
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe Suzuki–Miyaura reaction as a tool for modification of phenoxyl-nitroxyl radicals of the 4H-imidazole N-oxide series†
Yury A. Ten a,
Oleg G. Salnikovb,
Svetlana A. Amitinaa,
Dmitri V. Stassbc,
Tatyana V. Rybalovaab,
Maxim S. Kazantsevab,
Artem S. Bogomyakovd,
Evgeny A. Mostovichab and
Dmitrii G. Mazhukin*ab
a,
Oleg G. Salnikovb,
Svetlana A. Amitinaa,
Dmitri V. Stassbc,
Tatyana V. Rybalovaab,
Maxim S. Kazantsevab,
Artem S. Bogomyakovd,
Evgeny A. Mostovichab and
Dmitrii G. Mazhukin*ab
aNN Vorozhtsov Novosibirsk Institute of Organic Chemistry SB RAS, 9 Acad. Lavrentyeva Ave., Novosibirsk, 630090, Russia, d-mazhukin@yandex.ru
bNovosibirsk State University, 2 Pirogova Str., Novosibirsk, 630090, Russia
cVoevodsky Institute of Chemical Kinetics and Combustion SB RAS, 3 Institutskaya Str., Novosibirsk, 630090, Russia
dInternational Tomography Center SB RAS, 3A Institutskaya Str., Novosibirsk, 630090, Russia
First published on 20th July 2018
Abstract
2-(3,5-Di-tert-butyl-4-hydroxyphenyl)-5-(4-iodophenyl)-4,4-dimethyl-4H-imidazole 3-oxide reacts with phenylboronic acid and its substituted derivatives in a cross-coupling reaction of the Suzuki–Miyaura type to form 5-biphenyl derivatives of 4H-imidazole-N-oxide. Interaction of the same compound with B2(pin)2 in the presence of PdCl2(PPh3)2 proceeds through the formation of intermediate 1,3,2-dioxoborolane and leads to the product of homocoupling: biphenyl-bis(imidazole). Oxidation of the resultant imidazoles with lead dioxide quantitatively yields stable conjugated phenoxyl-nitroxyl mono- and diradicals, which are of interest as electroactive paramagnetic materials. The crystal structure of the monoradical, 2,6-di-tert-butyl-4-[1-oxido-4-(biphenyl-4-yl)-5,5-dimethyl-1H-imidazole-2(5H)-ylidene]cyclohex-2,5-dienone, its magnetic susceptibility, EPR spectra of the obtained hybrid radicals in solution, and cyclic voltammetry characteristics of 4H-imidazoles were studied.
Introduction
The cross-coupling reaction of the Suzuki–Miyaura type1 can be undoubtedly considered as one of the fundamental methods for constructing aromatic and heteroaromatic ensembles in the synthesis of biologically active compounds for pharmaceutical chemistry2 as well as in the chemistry of new materials (preparation of components of optoelectronic devices3 and liquid-crystalline semiconducting oligomers4) and in organic photovoltaics.5 Recent research on the Suzuki–Miyaura reaction includes extensive studies of various substrates involved in this process (for example, low-reactive aryl chlorides, aryl fluorides, or tosylates),6 a wide range of reagents (boronic acids and their derivatives),7 and several generations of palladium catalysts with different ligand environments.8Continuing the research on the reactivity of new stable radicals, i.e., hybrid phenoxyl-nitroxides of the 4H-imidazole N-oxide series (Chart 1),9 we turned our attention to the possibility of modifying the para-haloaryl group at the 5th position of the heterocycle using Pd-catalyzed cross-coupling reactions, in particular, the Suzuki–Miyaura process.
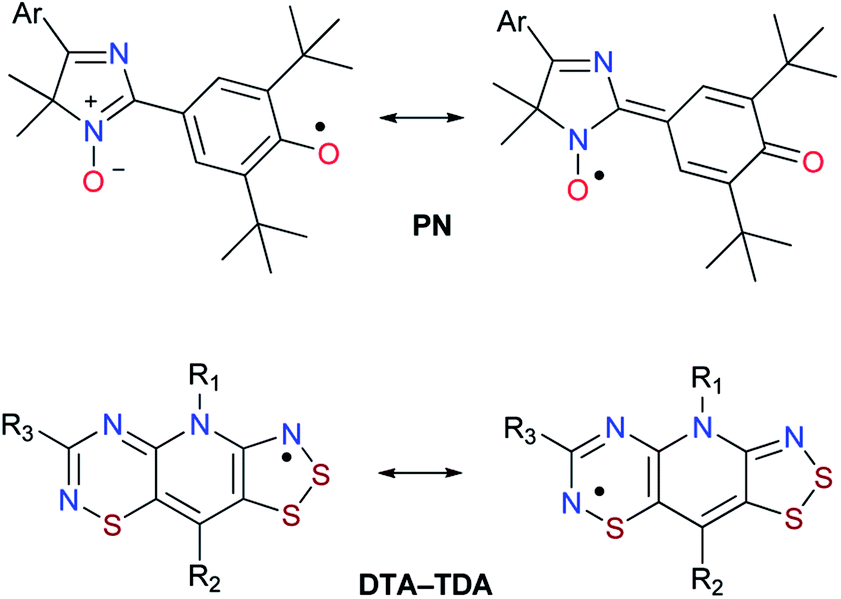 | ||
| Chart 1 Examples of stable hybrid radicals: 4H-imidazole N-oxide based phenoxyl-nitroxides (PN); 1,2,3-dithiazolo-1,2,4-thiadiazinyls (DTA-TDA). | ||
On the one hand, the introduction of an electron-withdrawing type of substituent possessing an −M-effect into the para-position of the 5-aryl moiety should lead to the additional stabilization of the radical center owing to further distribution of the electron density of an unpaired electron over the conjugated system. On the other hand, further functionalization of hybrid radicals means developing their practical applicability because of creation of new promising dyadic and triadic molecular systems.
While developing fundamentally new smart devices based on fully organic materials, researchers have become more interested in the design and construction of dyads and triads of DA (donor–acceptor) or ADA (acceptor–donor–acceptor) types, where the group containing a stable radical moiety plays the role of the acceptor.10 These systems can be considered molecular switches (transforming from a neutral state to a zwitterionic or diradical form in solution under the influence of external stimuli, i.e., temperature, pressure, or light), and some of them can have properties of conductors in a solid form.11 Typical examples of such dyads and triads are shown in Chart 2.
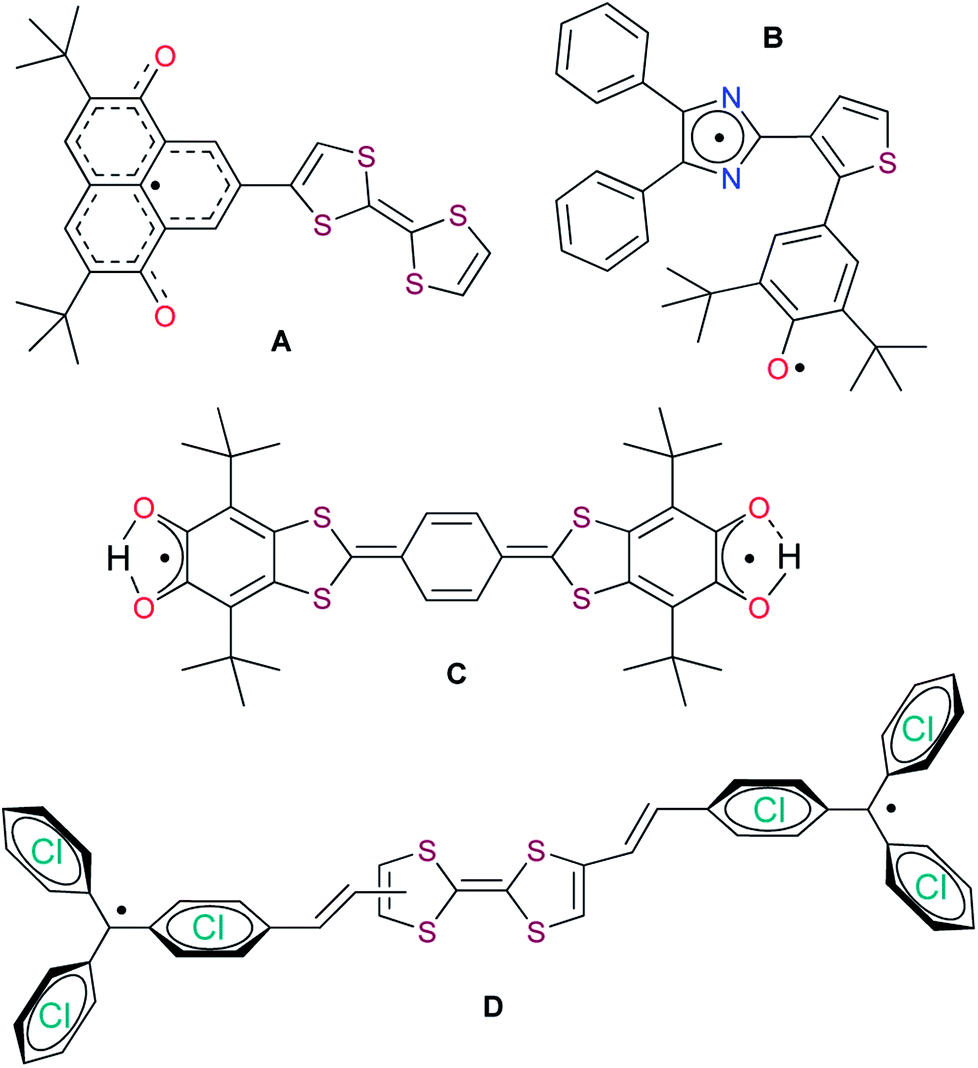 | ||
| Chart 2 Some electroactive and photoactive dyads and triads based on stable radicals: oxophenalyloxyl (A),10i phenoxyl-imidazolyl (B),10d,f di-tert-butyl-ortho-semiquinonyl (C),10e,g and trityl (D).10b | ||
As for stable hybrid radicals, only a few representatives of this type of paramagnetics were synthesized by scientists in the last decade. For instance, resonance-stabilized planar 1,2,3-dithiazolo-1,2,4-thiadiazinyl12a (Chart 1) and related radicals12b possess highly delocalized and easily tuned spin distributions. Therefore, the wide range of physical and chemical properties of thiazyl-based radicals make them useful building blocks for magnetic and conductive materials as well as coordinating ligands.
