
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorroboration of Zn(II)–Mg(II)-tertiary structure interplays essential for the optimal catalysis of a phosphorothiolate thiolesterase ribozyme†
Tzu-Pin Wang *ab,
Yu-Chih Su‡
a,
Yi Chen‡a,
Scott Severancec,
Chi-Ching Hwangd,
Yi-Ming Lioua,
Chia-Hui Lua,
Kun-Liang Lina,
Rui Jing Zhua and
Eng-Chi Wanga
*ab,
Yu-Chih Su‡
a,
Yi Chen‡a,
Scott Severancec,
Chi-Ching Hwangd,
Yi-Ming Lioua,
Chia-Hui Lua,
Kun-Liang Lina,
Rui Jing Zhua and
Eng-Chi Wanga
aDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung, 80708, Taiwan. E-mail: tzupinw@kmu.edu.tw; Fax: +886-07-312-5339; Tel: +886-07-312-1101, ext. 2756
bKaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, 80708, Taiwan
cDepartment of Molecular and Cellular Sciences, Liberty University College of Osteopathic Medicine, Lynchburg, Va 24515, USA
dDepartment of Biochemistry, Kaohsiung Medical University, Kaohsiung, 80708, Taiwan
First published on 21st September 2018
Abstract
The TW17 ribozyme, a catalytic RNA selected from a pool of artificial RNA, is specific for the Zn2+-dependent hydrolysis of a phosphorothiolate thiolester bond. Here, we describe the organic synthesis of both guanosine α-thio-monophosphate and the substrates required for selecting and characterizing the TW17 ribozyme, and for deciphering the catalytic mechanism of the ribozyme. By successively substituting the substrate originally conjugated to the RNA pool with structurally modified substrates, we demonstrated that the TW17 ribozyme specifically catalyzes phosphorothiolate thiolester hydrolysis. Metal titration studies of TW17 ribozyme catalysis in the presence of Zn2+ alone, Zn2+ and Mg2+, and Zn2+ and [Co(NH3)6]3+ supported our findings that Zn2+ is absolutely required for ribozyme catalysis, and indicated that optimal ribozyme catalysis involves the presence of outer-sphere and one inner-sphere Mg2+. A survey of the TW17 ribozyme activity at various pHs revealed that the activity of the ribozyme critically depends on the alkaline conditions. Moreover, a GNRA tetraloop-containing ribozyme constructed with active catalysis in trans provided catalysis and multiple substrate turnover efficiencies significantly higher than ribozymes lacking a GNRA tetraloop. This research supports the essential roles of Zn2+, Mg2+, and a GNRA tetraloop in modulating the TW17 ribozyme structure for optimal ribozyme catalysis, leading also to the formulation of a proposed reaction mechanism for TW17 ribozyme catalysis.
Introduction
Catalytic RNAs (ribozymes) are versatile and powerful catalysts1,2 and frequently are used with metal ions in diverse catalytic strategies.2 The involvement of metal ions in ribozyme catalysis is intimately associated with the fundamental structure of RNA. At physiological pH, RNA molecules are essentially polyelectrolytes laden with negatively charged phosphate groups. To counteract electrostatic repulsion due to the phosphate moieties, RNA typically sequesters metal cations for proper folding—a process in which metal ions can specifically bind RNA ligands to further stabilize RNA structures.3 Additionally, RNA can specifically recruit divalent metal ions to play a direct role in the chemical mechanism of ribozyme catalysis.3,4 Conversely, some small nucleolytic ribozymes perform catalysis in the presence of molar concentrations of monovalent metal ions and in the absence of divalent metal ions.2,4–6 The catalytic abilities of these ribozymes can be attributed to the proton transfer involved in general acid–base catalysis7,8 and the presence of a disproportionate amount of interacting ions in the ion atmosphere surrounding nucleic acids.9–13 However, the detection of ribozyme catalysis at high concentrations of monovalent cations does not necessarily preclude a critical function for divalent cations in ribozyme catalysis because divalent metal ions are not required to directly contact any of the functional groups in the active site of a ribozyme in order to play an essential catalytic role.4 Thus, gaining a comprehensive understanding of the diverse mechanisms of ribozyme catalysis requires knowledge of the specific roles of divalent metal ions in catalysis-active ribozyme structures.Among the divalent metal ions, Mg2+ is capable of specifically binding catalytic RNA by either direct inner-sphere coordination or indirect outer-sphere coordination in the presence of water ligands.4 Replacing Mg2+ with other metal ions can drastically alter the catalytic activity of ribozymes. Besides Mg2+, the Zn(II) ion also commonly participates in catalysis in biological systems and is the only divalent metal cofactor present in all six enzyme classes of the International Union of Biochemistry.14–17 Compared to Mg2+, Zn2+ has a higher phosphate monoester affinity but seldom plays a prominent role in ribozyme catalysis. This is likely due to a low concentration of free Zn2+ in vivo16 and the lower binding affinities of metal ions to ribozymes than to proteins.4 Nevertheless, identification of natural and nonnatural Zn2+-assisted or Zn2+-dependent nucleic acid enzymes could provide insights into the plausible functions of zinc in ribozyme catalysis.18
In addition to the integral roles of metal ions in RNA folding and its catalytic function, the tertiary RNA structure and function can be further supported by long-range interactions between RNA hairpins capped by GNRA (where N represents any nucleotide; R is a purine) tetraloops and their receptors,19–21 including RNA motifs19,22 and proteins.23 GNRA tetraloops were first identified in ribosomal RNA (rRNA) and are frequently used to cap RNA duplexes in rRNA.24 Thus, GNRA tetraloop–receptor interactions can be critical to optimizing the conformations and catalytic functions of ribozymes.
We previously described an in vitro selection of a novel ribozyme called the TW17 ribozyme, which catalyzes the hydrolysis of a phosphorothiolate thiolester bond in the presence of Zn2+ (Scheme 1).18 TW17 ribozyme catalysis demonstrated the indispensable role of Zn2+ as a cofactor and the requirement for the concomitant presence of Zn2+ and Mg2+ for the catalytic activity of this ribozyme. Herein, we describe the syntheses of guanosine α-thio-monophosphate (6), the substrate (18b) used in the in vitro selection of the TW17 ribozyme, and the additional substrates that proved to be essential in elucidating the roles of Zn2+ and Mg2+ in the chemical mechanism of ribozyme catalysis. In addition, pH titration of the ribozyme catalysis unveiled the requirement for alkaline conditions in TW17 ribozyme catalysis. Finally, the importance of a GNRA tetraloop for optimal ribozyme catalysis was supported by enhanced ribozyme catalysis in a GNRA tetraloop-retaining TW17 ribozyme construct with active catalysis in trans. Such critical information enabled the development of a proposed reaction mechanism of TW17 ribozyme catalysis in which the hydrolysis reaction is accelerated by an inner-sphere-coordinated zinc(II) hydroxide in the ribozyme under alkaline conditions. This finding is reminiscent of the many catalytically important roles that Zn2+ ions play in zinc-dependent metalloenzymes14,15,17,25 and thus suggests that ribozymes and protein enzymes could exploit similar zinc chemistry to effect chemical catalysis.
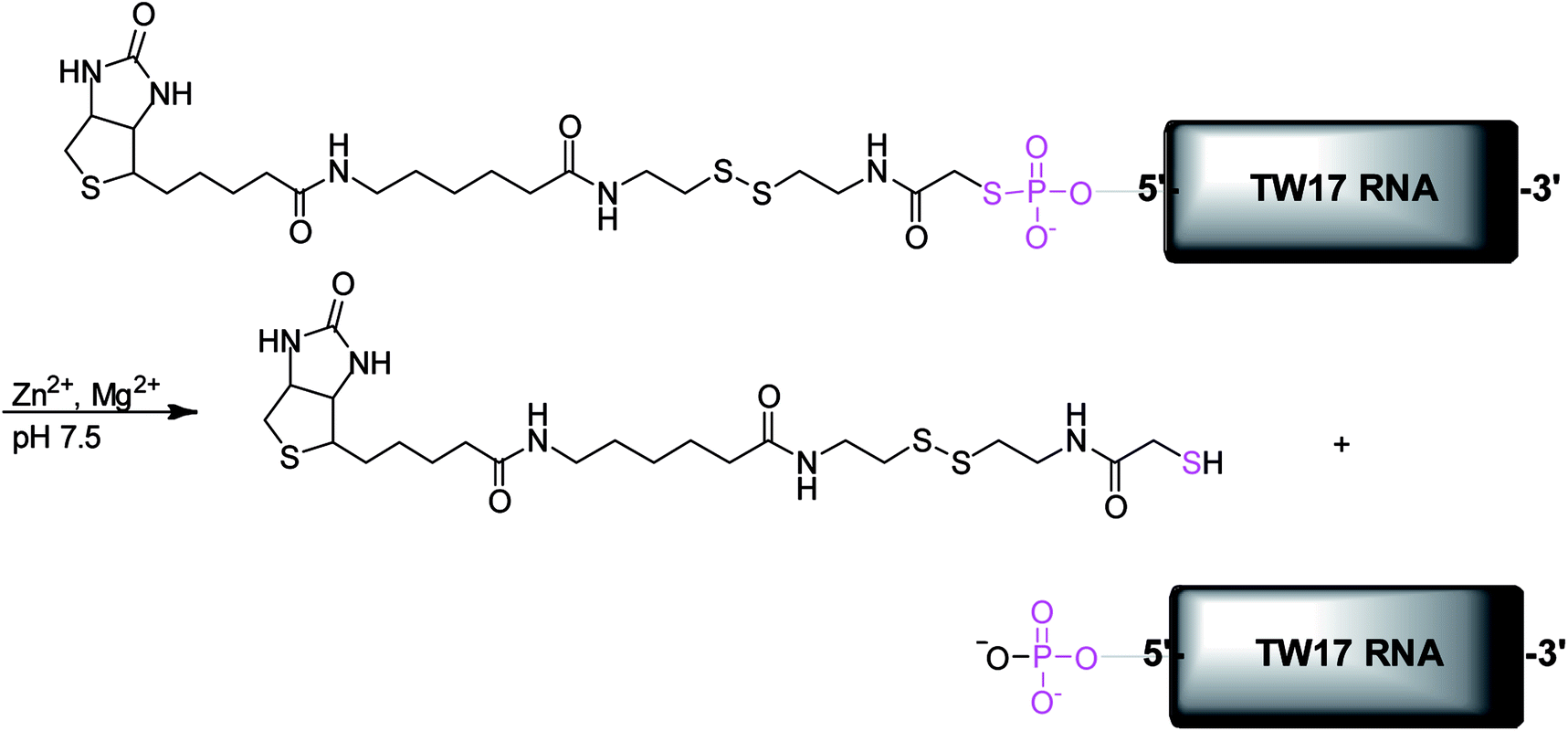 | ||
| Scheme 1 The phosphorothiolate thiolester hydrolysis reaction catalyzed by the TW17 ribozyme. | ||
Results
Synthesis of GMPS salts and substrates for selecting the TW17 ribozyme and for deciphering the ribozyme substrate specificity
We initiated this study by synthesizing the more water-soluble ammonium salt of guanosine α-thio-monophosphate (NH4–GMPS, 6; Scheme 2). GMPS was incorporated into the 5′ end of RNA during round-off transcription by T7 RNA polymerase, which subsequently enabled the coupling of small substrate molecules to GMPS-primed RNA. The afforded substrate-RNA pool paved the way for enriching and selecting active RNA sequences, including the TW17 ribozyme.18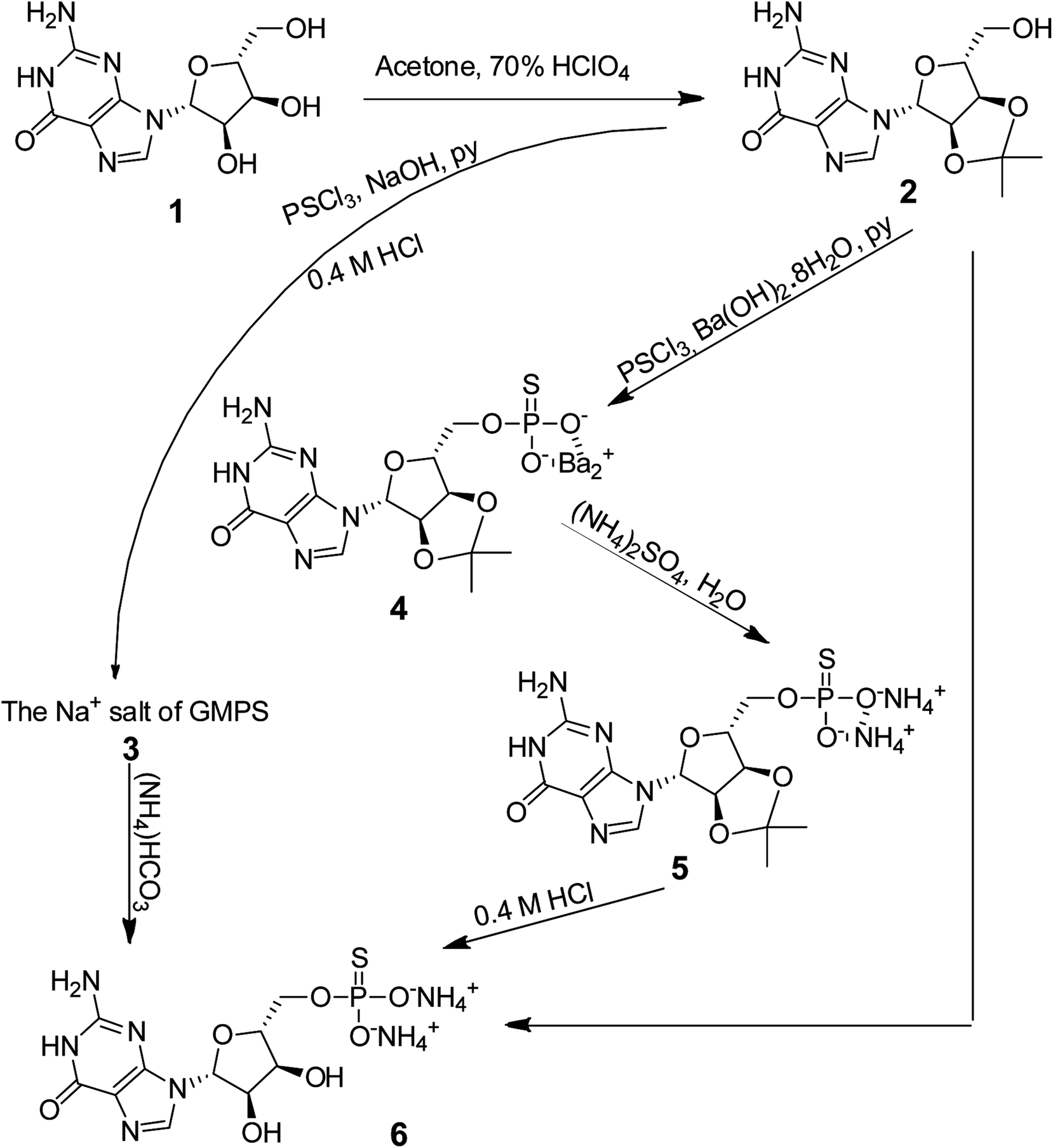 | ||
| Scheme 2 Synthesis of the ammonium salt of guanosine α-thio-monophosphate (6). Three routes for the synthesis of 6 were explored in this study. Only the route sequentially synthesizing 4 and 5 from 2 could produce 6 with a high yield (see text). | ||
The synthesis of GMPS has been reported previously but often involving tedious and unconventional procedures.26–31 We synthesized 6 from 1 by first protecting the 2′ and 3′ hydroxyl groups with acetone through ketal formation to afford 2 with an excellent yield (98%, Scheme 2).32 We initially attempted to directly synthesize 6 by thiophosphorylating 2, producing an ammonium salt in the presence of NH4OH, and finally, removing the acetone protection group to attain the desired 6 (the “⌋”-shaped arrow in Scheme 2). However, the synthesis of 6 was fruitless using this approach, primarily due to difficulties in recovering 6 from the reaction mixture. We also explored synthesizing 6 by thiophosphorylating 2, acquiring a sodium salt in the presence of NaOH and then deprotecting acetone to yield 3 (the sodium salt of GMPS; the curved arrow in Scheme 2), and finally subjecting 3 to an ion exchange reaction to obtain 6 in sequence. However, the yield of the reaction from 2 to 4 was too low (25%) to make the synthesis of 6 practical.
