity of the base catalyst in this reaction is unusually important and extends far beyond a simple consideration of basicity. Reactions in the presence of other bases as potential catalysts, such as NaOH, Li2CO3, Ca(OH)2 and strongly basic amines TEA, did not proceeded (entries 7–10). Aim to confirm if this transformation is solubility controlled, NaOH, Li2CO3, Ca(OH)2 and TEA were chosen for reacting in DMSO (entries 11–14). The experiment results showed that NaOH, and Li2CO3 could catalyze the transesterification reaction to give desired product in DMSO, whereas Ca(OH)2 and TEA could not. One could imagine that the transformation just is a base catalyzed transesterification, but non-basic alkali metal salts, including KI, CsI and NaI, could catalyze the transesterification reaction smoothly (entries 15–17). Therefore, the alkali metal ion may be involved in reaction process via C(acyl)–O activation of aryl esters. Notably, a reaction in the absence of catalyst did not proceed (entry 18). The use of other solvents, such as DMF, DMSO and H2O, led to the formation of 3aa in lower yields (entries 19–21). Both a decrease and increase in the reaction temperature (40 °C and 80 °C) reduced the yield of 3aa (see the ESI†).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.17 mmol), catalyst (10 mol%), solvent (2 mL), 60 °C, 48 h.b Isolated yield.c React at 120 °C, 24 h. | ||||
| 1 | Pd/γ-Al2O3 | K2CO3 | 1,4-Dioxane | 99 |
| 2 | K2CO3 | — | 1,4-Dioxane | Quant |
| 3 | K3PO4 | — | 1,4-Dioxane | 81 |
| 4 | KOtBu | — | 1,4-Dioxane | 79 |
| 5 | KOH | — | 1,4-Dioxane | 70 |
| 6 | Cs2CO3 | — | 1,4-Dioxane | 93 |
| 7 | NaOH | — | 1,4-Dioxane | NP |
| 8 | Li2CO3 | — | 1,4-Dioxane | NP |
| 9 | Ca(OH)2 | — | 1,4-Dioxane | NP |
| 10 | NEt3 | — | 1,4-Dioxane | NP |
| 11 | NaOH | — | DMSO | 48 |
| 12 | Li2CO3 | — | DMSO | 77 |
| 13 | Ca(OH)2 | — | DMSO | NP |
| 14 | NEt3 | — | DMSO | NP |
| 15 | KI | — | DMSO | 67c |
| 16 | CsI | — | DMSO | 55c |
| 17 | NaI | — | DMSO | 53c |
| 18 | — | — | 1,4-Dioxane | NP |
| 19 | K2CO3 | — | DMF | 93 |
| 20 | K2CO3 | — | DMSO | 94 |
| 21 | K2CO3 | — | H2O | 78 |
The above results indicate that K2CO3 is a competent catalyst for the transesterification reaction. We next proceeded to evaluate the scope of pyridin-2-yl ester partner under the reaction conditions I (Table 1, entry 2) and found that benzoates, naphthoate, thiophene-2-carboxylate, 1H-indole-2-carboxylate and cinnamate readily undergo the transesterification providing phenyl esters 3ba–3ta in moderate to high yields (see Scheme 2). Benzoates with various substitution patterns at the acyl moiety were screened. The reaction proceeded