Depending on the kinetic stability of radical types, there are two fundamentally different methods for their functionalization. The first method represents the interaction of their diamagnetic precursors directly along a reactive group with various nucleophilic or electrophilic reagents, and the radical center is generated at the final stage, when the product is treated with an oxidizing agent (or an excess of an electrophilic reagent). The second method consists of treating the stable radical with a mild selective reagent whose redox potential cannot change the paramagnetic status of the substrate. In this respect, a study on Pd-catalyzed C–C cross-coupling reactions (particularly, the Suzuki–Miyaura process) involving various types of persistent radicals was carried out here under the conditions of both methods.
For example, thermally unstable nitroxyl radicals stabilized by nitroxyl group conjugation with the benzene nucleus (t-butylaryl, tetrahydroquinolinyl, [3,1]-benzoxazinyl, and dihydroacridinyl type) were introduced into the Suzuki–Miyaura reaction in the form of their stable precursors, namely, secondary amines. After formation of the cross-coupling products, the latter were transformed into target nitroxides with m-CPBA in a yield of 37–70%.13 At the same time, more robust iodoarene derivatives of the benzotriazinyl radical (Blatter's radical),14a–c verdazyl radical,14d and iso-indolinoxyls14e readily reacted in cross-coupling with either arylboronic acids or aryltriolborate salts, which in contrast to parent acids possess greater reactivity. Depending on the number of iodoarene groups in these radicals, mono-, bis-, and tris-cross-coupling products were obtained without losing a radical center. The yields ranged from 97% for the mono-cross-coupling product to 67% for bis- and 52% for the tris-phenyl-substituted compound. As to the cyclic nitroxide based on an imidazole nucleus, to the best of our knowledge, there is only one report about the Suzuki–Miyaura reaction involving nitronyl nitroxyl radicals containing an ortho(meta)-iodophenyl group at the C-2 atom of the heterocycle. The authors explained a rather low yield of cross-coupling products in this reaction (40–43% for the ortho-iodophenyl derivative and 44% for the meta-iodophenyl derivative) by competitive destruction of a nitronyl nitroxide occurring at the elevated temperature under typical conditions of the Suzuki process (80 °C, 2 h).15
Accordingly, it was interesting to find out how hybrid phenoxyl-nitroxide radicals would behave under the conditions of the Suzuki–Miyaura reaction and whether they would be sufficiently stable to retain the paramagnetic center. Besides, Pd-catalyzed cross-coupling reactions involving 5-(haloaryl)-4H-imidazole derivatives and their N-oxides are not mentioned in any databases.
Results and discussion
To conduct cross-coupling experiments, the para-iodoarene substituted derivative of 4H-imidazole N-oxide was chosen as a model substrate because solely iodo derivatives show the greatest reactivity among other haloaryl compounds and produce quantitative yields of biphenyls under the classical Suzuki reaction conditions.A diamagnetic precursor of the hybrid radical, key 5-(p-iodophenyl)substituted 4H-imidazole 3-oxide 1, was synthesized in 6 stages based on Friedel–Crafts acylation of an iodobenzene excess with iso-butyric acid chloride followed by bromination of 4-iodo-iso-butyrophenone 2, treatment of bromo compound 3 with hydroxylamine, and hydrolysis of the formed 2-hydroxylamine oxime 4 by boiling with concentrated hydrobromic acid. Condensation of the obtained salt of 2-hydroxylamino ketone 5 with 4-hydroxy-3,5-di-tert-butylbenzaldehyde 6 in the presence of ammonium acetate led to 2,4-diaryl-2,5-dihydroimidazole 7 with a good yield, and oxidation of 7 with air oxygen in the presence of Cu(OAc)2 or a manganese dioxide excess (the second method is preferable because the yield of 4H-imidazole N-oxide is quantitative in this case) gave compound 1 (Scheme 1). Hybrid phenoxyl nitroxyl radical 8 was formed with a yield of more than 90% when imidazole 1 was treated with a threefold excess of PbO2.