Consequently, we decided to develop a more complicated but potentially more effective method to synthesize 6 (i.e., via the central route in Scheme 2). We first thiophosphorylated 2 in the presence of barium hydroxide, resulting in the formation of 4 with a good yield (75%). The barium ion in 4 was then quantitatively exchanged with an ammonium ion to afford 5 (98%), which was then subjected to removal of the acetone protection group and purification using a DEAE Sepharose Fast Flow column, sequentially, to produce the desired 6 (64%). Analysis revealed that 6 was free of contamination by the ammonium salt of the potential guanosine monophosphate (GMP) by-product. Specifically, the 31P NMR spectrum of 6 indicated the presence of GMPS (δ = 43.65) without detecting GMP (δ = 0) (ESI,† page S24). These results clearly demonstrated that 6 was effectively synthesized and that our method overcame the previously reported oxygen–sulfur exchange problem of the thiophosphate moiety in nucleotides.30,33 The overall yield of 6 from 1 using our synthesis approach was 46%. Compound 6 was characterized and confirmed by both 1H and 13C NMR spectroscopy, while the molecular mass was confirmed by HRMS.
Successfully acquiring 6 allowed us to focus on synthesizing the ribozyme substrate indispensable to the ribozyme selection. The evolution and selection of ribozymes from the artificial RNA pool initially primed with 6 at the 5′ head were achieved by covalently tagging the molecules in the GMPS-primed RNA pool with a biotin-derivatized substrate through a phosphorothiolate thiolester bond (Scheme 3). Active RNA sequences could be separated from inactive sequences in the substrate-RNA pool by taking advantage of the high affinity interaction between biotin and streptavidin.18 To expedite the in vitro selection of the ribozymes, we prepared the required substrate-RNA pool by dissecting the biotin-containing substrate 18b (Scheme 4) into two segments. Each segment was covalently linked to the GMPS-primed random artificial RNA pool sequentially (Scheme 3B). Specifically, the 18b-RNA conjugates were synthesized by initially coupling the GMPS-primed RNA pool to the pro-substrate 10b and subsequently biotinylating the product with commercially available sulfo-NHS-LC-biotin to generate the 18b-RNA pool used in each in vitro selection cycle.
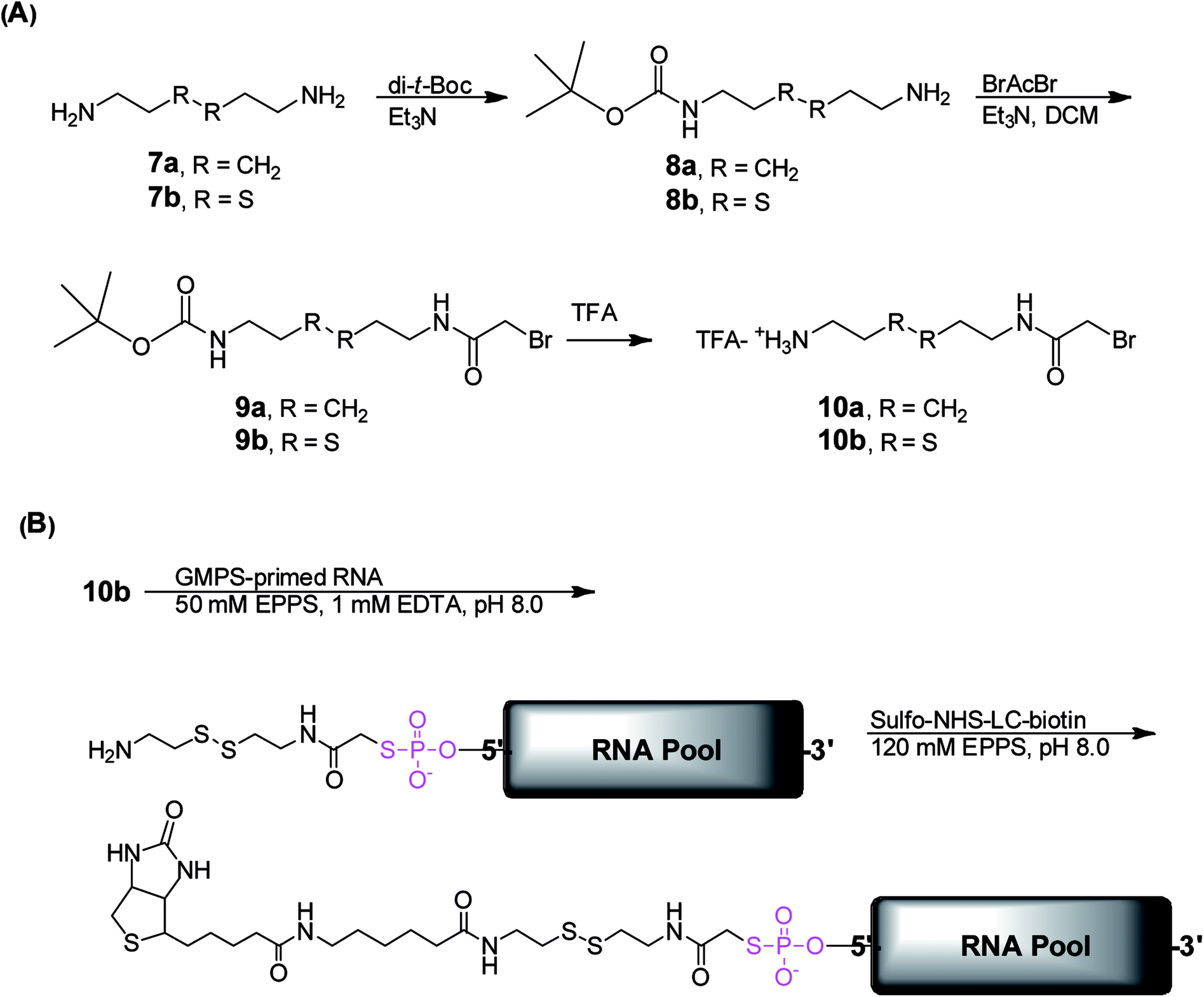 | ||
| Scheme 3 Synthesis of substrates and substrate-RNA conjugates. (A) Synthesis of the substrates required for the in vitro selection of the TW17 ribozyme and the determination of the substrate specificity of the TW17 ribozyme. (B) Preparation of the substrate-RNA conjugates for in vitro evolution and selection of the TW17 ribozyme. | ||
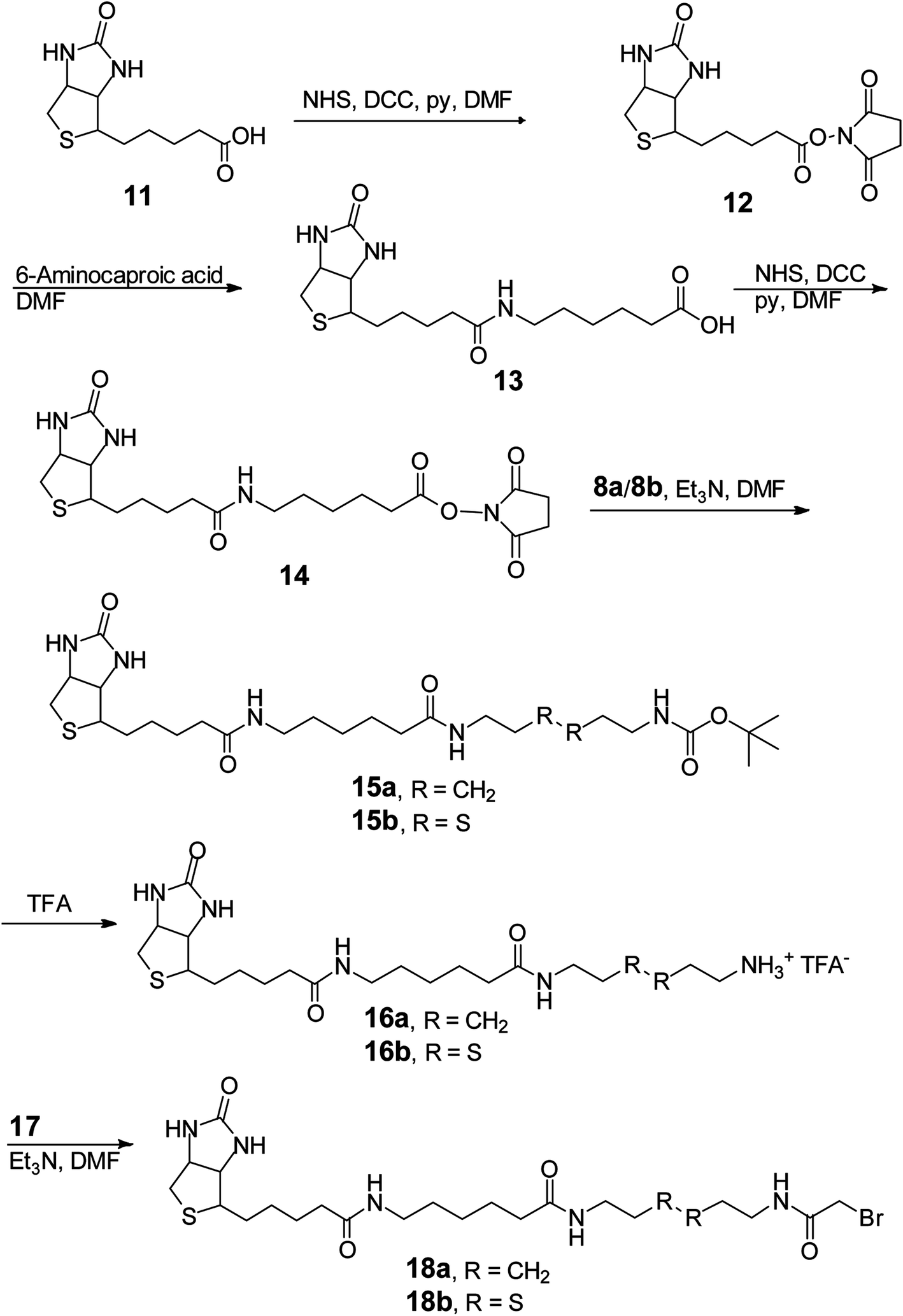 | ||
| Scheme 4 Synthesis of the substrates utilized in studying the Zn2+-dependent hydrolytic mechanism of the TW17 ribozyme. | ||
The effective synthesis of 10b was thus required to prepare the pool of 18b-RNA conjugates. We explored a single-reaction approach to synthesize 10b by reacting bromoacetyl bromide (BrAcBr) with a molar excess of cystamine (7b). TLC and NMR analyses indicated that the reaction products were too complicated to resolve and gave no evidence for the production of 10b. We reasoned that, in the presence of a molar excess of 7b, nucleophilic substitution reactions between the two amino groups in 7b and the bromine in both the carbonyl carbon and the less reactive methylene carbon of BrAcBr might have resulted in polymerization products that we did not further characterize. Therefore, we modified the synthesis strategy for 10b by utilizing N-tert-butoxycarbonyl (Boc) to protect one of the amino groups in 7b to produce 8b (39%).34 This was followed by the acylation of 8b in the presence of a molar excess of BrAcBr and subsequent TFA deprotection to obtain 10b (40%; Scheme 3A). The successful synthesis of 10b ultimately led to the in vitro selection of the TW17 ribozyme.18
In our attempt to characterize the substrate specificity of the ribozyme catalysis to determine the chemical reaction mechanism accelerated by the TW17 ribozyme, we realized that the organic synthesis of 18b was imperative to achieve this aim. The successful synthesis of 18b (Scheme 4) was attained in the following manner. Biotin (11) was converted to 14 using a previously published method.35 Compound 14 was then reacted with 8b to acquire 15b (81.5%). The TFA deprotection of 15b produced 16b, which was then subjected to amidation with 17 to produce the desired 18b (20.6%). The structure of 18b was confirmed by 1H and 13C NMR spectroscopy. Moreover, 18b was able to be effectively linked to the TW17 ribozyme and to support the expected ribozyme catalysis.18
However, as indicated previously,18 the presence of multiple functional groups in 18b, the phosphorothiolate thiolester bond between 18b and the TW17 ribozyme, and the numerous phosphodiester linkages in the TW17 ribozyme hampered the straightforward determination of the type of chemical reactions catalyzed by the TW17 ribozyme. Consequently, we performed systematic TW17 ribozyme catalysis studies in order to rule out reactions not catalyzed by the TW17 ribozyme. We utilized 18a, a structural analog of 18b lacking the internal disulfide bond, to demonstrate that the TW17 ribozyme was not a disulfide reductase RNA. The synthesis of 18a was very similar to that of 18b, except that the reactant 8b was replaced by 8a (Schemes 3 and 4). In addition, the intermediate 16a in the 18a synthesis process was obtained by following reported methods.35 The amidation reaction between 16a and 17 resulted in the synthesis of 18a (58.5%). The acquired 18a was later coupled to the TW17 ribozyme, and the resulting 18a-ribozyme conjugate exhibited catalytic activity identical to that of the TW17 ribozyme appended with 18b.18 Therefore, we ruled out the possibility that the TW17 ribozyme functions as a disulfide reductase RNA.