smoothly not only for the benzoates bearing an electron-donating group (Me, nBu, tBu, OMe) but also for those substrates having electron-withdrawing substituents (F, Cl, I, NO2, CN) on the para-, meta- or ortho-position. Thereinto, some substrates including pyridin-2-yl 3-chlorobenzoate 1i, pyridin-2-yl 4-cyanobenzoate 1m, pyridin-2-yl 3-nitrobenzoate 1o and pyridin-2-yl 2-chloro-4-nitrobenzoate 1p, showed low activity and 20 mol% of the catalyst and higher temperature (reaction conditions II) were required for reaction. The apparent electronic effect or steric effect was not founded in these benzoates. Using pyridin-2-yl thiophene-2-carboxylate 1r as a heteroaromatic carboxylate resulted in desired phenyl thiophene-2-carboxylate 3ra in excellent yield. But hardly conditions (reaction conditions II) were required for pyridin-2-yl 1H-indole-2-carboxylate 1s to furnish the products. Next, we consider if aliphatic acid ester is suitable for the transesterification reaction. The experiment results showed that saturated aliphatic acid ester, pyridin-2-yl 3-phenylpropanoate 1u, could not perform the transesterification reaction but unsaturated aliphatic acid ester 1t performed the reaction expediently albeit in comparatively lower yield under the conditions II.
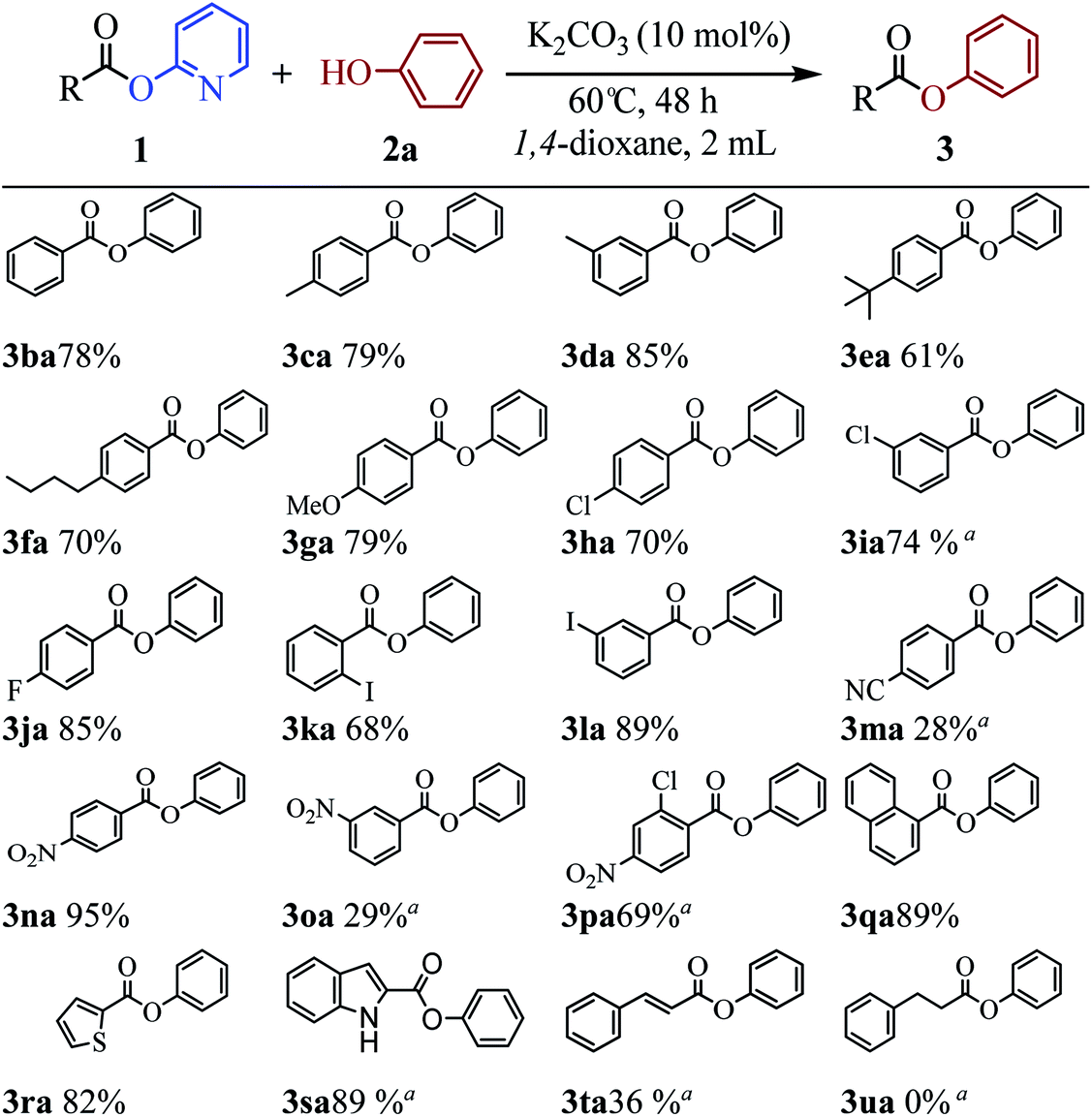 | ||
| Scheme 2 K2CO3-catalyzed transesterification of various pyridin-2-yl esters 1 with phenol 2a. The reaction conditions I: 1 (0.1 mmol), 2a (0.17 mmol), K2CO3 (10 mol%), 1,4-dioxane (2 mL), 60 °C, 48 h; yields shown are for the isolated products. aReaction conditions II: K2CO3 (20 mol%), 120 °C. | ||
The scope of the phenol partner was likewise substantial, affording products including phenyl esters 3ab–3ak, naphthyl esters 3al, 3am and quinolinyl ester 3an under the reaction conditions I (see Scheme 3). Many phenols, including 4-methylphenol 2b, 4-chlorophenol 2c, 2,3-dichlorophenol 2k and naphthalen-2-ol 2m, react with 1a to achieve quantitatively transesterification. Substrates containing sensitive functional groups such as 4-iodophenol 2d and 4-hydroxybenzaldehyde 2h are tolerated without any undesired side reactions. These results indicated that pyridyloxy is a good leaving group for the potassium catalyzed transesterification reaction.
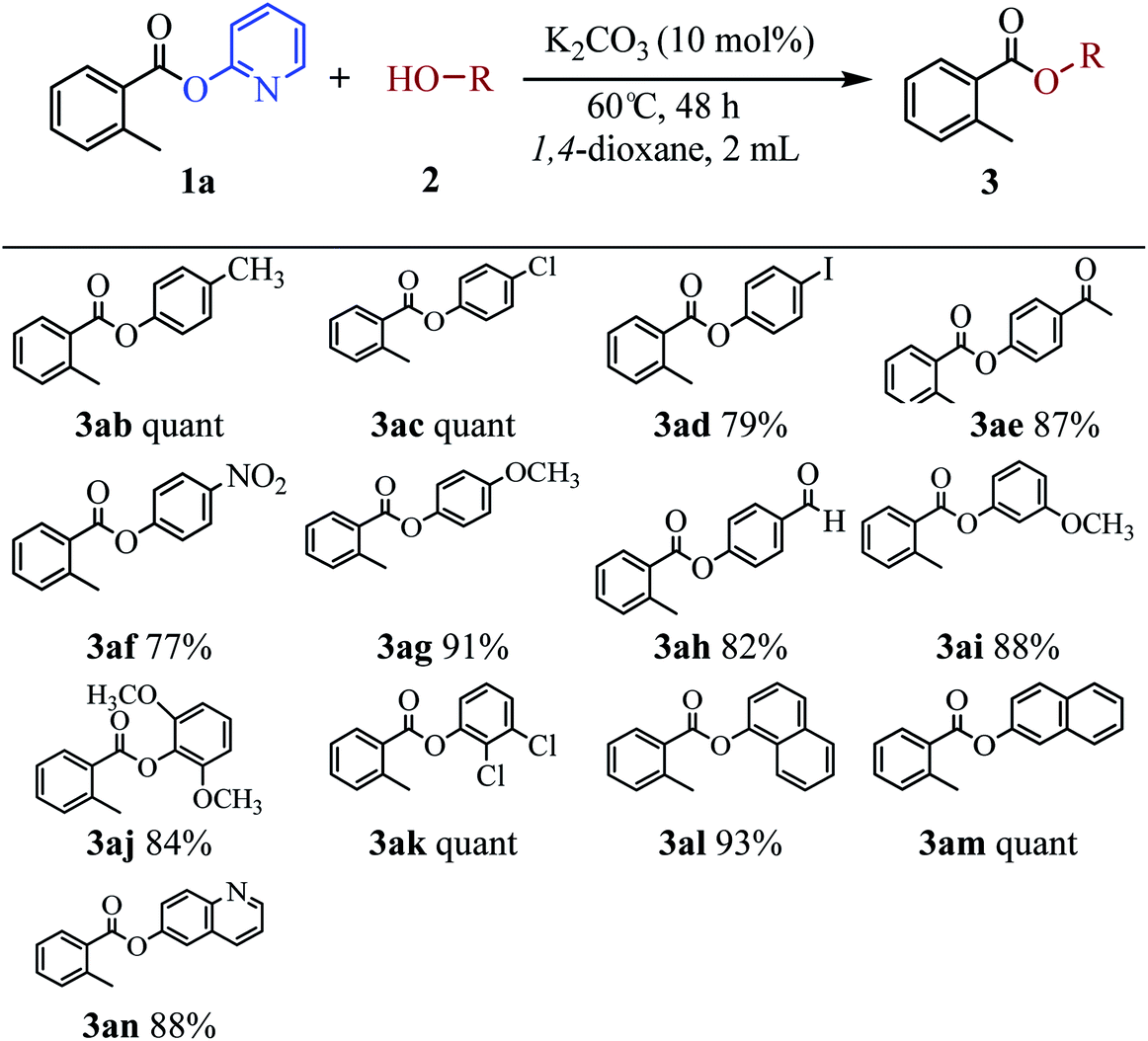 | ||
| Scheme 3 K2CO3-catalyzed transesterification of various phenols 2 with pyridin-2-yl 2-methylbenzoate 1a. The reaction conditions I: 1 (0.1 mmol), 2a (0.17 mmol), K2CO3 (10 mol%), 1,4-dioxane (2 mL), 60 °C, 48 h; yields shown are for the isolated products. | ||
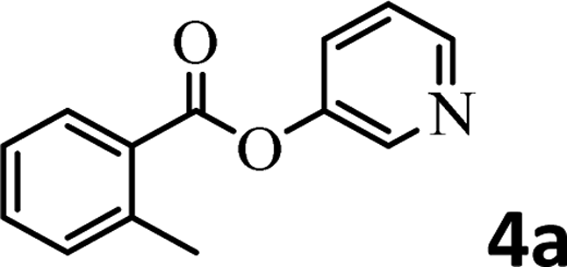
