
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiplex real-time PCR assay combined with rolling circle amplification (MPRP) using universal primers for non-invasive detection of tumor-related mutations†
Jian Gong a,
Yishuai Lib,
Ting Linc,
Xiaoyan Fengc and
Li Chu*ad
a,
Yishuai Lib,
Ting Linc,
Xiaoyan Fengc and
Li Chu*ad
aHebei Medical University, Hebei 050017, China. E-mail: gongjian2001@126.com
bDepartment of Thoracic Surgery, Hebei Chest Hospital, Hebei 050000, China. E-mail: liyishuai66@126.com
cApexbio Biotech Co., LTD, Beijing 100176, China
dDepartment of Pharmacology, Hebei University of Chinese Medicine, Hebei 050200, China. E-mail: chuli0614@126.com
First published on 1st August 2018
Abstract
With the continuous development and application of targeted drugs, it is particularly desirable to find a non-invasive diagnostic approach to screen patients for precision treatment. Specifically, detection of multiple cancer-related mutations is very important for targeted therapy and prediction of drug resistance. Although numerous advanced PCR methods have been developed to discriminate single nucleotide polymorphisms, their drawbacks significantly limit their application, such as low sensitivity and throughput, complicated operations, and expensive costs. In order to overcome these challenges, in this study, we developed a method combining multiplex and sensitive real-time PCR assay with rolling circle amplification. This allows specific and sensitive discrimination of the single nucleotide mutation and provides convenient multiplex detection by real-time PCR assay. The clinical potential of the MPRP assay was further demonstrated by comparing samples from 8 patients with a digital PCR assay. The coincident results between these two methods indicated that the MPRP assay can provide a specific, sensitive, and convenient method for multiplex detection of cancer-related mutations.
Introduction
Tumorigenesis has been reported to result from the accumulation of multiple genetic mutations.1 Understanding the genetic context of cancer is of upmost importance for precision treatment of patients. Non-small cell lung cancer (NSCLC), for example, has an EGFR mutation rate as high as 30–60% in East Asian populations. The phase III study of IPASS showed a significant clinical benefit of using EGFR tyrosine kinase inhibitor (TKI) in patients with positive EGFR mutations compared to traditional chemotherapy and in patients with negative EGFR mutations.2 Therefore, detection of cancer-related mutations, especially driver mutations, is significantly beneficial for the screening of patients for targeted therapy and the early detection of drug resistance. This is termed companion diagnostics.3 Since 2011, authorities such as ASCO and NCCN have successively established norms and guidelines for the detection of driver mutations such as EGFR and BRAF. Authorities reached an agreement that all patients receiving targeted drug treatment such as with an EGFR TKI or a BRAF inhibitor need to undergo molecular detection of EGFR, BRAF, etc.4–6Therefore, developing a multiplex, sensitive and convenient method to detect tumor-related mutations is an urgent need for clinical precision treatment. Liquid biopsies of blood or urine were recently established to be used for the diagnosis of cancer and other diseases. The clinical application of cell-free DNA, which contains the genetic information of tumors, took center stage.7 However, the low amount and shorter length of cell-free DNA limited the effective detection of tumor-related mutations.8 Moreover, sensitive detection of the mutation under the highly abundant background of wild-type DNA remains difficult. At present, several real-time PCR methods such as ARMS,9,10 COBAS,11,12 etc., have been developed to detect this mutation. Unfortunately, low sensitivity and false-positive results have limited their clinical application. Due to the increased number of approved targeted medications and the subsequent increase in DNA targets that need to be detected, more input samples need to be prepared. Excitingly, the padlock RCA method has been established to specifically identify single nucleotide polymorphisms (SNP).13 After complete annealing to the target DNA strand, two ends of the padlock probe can be ligated to circularize. A single mismatched base present at the 3′ terminal of the probe will abolish the ligation, thus ensuring high specificity in DNA and RNA detection. Then, rolling circle amplification (RCA), which is based on rolling circle replication of circular DNA pathogens in nature, was carried out to achieve exponential amplification of the circularized DNA.14,15 Combined with RCA technology, padlock RCA can detect SNP from 1 ng (300 copies) of genomic DNA and the accuracy rate of SNP genotyping results can reach 100%.16 However, the effectiveness of ligation and the stability of the reaction conditions can still be problematic. On the other hand, some methods require the removal of the residual probe which cannot hybridize to the target template, thereby increasing the complexity of detection.17 Hence, padlock RCA-based assay needs further improvement.
In this study, we presented the MPRP method to combine padlock RCA assay and real-time PCR technology to detect multiplex tumor-related mutations. We found that this combined method can specifically and sensitively distinguish the genetic mutation and allow convenient multi-detection of tumor-related mutations by real-time PCR. Furthermore, we demonstrated the clinical applicability of this combined method by multi-detection of EGFR mutations L858R and T790M and BRAF mutation V600E in patients' plasma.