We synthesized 10a to determine whether the TW17 ribozyme could function as an amidase RNA by hydrolyzing the second amide bond (relative to the phosphorothiolate ester bond) in the 18-TW17 ribozyme conjugate. The synthesis of 10a was straightforward and analogous to the synthesis scheme for 10b except that 7a was substituted for 7b (Scheme 3A). The yield from 8a to 10a was 35.5%. Compound 10a was again effectively conjugated to the TW17 ribozyme to give the 10a-TW17 ribozyme conjugate, which showed a significantly different migration from that of the TW17 ribozyme catalysis product in high-resolution urea-PAGE.18 This finding excluded the possibility that the ribozyme was an amidase RNA that hydrolyzed the indicated amide linkage. Finally, the additional chemical and biochemical characterization of TW17 ribozyme catalysis confirmed the conclusion that the ribozyme was a Zn2+-dependent phosphorothiolate thiolesterase RNA (Scheme 1).18 Because 18a was synthesized with a higher yield than that of 18b, we chose 18a as the substrate to be coupled to the TW17 ribozyme and to yield the 18a-ribozyme conjugate for all the future kinetic studies of TW17 ribozyme catalysis. The decision was further supported by the fact that, as previously described, the TW17 ribozyme demonstrated almost identical catalytic activity when covalently linking the ribozyme to either 18a or 18b.18
Characteristics of metal cofactors in TW17 ribozyme catalysis
Our previous study revealed that TW17 ribozyme catalysis was Zn2+-dependent and was more efficient in the presence of both Zn2+ and Mg2+.18 To gain insights into the intricate roles that divalent metal cations play in TW17 ribozyme activity, we analyzed ribozyme catalysis reactions in the presence of various concentrations of Zn2+, Mg2+, or [Co(NH3)6]3+. We were especially interested in obtaining information about the roles that Zn2+, inner-sphere Mg2+, and outer-sphere Mg2+ might play in sustaining optimal TW17 ribozyme catalysis. Moreover, we exploited classical Hill plot analyses to reveal correlations between the zinc concentrations and TW17 ribozyme catalysis. Early ribozyme studies utilized a similar approach of using the Hill equation to characterize discrete and independent sites for ion binding in ribozymes, and, typically, the data fitted to the Hill equation moderately well.36,37 Subsequent studies, however, indicated that the parameters from Hill analyses of metal ion-RNA titrations in earlier ribozyme research were derived from a simplified ligand-binding model of the metal ion-RNA association, which poorly represented the actual environment of the ribozyme.12,13,38 The problem of inappropriately applying the Hill equation to estimate the number of ligand-binding sites in biomacromolecules has also been explicated.39 We thus did not attempt to determine discrete zinc binding sites in the TW17 ribozyme under predefined ribozyme catalysis conditions. Instead, our Hill analyses on data from the zinc titration studies of TW17 ribozyme catalysis were performed to correlate the catalytic activity with the properties of the specifically associated Zn2+ and Mg2+ in the ion atmosphere surrounding the TW17 ribozyme.We first measured kobs values of TW17 ribozyme catalysis as a function of Zn2+ concentrations in the presence of a constant Mg2+ value of 100 mM in order to better understand the indispensable role of Zn2+ in the ion atmosphere of the ribozyme. A concentration of 100 mM Mg2+ was implemented because this was the Mg2+ concentration used in the standard condition of our original TW17 ribozyme study.18 The TW17 ribozyme showed insignificant catalytic activity in Zn2+ concentrations below 125 μM, but exhibited a steady increase of kobs when the Zn2+ concentrations were increased incrementally above 125 μM (Fig. 1A). Hill analyses of kobs from each Zn2+ concentration indicated that the ribozyme had a Hill coefficient (h) of 3.9, a dissociation constant (Kd) of 520 ± 4 μM, and a kobs,max of 6.7 × 10−2 ± 0.1 × 10−2 min−1 (Table 1). The results suggested that each TW17 ribozyme molecule required a minimum of 3.92 equivalences of Zn2+ specifically associated in the [Mg2+] (= 100 mM)-containing ion atmosphere of the ribozyme for proper catalysis. The large average Kd of 520 μM likely reflects the dynamic nature of specifically associated Zn2+ in the ion atmosphere of the TW17 ribozyme. In addition, the presence of Zn2+ had a positive and cooperative effect on TW17 ribozyme catalysis. Moreover, the kobs of the ribozyme more than doubled when the Zn2+ concentrations were increased from the 0.5 mM used in standard ribozyme catalysis to 1.25 mM. Since the deviation from the maximum kobs (kobs,max = 6.7 × 10−2 ± 0.1 × 10−2 min−1 in Table 1) was not substantial, however, we maintained a Zn2+ concentration of 0.5 mM in all subsequent metal titrations in order to compare the current results with our previously published TW17 ribozyme catalysis findings.18
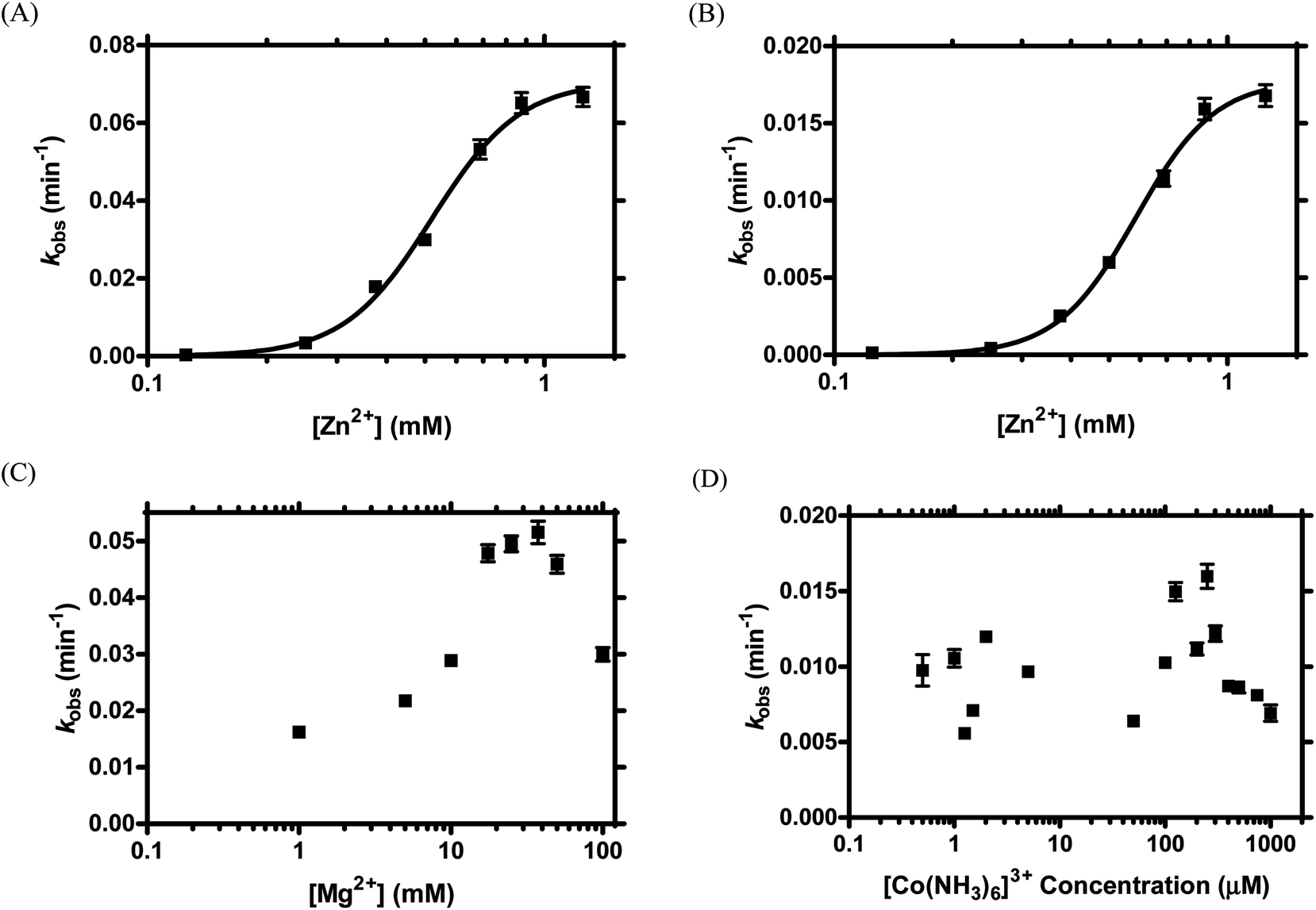 | ||
| Fig. 1 Metal titration in TW17 ribozyme catalysis to demonstrate the absolute requirement for Zn2+ and the critical role of inner- and outer-sphere Mg2+ association for optimal TW17 ribozyme catalysis. (A) The Zn2+-associated ion atmosphere surrounding the TW17 ribozyme was titrated by measuring the kobs of ribozyme catalysis under various Zn2+ concentrations (0.125–1.25 mM) and with a constant [Mg2+] = 100 mM. (B) Zn2+ in the ion atmosphere encompassing the TW17 ribozyme was titrated by measuring the kobs of ribozyme catalysis under varying Zn2+ concentrations (0.125–1.25 mM) in the absence of Mg2+. Each kinetic study of TW17 ribozyme catalysis was performed according to the standard reaction condition (see Experimental section) except that the standard concentrations of divalent metal ions were replaced by the specified concentrations of each divalent metal ion as stated above. Data from (A) and (B) were non-linearly fitted into the Hill equation (see Experimental section) to obtain the Hill coefficient (h), maximum kobs (kobs,max), and average dissociation constant (Kd). (C) The association of Mg2+ in the ion atmosphere of the TW17 ribozyme was studied by determining the kobs of ribozyme catalysis under varying Mg2+ concentrations (1–100 mM) and with a constant [Zn2+] of 0.5 mM. (D) Effects of [Co(NH3)6]3+ concentrations in TW17 ribozyme catalysis were determined by measuring the kobs of the hydrolytic reaction in the presence of 0.5 mM Zn2+. Each data point was the mean value of the determined kobs in three titration experiments, with the standard deviation represented by an up and down bar. Standard deviation bars for some data points are too small to be visible in the graph. | ||
We also exploited metal titration studies to determine the contribution to optimal TW17 ribozyme catalysis by the Mg2+ specifically associated with the ribozyme in the ion atmosphere. We reasoned that, by measuring the kobs of ribozyme catalysis as a function of Zn2+ concentrations in the absence of Mg2+, we could unveil the requirements of not only Zn2+ but also Mg2+ for optimal TW17 ribozyme activity. As before, the TW17 ribozyme had no catalytic activity at Zn2+ concentrations below 125 μM; above 125 μM, the kobs increased steadily and directly with a rise in Zn2+ concentration (Fig. 1B). For example, 0.5 mM Zn2+ itself was sufficient for TW17 ribozyme catalysis (kobs,0.5 mM Zn2+ = 0.005 min−1; Fig. S1†). The results again confirmed the obligatory presence of Zn2+ for TW17 ribozyme catalysis. Hill analyses of the kobs from each Zn2+ concentration produced values for h of 4.3, Kd of 580 ± 20 mM and a kobs,max of 1.8 × 10−2 ± 0.1 × 10−2 min−1 (Table 1).
The results obtained from Zn2+ titration in the presence and absence of 100 mM Mg2+ (Fig. 1A, B, and Table 1) clearly indicated that the specifically associated Zn2+ in the ion atmosphere of the TW17 ribozyme had similar values of h and Kd in the absence and presence of Mg2+. The value of kobs,max, however, showed over a threefold decrease (from 6.7 × 10−2 ± 0.1 × 10−2 min−1 to 1.8 × 10−2 ± 0.1 × 10−2 min−1) when Mg2+ was removed from TW17 ribozyme catalysis. These results strongly support the previous findings18 that Mg2+ is not absolutely required for the catalytic activity of the TW17 ribozyme but is essential for optimal ribozyme catalysis.