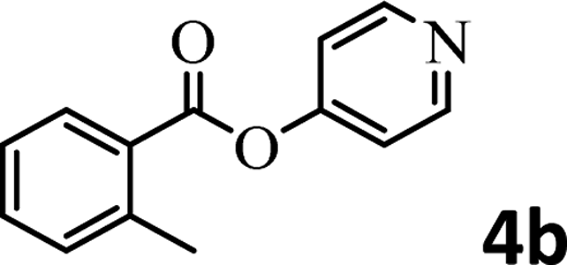
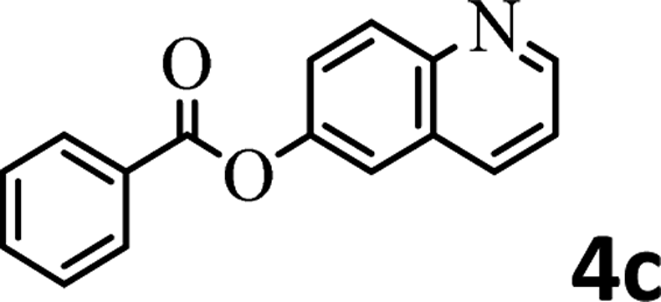
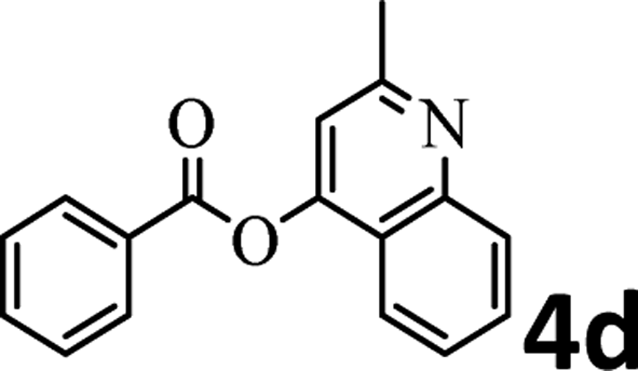
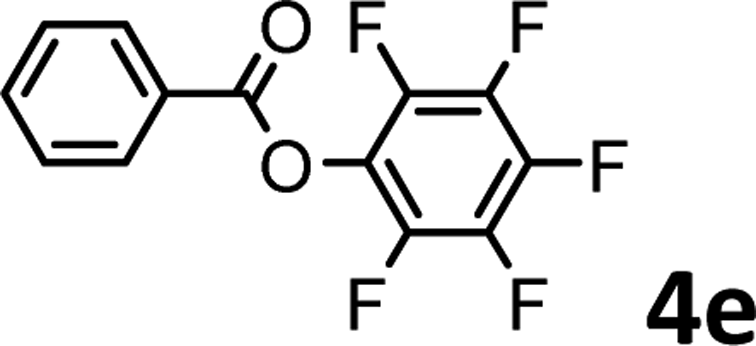
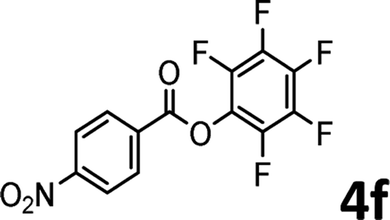
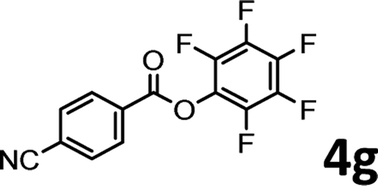
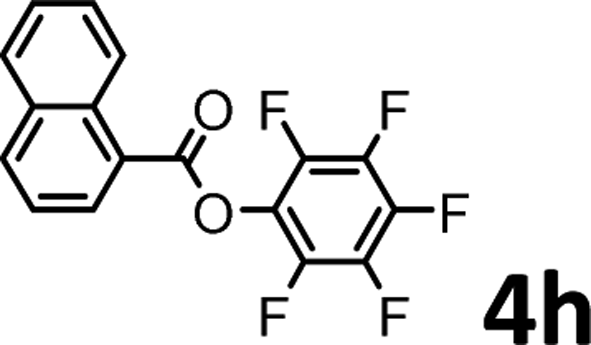
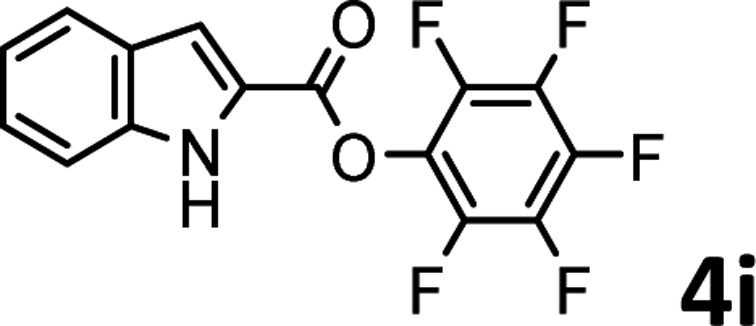
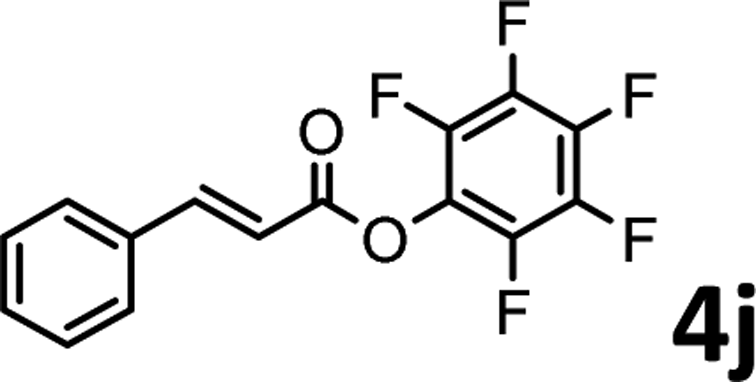
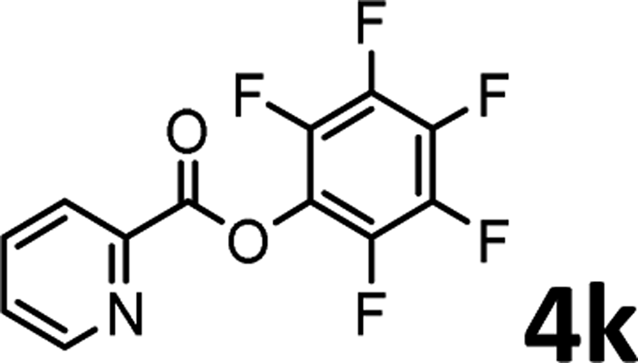
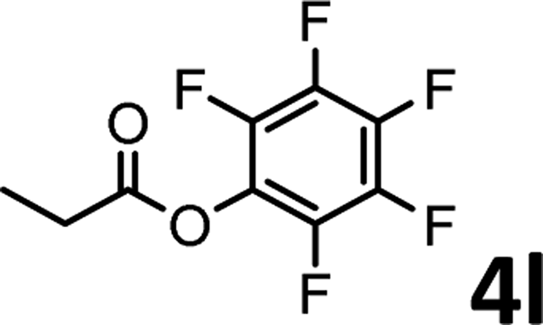
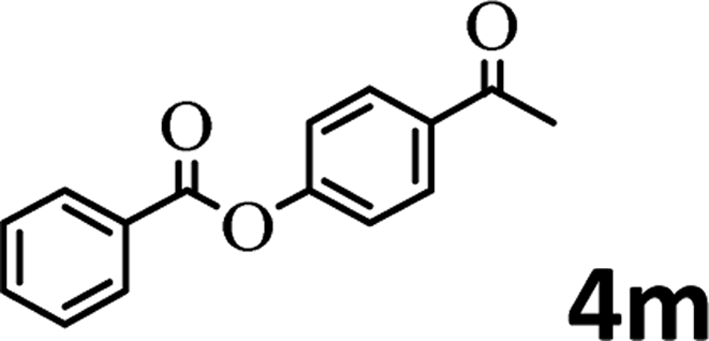
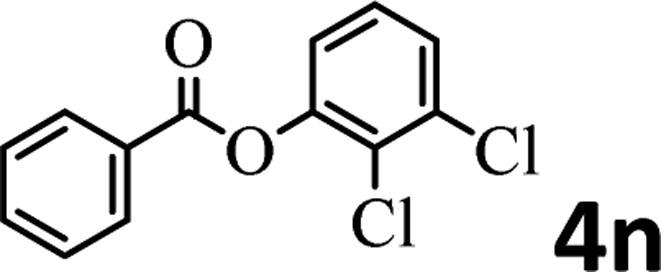
To demonstrate the synthetic utility of the method, other aryl or heteroaryl esters were used instead of pyridin-2-yl esters to react with phenols. In order to verify that the ortho-directed effect of nitrogen on C(acyl)–O bond cleavage of ester, pyridin-3-yl 2-methylbenzoate 4a and pyridin-4-yl 2-methylbenzoate 4b were used to react with phenol 2a (see Table 2, entries 1 and 2). Under the condition II, desired product 3aa was isolated in the yields of 55% and 84%, respectively. These results indicated there does not exist the chelating effect of nitrogen with potassium and electron-withdrawing effect (para- > meta-) of nitrogen is favorable for the C(acyl)–O bond cleavage. When quinolin-6-yl benzoate 4c or 2-methylquinolin-4-yl benzoate 4d was used as substrate, the same transesterification product 3ba was isolated in the yield of 37% or 83%, respectively (entries 3 and 4). The tremendous difference of yields further confirmed that electron-deficient heteroaryloxy group is easily exchanged for phenoxy group. What is more exciting is that, catalyzed by K2CO3, various perfluorophenyl esters 4e–4j performed the transesterification reaction with phenol 2a to give the desired esters in excellent yields (entries 5–10). Our experiments indicated that electron-withdrawing groups on the acyl fragment of the aryl ester 4f, 4g are unbeneficial to this reaction (entries 6 and 7). Interestingly, starting from perfluorophenyl picolinate 4k, which is more active in palladium catalyzed C(acyl)–O activation reactions in our previous work,15 the transesterification reaction was inhibited (entry 11). The presence of nitrogen in this position likely shuts down reactivity due to presumed coordination to potassium. It is believed that perfluorophenyl group is a very strong electron-withdrawing group and facilitate the C(acyl)–O bond cleavage in the presence of base,14b but when perfluorophenyl propionate 4l was used as substrate the transesterification reaction did not proceed (entry 12). This was also the case in the reaction of pyridin-2-yl 3-phenylpropanoate 1u (see Scheme 2, 3ua). These examples indicated that the