Results and discussion
Detection of cancer-related mutations helps identify patients who can greatly benefit from targeted therapy through improving treatment outcomes and reducing healthcare expenditures. The traditional way to detect cancer-related mutations involves a tissue biopsy, through which a sufficient sample is necessary.18 However, some patients cannot undergo surgery or puncture to obtain the quantity necessary for genetic testing. Recently, liquid biopsies, especially those which contain cell-free DNA samples, provide a convenient way for the detection of genetic mutations. Its advantage lies in the ability to reduce the risk of biopsy through non-invasive sampling and effectively prolonging the survival of patients. At present, an increasing number of drug targets have been developed, which makes it highly attractive to clinicians to be able to detect multiplex genetic mutations in patients in order to achieve the best clinical outcome. Hence, this non-invasive and multiplex detection approach of tumor-related mutations has great promise in clinical cancer therapies. Although numerous efforts have been made towards the development of new technologies to detect genetic mutations, including ARMS,9,10 COBAS,11,12 BEAMing technology and digital PCR, their applications are significantly limited by low sensitivity, false-positive results, complicated operations, low throughput, expensive chips and closed reagent.19–21 In the current study, we combined the padlock probe technology with multiplex real-time PCR to detect genetic mutations, and demonstrated the feasibility, great specificity and high sensitivity of the assay.Principle of the MPRP assay
The principle of the MPRP assay for the detection of gene mutations was illustrated in Fig. 1. To summarize, the padlock probe was composed of 3 parts: the H5 region, the link region, and the H3 region. The mismatched base was designed at the 3′ end of the H3 region (Table 1). The sequences of the link region within different padlock probes were the same, thus the primers uniF and uniR in this sequence are universal for RCA and real time PCR reactions. The length of the H3 region should be shorter than the H5 region to improve the specificity of subsequent ligation. The 5′–PO4 group and 3′–OH group that were modified to the end of the padlock probe can be specifically linked to form a DNA ring in the presence of HiFi Taq DNA ligase, when the padlock probe is completely complementary to the target DNA at the H5 and H3 regions. Otherwise, the ligation cannot occur if mismatched base pairs exist at the 3′ end of the H3 region. After the circularization of the padlock probe, exonuclease I and exonuclease III were added to digest the ssDNA and dsDNA, respectively. The RCA reaction was followed to amplify the circularized DNA ring, and finally the product was detected for mutant discrimination by multiplex real time PCR. The abundance of each target DNA is negatively correlated with its Ct value obtained from quantitative real-time PCR.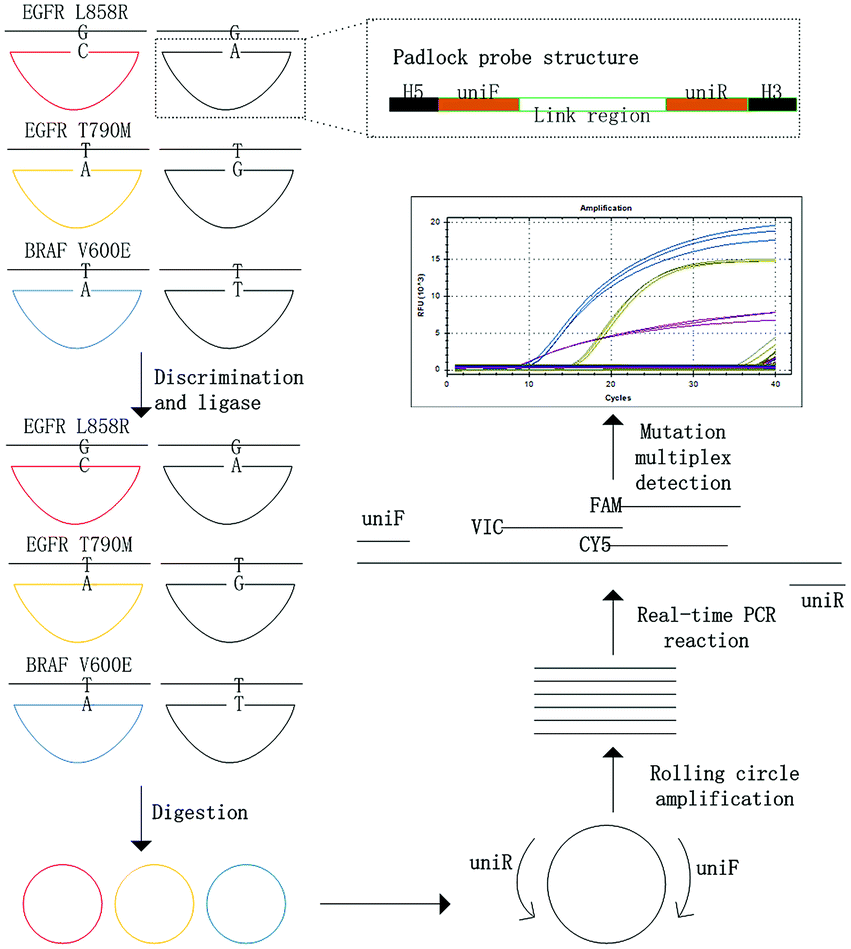 | ||
| Fig. 1 Schematic illustration of MPRP assay for multiple mutant detection. | ||
| Name | Sequence (5′–3′) | |
|---|---|---|
| Padlock probe | pEG858A | 5′–P-gcccaaaatctgtgatcttgaacataggtctcagtccagccattttagccgttcctcaca tcagactcgcactcgttagcaatcactttttacccagcagtttggcca-OH–3′ |
| pEG858C | 5′–P-gcccaaaatctgtgatcttgaacataggtctcagtccagccattttagccgttcctca catcagactcgcactcgttagcaatcactttttacccagcagtttggccc–OH-3′ | |