We further characterized the specific association of Mg2+ in the ion atmosphere surrounding the TW17 ribozyme by measuring the kobs of TW17 ribozyme catalysis as a function of varying Mg2+ concentrations in the presence of a constant concentration of 0.5 mM Zn2+. It is known that both inner-sphere and outer-sphere Mg2+ have functional roles in ribozyme catalysis.4,5 In the current study, the addition of 0.5 mM Zn2+ to the Mg2+ titration was crucial to understand the dedicated roles of Mg2+ in TW17 ribozyme catalysis because we had previously shown that Mg2+ alone cannot support ribozyme catalysis.18 The TW17 ribozyme displayed catalytic activity due to the Zn2+ background only when the Mg2+ concentration was below 1 mM but displayed a continuous increase of kobs when the Mg2+ concentration was greater than 1 mM and up to 37.5 mM, as the concentration where the TW17 ribozyme attained the maximum catalysis (kobs = 0.052 min−1; Fig. 1C). The results were consistent with the conclusion that Mg2+ is required for optimal TW17 ribozyme catalysis. The activity of the ribozyme, however, steadily declined when Mg2+ concentrations were increased beyond 37.5 mM. For example, the kobs of the TW17 ribozyme decreased from 0.046 min−1 to 0.030 min−1 when the Mg2+ concentration was increased from 50 mM to 100 mM, which is the Mg2+ concentration used in standard ribozyme catalysis.18 The decrease in activity was likely the result of the excess Mg2+ disturbing the TW17 ribozyme structure and impeding effective ribozyme catalysis. We decided to obtain kinetic data using the suboptimal concentration of 100 mM Mg2+ in TW17 ribozyme catalysis as it still bestowed 65% of the normalized maximum kobs and allowed us to directly compare the results to those of previous ribozyme studies.18
We also performed [Co(NH3)6]3+ titration of the reaction catalyzed by the TW17 ribozyme to differentiate the relative importance of inner- and outer-sphere Mg2+ in sustaining optimal ribozyme catalysis. In aqueous solutions, Mg2+ can coordinate with six water molecules to form [Mg(H2O)6]2+, in which the hexahydrated Mg2+ complex can electrostatically attract appropriate functional groups in RNA by outer-sphere coordination.4 Exchange-inert [Co(NH3)6]3+ is very similar in size to [Mg(H2O)6]2+ and has been shown to serve as a substitute for [Mg(H2O)6]2+ for supporting ribozyme catalysis.4,5 Indeed, we observed a [Co(NH3)6]3+-dependent increase of TW17 ribozyme activity when including 50 μM [Co(NH3)6]3+ to the ribozyme catalysis reaction previously performed in the presence of only 0.5 mM of Zn2+ (lanes 4 and 8 in Fig. S2†). In addition, [Co(NH3)6]3+ alone did not result in any detectable TW17 ribozyme catalytic activity, which was consistent with the results and suggested a secondary and structural role of Mg2+ in ribozyme catalysis.18 When acquiring the kobs of TW17 ribozyme catalysis as a function of [Co(NH3)6]3+ concentrations in the presence of a constant concentration of 0.5 mM Zn2+, we obtained values for kobs that unexpectedly fluctuated between 0.006 min−1 and 0.016 min−1 for ribozyme catalysis when titrating [Co(NH3)6]3+ from 0.5 μM to 1000 μM (Fig. 1D). Moreover, these values of kobs were all smaller than the kobs,max value of 0.052 min−1 obtained from the Mg2+ titration of TW17 ribozyme catalysis (Fig. 1C), which indicates the insufficiency of the combination of [Co(NH3)6]3+ and Zn2+ for appropriate TW17 ribozyme catalysis. Consequently, the results of the Zn2+-only titration (Fig. 1B), and also of the Mg2+ and [Co(NH3)6]3+ titrations in the presence of [Zn2+] = 0.5 mM (Fig. 1C and D) indicated that both inner-sphere and outer-sphere Mg2+ are equally required for optimal TW17 ribozyme catalysis.
TW17 ribozyme catalysis is facilitated in alkaline conditions
Similar to protein enzymes for functional group transfer reactions, the catalytic mechanisms of ribozymes generally involve proton transfer in general acid–base catalysis to achieve a rate enhancement.7,8 Understanding the acid–base chemistry of ribozymes is essential to comprehending the intricate reaction mechanisms of ribozyme catalysis. Consequently, we performed TW17 ribozyme catalysis in varying pH conditions to evaluate the influence of pH on ribozyme catalysis and to shed light on the detailed catalysis mechanism of the ribozyme (Fig. 2). The TW17 ribozyme exhibited barely any detectable activity (kobs ∼0) when the pH of the reaction was below 6.5. The kobs gradually increased as the pH of the reaction increased from 6.5 to 7.4, at which point the TW17 ribozyme displayed a drastic increase in kobs. The TW17 ribozyme had a similar kobs at all pH levels between 7.5 and 8.0. The TW17 ribozyme, however, gave a significant fluctuation of the kobs values when the pH of the reaction mixture was higher than 8.0. Since an increase in the catalytic rates in alkaline conditions is one of the characteristics in the mechanism of general base catalysis, the pH titration data thus suggested that TW17 ribozyme catalysis could involve a general base. However, the current pH titration results deviate significantly from those typically observed with general base catalysis40 and prevented the fitting of the kobs data of TW17 ribozyme catalysis at each pH in order to acquire the values of pKa for a general base and the kobs,max in the presence of the general base. Nevertheless, the TW17 ribozyme unequivocally demonstrated the ability to accelerate catalysis in alkaline conditions.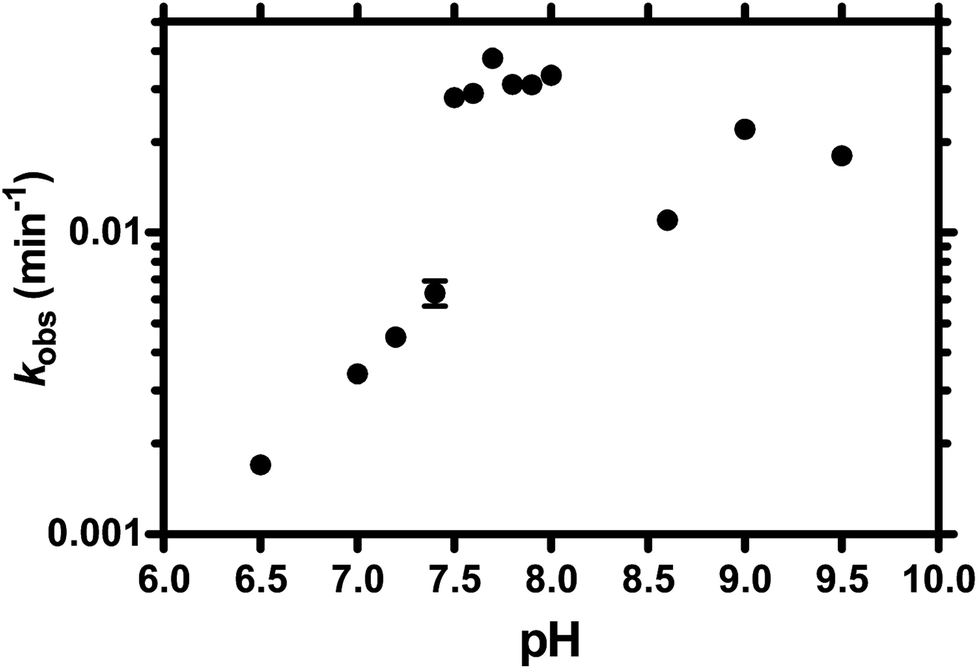 | ||
| Fig. 2 Evidence for the requirement for alkaline conditions in TW17 ribozyme catalysis. Reactions were carried out under the conditions provided in the Experimental section. The values of kobs measured in the presence of buffer solutions with specific pH values were determined as before. Each data point was the mean value of analyses performed in triplicate, with the standard deviation represented by an up and down bar. The standard deviation bars for some data points are too small to be visible. | ||
Additional base-pairings between substrate-bearing and catalytic RNA molecules in trans-acting TW17 ribozyme catalysis systems unable to enhance their binding affinities
The metal titration studies (Fig. 1) provided critical information concerning the appropriate Zn2+ and Mg2+ concentrations for optimal TW17 ribozyme catalysis, and suggested a strategy to improve the catalysis of a previously reported trans-acting TW17 ribozyme system with KM = 1.77 ± 1.20 μM, kcat = 1.65 ± 0.80 min−1, and kcat/KM = 1.6 × 104 ± 1.3 × 104 s−1 M−1.18 The trans-acting TW17 ribozyme system consisted of the substrate-bearing S1–18 RNA (designated as TW17S-1 RNA in this study) and the catalysis-active TW1722–87 ribozyme RNA (renamed as TW17C-1 RNA here), and exploited 6–8 base-pairings (bp) in the P1 region to form a complex between these two RNA molecules (Fig. S3†).18 We reasoned that the trans-acting TW17 ribozyme system might confer better catalytic efficiency in the presence of the appropriate Zn2+ and Mg2+ concentrations required under optimal reaction conditions. In addition, the catalytic efficiency of the trans-acting TW17 ribozyme system might be further improved if the affinity between the substrate-bearing RNA and the catalytic core RNA in the trans-acting system could be increased. We thus decided to first modify the RNA structures in order to increase both the substrate–catalyst RNA affinity and the catalytic efficiency in trans-acting TW17 ribozyme systems. Greater catalytic efficiency in a trans-acting TW17 ribozyme system with modified RNA structures could then result from amending the optimal Zn-dependent reactions in the presence of appropriate Zn2+ and Mg2+ concentrations.New trans-acting TW17 ribozyme systems utilizing modified RNA structures were obtained by increasing the numbers of bp in the P1 helix through site-directed mutagenesis and sequence insertions in this region (Fig. S3†). The syntheses of the RNA molecules in the different trans-acting TW17 ribozyme systems are described in Tables S1 and S2.† We initially expected that an incremental increase in the number of bp in the P1 helix would promote the binding of substrate-bearing RNA to catalysis-active RNA and eventually increase the catalytic efficiencies in the trans-acting TW17 ribozyme systems. However, analysis of Langmuir binding isotherms as described in Table S3† revealed that the additional bp in the P1 helix never translated to a higher binding affinity—namely, a lower Kd and a more negative ΔGbinding—in the structure-modified trans-acting TW17 ribozyme systems (Table S4†). On the contrary, more bp actually decreased the substrate–catalyst RNA binding affinities in almost all of the studied trans-acting TW17 ribozyme systems (Table S4†). Thus, fine-tuning bp in the P1 region is not a legitimate approach to improving the catalytic efficiency in trans-acting TW17 ribozyme systems.
GNRA tetraloop motifs essential to optimal trans-phosphorothiolate thiolesterase activity of the TW17 ribozyme
The incoherent correlation between the number of bp in the P1 region and ribozyme catalysis of trans-acting TW17 ribozyme systems led us to further scrutinize the structure of the cis-acting TW17 ribozyme (Fig. S4†)18 and to evaluate the importance of a GAGA tetraloop (L1 in Fig. S4†) for capping the P1 helix in the RNA. The GAGA tetraloop was dissected in the original experiments involving the trans-acting TW17 ribozyme system (the TW17S-1 RNA + TW17C-1 RNA system) and was further disrupted in later experiments using the trans-acting systems for Langmuir isotherm analysis (Fig. S3†). The GAGA tetraloop belongs to a previously described family of GNRA tetraloops.19–21 Thus, the structure of the GAGA tetraloop might engage in a long-range interaction with a corresponding receptor RNA motif and might act to stabilize the tertiary structure of the TW17 ribozyme. This stabilization could be essential for optimal catalysis of the ribozyme. We therefore constructed two GAGA tetraloop-containing trans-acting TW17 ribozyme systems (Fig. 3A and B) and evaluated whether the presence of the GAGA tetraloop improved the catalytic efficiency of the ribozyme.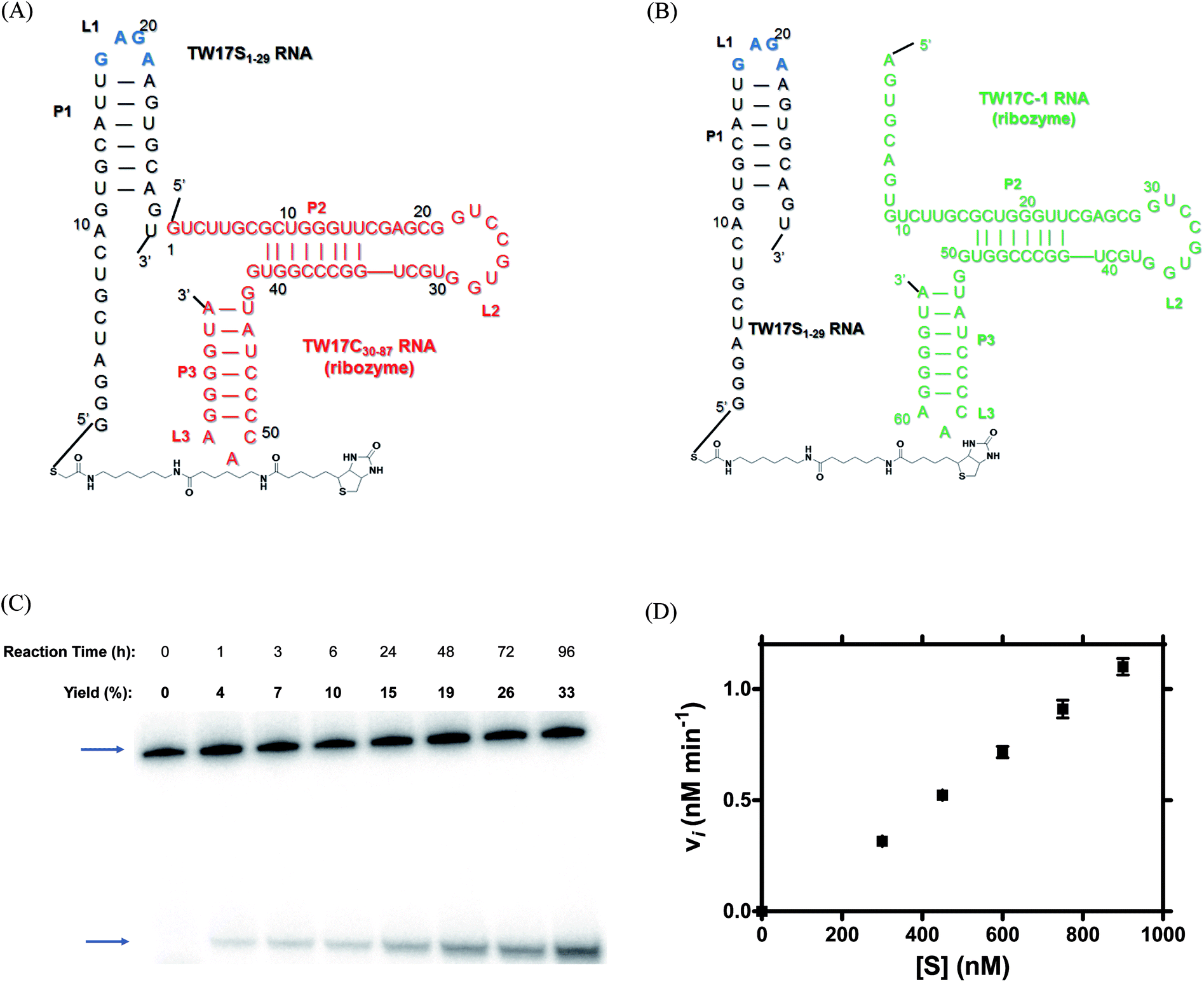 | ||
| Fig. 3 Secondary structures of two GAGA tetraloop-containing trans-acting TW17 ribozyme systems and multiple substrate turnover kinetics of one of the GAGA tetraloop-containing trans-acting TW17 ribozyme systems. (A) The trans-acting ribozyme system consisted of the TW17S1–29 RNA (highlighted in black) and TW17C30–87 RNA (highlighted in red). (B) In the trans-acting TW17S1–29 RNA–TW17C-1 RNA system, the TW17C-1 RNA (highlighted in green) was substituted for the TW17S30–87 RNA used in (A). The GAGA tetraloop is highlighted in blue within the structure of the TW17S1–29 RNA in both (A) and (B). In addition, the substrate 18a is covalently linked to the 5′ end of the TW17S1–29 RNA. (C) Multiple substrate turnover reactions of the trans-acting TW17S1–29 RNA-TW17C-1 system in the presence of the Zn2+ concentration of 0.5 mM and Mg2+ concentration of 37.5 mM were analyzed by SAv gel-shift assay in 20% urea-PAGE. The catalysis in the multiple substrate turnover reactions was measured by including 300 nM of the 32P-labeled 18a-TW17S1–29 RNA and 30 nM of the TW17C-1 RNA in the reactions. The image was acquired using an Amersham Typhoon PhosphorImager system and was quantified by ImageQuant software. The top blue arrow in the image represents the location of the SAv-retarded 32P-labeled 18a-TW17S1–29 RNA conjugate; the bottom blue arrow indicates the migration of the 32P-labeled TW17 ribozyme-catalyzed reaction product. The initial velocity (vi) was acquired by measuring the slope of the time-course curve from 0 to 1 h in the reaction, and was determined to be 0.2 nM min−1 for the reaction. The reaction afforded a product yield of 33% after 4-d catalysis. (D) Multiple substrate turnover in the trans-acting TW17S1–29 RNA-TW17C-1 system was studied in the presence of a constant 30 nM of the catalytic TW17C-1 ribozyme and an excess of the 32P-labeled 18a-S1–29 RNA conjugate concentrations, ranging from 300 to 900 nM. Initial velocity data for each 18a-S1–29 RNA conjugate concentration were acquired by measuring the slope of the time-course curve within the first 30 min of the reaction. Each data point in the figure is the average of two separate measurements. | ||
With no requirement to further modify the structure of the TW17 ribozyme, except for dividing the cis-acting ribozyme into two RNA fragments, we first determined the optimal metal concentrations for TW17 ribozyme catalysis, because the previous experiments with the trans-acting TW17 ribozyme system yielded poor catalytic efficiency under the standard reaction condition of [Zn2+] = 0.5 mM and [Mg2+] = 100 mM.18 Metal titration studies had already indicated that the cis-acting TW17 ribozyme performed better in the presence of either [Zn2+] = 1.25 mM or [Mg2+] = 37.5 mM (Fig. 1). Since the cis-acting TW17 ribozyme was easier to synthesize and had demonstrated more efficient catalysis than the original trans-acting TW17 ribozyme system, we again exploited the cis-acting RNA to acquire appropriate metal concentration combinations for improved TW17 ribozyme catalysis. We studied TW17 ribozyme catalysis in the presence of the following three different metal concentration combinations: (1) [Zn2+] = 1.25 mM and [Mg2+] = 100 mM; (2) [Zn2+] = 0.5 mM and [Mg2+] = 37.5 mM; (3) [Zn2+] = 1.25 mM and [Mg2+] = 37.5 mM. Fig. S5† clearly shows that the TW17 ribozyme, in the presence of a Zn2+ concentration of 0.5 mM and a Mg2+ concentration of 37.5 mM, produced the highest catalytic efficiency among the three by providing a pseudo first-order rate constant kobs of 0.041 min−1, which was 0.46-fold higher than the kobs value of 0.028 min−1 previously obtained in the standard reaction of TW17 ribozyme catalysis.18 The Zn2+ concentration of 0.5 mM and the Mg2+ concentration of 37.5 mM resulted in optimal catalysis of the TW17 ribozyme and were thus used in subsequent kinetic studies of the trans-acting TW17 ribozyme systems.