acyl fragment of the aryl ester strongly influenced the reaction outcome and what kind of picolinate or aliphatic acid esters is not suitable for the new transesterification reaction. Further experiment confirmed that 4-acetylphenyl benzoate 4m and 2,3-dichlorophenyl benzoate 4n as well as perfluorophenyl benzoate (‘activated ester’)14b performed the transesterification reaction expediently to give the 3ba in moderate yields (entries 13 and 14). These results further expanded the substrates scope of new transesterification reaction to all of aryl esters. Alkyl esters, even strong electron-deficient alkyl ester-2,2,2-trifluoroethyl benzoate 4o, could not react with 2a to afford desired product (entry 15). The result indicated that (hetero)aromatic ring in alkoxy part of ester 1 is necessary in the novel transesterification reaction and could control the C(acyl)-O bond cleavage with the aid of potassium.
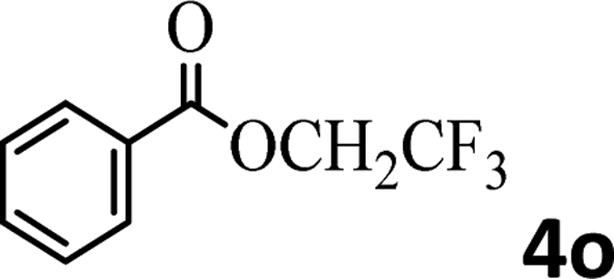
In order to get a better understanding about the essence of this transesterification reaction, we surveyed the reactivity of various substituted phenyl benzoates with various phenols under reaction conditions II (Table 3). Because the reactivity and stability of C(acyl)–X species can be crudely approximated by the pKa of their leaving groups, we refer to the pKa data (spectrophotometric method, H2O) of corresponding phenols. As shown in Table 3, the electron-withdrawing effect let to lower pKa value of phenols and the reactivity of esters is higher at stronger electron-withdrawing effect in the (O–Ar) fragment of the aryl esters. The 4-nitrophenyl benzoate showed the best reaction activity in the transesterification with all of phenols. Conversely, the transesterifications of other benzoates with 4-nitrophenol were difficult. Although the electron-withdrawing effect of 4-nitrophenyl is lower than perfluorophenyl (perfluorophenol, pKa = 5.49), 4-nitrophenyl benzoate react with phenol to achieve quantitative transesterification. Interestingly, although naphthalen-2-ol and phenol have nearly equal pKa value, naphthalen-2-yl benzoate could also react with phenol to give the transesterification product in 89% yield, while 4-cholorophenyl benzoate could not react with phenol. And, when naphthalen-2-yl benzoate react with 4-methoxyphenol, the yield of desired product is much higher than 4-nitrophenyl benzoate's and reach to quantitative yield. These results could not be explained by a simple nucleophilic substitution reaction process. In addition, phenoxy group of phenyl benzoate could be exchanged for 4-methoxyl phenol to give the transesterification product in 54% yield.