| pEG790G | 5′–P-tgatgagctgcacggtggaggacataggtctcagtccagccattttagccgttcctc acatcagactcgcactcgttagcaatcactttttcagccgaagggcatgagctgcg–OH-3′ | |
| pEG790A | 5′–P-tgatgagctgcacggtggaggacataggtctcagtccagccattttagccgttcctca catcagactcgcactcgttagcaatcactttttcagccgaagggcatgagctgca–OH-3′ | |
| pBR600A | 5′–P-ctgtagctagaccaaaatcacctatacataggtctcagtccagccattttagccgttcct cacatcagactcgcactcgttagcaatcactttttggacccactccatcgagatttca–OH-3′ | |
| pBR600T | 5′–P-ctgtagctagaccaaaatcacctatacataggtctcagtccagccattttagccgttcct cacatcagactcgcactcgttagcaatcactttttggacccactccatcgagatttct–OH-3′ | |
| Template DNA for padlock probe | tBR600T | 5′-ataggtgattttggtctagctacagtgaaatctcgatggagtgggtcccat-3′ |
| tBR600A | 5′-ataggtgattttggtctagctacagagaaatctcgatggagtgggtcccat-3′ | |
| tEG790C | 5′-ctcacctccaccgtgcagctcatcacgcagctcatgcccttcggctgcctc-3′ | |
| tEG790T | 5′-ctcacctccaccgtgcagctcatcatgcagctcatgcccttcggctgcctc-3′ | |
| tEG858T | 5′-agcatgtcaagatcacagattttgggctggccaaactgctgggtgcggaagagaa-3′ | |
| tEG858G | 5′-agcatgtcaagatcacagattttgggcgggccaaactgctgggtgcggaagagaa-3′ | |
| Digital PCR primers | 858F | 5′-gcagcatgtcaagatcacagatt-3′ |
| 858R | 5′-cctccttctgcatggtattctttct-3′ | |
| 790F | 5′-gcctgctgggcatctg-3′ | |
| 790R | 5′-tctttgtgttcccggacatagtc-3′ | |
| BF | 5′-acctcagatatatttcttcatg-3′ | |
| BR | 5′-ccagacaactgttcaaac-3′ | |
| PCR probes | P858M | 5′-FAM-agtttggcccgcccaa-MGB-3′ |
| P858W | 5′-VIC-agtttggccagcccaa-MGB-3′ | |
| P790W | 5′-VIC-tgagctgcgtgatga-MGB-3′ | |
| P790M | 5′-CY5-tgagctgcatgatga-MGB-3′ | |
| PBW | 5′-CY5-tcgagatttcactgtagct-MGB-3′ | |
| PBM | 5′-VIC-tcgagatttctctgtagct-MGB-3′ | |
| Universal primers | uniF | 5′-tggctggactgagacctatgt-3′ |
| uniR | 5′-agccgttcctcacatcagac-3′ |
Optimization, specificity and sensitivity of MPRP assay for mutation detection
In this study, padlock probe technology was used to discriminate the specific mutation, which has been demonstrated in microbiological and microRNA detection because of its high sensitivity and specificity for single nucleotide discrimination resolution.22,23 Only the perfect matched padlock probe could be ligated and circularized. Compared with oligonucleotide gap-fill ligation, one step of ligation in our study not only acquired high specificity, but also avoided the ligation events occurring at the 5′-end of the wrong padlock gap probe, which occupied the position and decreased the efficiency of ligation.24The successful circularization and specific discrimination of padlock probes depends on the activity of DNA ligase and the optimization of the reaction. T4 DNA ligase, which is one of the most popular DNA ligases used for joining the 5′ phosphate and 3′ hydroxyl terminal of duplex DNA or RNA,25 has the capability of SNP discrimination in RNA, but no capacity of SNP detection in DNA.24,26 HiFi Taq DNA ligase, which demonstrates strong capability of SNP discrimination in DNA, is quite suitable for the padlock probe.27
Moreover, a couple of factors affecting the padlock RCA reaction for SNP discrimination have been reported.28–30 Ligase temperature is one of the most important factors for the efficiency of ligation, which can be predicted using the Thermostable Ligase Reaction Temperature Calculator v0.8.4 tool from NEB (http://ligasecalc.neb.com/#!/ligation). In our experiment, 43 °C was selected as the ligase temperature. Moreover, the bases at the 3′ end of the padlock probe were LNA modified to enhance the base stacking of perfectly matched base pairs and decrease the stacking stability of the mismatched pairs.31 The optimal concentration of the padlock probe for padlock RCA was calculated to be 0.1 pM according to the Ct value of template DNA determined by real time PCR, which was chosen in the following experiments (Fig. S1†). In addition, the stability of the padlock RCA reaction was further improved by endonuclease digestion and betaine solution as previously reported.17,32