Time-course studies of TW17 ribozyme catalysis by the GAGA tetraloop-containing trans-acting systems (Fig. 3A and B) again confirmed that TW17 ribozyme catalysis in the presence of [Zn2+] = 0.5 mM and [Mg2+] = 37.5 mM was more efficient than when performed under the standard reaction condition (Fig. 3C and S6†). Additionally, the trans-acting TW17S1–29 RNA–TW17C-1 RNA system unexpectedly demonstrated more efficient catalysis than the TW171–29 RNA–TW17C30–87 RNA system in all of the reaction conditions studied here. We initially assumed that the 5′ terminal segment in the TW17C-1 RNA might base-pair with the P1 helix in the TW17S1–29 RNA to destabilize the GAGA motif, leading to impaired TW17 ribozyme catalysis. The results shown in Fig. 3C and S6† thus suggest that destabilization of the secondary structure of the P1 helix might be required for optimal TW17 ribozyme catalysis. As the trans-acting TW17S1–29 RNA–TW17C-1 RNA system in the presence of the Zn2+ concentration of 0.5 mM and the Mg2+ concentration of 37.5 mM provided the largest initial velocity (vi) of 0.2 nM min−1 and a 4 day yield of 33% (Fig. 3C), we decided to use these conditions in further studies of the multiple turnover catalysis.
The multiple substrate turnover capacity of the trans-acting TW17S1–29 RNA–TW17C-1 ribozyme system was determined by measuring the vi of the trans-acting TW17 ribozyme catalysis system under varied concentrations of the substrate-RNA (18a-TW17S1–29 RNA; 300–900 nM), a constant ribozyme (the TW17C-1 ribozyme) concentration (30 nM), and constant substrate excess. Initial velocity (vi) versus substrate concentration ([S]) of the 18a-TW17S1–29 RNA were plotted (Fig. 3D) but did not provide reasonable values for the essential kinetic parameters, including for KM and kcat for the in trans TW17 ribozyme catalysis system. Specifically, when performing nonlinear curve fitting of the data to the Michaelis–Menten equation, we obtained very large KM and kcat values, with their standard errors even larger (results not shown). The acquisition of unrealistic values of KM and kcat could have been predicted from Fig. 3D, which clearly indicates that the range of substrate concentrations utilized was not broad enough to accurately determine the kinetic constants for the in trans TW17 ribozyme catalysis system in this study. Nevertheless, the trans-acting TW17S1–29 RNA–TW17C-1 ribozyme system provided values of vi and product yields (Fig. 3 and S6†) significantly superior to those acquired from the original in trans TW17 ribozyme catalysis system (vi of 0.027 nM min−1 and 24 h yield of 5%)18 under the same concentrations of substrate and catalyst RNA molecules. This finding clearly demonstrates the requirement for the GAGA tetraloop in the TW17 ribozyme for optimal catalysis and provides essential information for designing additional catalysis-efficient multiple substrate turnover trans-acting systems for the ribozyme.
Discussion
We successfully synthesized and exploited 6 and 18b to select the TW17 ribozyme from an artificial RNA pool (Schemes 2–4). In addition, we meticulously synthesized 10a, 10b, and 18a because these compounds were essential for characterizing the substrate specificity of the TW17 ribozyme (Schemes 3 and 4). The effective synthesis of 6 was significant because the compound provided the critical molecular handle to link small substrate molecules, including 10 and 18, to the TW17 ribozyme. We developed an improved strategy to synthesize 6 from 1 with an overall yield of 46% by avoiding the use of the troubling trialkyl phosphate solvent, which complicates the reaction with thiophosphoryl chloride and gives rise to undesirable oxygen/sulfur exchange products.30 The successful high-yield synthesis of 6 provided a crucial impetus for the discovery of the TW17 ribozyme in addition to a straightforward approach to specifically modify RNA at the 5′ termini.The synthesis of 18a was also important in this study as the structure of 18a differs from that of 18b—the substrate used in the in vitro selection of the TW17 ribozyme—by only a disulfide bond not specifically cleaved by ribozyme catalysis.18 The current study demonstrated that the synthesis of 18a provides a much higher yield (54.9% from 14 to 18a) than the synthesis of 18b (16.8% from 14 to 18b), while both compounds afforded the TW17 ribozyme conjugates with identical catalytic activity. Ready access to 18a provided a key advantage when synthesizing the TW17 ribozyme-18a conjugate for studying the kinetics of ribozyme catalysis and for understanding the intricate roles of divalent metal ions and tertiary interactions in ribozyme catalysis.
We investigated the critical roles of Zn2+ and Mg2+ in TW17 ribozyme catalysis by performing metal titration during ribozyme kinetics to gain insights into the simultaneous interactions of Zn2+ and Mg2+ with the ribozyme in the ion atmosphere and to optimize the hydrolytic reaction (Table 1). The results from the Zn2+ titration of TW17 ribozyme catalysis in the presence of a constant concentration of 100 mM Mg2+ indicated that the ribozyme absolutely requires specifically associated Zn2+ ions in the ion atmosphere to accelerate phosphorothiolate thiolester hydrolysis (Fig. 1A, Scheme 1 and Table 1). The role of the specifically associated Zn2+ in the TW17 ribozyme can be inferred from several Zn protein enzyme studies. Hydrolytic reactions catalyzed by many modern zinc-dependent protein hydrolases require the catalytic involvement of zinc ions that are often bound to proteins through inner-sphere coordination.14,15,17,25 Inner-sphere zinc in these proteins is commonly coordinated by thiolates in the side chains of cysteine residues. Moreover, NMR and XAS analyses of RNase P revealed that zinc ions employ inner-sphere coordination with one or more RNA ligands in the ribozyme.41 The results of these studies support the idea that Zn2+ ions crucial to TW17 ribozyme catalysis could be specifically bound to the ribozyme by inner-sphere coordination in the ion atmosphere. Further, the sulfur moiety in GMPS could be one of the ligands that coordinate Zn2+ in the 18a-TW17 ribozyme conjugate. We could not determine the coordination numbers or the geometries of the Zn2+ cations specifically associated with the TW17 ribozyme. The flexible coordination number (between four and six) and geometry (tetrahedral, trigonal bipyramidal, square pyramidal, and octahedral) of Zn2+ in zinc-dependent protein metalloenzymes, however, suggest similar possibilities for Zn2+ in the TW17 ribozyme.14,15,17,25 We believe that one of the essential Zn2+ ions in the TW17 ribozyme active site coordinates with the ribozyme by four-folded inner-sphere interactions and uses the phosphorothiolate sulfur as one of its ligands (vide infra). However, advanced structural studies of the TW17 ribozyme are required to elucidate the coordination spheres and geometries of the specifically bound Zn2+ in the ribozyme.
In addition to the indispensable role of Zn2+ in TW17 ribozyme catalysis, the ribozyme also requires the inner-sphere and outer-sphere Mg2+ in the ion atmosphere for optimal catalysis. Titration studies of systematically changing Zn2+ concentrations in the absence of Mg2+ and of varying Mg2+ concentrations in the presence of a constant concentration of 0.5 mM Zn2+ in TW17 ribozyme catalysis provided evidence that the ribozyme could prime its structure for optimal catalysis in the presence of Mg2+ (Fig. 1B, C and Table 1) in the ion atmosphere. The importance of specifically associated outer-sphere Mg2+ for maintaining the TW17 ribozyme structure for optimal ribozyme catalysis was indicated by our [Co(NH3)6]3+ titration studies of ribozyme kinetics (Fig. 1D and S2†). We unequivocally demonstrated that the presence of [Co(NH3)6]3+ enhanced the catalytic activity of the ribozyme (Fig. S2†). It is noted that a similar kobs could be obtained when using [Co(NH3)6]3+ concentrations nearly two orders of magnitude lower than that of Mg2+. This result is as expected because [Co(NH3)6]3+ has a larger charge than Mg2+ and, therefore, a greater capacity to stabilize the ribozyme structure.41 Incrementally increasing the [Co(NH3)6]3+ concentration in the presence of 0.5 mM Zn2+, however, never resulted in wild-type catalytic activity for the TW17 ribozyme (kobs = 0.028 min−1; Fig. 1D). These results argue against a catalytic role of outer-sphere Mg2+ in TW17 ribozyme catalysis and support the conclusion that the main function of the outer-sphere Mg2+ is to stabilize the ribozyme structure for effective catalysis. Alternatively, the importance of specifically associated inner-sphere Mg2+ in the ion atmosphere of the TW17 ribozyme was also supported by the [Co(NH3)6]3+ titration study of ribozyme catalysis in the presence of a constant concentration of 0.5 mM Zn2+ (Fig. 1D and S2†). As discussed above, additions of any concentration of [Co(NH3)6]3+ never restored the wild-type activity of TW17 ribozyme catalysis. Moreover, the results of TW17 ribozyme catalysis titrated by Zn2+ exclusively also revealed that Zn2+ alone could also not fully recover the wild-type activity of the ribozyme (Fig. 1B). Consequently, the TW17 ribozyme demands the presence of both inner-sphere and outer-sphere Mg2+ in the ion atmosphere to modulate the ribozyme structure and to attain optimal ribozyme catalysis.
Titration studies of TW17 ribozyme catalysis at various pH levels provide crucial evidence indicating a requirement for alkaline conditions in ribozyme catalysis (Fig. 2). It is possible that TW17 ribozyme catalysis follows a general base mechanism. The current pH titration study, however, did not provide conclusive evidence to support the presence of a general base and to determine the values of pKa and kobs,max for the general base. Nevertheless, we currently favor the hypothesis that hydrated Zn2+ plays the role of a general base to coordinate with the TW17 ribozyme by inner-sphere interactions at the active site. Deprotonation of a coordinated water ligand in the hydrated Zn2+ is thus indispensable for it to assume the proposed role of a general base. However, since solvated Zn(aq)2+ has a pKa of ∼9,42 it implies that less than 90% of Zn(aq)2+ will be deprotonated at the optimal pH for ribozyme catalysis (pH 7.5–8.0; Fig. 2). Consequently, for effective TW17 ribozyme catalysis, the hydrated Zn2+ in the ribozyme must increase the acidity of a water ligand and decrease its pKa from 9. The feasibility of an increase in the acidity of the hydrated Zn2+ in the TW17 ribozyme is again supported by a previous study demonstrating the deprotonation process of a coordinated water ligand from Zn(MeOPS)(aq) to Zn(MeOPS)(OH)−.43 Significantly, the obtained pKa for the deprotonation process of Zn(MeOPS)(aq) was 6.9 ± 0.2 and the considerable shift in acidity was attributed to the effects of the sulfur ligand and reduction of the Zn2+ coordination number from 6 to 4. We believe that the hydrated Zn2+ in the TW17 ribozyme also exploits four-folded inner-sphere coordination with the GMPS sulfur as one of its ligands to dramatically decrease the pKa of a coordinated water ligand from 9 in this study. However, we do not rule out the possibility that one or more of the ribozyme nucleotides, such as guanine, acts as a general base to facilitate the proton transfer with the hydrated Zn2+.8 Nevertheless, the deprotonated form of the hydrated Zn2+ specifically bound to the TW17 ribozyme active site is well positioned in the alkaline conditions essential to ribozyme catalysis. Interestingly, modern zinc-dependent protein hydrolases also take advantage of the same zinc chemistry to catalyze hydrolytic reactions by using zinc ions to coordinate a nucleophilic water ligand and to lower its pKa.14,15,17,25 The current research is thus the first study to raise the possibility that catalytic RNA and protein enzymes could make use of the same fundamental properties of zinc coordination chemistry to accelerate hydrolysis reaction rates.