|
|||||
|---|---|---|---|---|---|
| Ester | Phenol | ||||
|
|
|
|
|
|
| pKa = 7.17b | pKa = 9.42 | pKa = 9.93 | pKa = 9.99 | pKa = 10.21 | |
| a Reaction conditions: ester (0.1 mmol), phenol (0.17 mmol), K2CO3 (20 mol%), 1,4-dioxane (2 mL), 120 °C, 48 h, isolated yield.b pKa data (spectrophotometric method, H2O) was cited from Internet Bond-energy Databank (iBonD), Home Page. http://ibond.chem.tsinghua.edu.cn. | |||||
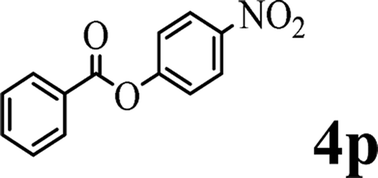 |
— | ✓ (81%) | ✓ (79%) | ✓ (quant) | ✓ (54%) |
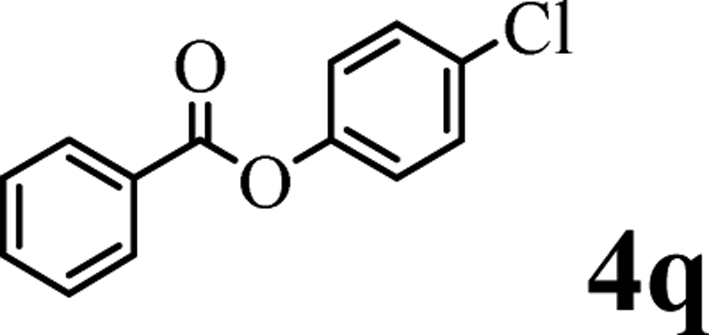 |
Trace | — | <20% | ✗ | ✓ (68%) |
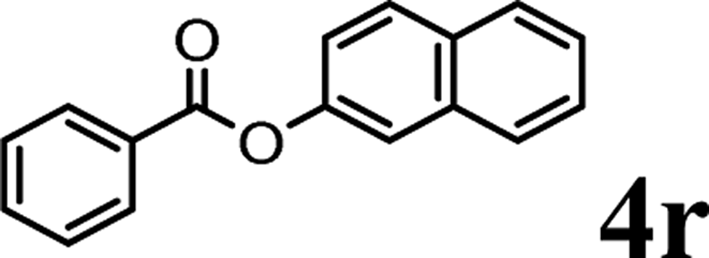 |
Trace | <20% | — | ✓ (89%) | ✓ (quant) |
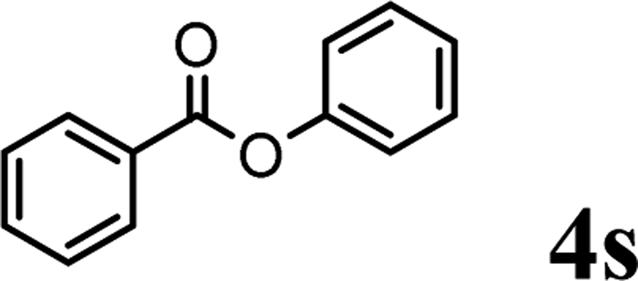 |
Trace | ✗ | Trace | — | ✓ (54%) |
 |
Trace | ✗ | ✗ | Trace | — |
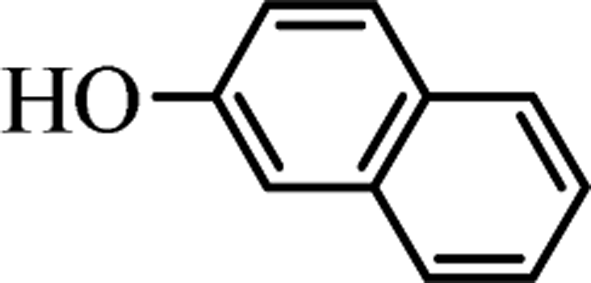
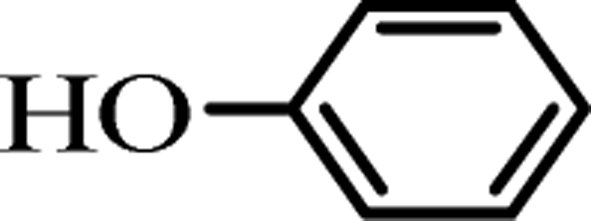
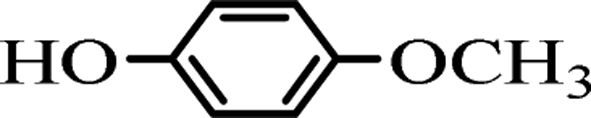
To investigate the practical application of this newly developed transesterification in organic synthesis, we conducted a 10 gram-scale reaction of 1a (10 g) with 2a in the presence of only 10 mol% of the K2CO3 catalyst, and isolated the desired product 3aa in 99% yield. As we can see, even though the reaction scale was magnified up to 470 times, ideal synthetically yields could be still obtained.
The core issue of alkali metal bases-catalyzed reaction mechanism is if the alkali metal ion participates in reaction process by means of coordination effect as same as transition metals. Houk and coauthors suggest two plausible mechanism – one ionic involving K+ and tBuO− ions, the other a neutral heterolytic mechanism involving the [KOtBu]4 tetramer – for KOtBu catalyzed C–H silylation of heteroarenes, based on a combination of empirical evidence and DFT calculations.23 But the mechanistic details of the potassium carbonate-catalyzed transesterification are not well understood at this point. Nevertheless, a number of experiments were conducted to gain insight into the underlying manifolds involved (see Scheme 4). The model reaction proceeded well in the presence of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) to produce the desired product 3aa in quantitative yield. This result indicated that a radical process might not be involved in the present transformation. We also studied the effect of potassium chelating agents in the transesterification reaction to investigate the importance of the cation in the catalysis. When 18-crown-6 was added to model reaction using K2CO3 as the catalyst, quantitative transesterification was still observed, suggesting either that ineffective chelation of the metal ion had occurred or that the cation was not necessary to the reactivity in this particular case. Furthermore, the reaction also proceeded well in argon atmosphere, which may exclude the participation of molecular oxygen in this transformation. To identify the byproduct, which was formed by the cleavage of the C–O bond of esters, the model reaction solution was detected with GCMS after the end of the reaction (see the ESI†). The GC area% data showed that along with the desired product 3aa, a less amount of 2-pyridone was obtained, which indicated that the alkoxy group of esters eliminated to phenols in this reaction. Likewise, the GC-MS analysis of reaction of naphthalen-2-yl benzoate with phenol illustrated that naphthol was substituted by phenol in the reaction. The isolated phenols could react with carboxylic acids to synthesize esters for resource recycling.