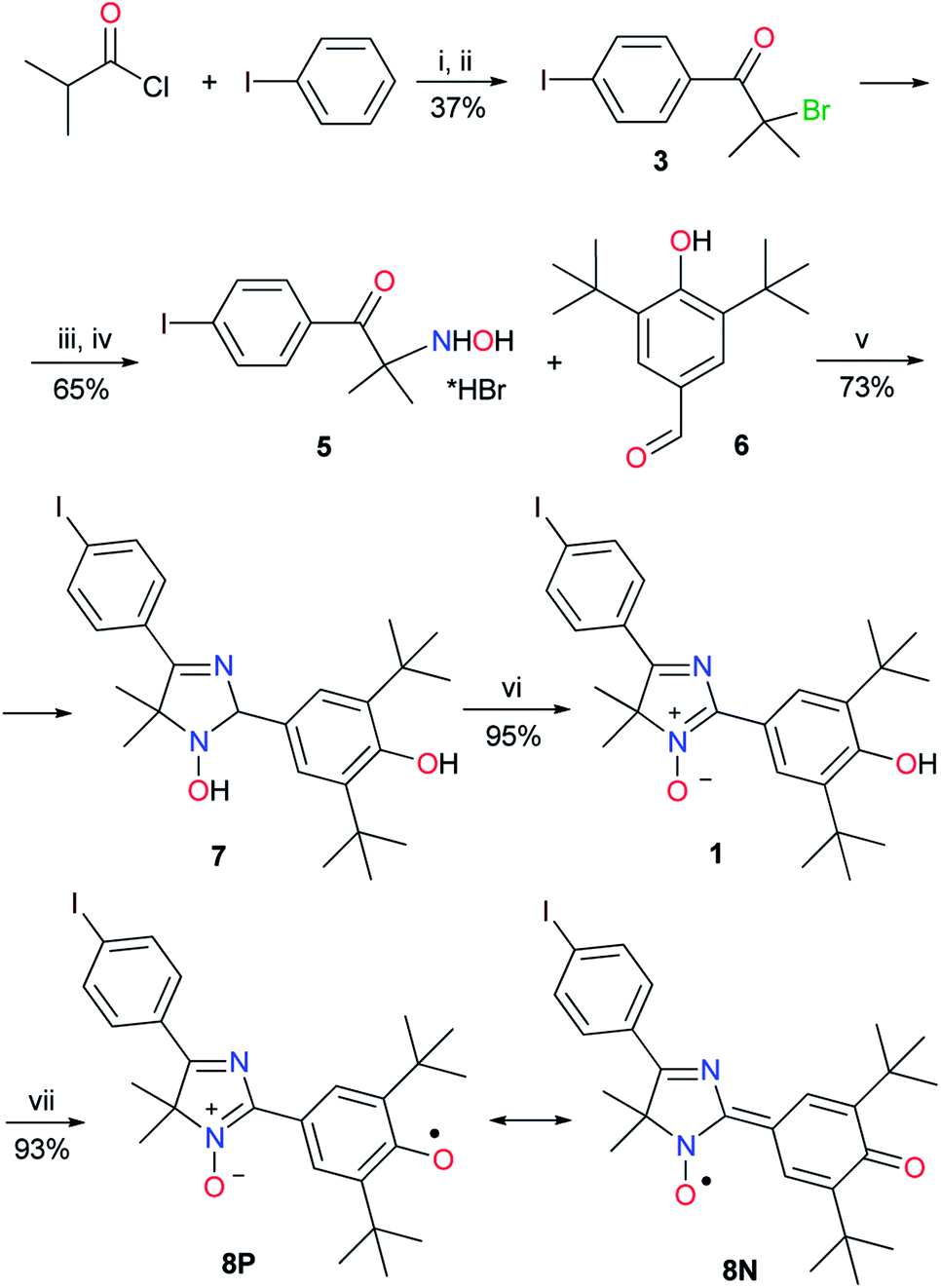 | ||
| Scheme 1 Reagents and conditions: (i) AlCl3, 45 °C, 15 h; (ii) Br2, Et2O/dioxane, room temperature (rt), 2 h; (iii) NH2OH × HCl, NaOH, MeOH/H2O, 65 °C, 7 h; (iv) 48% HBr, Δ, 2 h; (v) NH4OAc, MeOH, rt, 6 h; (vi) MnO2, CHCl3, rt, 7 h; and (vii) PbO2, CHCl3, rt, 2 h. | ||
At first, this radical 8 was introduced into the typical Suzuki cross-coupling reaction under standard classical conditions (arylboronic acid 9d, Pd[(PPh3)4] (5 mol%), K2CO3, a two-phase system (toluene–water), 110 °C, argon atmosphere). Heating the mixture for 24 h caused the solution to lose its dark brown color typical for the conjugated radical, then disappearance of the EPR signal and formation of a single product, bright yellow diamagnetic 5-biphenyl-4H-imidazole N-oxide 10d, in a quantitative yield. Oxidation of the obtained compound with lead dioxide in chloroform smoothly led to corresponding stable phenoxyl-nitroxyl radical 11d. Thereafter, its immediate precursor, phenolic imidazole 1, was introduced into the cross-coupling reaction to reduce the total number of steps necessary to synthesize new biphenyl derivatives of the hybrid radical. Thus, compound 1 was involved in the Suzuki–Miyaura reaction with various arylboronic acids possessing both donor and acceptor groups (Scheme 2). Regardless of the nature of substituents in the reagent and its steric features, conversion of imidazole 1 was complete, and cross-coupling products 10a–d were obtained in good yields (66–85%). Apparently, the presence of a sterically hindered phenolic group in the substrate did not inhibit the cross-coupling process, nor did it have a significant effect on the reaction yields. It should be noted that transformation of biphenyl derivatives of 10a–d into phenoxyl-nitroxide radicals starts even when their solutions are kept in ambient air, whereas oxidation of 4H-imidazoles 10a–d with lead dioxide ends within a few hours, and hybrid radicals 11a–d form quantitatively (Scheme 2).
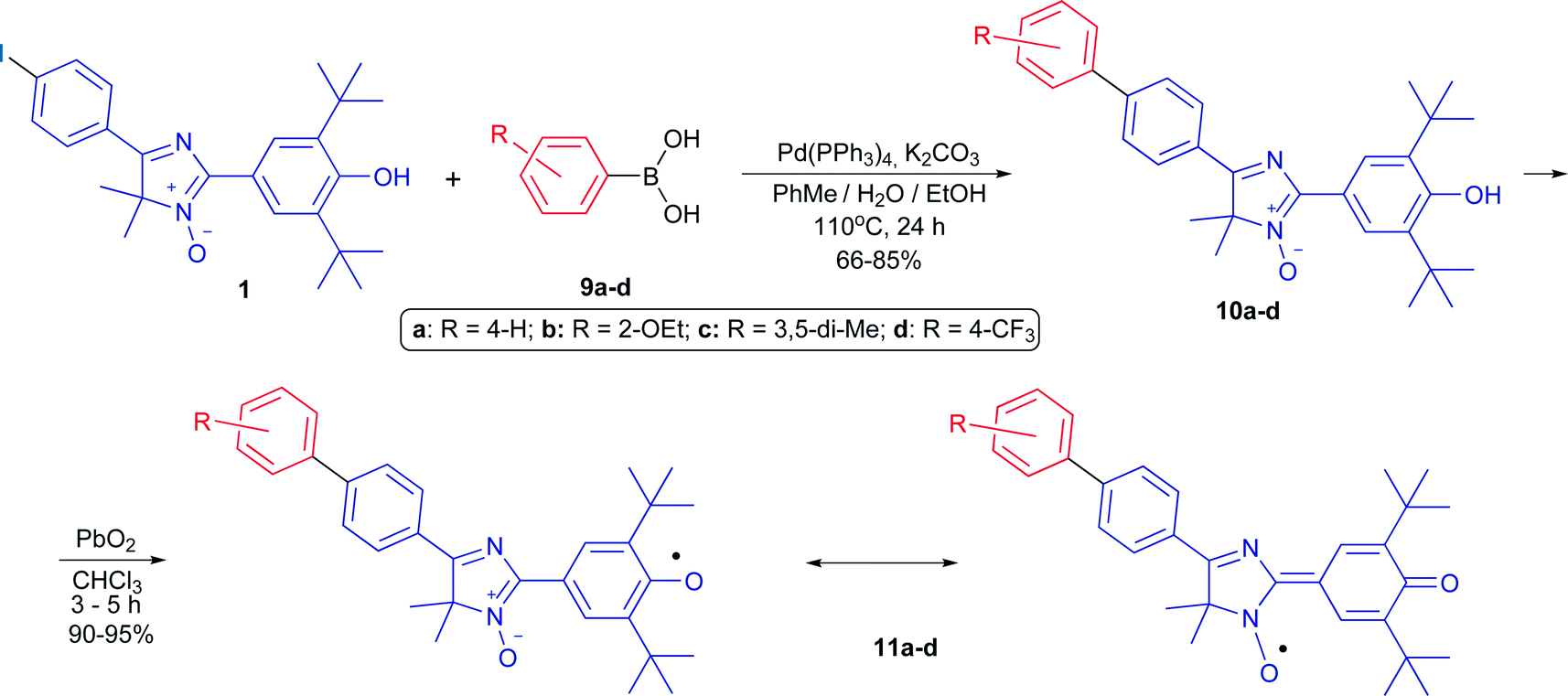 | ||
| Scheme 2 The Suzuki cross-coupling reaction of 4H-imidazole N-oxide 1 with substituted arylboronic acids and the synthesis of biphenyl derivatives of hybrid phenoxyl-nitroxide. | ||
Cyclic voltammograms of the 4H-imidazoles 10a–d are presented in Fig. 1. All the compounds show similar redox behavior consisting of irreversible oxidation and reduction features. Apparently, oxidation of starting phenol-imidazole 10 proceeds via irreversible step-by-step formation of galvinoxyl radical I, quinoid cation II, and dication radical III, respectively (Scheme S1 in ESI†). The reduction step consists only of an irreversible peak, which corresponds to the formation of anion-radical IV. Oxidation and reduction peaks are listed in Table S1, ESI.†
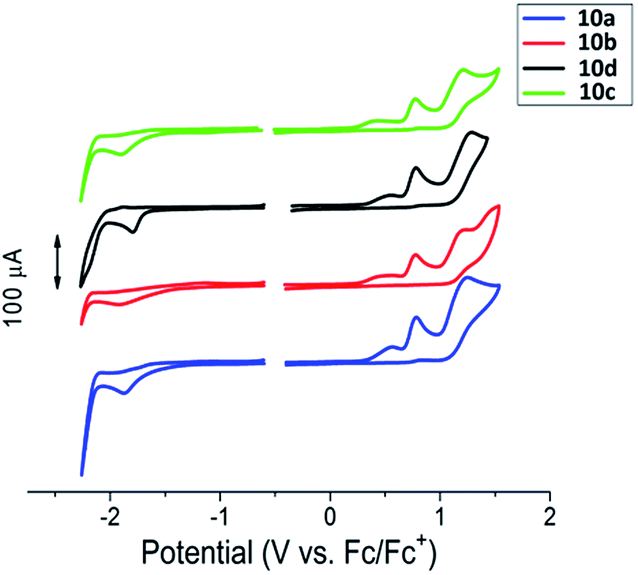 | ||
| Fig. 1 Cyclic voltammograms for cross-coupling products, 4H-imidazoles 10a–d in a CH3CN solution. Potentials calibrated vs. Fc/Fc+. | ||
EPR spectra of all the obtained hybrid radicals contain a set of 21 lines, characterized by a triplet form consisting of 7 lines in each segment due to the interaction of the unpaired electron with two nonequivalent nitrogen nuclei (with the isotropic hyperfine splitting (hfs) constants AN1 = 0.541 mT and AN2 = 0.061 mT, respectively) and with two nonequivalent protons in the molecular quinone fragment (hfs constants AH1 = 0.161 mT, AH2 = 0.149 mT) (Fig. 2 and S2–S4 in ESI†). The hfs constants for “distant” protons of the biphenyl moiety are negligible, indicating the absence of noticeable spin density delocalization on the aryl substituent at the C-4 position of the heterocycle.