In addition to the essential functions of Zn2+ and Mg2+ in TW17 ribozyme catalysis, tertiary interactions and partial destabilization of the secondary structure elements in the ribozyme also play distinct roles in the optimal catalytic activity of the TW17 ribozyme. For example, GAGA tetraloop–receptor interaction in the TW17 ribozyme is absolutely required for optimal ribozyme catalysis (Fig. 3 and S6†). Moreover, structural perturbation of the P1 stem, capped by the GAGA tetraloop in the TW17 ribozyme, may promote the ribozyme's sampling of active conformations in order to perform optimal catalysis. The requirement of the partially destabilized P1 stem for the GAGA tetraloop–receptor interaction also substantiates the peculiar observation that incorporating additional base pairs in the P1 stem of the TW17 ribozyme has adverse effects on the interactions of both the substrate-bearing and catalytic TW17 RNA molecules (Table S4†). Decreased stability in the secondary structure elements in order to form a tertiary RNA structure has been noted in cooperative RNA folding in the presence of molecular crowders.44
We propose the following reaction mechanism of TW17 ribozyme catalysis based on key insights gained from both the metal and pH titration experiments and also from the secondary and tertiary interaction studies of ribozyme catalysis (Scheme 5). The mononuclear divalent metal ion center of the TW17 ribozyme is proposed to be the deprotonated form of the hydrated Zn2+ (green in Scheme 5) coordinating with the ribozyme by four-folded inner-sphere interactions. In addition, the folding of the TW17 ribozyme to form the proper structure of the catalytic site requires the presence of specifically associated inner-sphere and outer-sphere Mg2+ in the ion atmosphere (blue-colored “M2+” in Scheme 5). Finally, GAGA tetraloop–receptor interaction (cyan-colored curved double arrow in Scheme 5) is also indispensable, and tunes the TW17 ribozyme structure for optimal catalysis.
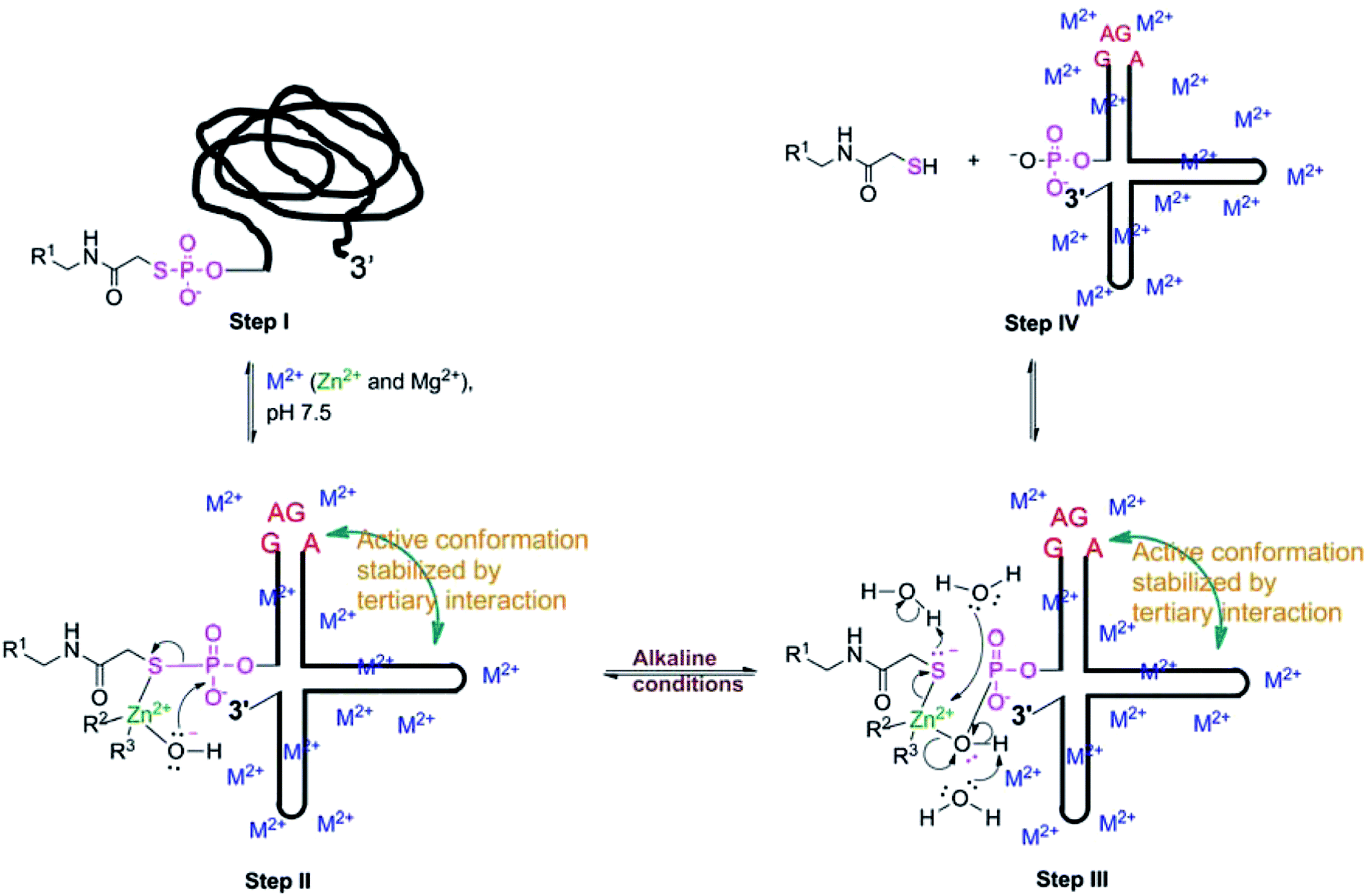 | ||
| Scheme 5 The proposed mechanism of optimal TW17 ribozyme catalysis. | ||
In the proposed reaction mechanism of TW17 ribozyme catalysis in Scheme 5, the catalytic cycle begins at Step I with the ribozyme structure having no catalytic activity. In the presence of the optimal concentrations of Zn2+ and Mg2+ ([Zn2+] = 0.5 mM and [Mg2+] = 37.5 mM), the TW17 ribozyme, aided by the GAGA tetraloop–receptor interaction, folds into the most active conformation (Step II in Scheme 5). The appropriate active structure in alkaline conditions allows the Zn2+-coordinated hydroxide ion to initiate a nucleophilic attack on the adjacent electron-deficient phosphorus in the phosphorothiolate to form a square planar transition state structure which is different from the trigonal bipyramidal structure typically found in self-cleaving ribozyme reactions.5 The instability of the transition state structure leads to the breaking of the phosphorothiolate thiolester bond as well as the linkage between the mononuclear Zn2+ and hydroxide (Steps III and IV in Scheme 5). The rapid ligand exchange effected by the properties of Zn2+ facilitates the departing hydroxide ligand in the mononuclear Zn2+ to be simultaneously replaced by a water ligand. Protonation of the thiolate and deprotonation of a water ligand in the mononuclear Zn2+ center are likely achieved by water molecules surrounding the TW17 ribozyme. More structural analysis of the TW17 ribozyme is required to further correlate the results from biochemical studies of ribozyme kinetics with the proposed catalytic mechanism to better comprehend the unique roles of divalent metal ions in ribozyme catalysis.
Conclusion
We extensively characterized the crucial roles of Zn2+, Mg2+, and a GAGA tetraloop–receptor interaction in TW17 ribozyme catalysis and confirmed the importance of these concurrent interactions in optimizing the hydrolytic reactions catalyzed by the ribozyme. We accomplished this characterization in four ways. First, we successfully synthesized 6 and 18b, which are each required for the in vitro selection of the TW17 ribozyme, a Zn2+-dependent phosphorothiolate thiolesterase RNA (Scheme 1). Three additional compounds—10a, 10b and 18a—were also synthesized and proved to be essential for delineating the chemical mechanism of TW17 ribozyme catalysis and elucidating the critical functions of Zn2+, Mg2+, and the tertiary interactions in the hydrolytic reaction. The effective organic synthesis of 6 and 18a with good yields permitted the production of ample amounts of the 18a-TW17 ribozyme conjugate, which was necessary to apprehend the Zn2+-dependent catalytic mechanism of the ribozyme. Second, our metal titration studies of TW17 ribozyme catalysis provided strong evidence that the ribozyme requires specifically associated Zn2+ and outer-sphere and inner-sphere equivalences of Mg2+ in the ion atmosphere of the ribozyme for optimal catalysis. Third, we demonstrated a requirement for alkaline conditions for TW17 ribozyme catalysis by measuring the ribozyme activity under various pH conditions. Fourth, we provided substantial evidence that the tertiary structure formation contributed by the GAGA tetraloop–receptor interaction in the TW17 ribozyme was vital for optimal ribozyme catalysis.Critical information obtained from the current study leads us to propose that optimal TW17 ribozyme catalysis is a Zn2+-dependent catalytic mechanism, requiring the presence of Mg2+ and a GAGA tetraloop–receptor interaction. We believe that a hydrated Zn2+ ion specifically bound to the TW17 ribozyme has one of its water ligands deprotonated in alkaline conditions and this is essential for ribozyme catalysis. Significantly, it has been well documented that a hydrated zinc plays a similar role in Zn-dependent protein hydrolases involved in group transfer reactions, including hydrolytic reactions. Our current studies of the hydrolytic reaction mechanism catalyzed by the TW17 ribozyme thus suggest that both ribozymes and protein enzymes employ the same zinc coordination chemistry to accelerate the rates of group transfer reactions. Metabolic processes in the primordial RNA world may have taken advantage of the chemical properties of zinc similar to those extensively employed in the catalytic biological reactions of modern living organisms.
Experimental section
Most reagent-grade chemicals were purchased from commercial sources (Sigma-Aldrich, Acros, Alfa Aesar, Mallinckrodt Baker, and Merck KGaA), and further purified, if necessary. In addition, sulfo-NHS-LC-biotin was acquired from Pierce (Rockford, IL, USA). DEAE Sepharose Fast Flow was purchased from GE Healthcare (Taipei, Taiwan). 1H and 13C NMR spectra were recorded using either a Varian 200 or 400 MHz spectrometer at Kaohsiung Medical University, Taiwan (KMU). NMR samples were prepared in CD3OD, C2D6OS, CDCl3, or D2O; chemical shifts of 1H signals were reported in parts per million downfield from TMS. 13C signals were given in parts per million based on the internal standard of each deuterated solvent. 31P signals were reported as parts per million downfield from 85% H3PO4. ESI high-resolution mass spectra were acquired using a Bruker APEX II Fourier-transfer mass spectrometer (FT-MS) at the Department of Chemistry, National Sun Yat-Sen University, Taiwan. FAB high-resolution mass spectra were produced by a JEOL JMS-700 instrument at the Department of Chemistry, National Cheng Kung University, Taiwan.Synthesis of the ammonium salt of guanosine α-thio-monophosphate (6)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 15 min) into two fractions: a yellowish supernatant and a light-yellow precipitate. The supernatant fraction was worked up three times in the following manner: two volumes of 95% ethanol were added to promote precipitation, followed by centrifuging two times at 13000 rpm for 15 min to separate the newly acquired precipitate from the supernatant, collecting and pooling the precipitate with the precipitate fraction, and keeping the remaining supernatant for the next workup by repeating the above procedures. The pooled precipitate fraction was washed sequentially with water and pyridine, and ultimately centrifuged at 13000 rpm for 15 min to separate the precipitate from the supernatant. The afforded supernatant was later worked up twice by the 95% ethanol precipitation procedures described above to provide a precipitate requiring no further purification. All the precipitates were pooled a second time, washed by acetone, and lyophilized to afford 4 (1.3 g; 75%) as a light-yellow solid. 1H NMR (400 MHz) (D2O) δ: 8.10 (s, 1H), 6.03 (d, 1H), 5.30 (dd, 1H), 5.19 (dd, 1H), 4.468 (m, 1H), 3.95 (m, 2H), 1.58 (s, 3H), 1.37 (s, 3H). The 13C NMR spectrum of 4 is also not available due to limited solubility of the compound in D2O. HRMS (FAB) calculated for C13H17O7N5PSBa, [M + H]+ 555.9639 (calcd), 555.9640 (found).000 rpm, 15 min) to afford the desired supernatant. The acquired aqueous phase was mixed with 4–5 volumes of acetone and centrifuged at 13000 rpm for 15 min to ensure efficient recovery of the precipitate, which was subsequently lyophilized to afford 5 (0.4 g, 0.88 mmol; 98%) as a white solid. The 1H spectrum of 5 was also identical to that of 4. Again, acquisition of the 13C NMR spectrum and HRMS data of 5 was not feasible due to the limited solubility of the compound in D2O.000 rpm for 15 min to separate the precipitate from the remainder of the solution, at which point the supernatant was removed and the precipitate was washed twice with acetone. The precipitate was lyophilized to afford a crude ammonium salt of O-[(2R,3S,4R,5R)-5-(2-amino-6-oxo-1,6-dihydro-purin-9-yl)-3,4-dihydroxy-tetrahydro-furan-2-ylmethyl]ester (6; 0.30 g) as a light-yellow solid.