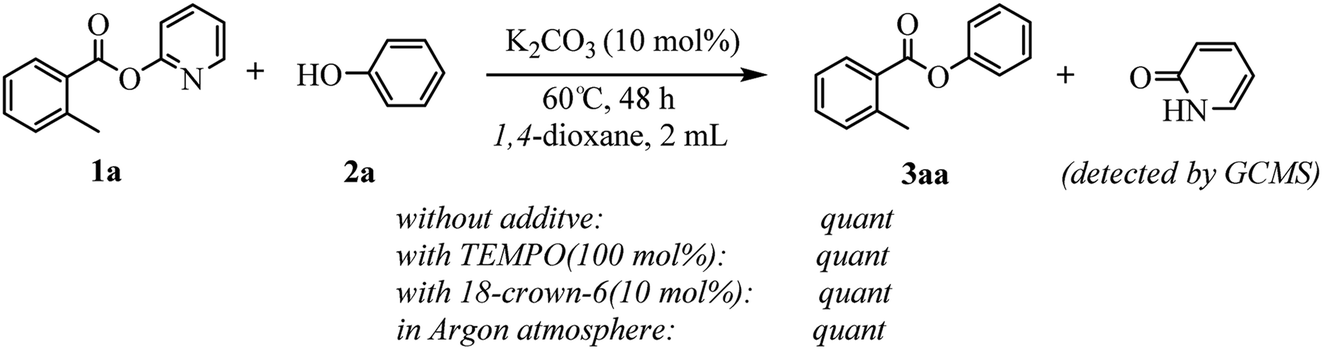 | ||
| Scheme 4 Control experiments to elucidate the reaction pathways. | ||
Because K2CO3 dissolves in reaction solvent, we attempted to look for mechanism's evidence by means of heterogeneous approach. The filtrated used K2CO3 catalyst (after reaction) was studied by transmission electron microscopy (TEM), energy dispersive spectroscopy (EDS) and solid state NMR. The morphology of used K2CO3catalyst was characterized by TEM. As shown in Fig. 1a and b, used K2CO3 catalyst had narrow size distribution and the HRTEM images depicted clearly visible lattice fringes that evince the formation of crystalline K2CO3. Aim to confirm whether coordination effect between K2CO3 and reactants existed, the EDSs of fresh and used catalysts were compared (Fig. 1c and d). The EDS data showed there is a remarkably improvement of carbon content of catalyst after reaction. Not only that, the strong 1H signals were detected in the solid state 1H NMR spectra of used catalyst (see the ESI†). These results indirectly proved that K2CO3 may be catalyzed the novel transesterification reaction via coordination effect and not just a base.
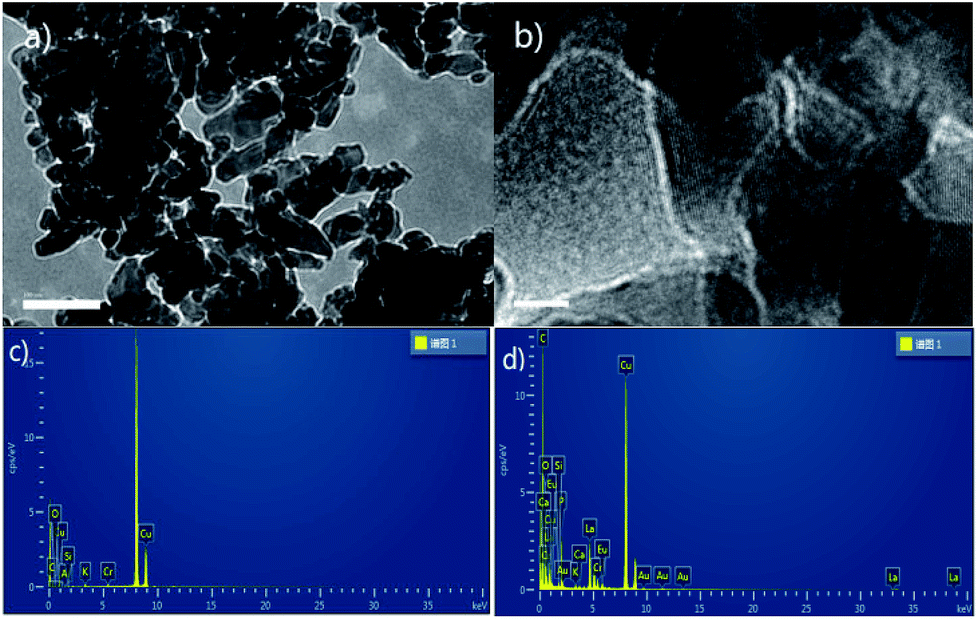 | ||
| Fig. 1 (a) 200 nm TEM images of used catalyst; (b) 10 nm HRTEM images of used catalyst; (c and d) the EDS of fresh and used catalysts. | ||
On the basis of experiments and previous reported literature, the possible mechanism and intermediates I–V are postulated as follows (see Scheme 5). Initially, the “π-rich” two aryl (or heteroaryl) group of ester 1 interacts with K+ to form a cation–π complex I.23 It might explain why saturated aliphatic acid esters is not suitable for the new transesterification reaction. The C(acyl)–O cleavage occurs through a three-centered transition state II to give the potassium intermediate III.14a The phenol 2 attacked the acyl carbon to form intermediate IV. Subsequently, the alkoxide attack on hydrogen cation resulted in the other phenol (byproduct), generating intermediate V. Electron transfer of oxygen anion resulted in the desired ester 3, and regenerated K2CO3.