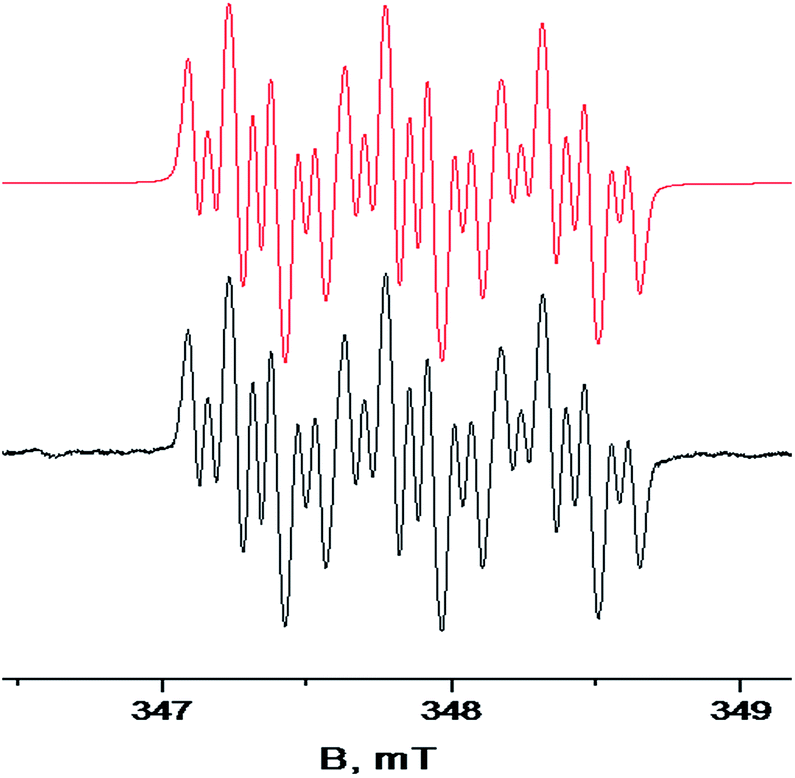 | ||
| Fig. 2 An ESR spectrum of 4-biphenyl-substituted hybrid radical 11a, recorded at 20 °C in a degassed toluene solution; the black curve is the experimental spectrum, the red curve is its mathematical reconstruction; giso = 2.0059, AN1 = 0.541 mT, AN2 = 0.061 mT, AH1 = 0.161 mT, AH2 = 0.149 mT. | ||
We succeeded in growing suitable single crystals of unsubstituted biphenyl derivative 11a for X-ray crystallographic analysis (Fig. 3). The best solvent for these hybrid radicals seemed to be dipolar aprotic acetonitrile having a high dielectric constant. It should be noted that if solutions of radicals are kept long in a nonpolar solvent at room temperature, then these radicals partially decompose.
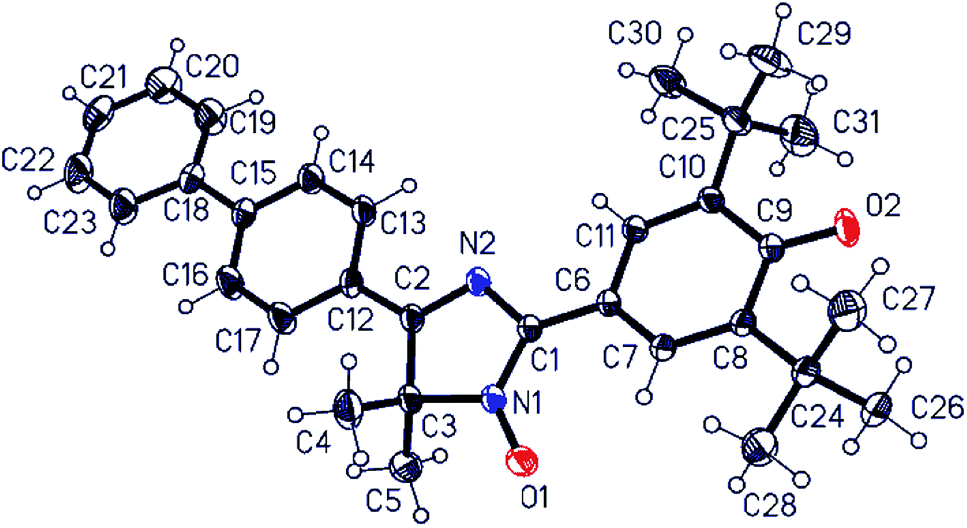 | ||
| Fig. 3 Molecular structure (an ORTEP diagram with 30% ellipsoid probability) and atom numbering of hybrid phenoxyl-nitroxide 11a. Selected bond lengths (Å): N1–O1 1.270(3), N1–C1 1.354(3), N2–C1 1.395(3), N2–C2 1.296(3), C2–C3 1.511(4), N1–C3 1.502(3), C1–C6 1.409(3), C6–C7 1.420(3), C7–C8 1.3573, C8–C9 1.477(4), C9–O2 1.246(3), C2–C12 1.464(3), and C15–C18 1.489(4). Valence angles (°): N1–C1–N2 109.6(2), C1–N1–C3 110.2(2), C1–N2–C2 109.3(2), and C2–C3–N1 98.1(2). Torsion angles (°): N1–C1–C6–C7 −6.6(4), N2–C2–C12–C13 −1.5(4), and C14–C15–C18–C19 8.0(5). | ||
Among notable features of the molecular structure of radical 11a are characteristic lengths of the C–O bond (1.246(3) Å) and C–C bond connecting imidazole and quinoid rings (1.409(3) Å). The first one is equal to the same C–O bond of 2,4,6-tri-tert-butylphenoxyl radical16 and very slightly differs from that of the C–O bond in related biradicaloids (1.252 Å (ref. 17a) and 1.241 Å,17c respectively) but significantly differs from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length in diamagnetic 3,5,3′,5′-tetra-tert-butyl-4,4′-diphenoquinone (1.228 Å).18 The second one is the typical “one-and-half” bond with the length close to that of the “bridge bond” CC (1.408 Å) of conjugated phenoxyl radical galvinoxyl19 and the CC bond (1.403 Å) between phenoxyl and heterocyclic moieties in biradicaloid QBT.17c The data above suggest that a radical molecule should exist mostly in the phenoxyl form in a solid state. Although the imidazole and both biphenyl rings are plane within the margin of experimental error, the quinoid ring is slightly distorted [±0.040(3) Å], with C9 and O2 atoms deviating from the quinoid ring plane by 0.051(3) and 0.186(2) Å, respectively. The tetracyclic frame of 11a is virtually flat; for example, dihedral angles between imidazole and adjacent rings are 5.8° for the phenoxyl ring and 1.2° for an aryl unit of biphenyl. The torsion angle between ring planes in the biphenyl moiety is 7.2°. Such planarity of the tetracyclic moiety is appropriate for formation of 0D supramolecular unity, namely a dimer of molecules, via an interaction of plane π-systems (Fig. 4 and Table S2 in ESI†) through “head-to-head” stacking. The molecular dimers in turn interact with the nearest ones via weak C17–H⋯O2 hydrogen bonds (Table S2 in ESI†) binding them along two directions into layers parallel to plane (1, 0, −2). There are no short interatomic distances between the layers.
O bond length in diamagnetic 3,5,3′,5′-tetra-tert-butyl-4,4′-diphenoquinone (1.228 Å).18 The second one is the typical “one-and-half” bond with the length close to that of the “bridge bond” CC (1.408 Å) of conjugated phenoxyl radical galvinoxyl19 and the CC bond (1.403 Å) between phenoxyl and heterocyclic moieties in biradicaloid QBT.17c The data above suggest that a radical molecule should exist mostly in the phenoxyl form in a solid state. Although the imidazole and both biphenyl rings are plane within the margin of experimental error, the quinoid ring is slightly distorted [±0.040(3) Å], with C9 and O2 atoms deviating from the quinoid ring plane by 0.051(3) and 0.186(2) Å, respectively. The tetracyclic frame of 11a is virtually flat; for example, dihedral angles between imidazole and adjacent rings are 5.8° for the phenoxyl ring and 1.2° for an aryl unit of biphenyl. The torsion angle between ring planes in the biphenyl moiety is 7.2°. Such planarity of the tetracyclic moiety is appropriate for formation of 0D supramolecular unity, namely a dimer of molecules, via an interaction of plane π-systems (Fig. 4 and Table S2 in ESI†) through “head-to-head” stacking. The molecular dimers in turn interact with the nearest ones via weak C17–H⋯O2 hydrogen bonds (Table S2 in ESI†) binding them along two directions into layers parallel to plane (1, 0, −2). There are no short interatomic distances between the layers.