000 rpm, 15 min) into two fractions: a yellowish supernatant and a light-yellow precipitate. The supernatant fraction was worked up three times in the following manner: two volumes of 95% ethanol were added to promote precipitation, followed by centrifuging two times at 13000 rpm for 15 min to separate the newly acquired precipitate from the supernatant, collecting and pooling the precipitate with the precipitate fraction, and keeping the remaining supernatant for the next workup by repeating the above procedures. The pooled precipitate fraction was washed sequentially with water and pyridine, and ultimately centrifuged at 13000 rpm for 15 min to separate the precipitate from the supernatant. The afforded supernatant was later worked up twice by the 95% ethanol precipitation procedures described above to provide a precipitate requiring no further purification. All the precipitates were pooled a second time, washed by acetone, and lyophilized to afford 4 (1.3 g; 75%) as a light-yellow solid. 1H NMR (400 MHz) (D2O) δ: 8.10 (s, 1H), 6.03 (d, 1H), 5.30 (dd, 1H), 5.19 (dd, 1H), 4.468 (m, 1H), 3.95 (m, 2H), 1.58 (s, 3H), 1.37 (s, 3H). The 13C NMR spectrum of 4 is also not available due to limited solubility of the compound in D2O. HRMS (FAB) calculated for C13H17O7N5PSBa, [M + H]+ 555.9639 (calcd), 555.9640 (found).000 rpm, 15 min) to afford the desired supernatant. The acquired aqueous phase was mixed with 4–5 volumes of acetone and centrifuged at 13000 rpm for 15 min to ensure efficient recovery of the precipitate, which was subsequently lyophilized to afford 5 (0.4 g, 0.88 mmol; 98%) as a white solid. The 1H spectrum of 5 was also identical to that of 4. Again, acquisition of the 13C NMR spectrum and HRMS data of 5 was not feasible due to the limited solubility of the compound in D2O.000 rpm for 15 min to separate the precipitate from the remainder of the solution, at which point the supernatant was removed and the precipitate was washed twice with acetone. The precipitate was lyophilized to afford a crude ammonium salt of O-[(2R,3S,4R,5R)-5-(2-amino-6-oxo-1,6-dihydro-purin-9-yl)-3,4-dihydroxy-tetrahydro-furan-2-ylmethyl]ester (6; 0.30 g) as a light-yellow solid.The obtained crude 6 (0.30 g) was dissolved in 0.1 M ammonium bicarbonate (pH 8.0; 25 mL), loaded onto a DEAE Sepharose Fast Flow (GE Healthcare) column, and eluted with 0.1 M ammonium bicarbonate isocratically. The eluate in each fraction was diluted 100-fold and the absorbance of each fraction was measured at 253 nm. Fractions with high A253 values (>10) were pooled and concentrated by either lyophilizing or reducing the pressure in vacuo. The acquired solid was resuspended in 1.5 mL of water; the resuspended solid was subsequently subjected to four cycles of the addition of 95% of ethanol (20 mL), centrifugation at 13000 rpm for 15 min, and collection of the precipitate in sequence. The final precipitate was lyophilized to acquire 6 (0.23 g, 0.56 mmol; 64%) as a white solid. 1H NMR (400 MHz) (D2O) δ: 8.16 (s, 1H, NH), 5.82 (d, 1H), 4.43 (dd, 1H), 4.25 (m, 1H), 3.96 (m, 1H). 13C NMR (100.67 MHz) (C2D6OS) δ: 159.1, 154.1, 151.9, 138.0, 116.2, 86.8, 84.7, 74.2, 71.0, 64.0, 64.0. 31P NMR (161.92 MHz) (D2O) δ: 43.65 (s, 1P). HRMS (FAB) calculated for C10H21O7N7PS, [MH+] 414.0961 (calcd), 414.0962 (found).
Synthesis of substrates for TW17 ribozyme catalysis
The syntheses of 8a, 12, 13, 14, 15a, and 16a have been reported previously.35 However, the detailed procedures for the syntheses of 8b, 9a, 9b, 10a, 10b, 15b, 16b, 17, 18a, and 18b are described as follows.:2) and loaded onto a pre-equilibrated silica column. The products were sequentially separated by eluting with solutions of 1:2, 1:1, and 2:1 ratios of EA to hexane. Fractions containing the desired product were pooled and evaporated under reduced pressure to obtain 9a (0.14 g; 35.5%). 1H NMR (400 MHz) (CDCl3) δ: 6.71 (s, 1H, NHCOCH2), 4.66 (s, 1H, OCONH), 4.54 (s, 1H, CONH), 3.88 (s, 2H, COCH2Br), 3.28 (q, 2H, CH2NH), 3.13 (q, 2H, CH2NH), 1.34–1.56 (s, 9H, CH3; m, 8H, CH2). 13C NMR (100.67 MHz) (CDCl3) δ: 165.45, 156.02, 40.67, 40.59, 40.54, 29.87, 28.85, 28.41, 27.15, 26.91. HRMS (ESI) calculated for C8H18BrN2O, [M + H]+ 237.0602 (calcd), 237.0601 (found).:7) solution and loaded onto a pre-equilibrated silica column. The products were sequentially separated by eluting with solutions of 3:7, 2:3, and 1:1 ratios of EA to hexane. Fractions containing 9b product were pooled, dried over MgSO4, and evaporated under reduced pressure to acquire 9b (0.11 g; 40.2%). 1H NMR (400 MHz) (CDCl3) δ: 7.19 (s, 1H, NHCOCH2Br), 4.99 (s, 1H, 1H, NHCO2), 3.90 (s, 2H, COCH2Br), 3.63 (q, 2H, CH2NHCO2), 3.46 (q, 2H, CH2NHCOCH2), 2.86 (t, 2H, CH2CH2NHCO2), 2.80 (t, 2H, CH2CH2NHCOCH2), 1.45 (s, 9H, CH3). 13C NMR (100.67 MHz) (CDCl3) δ: 165.99, 155.90, 79.74, 39.43, 38.84, 37.98, 37.61, 28.91, 28.37. HRMS (ESI) calculated for C11H21BrN2O3S2, [M + Na]+ 395.0075 (calcd), 395.0073 (found).:1, 9:1, and 4:1 ratios of DCM/MeOH, dried over MgSO4, filtered, and evaporated under reduced pressure to provide 15b (0.45 g; 81.5%) as a white solid. 1H NMR (400 MHz) (C2D6OS) δ: 7.97 (s, 1H, CONH), 7.76 (s, 1H, CONH), 7.02 (s, 1H, CONH), 6.45 (s, 1H, CONH), 6.39 (s, 1H, CONH), 4.33 (t, 1H, CHN), 4.14 (t, 1H, CHN), 3.20 (t, 2H, CH2NH), 3.11 (dd, 1H, CHS), 3.01 (t, 2H, CH2NH), 2.83 (d, 1H, CHHS), 2.78 (t, 4H, CH2S), 2.74 (d, 1H, CHHS), 2.06 (t, 4H, CH2CO), 1.22–1.62 (m, 21H). 13C NMR (100.67 MHz) (C2D6OS) δ: 172.26, 171.81, 162.73, 155.54, 77.82, 61.05, 59.20, 55.45, 38.31, 37.84, 37.54, 37.37, 35.80, 35.31, 35.23, 30.79, 28.99, 28.22, 28.04, 26.12, 25.35, 24.99. HRMS (ESI) calculated for C25H45N5O5S3, [M + Na]+ 614.2480 (calcd), 614.2477 (found).Bromo-acetic acid 2,5-dioxo-pyrrolidin-1-yl ester (17)
Bromoacetyl bromide (2.32 g, 11.5 mmol) was added in a dropwise manner to 15 mL of a pyridine (0.91 g, 11.5 mmol)-containing DCM solution (15 mL) while steadily stirring. After 5 min at rt, the solution became cloudy with the formation of a white precipitate. At this point in the reaction, N-hydroxysuccinimide (NHS; 1.32 g, 11.5 mmol) was slowly added to the solution. The reaction was allowed to proceed for an additional 30 min at rt while the turbidity of the solution gradually decreased and the solution turned clear. The reaction products were extracted with a saturated solution of sodium bicarbonate twice, 1 M HCl twice, and a saturated sodium chloride solution once. The final organic phase was dried over MgSO4, filtered, and evaporated under reduced pressure to give 17 (1.42 g; 52.6%) as a brown solid. 1H NMR (400 MHz) (CDCl3) δ: 4.10 (s, 2H, CH2Br), 2.87 (s, 4H, CH2 of NHS). 13C NMR (100.67 MHz) (CDCl3) δ:168.43, 162.94, 25.53, 21.13. HRMS (ESI) calculated for C6H6O4NBrNa, [M + Na]+ 257.9378 (calcd), 257.9380 (found).:1) solution and loaded onto a pre-equilibrated silica column. The products were sequentially separated by eluting with solutions of 19:1, 9:1, 6:1, and 4:1 ratios of DCM/MeOH, dried over MgSO4, filtered, and evaporated under reduced pressure to obtain 18a (0.13 g; 58.5%) as a white solid. 1H NMR (400 MHz) (CD3OD) δ: 4.52 (t, 1H, CHN), 4.34 (t, 1H, CHN), 3.85 (s, 2H, CH2Br), 3.22 (t, 6H, CH2NH), 3.22 (dd, 1H, CHS), 2.96 (d, 1H, CHHS), 2.74 (d, 1H, CHHS), 2.22 (t, 4H, CH2CO), 1.40–1.80 (m, 20H). 13C NMR (100.67 MHz) (CD3OD) δ: 176.05, 176.00, 169.35, 166.05, 63.38, 61.62, 57.01, 47.91, 41.05, 40.78, 40.21, 36.99, 36.82, 30.31, 30.12, 30.05, 29.78, 29.50, 28.84, 27.55, 27.46, 26.93, 26.74. HRMS (ESI) calculated for C24H42BrN5O4S, [M + Na]+ 598.2038 (calcd), 598.2040 (found).:1 DCM/MeOH solution and loaded onto a pre-equilibrated silica column. The products were sequentially separated by eluting with solutions of 19:1, 9:1, 6:1, and 4:1 ratios of DCM/MeOH, dried over MgSO4, filtered, and evaporated under reduced pressure to yield 18b (0.047 g; 20.6%) as a white solid. 1H-NMR (400 MHz) (CD3OD) δ: 4.50 (t, 1H, CHN), 4.32 (t, 1H, CHN), 3.85 (s, 2H, CH2Br), 3.51 (t, 4H, CH2NH), 3.22 (dd, 1H, CHS), 3.19 (t, 2H, CH2NH), 2.93 (d, 1H, CHHS), 2.84 (t, 4H, CH2S), 2.71 (d, 1H, CHHS), 2.21 (t, 4H, CH2CO), 1.31–1.75 (m, 12H). 13C-NMR (100.67 MHz) (CD3OD) δ: 176.31, 176.00, 173.95, 164.80, 63.38, 61.63, 57.00, 41.05, 40.20, 39.95, 39.45, 38.54, 37.45, 36.90, 36.83, 30.11, 29.77, 29.49, 27.53, 26.91, 26.59. HRMS (FAB) calculated for C22H39O4N5BrS3, [M + H]+ 612.1348 (calcd), 612.1348 (found).TW17 ribozyme synthesis and purification
Both the 5′ GMPS-primed TW17 RNA (87-mer; 5′-GGGAUCGUCAGUGCAUUGAGAAGUGCAGUGUCUUGCGCUGGGUUCGAGCGGUCCGUGGUGCUGGCCCGGUGGUAUCCCCAAGGGGUA-3′) and the TW17 RNA body-labeled with32 P were prepared according to the previously reported method.18 Radio-labeled TW17 ribozyme substrate conjugates and their reaction products were analyzed using either urea-polyacrylamide gel electrophoresis (urea-PAGE) or streptavidin (SAv) gel-shift assays in urea-PAGE, visualized, and quantified by an Amersham Typhoon PhosphorImager at KMU. All the aqueous solutions were prepared from diethyl pyrocarbonate (DEPC)-treated water to prevent RNA degradation.General kinetics of TW17 ribozyme catalysis
The procedures for measuring the kinetics of TW17 ribozyme catalysis under a standard condition have been reported previously18 but are briefly described below. The 32P-body-labeled TW17 ribozyme was initially dissolved in EK buffer (50 mM EPPS, 500 mM KCl, pH 7.5), heated at 95 °C for 1 min, centrifuged, and then cooled at rt for 10 min to allow the RNA to refold in the absence of metal ions. Mg2+ and Zn2+ were then added sequentially in 5 min intervals to the RNA solution to commence TW17 ribozyme catalysis at rt. The reaction mixture for the standard TW17 ribozyme catalysis contained 1 μM of the ribozyme, 100 mM MgCl2, 0.5 mM ZnCl2, and 0.24% Triton X-100 in EK buffer. Aliquots were removed from the reaction mixture at indicated times during the course of the reaction and purified twice by ethanol precipitation. The purified samples were resolved by SAv gel shift assays utilizing 8% urea-PAGE and analyzed by an Amersham Typhoon PhosphorImager system. The resulting data were fitted into a single-exponential equation for first-order kinetics F(t) = F0 + Fmax(1 − e−kobst) [F(t), percent cleavage of the reactant at a specific time point t] to acquire the first-order rate constant kobs (GraphPad, La Jolla, CA, USA).Metal titration
The metal titration experiments were performed by following the procedures stated in the previous section except that the divalent metal ions were replaced by different combinations and concentrations of Zn2+, Mg2+, or [Co(NH3)6]3+. Solid magnesium chloride was dissolved in DEPC-treated water to prepare a 2 M Mg2+ stock solution, which was stored at −20 °C. A 200 mM Zn2+ stock solution was prepared by dissolving ZnCl2(s) in DEPC-treated water and stored at −20 °C. The stock solution of 40 mM [Co(NH3)6]3+ was similarly obtained by dissolving [Co(NH3)6]Cl3(s) in DEPC-treated water and storing at −20 °C. Analyses of Zn2+ titration in the presence of 100 mM Mg2+ were performed by substituting various concentrations of Zn2+ (0.13–10 mM) for 0.5 mM Zn2+ in the kinetic analysis reaction mixtures. For kinetic analyses of ribozyme catalysis in the absence of 100 mM Mg2+, several concentrations of Zn2+ ranging from 0.13 to 1.88 mM were substituted for the 0.5 mM Zn2+ used in the standard reaction mixtures. Alternatively, in the presence of 0.5 mM Zn2+, the 100 mM Mg2+ in the standard kinetic condition was replaced by several concentrations of Mg2+ (1–100 mM), and TW17 ribozyme catalysis was measured. Finally, catalysis measurements analyzing [Co(NH3)6]3+ titration in the presence of 0.5 mM Zn2+ were performed by replacing the 100 mM Mg2+ required for the standard kinetic condition in TW17 ribozyme catalysis with concentrations of [Co(NH3)6]3+ between 1 and 500 μM. In each kinetic analysis, the duration of the reactions ranged from 3 to 24 h in order to ensure the first-order reaction reached a maximal product yield. Each kobs value was acquired by fitting data into the single-exponential equation for the first-order kinetics described previously [general kinetics of TW17 ribozyme catalysis]. A reported rate constant was determined by calculating the average of the rates from triplicate experiments accompanied with the standard deviations. The obtained values of kobs from concentrations of substituted Zn2+ ions {[Mn+]}, were fitted into the Hill equation kobs = {[(kobs)max][Mn+]h}/{Kd + [Mn+]h} to attain Kd (average dissociation constant for a TW17 ribozyme–Mn+ complex at equilibrium) and the Hill coefficient h, which was calculated as the minimum Mn+ equivalence essential to TW17 ribozyme catalysis (GraphPad).pH-dependent kinetics