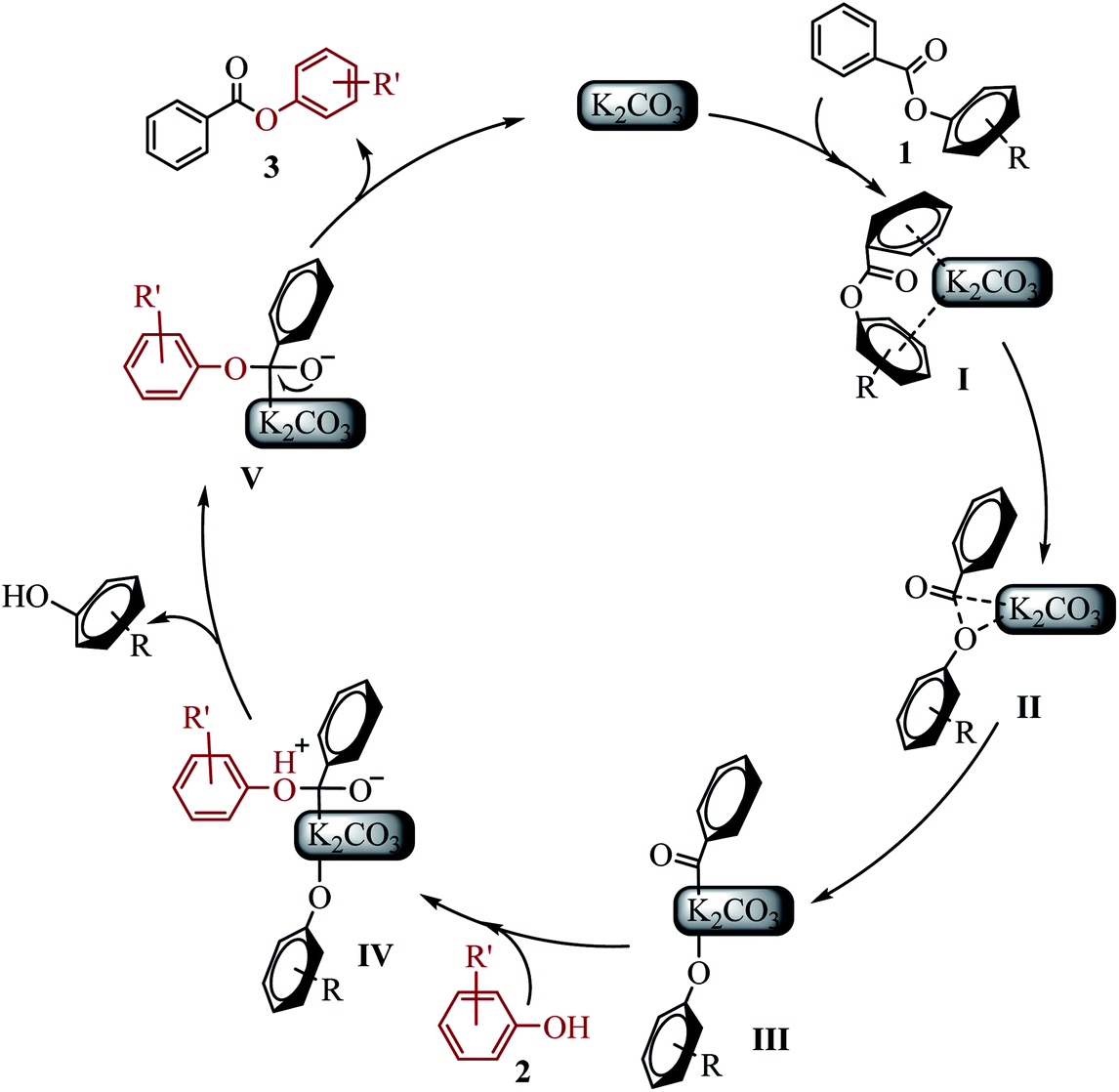 | ||
| Scheme 5 Proposed preliminary mechanism. | ||
Experimental
Preparation of aryl esters (1a–u; 4a–o)
A mixture of carboxylic acid (10 mmol), 2-pyridone or phenol derivative (10 mmol), DMAP (4-(dimethylamino)pyridine, 1 mmol), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiamide hydrochloride (EDC·HCl, 10 mmol) in THF (50 mL) was stirred overnight at 25 °C. The resulting mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl ether/petroleum ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3–1:10 as eluent) to afford a corresponding (hetero)aryl esters.
:3–1:10 as eluent) to afford a corresponding (hetero)aryl esters.
General procedure for K2CO3-catalyzed transesterification
In a typical reaction, aryl ester (0.10 mmol), phenol (0.17 mmol), catalyst (0.01 mmol or 0.02 mmol), and the solvent 1,4-dioxane (2.0 mL) were charged in a 25 mL oven dried reaction tube. Reaction was carried out 60 °C or 120 °C for 48 h in an oil bath under air condition. After being cooled to room temperature, the reaction solution was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1:5–1:15 as an eluent) to afford the desired product 3. All the products were also confirmed by comparing the 1H NMR and 13C NMR data with authentic samples.
10 gram-scale synthesis of 3aa
In a 10 gram-scale reaction, pyridin-2-yl 2-methylbenzoate 1a (10 g, 47.0 mmol), phenol 2a (7.51 g, 79.9 mmol), K2CO3 (0.65 g, 4.7 mmol), and the solvent (150 mL) were charged in a 250 mL ovendried round flask. Reaction was carried out 60 °C for 48 h in an oil bath under air condition. After being cooled to room temperature, the reaction solution was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1:5–1:15 as an eluent) to afford the desired product 3aa (9.85 g, 99% yield).
Conclusions
In conclusion, we have disclosed an alkali metal catalyzed transesterification of various heteroaryl esters or aryl esters with phenols. The chemistry proceeds under mild conditions and enables the direct synthesis of a wide array of useful aryl esters, with high tunability in both the ester and phenol. In general, the pyridyloxy and electron-deficient phenoxy of the aryl esters are easily exchanged by electron-rich phenols. But some examples could not be explained by a simple nucleophilic substitution reaction process. Being insoluble in many of the solvent systems, K2CO3 offers the advantages of ease of recovery from the reaction medium by mere filtration and reuse of the recovered material. This methodology meets the requirement of the ideal transesterification, which is to achieve quantitative yield with the reactants in a ratio near 1:1 by use of mild, recyclable catalysts without requiring any technology to remove any coproduct. Detailed mechanistic studies by computational and experimental methods are underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by National Science Foundation of China (21462031), Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (NJYT-17-A22).Notes and references
- (a) J. Otera and J. Nishikido, Esterification: Methods, Reactions, and Applications, Wiley, Hoboken, NJ, 2009, p. 1 CrossRef; (b) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, New York, 1999, vol. 1, pp. 1–2640 Search PubMed; (c) J. D. Nguyen, E. M. D'Amato, J. M. Narayanam and C. R. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef PubMed.
- Some reviews on transesterifications: (a) J. Otera, Chem. Rev., 1993, 93, 1449 CrossRef; (b) G. A. Grasa, R. Singh and S. P. Nolan, Synthesis, 2004, 971 Search PubMed; (c) J. Otera, Acc. Chem. Res., 2004, 37, 288 CrossRef PubMed.
- (a) M. Kumar, S. Bagchi and A. Sharma, New J. Chem., 2015, 39, 8329 RSC; (b) S. Magens, M. Ertelt, A. Jatsch and B. Plietker, Org. Lett., 2008, 10, 53–56 CrossRef PubMed.
- Some papers on Lewis acids as catalysts for transesterifications: (a) D. P. Sheng and I. O. Kady, Appl. Catal., A, 2009, 365, 149 CrossRef; (b) J. W. J. Bosco and A. K. Saikia, Chem. Commun., 2004, 1116 RSC; (c) T. Ohshima, T. Iwasaki, Y. Maegawa, A. Yoshiyama and K. Mashima, J. Am. Chem. Soc., 2008, 130, 2944 CrossRef PubMed; (d) A. Solhy, J. H. Clark, R. Tahir, S. Sebti and M. Larzek, Green Chem., 2006, 8, 871 RSC.
- Some papers on organic and inorganic bases as catalysts for transesterification: (a) D. A. Watson, X. Fan and S. L. Buchwald, J. Org. Chem., 2008, 73, 7096 CrossRef PubMed; (b) V. Sridharan, M. Ruiz and J. C. Menéndez, Synthesis, 2010, 1053 Search PubMed; (c) S. R. Jagtap, M. D. Bhor and B. M. Bhanage, Catal. Commun., 2008, 9, 1928 CrossRef.