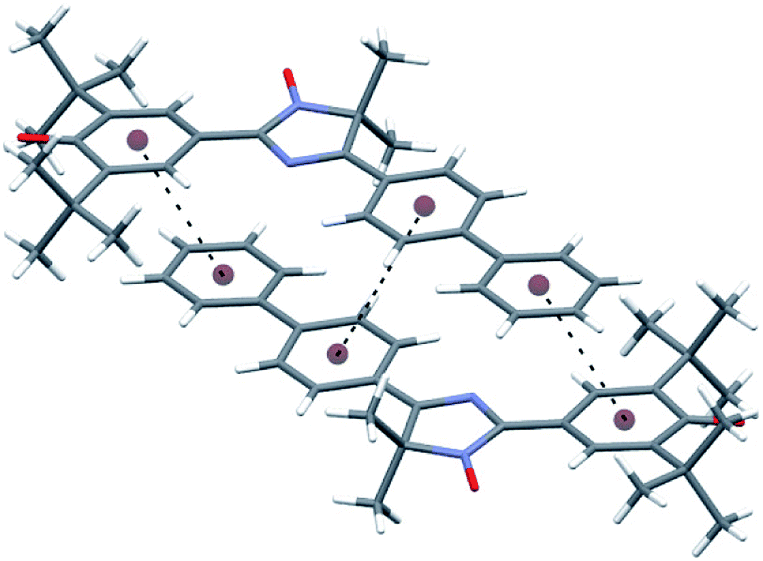 | ||
| Fig. 4 The pair of molecules of compound 11a associated by π–π interactions. | ||
Assessment of magnetic features of biphenyl radical 11a showed that at 300 K, μeff was 1.73 μB, in good agreement with the theoretical spin value μB (Fig. 5) for one paramagnetic center with spin S = 1/2 for a g-factor equal to 2. Intermolecular exchange interactions are weak.
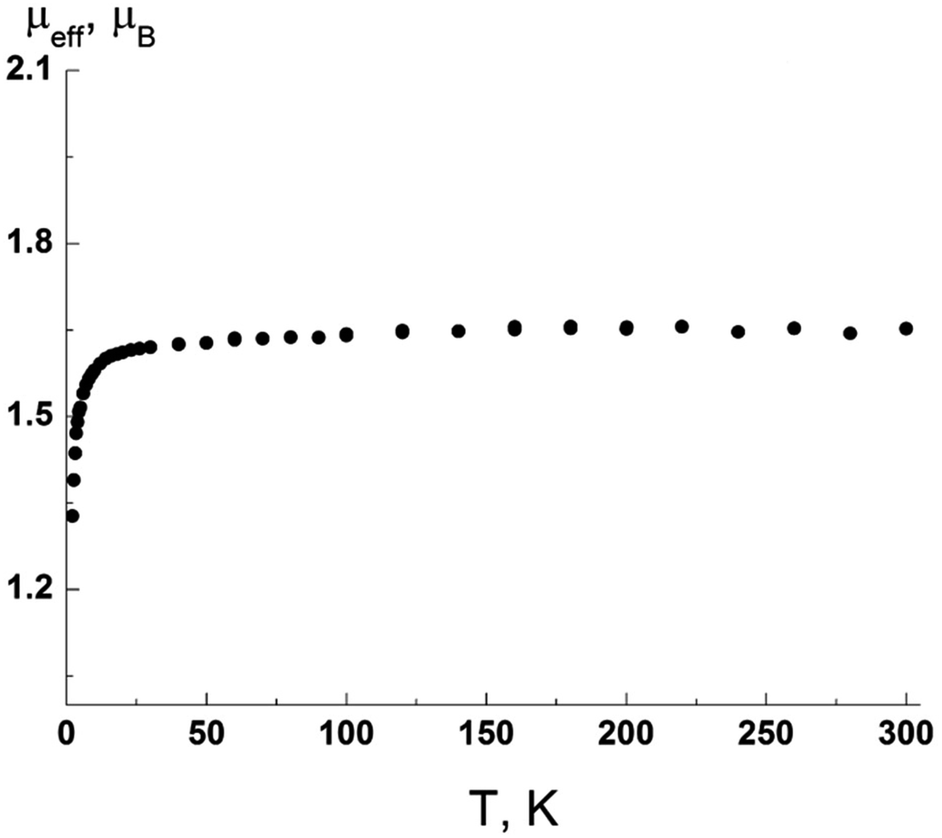 | ||
| Fig. 5 Experimental μeff (T) dependence for radical 11a. | ||
Having such a reactive substrate as imidazole 1, we were tempted to try it in the self-coupling reaction to obtain a symmetric biphenyl compound bearing 4H-imidazole moieties at positions C-4 and C-4′, i.e., to carry out a reaction of the following type: 2Ar–I + {M} → Ar–Ar + MI2. This approach would allow us to synthesize another interesting type of a paramagnetic substance, namely, a symmetrical hybrid diradical, which has not been known previously. Similar compounds, for example, molecules formed by quinonoidal residues conjugated with donor bridging fragments, switch to the biradicaloid state under the influence of external stimuli (e.g., temperature).17 As reported recently, they could substantially increase the power conversion efficiency of organic photovoltaic systems if used in small quantities (0.3–0.6 wt%) as additives.20 There is another class of stable diradicals, bis(nitronyl nitroxides), and their paramagnetic nuclei interact with each other through a rigid π-linker; they are of interest as weakly antiferromagnetically coupled spin-dimers with a singlet ground state that can be turned into a triplet state in a magnetic field.21
To obtain the desired diradical based on imidazole 1, we tried to employ the recently developed palladium-catalyzed version of the Ullmann-type reaction, which proceeds with iodoarene derivatives without phosphine ligands in the presence of palladium and potassium acetates under aerobic conditions.22 Nevertheless, under reductive coupling conditions described by those authors (10 mol% Pd(OAc)2, 5 equiv. of AcOK, Me2CO, 100 °C), imidazole 1 remained unchanged even after 24 h heating in a sealed ampoule.
Earlier, Higuchi and Kurth proposed practical one-pot synthesis of dimeric polyheterocycles, bis(terpyridines), which involves forming the corresponding boronic ester from 4′-(para-bromophenyl) terpyridine via the Miyaura reaction, followed by the Suzuki reaction of the appropriate bromo derivative under specially selected and controlled conditions (temperature, the solvent, base, quantity of the boron reagent, and Pd catalyst).23 We tested a similar tandem process involving tricyclic iodo derivative 1 under conditions that allowed us to avoid the use of strong bases. Indeed, the interaction of two equivalents of 4H-imidazole 1 with bis(pinacolato)diboron 12 and potassium acetate as a base in the presence of dichlorobis(triphenylphosphine)palladium in dimethylformamide (DMF) with heating to 110–115 °C resulted in formation of a dark orange precipitate of homocoupling product 13 (Scheme 3). This reaction is likely to proceed with the formation of an intermediate (2-aryldioxaborolane 14) which then reacts with an excess of substrate 1, leading to biphenyl derivative 13. Indeed, 1,3,2-dioxoborolane 14 was isolated as the dominant product in the temperature-controlled process via the interaction of 4H-imidazole 1 with a slight excess of B2(pin)2. Its structure was proved unambiguously by a combination of elemental analysis data and 1H, 13C, and 11B NMR spectra.