As was the case for the metal titration experiments, pH titration studies of TW17 ribozyme catalysis were also carried out by following previously described procedures [general kinetics for TW17 ribozyme catalysis] except that various pH buffer solutions were substituted for the EK buffer in the reaction mixtures. Buffers for the pH titration studies were MES (pH 5.5–6.5), PIPES (pH 6.5–7.5), or EPPS (7.5–8.5); each buffer also contained 500 mM KCl. As in the metal titration experiments, reaction times of 3 to 24 h were required to ensure that the first-order reaction reached a maximal product yield. Each kobs value was acquired by fitting data into the single-exponential equation for the first-order kinetics [general kinetics of TW17 ribozyme catalysis]. A reported rate constant accompanied by a standard deviation represented the average of rates obtained from the kinetic experiments performed in triplicate. Values of kobs obtained from TW17 ribozyme catalysis under different pH conditions were fitted into the equation for general base catalysis,40 kobs = [(kobs)max]/[1 + 10(pKa − pH)], but were unable to provide reasonable values of pKa for a general base and kobs,max in the presence of the general base.Preparation of RNA molecules for Langmuir isotherm study and trans-phosphorothiolate thiolesterase assay of trans-acting TW17 ribozyme systems
Ten bimolecular trans-acting TW17 ribozyme systems were prepared by following the previously reported methods18 and are described in Table S1 (ESI†). Binary RNA binding affinities in the bimolecular trans-acting TW17 ribozyme systems were determined for eight pairs of constructs: TW17S-1 RNA + TW17C-1 RNA, TW17S-2 RNA + TW17C-2 RNA, TW17S-3 RNA + TW17C-3 RNA, TW17S-4 RNA + TW17C-4 RNA, TW17S-5 RNA + TW17C-5 RNA, TW17S-6 RNA + TW17C-5 RNA, TW17S-1 RNA + TW17C-5 RNA, and TW17S-2 RNA + TW17C-3 RNA systems. In addition, the trans-phosphorothiolate thiolesterase activities of the two TW17 ribozyme systems, TW17S1–29 RNA + TW17C30–87 RNA and TW17S1–29 RNA + TW17C22–87 RNA, were measured. Primers required for the construction of the trans-acting TW17 ribozyme systems can be found in Table S2 (ESI†).Langmuir isotherm study to determine binding affinities in trans-acting TW17 ribozyme systems
The Langmuir isotherm study of the TW17S-1 + TW17C-1 system provides a generic description of the binding affinity experiments performed using trans-acting TW17 ribozyme systems. The 32P-body-labeled TW17S-1 RNA was dissolved in 4X EK buffer (200 mM EPPS, 2 M KCl, pH 7.5) in the presence of varying concentrations of Zn2+ (2.25–75 mM) to give the 0.14 μM TW17S-1 RNA solutions used in all the binding affinity assays (Table S3; ESI†). The concentration of Zn2+ in a 0.14 μM TW17S-1 solution was determined by multiplying 2.25 mM by the molar ratio of TW17C-1 RNA to TW17S-1 RNA. However, the [Zn2+] in the 0.14 μM TW17S-1 solutions was held constant at 2.25 mM when the molar ratio of TW17C-1 RNA to TW17S-1 RNA was less than 1. Conversely, the label-free TW17C-1 RNA was dissolved in 4X EK buffer in the presence of varied concentrations of Mg2+ (0–780 mM) to yield the 0–7.0 μM TW17C-1 RNA solutions (Table S3; ESI†) also essential for performing the binding affinity assays. The concentration of Mg2+ in the TW17C-1 solution was determined by multiplying 94 mM with the molar ratio of TW17C-1 RNA to TW17S-1 RNA. However, the TW17C-1 solutions always had a [Mg2+] equal to 94 mM when the molar ratio of TW17C-1 RNA to TW17S-1 RNA was less than one, and a Mg2+ concentration of 780 mM when the molar ratio of TW17C-1 RNA to TW17S-1 RNA was higher than 20 due to the limited solubility of MgCl2 when solutions with a [Mg2+] > 3 M were prepared.The acquired TW17S-1 RNA and TW17C-1 RNA solutions were boiled at 95 °C for 1 min, cooled at rt for 10 min, mixed in a 1:1 (v/v) ratio, and reacted at rt for 30 min. The afforded binding reaction products were separated using 10% non-denaturing PAGE at 4 W for 70 min and analyzed by an Amersham Typhoon PhosphorImager system. The non-denaturing PAGE apparatus was surrounded by ice during electrophoresis in order to reduce the effects of a temperature increase caused by heat generated during electrophoresis. Percent binding values (Y) from specific concentrations of TW17C-1 RNA ([TW17C-1 RNA]) were fitted according to the Langmuir isotherm equation {Y = (Ymax × [TW17C-1 RNA])/(Kd + [TW17C-1 RNA])} to attain Kd (the dissociation constant for the TW17S-1 RNA–TW17C-1 RNA complex at equilibrium) and Ymax, which is the maximum percent binding of TW17S-1 RNA to TW17C-1 RNA (GraphPad).
Trans-phosphorothiolate thiolesterase assay
Multiple substrate turnover analysis of the trans-phosphorothiolate thiolesterase reaction in the TW17S1–29 RNA + TW17C30–87 RNA and TW17S1–29 RNA + TW17C22–87 RNA systems was carried out according to procedures previously described.18 The initial velocity for each catalytic reaction in the presence of a specific concentration of the 32P-labeled S1–29 RNA-18a conjugate was studied by nonlinear least-square curve fitting to the Michaelis–Menten equation {v = (Vmax × [S1–29 RNA])/(KM + [S1–29 RNA])}, but were unable to provide reasonable values of KM, kcat, and kcat/KM (GraphPad).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Susan Fetzer for critically reading the manuscript. We also thank the Center for Research Resources and Development of KMU for technical and instrumental assistance. This work was supported with funding from the National Science Council of Taiwan (NSC 96-2113-M-037-004-MY2 and NSC 100-2113-M-037-008-) and the Ministry of Science and Technology of Taiwan (MOST 103-2113-M-037-010-) awarded to T.-P. W.References
- T. R. Cech, Cell, 2009, 136, 599–602 CrossRef PubMed.
- T. J. Wilson and D. M. J. Lilley, Science, 2009, 323, 1436–1438 CrossRef PubMed.
- W. E. Draper, E. J. Hayden and N. Lehman, Nucleic Acids Res., 2008, 36, 520–531 CrossRef PubMed.
- R. K. O. Sigel and A. M. Pyle, Chem. Rev., 2007, 107, 97–113 CrossRef PubMed.
- M. J. Fedor, Annu. Rev. Biophys., 2009, 38, 271–299 CrossRef PubMed.
- K. M. Brooks and K. J. Hampel, Biochemistry, 2011, 50, 2424–2433 CrossRef PubMed.
- D. M. J. Lilley, Biochem. Soc. Trans., 2017, 45, 683–691 CrossRef PubMed.
- E. A. Frankel, C. A. Strulson, C. D. Keating and P. C. Bevilacqua, Biochemistry, 2017, 56, 2537–2548 CrossRef PubMed.
- G. S. Manning, J. Chem. Phys., 1969, 51, 934–938 CrossRef.
- Y. Bai, M. Greenfeld, K. J. Travers, V. B. Chu, J. Lipfert, S. Doniach and D. Herschlag, J. Am. Chem. Soc., 2007, 129, 14981–14988 CrossRef PubMed.
- M. Gebala, G. M. Giambaşu, J. Lipfert, N. Bisaria, S. Bonilla, G. Li, D. M. York and D. Herschlag, J. Am. Chem. Soc., 2015, 137, 14705–14715 CrossRef PubMed.
- D. E. Draper, RNA, 2004, 10, 335–343 CrossRef PubMed.
- J. Lipfert, S. Doniach, R. Das and D. Herschlag, Annu. Rev. Biochem., 2014, 83, 813–841 CrossRef PubMed.
- D. S. Auld, BioMetals, 2001, 14, 271–313 CrossRef PubMed.
- B. L. Vallee and D. S. Auld, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 220–224 CrossRef.
- C. E. Outten and T. V. O'Halloran, Science, 2001, 292, 2488–2492 CrossRef PubMed.
- K. A. McCall, C.-C. Huang and C. A. Fierke, J. Nutr., 2000, 130, 1437S–1446S CrossRef PubMed.
- T.-P. Wang, Y.-C. Su, Y. Chen, Y.-M. Liou, K.-L. Lin, E.-C. Wang, L.-C. Hwang, Y.-M. Wang and Y.-H. Chen, Biochemistry, 2012, 51, 496–510 CrossRef PubMed.
- A. Furukawa, T. Maejima, S. Matsumura and Y. Ikawa, Biosci., Biotechnol., Biochem., 2016, 80, 1386–1389 CrossRef PubMed.
- L. Jaeger, F. Michel and E. Westhof, J. Mol. Biol., 1994, 236, 1271–1276 CrossRef PubMed.
- S. E. Butcher and A. M. Pyle, Acc. Chem. Res., 2011, 44, 1302–1311 CrossRef PubMed.
- J. Ishikawa, Y. Fujita, Y. Maeda, H. Furuta and Y. Ikawa, Methods, 2011, 54, 226–238 CrossRef PubMed.
- H. Furusawa, S. Fukusho and Y. Okahata, ChemBioChem, 2014, 15, 865–871 CrossRef PubMed.
- C. R. Woese, S. Winker and R. R. Gutell, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 8467–8471 CrossRef.
- M. Hernick and C. Fierke, Arch. Biochem. Biophys., 2005, 433, 71–84 CrossRef PubMed.
- E. Veliath, B. L. Gaffney and R. A. Jones, Nucleosides, Nucleotides Nucleic Acids, 2014, 33, 40–52 CrossRef PubMed.
- M. Strenkowska, P. Wanat, M. Ziemniak, J. Jemielity and J. Kowalska, Org. Lett., 2012, 14, 4782–4785 CrossRef PubMed.
- M. Olesiak, D. Krajewska, E. Wasilewska, D. KorczyŇski, J. Baraniak, A. Okruszek and W. J. Stec, Synlett, 2002, 0967–0971 CrossRef.
- E. J. Behrman, J. Chem. Res., 2000, 446–447 CrossRef.
- B. Fischer, A. Chulkin, J. L. Boyer, K. T. Harden, F. P. Gendron, A. R. Beaudoin, J. Chapal, D. Hillaire-Buys and P. Petit, J. Med. Chem., 1999, 42, 3636–3646 CrossRef PubMed.
- A. B. Burgin and N. R. Pace, EMBO J., 1990, 9, 4111–4118 CrossRef PubMed.
- B. Zhang, Z. Cui and L. Sun, Org. Lett., 2001, 3, 275–278 CrossRef PubMed.
- M. Oivanen, M. Ora, H. Almer, R. Stromberg and H. Lonnberg, J. Org. Chem., 1995, 60, 5620–5627 CrossRef.
- K. A. Jacobson, B. Fischer and X. D. Ji, Bioconjugate Chem., 1995, 6, 255–263 CrossRef PubMed.
- T.-P. Wang, Y.-J. Chiou, Y. Chen, E.-C. Wang, L.-C. Hwang, B.-H. Chen, Y.-H. Chen and C.-H. Ko, Bioconjugate Chem., 2010, 21, 1642–1655 CrossRef PubMed.
- X. Fang, T. Pan and T. R. Sosnick, Biochemistry, 1999, 38, 16840–16846 CrossRef PubMed.
- S. K. Silverman and T. R. Cech, Biochemistry, 1999, 38, 8691–8702 CrossRef PubMed.
- V. B. Chu, Y. Bai, J. Lipfert, D. Herschlag and S. Doniach, Curr. Opin. Chem. Biol., 2008, 12, 619–625 CrossRef PubMed.
- J. Weiss, FASEB J., 1997, 11, 835–841 CrossRef PubMed.
- S. Nakano, D. M. Chadalavada and P. C. Bevilacqua, Science, 2000, 287, 1493–1497 CrossRef PubMed.
- K. S. Koutmou, A. Casiano-Negroni, M. M. Getz, S. Pazicni, A. J. Andrews, J. E. Penner-Hahn, H. M. Al-Hashimi and C. A. Fierke, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2479–2484 CrossRef PubMed.
- H. Sigel and R. B. Martin, Chem. Soc. Rev., 1994, 23, 83–91 RSC.
- C. P. Da Costa, A. Okruszek and H. Sigel, ChemBioChem, 2003, 4, 593–602 CrossRef PubMed.
- K. A. Leamy, N. H. Yennawar and P. C. Bevilacqua, Biochemistry, 2017, 56, 3422–3433 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full details of supporting figures referenced in the text, NMR and MS spectra for some known and all the new compounds (2, 3, 6, 8b, 9a, 9b, 10a, 10b, 15b, 16b, 17, 18a, and 18b). See DOI: 10.1039/c8ra05083j |
| ‡ Authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2018 |