- Some papers on NHCs as catalysts for transesterifications: (a) G. A. Grasa, R. M. Kissling and S. P. Nolan, Org. Lett., 2002, 21, 3583 CrossRef; (b) G. W. Nyce, J. A. Lamboy, E. F. Connor, R. M. Waymouth and J. L. Hedrick, Org. Lett., 2002, 21, 3587 CrossRef; (c) T. Q. Zeng, G. H. Song and C. J. Li, Chem. Commun., 2009, 6249 RSC; (d) R. Singh, R. M. Kissling, M. A. Letellier and S. P. Nolan, J. Org. Chem., 2004, 69, 209 CrossRef PubMed.
- C. Yu, H. Huang, X. Li, Y. Zhang and W. Wang, J. Am. Chem. Soc., 2016, 138, 6956–6959 CrossRef PubMed.
- (a) L. Pilato, Phenolic Resins: A Century of Progress, Springer, Heidelberg, 1st edn, 2010 CrossRef; (b) M. Granda, C. Blanco, P. Alvarez, J. Patrick and R. Merendez, Chem. Rev., 2014, 114, 1608 CrossRef PubMed.
- (a) D. G. Yu, B. J. Li, S. F. Zheng, B. T. Guan, B. Q. Wang and Z. J. Shi, Angew. Chem., Int. Ed., 2010, 49, 4566 CrossRef PubMed; (b) Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C. J. Li, Angew. Chem., Int. Ed., 2015, 54, 14487 CrossRef PubMed; (c) H. Zeng, Z. Qiu, A. Dom&nguez-Huerta, Z. Hearne, Z. Chen and C. J. Li, ACS Catal., 2017, 7, 510 CrossRef.
- (a) H. Xu, K. Muto, J. Yamaguchi, C. Zhao, K. Itami and D. G. Musaev, J. Am. Chem. Soc., 2014, 136, 14834–14844 CrossRef PubMed; (b) L. Xu, B. J. Li, Z. H. Wu, X. Y. Lu, B. T. Guan, B. Q. Wang, K. Q. Zhao and Z. J. Shi, Org. Lett., 2010, 12, 4 CrossRef PubMed; (c) K. W. Quasdorf, X. Tian and N. K. Garg, J. Am. Chem. Soc., 2008, 130, 14422–14423 CrossRef PubMed; (d) C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236–2239 CrossRef PubMed; (e) R. Takise, K. Muto, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2014, 53, 6791–6794 CrossRef PubMed; (f) J. Cornella, E. P. Jackson and R. Martin, Angew. Chem., Int. Ed., 2015, 54, 4075–4078 CrossRef PubMed; (g) M. Tobisu and N. Chatani, Top. Curr. Chem., 2016, 374, 41 CrossRef PubMed; (h) J. Yang, T. Chen and H. B. Han, J. Am. Chem. Soc., 2015, 137, 1782–1785 CrossRef PubMed; (i) Y. Gu and R. Martín, Angew. Chem., Int. Ed., 2017, 56, 3187 CrossRef PubMed; (j) L. Guo, C. C. Hsiao, H. Yue, X. Liu and M. Rueping, ACS Catal., 2016, 6, 4438–4442 CrossRef.
- (a) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671–2678 RSC; (b) K. Amaike, K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 13573–13576 CrossRef PubMed; (c) K. Muto, J. Yamaguchi, D. G. Musaev and K. Itami, Nat. Commun., 2015, 6, 7508 CrossRef PubMed; (d) L. Guo, A. Chatupheeraphat and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 11810–11813 CrossRef PubMed; (e) H. F. Yue, L. Guo, S. C. Lee, X. Q. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972–3976 CrossRef PubMed; (f) X. H. Pu, J. F. Hu, Y. Zhao and Z. Z. Shi, ACS Catal., 2016, 6, 6692–6698 CrossRef; (g) Q. Q. Lu, H. Yu and Y. Fu, J. Am. Chem. Soc., 2014, 136, 8252–8260 CrossRef PubMed.
- R. Kakino, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2001, 74, 371–376 CrossRef.
- H. Tatamidani, F. Kakiuchi and N. Chatani, Org. Lett., 2004, 6, 3597–3599 CrossRef PubMed.
- (a) T. B. Halima, W. Zhang, I. Yalaoui, X. Hong, Y. Yang, K. N. Houk and S. G. Newman, J. Am. Chem. Soc., 2017, 139, 1311–1318 CrossRef PubMed; (b) T. B. Halima, J. K. Vandavasi, M. Shkoor and S. G. Newman, ACS Catal., 2017, 7, 2176–2180 CrossRef.
- (a) Y. S. Bao, M. Baiyin, B. Agula, M. L. Jia and Z. Bao, J. Org. Chem., 2014, 79, 6715 CrossRef PubMed; (b) Y. S. Bao, Z. Bao, B. Agula, M. Baiyin and M. L. Jia, J. Org. Chem., 2014, 79, 803 CrossRef PubMed; (c) Y. S. Bao, L. Wang, M. Jia, A. Xu, B. Agula, M. Baiyin and Z. Bao, Green Chem., 2016, 18, 3808–3814 RSC.
- (a) A. A. Toutov, W. B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef PubMed; (b) A. A. Toutov, K. N. Betz, D. P. Schuman, W. B. Liu, A. Fedorov, B. M. Stoltz and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 1668–1674 CrossRef PubMed; (c) V. Leich, T. P. Spaniol and J. Okuda, Organometallics, 2016, 35, 1179–1182 CrossRef.
- A. L. Rodriguez, T. Bunlaksananusorn and P. Knochel, Org. Lett., 2000, 2, 3285–3287 CrossRef PubMed.
- R. Gai, D. F. Back and G. Zeni, J. Org. Chem., 2015, 80, 10278–10287 CrossRef PubMed.
- R. Radhakrishan, D. M. Do, S. Jaenicke, Y. Sasson and G. K. Chuah, ACS Catal., 2011, 1, 1631–1636 CrossRef.
- Z. G. Chen, Y. Wang, J. F. Wei, P. F. Zhao and X. Y. Shi, J. Org. Chem., 2010, 75, 2085–2088 CrossRef PubMed.
- X. Li, X. Xu, P. Hu, X. Xiao and C. Zhou, J. Org. Chem., 2013, 78, 7343–7348 CrossRef PubMed.
- Y. F. Liang and N. Jiao, Angew. Chem., 2014, 126, 558–562 CrossRef.
- S. Banerjee, Y. Yang, I. D. Jenkins, Y. Liang, A. A. Toutov, W. Liu, D. P. Schuman, R. H. Grubbs, B. M. Stoltz, E. H. Krenske, K. N. Houk and R. N. Zare, J. Am. Chem. Soc., 2017, 139, 6880–6887 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental section and characterization data for the products, 1H NMR, 13C NMR spectra of the products. See DOI: 10.1039/c8ra04984j |
| This journal is © The Royal Society of Chemistry 2018 |