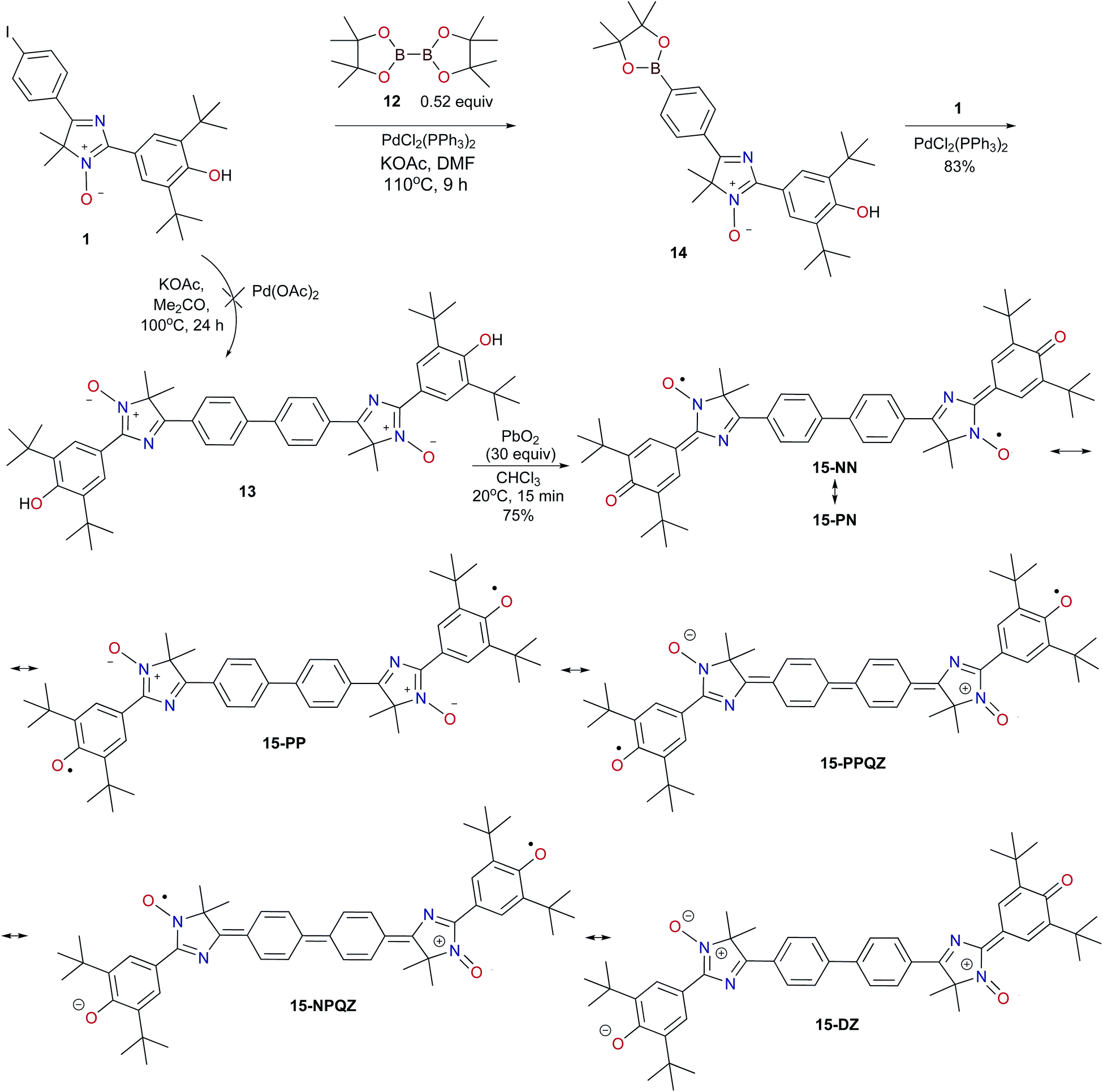 | ||
| Scheme 3 The Suzuki–Miyaura homocoupling reaction involving 4H-imidazole N-oxide 1 and preparation of symmetrical hybrid diradical 15. | ||
Obtained biphenyl bis(imidazole) 13 was poorly soluble in conventional organic solvents (alcohols, halocarbons, DMF, or DMSO); therefore, it can be isolated from the reaction mixture quantitatively. To separate the precipitated product from palladium compounds and inorganic impurities, we performed the Soxhlet hot extraction by boiling the precipitate with chloroform. Again, heating compound 13 in an oxygen atmosphere resulted in staining the solution with a dark orange color, as was the case for biphenyl imidazole derivatives 10. Evidently, the staining was due to the formation and accumulation of stable phenoxyl-nitroxyl radicals (TLC control). To prove the dimeric structure of the obtained product, we used NMR spectroscopy along with elemental analysis data.
Thus, signals typical for a symmetrical structure were observed in the spectra of bis(imidazole) 13 in a trifluoroacetic acid solution. For example, singlets of gem-methyl and t-butyl groups shifted downfield to 2.06 and 1.57 ppm, respectively, in the proton spectrum of compound 13. Protons of the biphenyl part represent the AA′BB′ system with doublets at 8.05 and 8.51 ppm. The 13C NMR spectrum of 13 is even more remarkable in this respect (downfield shifting of several signals). Thus, low-field signals at 166.0 and 194.3 ppm were attributed to carbon atoms respectively associated with nitrogen atoms of nitrone and imino groups of the heterocycle (which corresponds to the downfield shifting of these signals by 18–19 ppm). Apparently, the covalent interaction of bis(imidazole) 13 with trifluoroacetic acid contributes to the downfield shifting of signals of the 1H and 13C nuclei. CF3COOH is known to be capable of protonating a number of organic compounds, for instance, ketones, various imino derivatives, and cyclic nitrones, by shifting an appropriate carbon signal of the double CX bond in the 13C NMR spectrum into a low field from 1.5 to 27.5 ppm.24
The oxidation of biphenyl bis(imidazole) 13 with a large excess of lead dioxide (20–30 equiv.) in a dilute chloroform solution for 15–30 min resulted in complete conversion of bis(phenol) 13 and formation of hybrid diradical 15, which was isolated in a high yield by gentle evaporation of the reaction solution in a solid form as a dark brown powder. We found that compound 15 could be purified by flash chromatography on silica gel without any noticeable decomposition. The diradical is highly soluble in chlorinated solvents (methylene chloride, chloroform, and CCl4) but is virtually insoluble in nonpolar or protic/aprotic solvents (hexane, ether, ethanol, and acetone). The use of other oxidizing systems to transform bis(phenol) 13 (for example, DDQ or an aqueous alkaline solution of potassium ferricyanide) also led to the preferential formation of compound 15, but there was significant accumulation of impurities in this case. Isolated from chloroform, the diradical is a crystal solvate containing one molecule of the solvent. A peak at m/z 782.4 with the intensity of ∼4% corresponding to the [M + 2] ion (Fig. S5 in ESI†) is observed in the high-resolution mass spectrum of diradical 15. It is worth noting that all the previously obtained hybrid monoradicals in their high-resolution mass spectrum are characterized by an [M + 1] ion. Other characteristic signals in the mass spectrum of compound 15 are ions at m/z 766 (12%) and 750 (18%), corresponding to a sequential loss of one and two oxygen atoms by the diradical molecule.
It should be noted that this is the first example of formation of a stable dimeric phenoxyl-nitroxide that can apparently exist in various hybrid forms (15-NN, 15-NP, and 15-PP), including exotic biradical species with a zwitterionic structure whose charged parts are separated by the diphenoquinone linker (15-PPQZ and 15-NPQZ) as well as diamagnetic zwitterionic structure 15-DZ (Scheme 3).
Comparative cyclic voltammograms of radicals 8 and 15 are presented in Fig. 6. Iodine-substituted radical 8 yields two irreversible one-electron oxidation peaks at −0.48 and 0.76 V, which correspond to formation of the oxoammonium cation and radical dicationic species. Nonetheless, diradical 15 undergoes reversible two-electron oxidation at E1/2 = −0.63 V forming a dioxammonium feature (Scheme S2 in ESI†). Further oxidation of dioxammonium dication proceeds quasi-reversibly at 0.80 V.
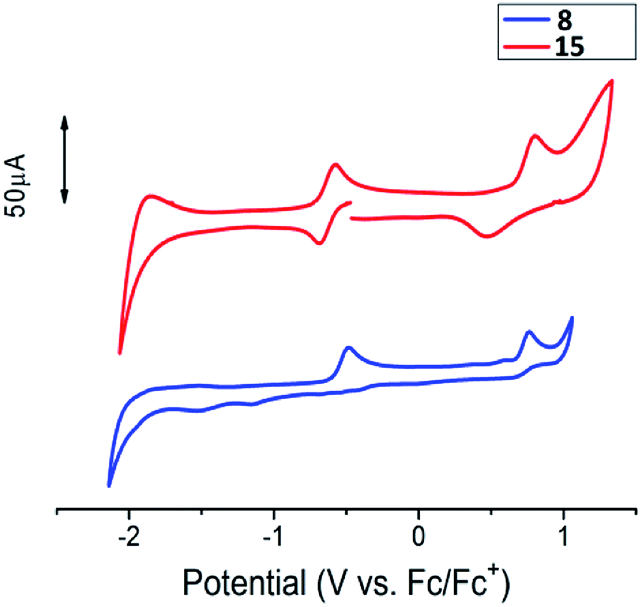 | ||
| Fig. 6 Cyclic voltammograms for monoradical 8 and diradical 15 in a CH3CN solution. | ||
The EPR spectral and structural characteristics of this unusual diradical and its magnetic analysis will be the subject of our future study and deserve a separate publication.
Conclusions
Thus, we showed that 5-(para-iodophenyl)-4,4-dimethyl-2-(3,5-di-tert-butyl-4-hydroxyphenyl)-4H-imidazole 3-oxide possessing high reactivity in Pd-catalyzed processes can readily react with various arylboronic acids as well as bis(pinacolato)diboron in one-pot tandem synthesis with formation of the corresponding biphenyl derivatives in high yields. The products can be easily oxidized to stable hybrid mono- and diradicals of the phenoxyl-nitroxyl series. We are planning to extend this approach to other types of cross-coupling (e.g., the Stille reaction25 and direct arylation of donor heterocycles26) to synthesize new dimeric conjugated diradicals, corresponding to A–D–A triads in which the ter(quater)phenyl or ter(quater)thiophene core will be the donor linker. Obviously, the 1,3,2-dioxoborolane derivative of 4H-imidazole 3-oxide synthesized here can be applied to obtain asymmetric hetero diradicals bearing different types of a paramagnetic nucleus,27 which are appealing as building blocks for functional materials, particularly, for creation of components of a quantum computer.Experimental
General
Analytical-grade reagents and solvents were used, and reactions were monitored by TLC. Column chromatography and TLC were performed using Acros silica gel 60A (0.035–0.070 mm) and Sorbfil PTLC-AF-UV 254 (Russia), respectively. Mass spectra were registered on a DFS high-resolution mass spectrometer. 1H NMR and 13C NMR spectra were recorded on Bruker AV-300, AV-400, DRX-500 spectrometers at 300/400/500 and 75/100/125 MHz, respectively, for 1–5% solutions of compounds in CDCl3, DMSO-d6, or CF3COOH (for bis(imidazole) 13). Residual proton signals from the deuterated solvents served as references [DMSO-d6 (2.50 ppm), CDCl3 (7.24 ppm), for 1H spectra]. 13C NMR chemical shifts are reported in reference to an undeuterated residual solvent [CDCl3 (76.9 ppm) or DMSO-d6 (39.4 ppm)]. The assignment of signals of carbon atoms in 13C NMR spectra of 4H-imidazole derivatives 1 and 10a–d was carried out on the basis of an earlier spectral study on the spectra of cyclic nitrones.28 Fourier transform infrared (FT-IR) spectra were acquired in KBr pellets on a Bruker Vector-22. The UV-Vis spectra were obtained for EtOH solutions using a Hewlett-Packard HP 8453 spectrophotometer. Raman spectrum of diradical 15 was recorded with a Bruker Optics SENTERRA microscope spectrometer equipped with a 785 nm direct diode laser with 1 cm−1 resolution and 25 mW laser power. The Raman shift range considered was between 170 and 3000 cm−1. Elemental analyses were performed on an automatic CNS analyzer Euro EA 3000. The melting points were determined on an FP 81 HT instrument, Mettler Toledo.Cyclic voltammetry measurements were conducted in a CH3CN solution on a computer-controlled P-8nano potentiostat/galvanostat (Elins, Russia) in combination with a three-electrode cell (Gamry); 0.1 M n-Bu4NPF6 was applied as a supporting electrolyte. Pt, a Pt wire, and Ag/AgCl served as a working, counter, and reference electrode, respectively. The reference electrode was calibrated by measurement of the redox potentials of ferrocene.
Arylboronic acids 9a–d, B2(pin)2 (bis(pinacolato)diboron) 12, and palladium catalysts were obtained from commercial sources (Acros Organics); 3,5-di-t-butyl-4-hydroxy-benzaldehyde 6 was synthesized according to a procedure described in the literature.29 All the other reagents were purchased from Acros Organics or Alfa-Aesar.
Synthesis of key 2-(3,5-di-t-butyl-4-hydroxyphenyl)-5-(4-iodophenyl)-4,4-dimethyl-4H-imidazole 3-oxide 1, its subsequent cross-coupling reactions, and oxidation of products are described in ESI.†
X-Ray structural analysis
The unit cell parameters and experimental intensities of radical 11a were measured at 296(2) K with a Bruker Kappa Apex II CCD diffractometer with Mo Kα radiation (λ = 0.71073 Å) and a graphite monochromator. The structure was solved by direct methods and refined in an anisotropic (isotropic for H) approximation using the SHELX-97 software suite.30 Positions of the hydrogen atoms were calculated and refined by means of the riding model. Analysis of the geometry and intermolecular interactions was carried out in the PLATON software.31Crystallographic data of 11a:‡ C31H35N2O2, FW = 467.61, monoclinic, P21/c a 11.5429(7), b 14.3317(8), c 16.995(1) Å, β 109.552(2)°, V 2649.3(3) Å3, Dcalc 1.172 g cm−3, μ(Mo-Kα) 0.073 mm−1, F(000) 1004.0, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 398 measured reflections θfull 31.19°, completeness 99.7% (θmax), 7602 independent (Rint 0.0558), 324 parameters, R1 0.0954 [for 5116 observed I > 2σ(I)], wR2 0.3367 (all data), GooF = 1.308, largest diff. peak and hole 0.414 and −0.728 e A−3.
398 measured reflections θfull 31.19°, completeness 99.7% (θmax), 7602 independent (Rint 0.0558), 324 parameters, R1 0.0954 [for 5116 observed I > 2σ(I)], wR2 0.3367 (all data), GooF = 1.308, largest diff. peak and hole 0.414 and −0.728 e A−3.
EPR spectrometry
Continuous-wave EPR spectra were acquired on a Bruker EMX spectrometer at room temperature in dilute (ca. 5 × 10−5 M) toluene solutions degassed by means of repeated freeze–pump–thaw cycles. At modulation 0.01 mT@100 KHz and MW power 2 mW, the spectra were recorded as single slow scans (ca. 3 h). Isotropic g values were determined using solid DPPH (2,2-diphenyl-1-picrylhydrazyl) as a standard and were found to be typical for cyclic nitroxides. The accuracy of determining hyperfine coupling constants and g values was estimated at 0.005 mT and 0.0001, respectively.Magnetic measurements
Magnetic susceptibility of a polycrystalline sample of 11a was measured with a Quantum Design MPMSXL SQUID magnetometer in the temperature range 2 to 300 K in a magnetic field of up to 5 kOe.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Russian Foundation for Basic Research (grants no. 15-03-02741 and 17-53-50043) and the Multi-Access Chemical Service Center SB RAS for spectral and analytical measurements.Notes and references
- (a) I. Maluenda and O. Navarro, Molecules, 2015, 20(5), 7528 CrossRef PubMed; (b) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58(48), 9633 CrossRef; (c) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef; (d) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513 CrossRef.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443 CrossRef PubMed.
- B. S. Nehls, F. Galbrecht, A. Bilge, D. J. Brauer, C. W. Lehmann, U. Scherf and T. Farrell, Org. Biomol. Chem., 2005, 3, 3213 RSC.
- G. Hu, S. P. Kitney, S. M. Kelly, W. Harrison and M. O'Neill, Liq. Cryst., 2017, 44(11), 1632 CrossRef.
- A. Krishna, A. V. Lunchev and A. C. Grimsdale, in Synthetic Methods for Conjugated Polymer and Carbon Materials, ed. M. Leclerc and J.-F. Morin, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2017, 2, pp. 59–87 Search PubMed.
- A. Littke, in Modern Arylation Methods, ed. L. Ackermann, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009, 2, pp. 25–67 Search PubMed.
- (a) H. Doucet, Eur. J. Org. Chem., 2008,(12), 2013 CrossRef; (b) D. G. Hall, Boronic Acids, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005 CrossRef.
- (a) P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497 CrossRef; (b) G. A. Molander and B. Canturk, Angew. Chem., Int. Ed., 2009, 48(49), 9240 CrossRef PubMed; (c) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41(11), 1461 CrossRef PubMed.
- Y. Ten, S. Amitina, A. Bogomyakov, Y. Gatilov, A. Lomanovich, I. Grigor'ev and D. Mazhukin, Book of Abstracts of 14-th International Conference on Molecule-based Magnets, Saint Petersburg, Russia, 2014 Search PubMed.
- (a) M. Souto, C. Rovira, I. Ratera and J. Veciana, CrystEngComm, 2017, 19, 197 RSC; (b) M. Souto, V. Lloveras, S. Vela, M. Fumanal, I. Ratera and J. Veciana, J. Phys. Chem. Lett., 2016, 7, 2234 CrossRef PubMed; (c) S. Vela, M. Souto, I. Ratera, C. Rovira and J. Veciana, J. Phys. Chem. A, 2016, 120, 10297 CrossRef PubMed; (d) T. Ikezawa, K. Mutoh, Y. Kobayashi and J. Abe, Chem. Commun., 2016, 52, 2465 RSC; (e) N. O. Chalkov, V. K. Cherkasov, G. A. Abakumov, A. G. Starikov and V. A. Kuropatov, Beilstein J. Org. Chem., 2016, 12, 2450 CrossRef PubMed; (f) H. Yamashita, T. Ikezawa, Y. Kobayashi and J. Abe, J. Am. Chem. Soc., 2015, 137, 4952 CrossRef PubMed; (g) N. O. Chalkov, V. K. Cherkasov, G. A. Abakumov, G. V. Romanenko, S. Y. Ketkov, I. V. Smolyaninov, A. G. Starikov and V. A. Kuropatov, Eur. J. Org. Chem., 2014, 4571 CrossRef; (h) J. Guasch, L. Grisanti, L. Lloveras, J. Vidal-Gancedo, M. Souto, D. C. Morales, M. Vilaseca, C. Sissa, A. Painelli, I. Ratera, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2012, 51, 11024 CrossRef PubMed; (i) S. Nishida, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 7277 CrossRef PubMed.
- M. Souto, H. Cui, M. Peña, V. G. Baonza, H. O. Jeschke, M. Tomic, R. Valentí, D. Blasi, I. Ratera, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2016, 138, 11517 CrossRef PubMed.
- (a) S. M. Winter, A. R. Balo, R. J. Roberts, K. Lekin, A. Assoud, P. A. Dube and R. T. Oakley, Chem. Commun., 2013, 49, 1603 RSC; (b) P. Vasko, J. Hurmalainen, A. Mansikkamäki, A. Peuronen, A. Mailman and H. M. Tuononen, Dalton Trans., 2017, 46, 16004 RSC.
- (a) H. Oka, H. Kouno and H. Tanaka, J. Mater. Chem., 2007, 17, 1209 RSC; (b) H. Oka, T. Tamura, Y. Miura and Y. Teki, J. Mater. Chem., 2001, 11, 1364 RSC; (c) H. Oka, T. Tamura, Y. Miura and Y. Teki, J. Mater. Chem., 1999, 9, 1227 RSC.
- (a) A. Bodzioch, M. Zheng, P. Kaszynski and G. Utecht, J. Org. Chem., 2014, 79, 7294 CrossRef PubMed; (b) Y. Takahashi, Y. Miura and N. Yoshioka, Chem. Lett., 2014, 43, 1236 CrossRef; (c) C. P. Constantinides, P. A. Koutentis and G. Loizou, Org. Biomol. Chem., 2011, 9, 3122 RSC; (d) Y. Miura, S. Ooshima and N. Yoshioka, Book of Abstracts of the 9-th Japanese-Russian International Workshop on Open Shell Compounds and Molecular Spin Devices, Awaji Island, Japan, 2015 Search PubMed; (e) K. E. Fairfull-Smith and S. E. Bottle, Eur. J. Org. Chem., 2008, 5391 CrossRef.
- C. Stroh, M. Mayor and C. Hänisch, Eur. J. Org. Chem., 2005, 3697 CrossRef.
- V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256 RSC.
- (a) H. Wei, L. Zhang, H. Phan, X. Huang, T. S. Herng, J. Zhou, W. Zeng, J. Ding, S. Luo, J. Wu and Z. Zeng, Chem.–Eur. J., 2017, 23, 9419 CrossRef PubMed; (b) K. Naoda, D. Shimizu, J. O. Kim, K. Furukawa, D. Kim and A. Osuka, Chem.–Eur. J., 2017, 23, 8969 CrossRef PubMed; (c) F. Tampieri, L. Colella, A. Maghsoumi, J. Marti-Rujas, E. Parisini, M. Tommasini, C. Bertarelli and A. Barbon, J. Phys. Chem. C, 2016, 120, 5732 CrossRef; (d) L. Colella, L. Brambilla, V. Nardone, E. Parisini, C. Castiglioni and C. Bertarelli, Phys. Chem. Chem. Phys., 2015, 17, 10426 RSC; (e) Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC; (f) D. Fazzi, E. V. Canesi, F. Negri, C. Bertarelli and C. Castiglioni, ChemPhysChem, 2010, 11, 3685 CrossRef PubMed.
- R. Shukla and R. Rathore, Synthesis, 2008, 3769 Search PubMed.
- H.-S. Sheu, C.-H. Lee, Y. Wang and L. Y. Chiang, J. Chin. Chem. Soc., 1994, 41, 797 CrossRef.
- (a) Z. Kan, L. Colella, E. V. Canesi, A. Vorobiev, V. Skrypnychuk, G. Terraneo, D. R. Barbero, C. Bertarelli, R. C. I. MacKenzie and P. E. Keivanidis, J. Mater. Chem. A, 2016, 4, 1195 RSC; (b) Z. Kan, L. Colella, E. V. Canesi, G. Lerario, R. S. S. Kumar, V. Bonometti, P. R. Mussini, G. Cavallo, G. Terraneo, P. Pattanasattayavong, T. D. Anthopoulos, C. Bertarelli and P. E. Keivanidis, Sol. Energy Mater. Sol. Cells, 2014, 120, 37 CrossRef.
- (a) K. Kolanji, P. Ravat, A. S. Bogomyakov, V. I. Ovcharenko, D. Schollmeyer and M. Baumgarten, J. Org. Chem., 2017, 82, 7764 CrossRef PubMed; (b) Y. B. Borozdina, E. Mostovich, V. Enkelmann, B. Wolf, P. T. Cong, U. Tutsch, M. Lang and M. Baumgarten, J. Mater. Chem. C, 2014, 2, 6618 RSC; (c) E. A. Mostovich, Y. Borozdina, V. Enkelmann, K. Removic-Langer, B. Wolf, M. Lang and M. Baumgarten, Cryst. Growth Des., 2012, 12, 54 CrossRef.
- M. Yu, R.-Y. Tang and J.-H. Li, Tetrahedron, 2009, 65, 3409 CrossRef.
- F. S. Han, M. Higuchi and D. G. Kurth, Org. Lett., 2007, 9, 559 CrossRef PubMed.
- (a) H. Günther, NMR Spectroscopy: basic principles, concepts, and applications in chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013 Search PubMed; (b) I. A. Grigor'ev, V. I. Mamatyuk, G. I. Shchukin, V. V. Martin and L. B. Volodarskii, Chem. Heterocycl. Compd., 1986, 861 (Khim. Geterotsikl. Soedin., 1986, 1065) CrossRef; (c) M. Allen and J. D. Roberts, Can. J. Chem., 1981, 59, 451 CrossRef.
- (a) C. Cordovilla, C. Bartolome, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040 CrossRef; (b) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 Search PubMed; (c) J. K. Stille, Angew. Chem., Int. Ed., 1986, 25, 508 CrossRef.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupre and M. Leclerc, Chem. Rev., 2016, 116, 14225 CrossRef PubMed.
- (a) Y. Takahashi, R. Matsuhashi, Y. Miura and N. Yoshioka, Chem.–Eur. J., 2018, 24, 7939 CrossRef PubMed; (b) Y. Takahashi, Y. Miura and N. Yoshioka, ChemPhysChem, 2018, 19, 175 CrossRef PubMed; (c) S. E. Tolstikov, E. V. Tretyakov, D. E. Gorbunov, I. F. Zhurko, M. V. Fedin, G. V. Romanenko, A. S. Bogomyakov, N. P. Gritsan and D. G. Mazhukin, Chem.–Eur. J., 2016, 22, 14598 CrossRef PubMed; (d) N. M. Gallagher, J. J. Bauer, M. Pink, S. Rajca and A. Rajca, J. Am. Chem. Soc., 2016, 138, 9377 CrossRef PubMed.
- I. A. Grigor'ev, I. A. Kirilyuk and L. B. Volodarskii, Chem. Heterocycl. Compd., 1988, 1355 (Khim. Geterotsikl. Soedin., 1988, 1640) CrossRef.
- S. Rinker, P. R. James, M. Neumann, O. Erpeldinger and F. Kraushaar, US Pat., 20100267992A1, 2010.
- (a) G. M. Sheldrick, SHELX-97 (Release 97-2), University of Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef PubMed.
- (a) A. L. Spek, PLATON (Version, 10M), Utrecht Univ., Netherlands, 2003 Search PubMed; (b) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical and spectroscopic data for all new compounds. Experimental and simulated ESR spectra for the new radicals, CV data for 4H-imidazole N-oxides. CCDC 1564701. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra05103h |
| ‡ Additional crystallographic data (CCDC 1564701) for hybrid radical 11a. |
| This journal is © The Royal Society of Chemistry 2018 |