In order to investigate the feasibility and specificity of the MPRP system, three padlock probes were utilized to detect three different target DNAs with point mutations in a Padlock RCA reaction. For example, probe pEG858A and pEG858C were perfectly matched with target tEG858T and tEG858G, respectively. Similarly, probe pEG790G and pEG790A were perfectly matched with target tEG790C and tEG790T, while probe pBR600A and pBR600T were perfectly matched with target tBR600T and tBR600A, respectively (Table 1). Finally, one pair of universal primers and three different probes labeled with different fluorescent moieties were used to perform multiplex real time PCR. It was shown in Table 2 that in the presence of mutant DNA and corresponding padlock probes, the Ct value of mutant DNA obtained from the MPRP assay was able to reach 9.74 ± 0.565, 7.99 ± 0.143 and 15.27 ± 0.243, respectively (Fig. S2†). No valid Ct value was observed with those padlock probes complementary to the wild type DNA. The results demonstrated the feasibility and specificity of the MPRP assay to detect multiplex DNA point mutations.
| Sample type | Padlock probe type | Average Ct value (mean ± standard error) |
|---|---|---|
| a ND: not detected.b The Ct value beyond 35 means that the data is unbelievable and can be considered as undetected. | ||
| EGFR L858R mutation (tEG858G) | L858R MU | 9.74 ± 0.565 |
| L858R WT | NDa | |
| L858R RCA NTC | ND | |
| L858R NTC | ND | |
| EGFR T790M mutation (tEG790T) | T790M MU | 7.99 ± 0.143 |
| T790M WT | 36.98 ± 0.660b | |
| T790M RCA NTC | ND | |
| T790M NTC | ND | |
| BRAF V600E mutation (tBR600A) | BRAF MU | 15.27 ± 0.243 |
| BRAF WT | ND | |
| BRAF RCA NTC | ND | |
| BRAF NTC | ND | |
Besides the specificity, sensitivity also affects the application potential of the SNP detection approach. To evaluate the sensitivity of the MPRP assay for mutant DNA detection, we prepared 0.05%, 0.1%, 1% and 5% of mutant standard DNA templates including the EGFR mutation L858R, T790M and BRAF mutation V600E, through dilution with wild type DNA according to the copy numbers. Then, the multiplex real-time PCR assay was performed to semi-quantify the level of mutant DNA, which was negatively associated with the Ct value. As was observed in Table 3, the corresponding Ct values of 0.1%, 1% and 5% of the EGFR T790M mutation were determined to be 31.72 ± 0.319, 25.39 ± 0.044 and 18.74 ± 0.458, respectively. Consistently, the Ct value of 0.1%, 1% and 5% of the EGFR L858R mutation was 34.95 ± 0.139, 17.69 ± 0.127 and 18.02 ± 0.234, respectively. The MPRP assay could detect 1% and 5% of the BRAF V600E mutation with Ct values of 20.85 ± 0.057 and 23.94 ± 0.063, respectively, but was not sensitive enough to detect the lower dilutions of the BRAF V600E mutation and the 0.05% of the two EGFR mutations (Fig. S3†).
| Gene type | Mutation ratio | Average Ct value (mean ± standard error) |
|---|---|---|
| a ND: not detected.b The Ct value beyond 35 means that the data is unbelievable and can be considered as undetected. | ||
| EGFR T790M | 5% | 18.74 ± 0.458 |
| 1% | 25.39 ± 0.044 | |
| 0.1% | 31.72 ± 0.319 | |
| 0.05% | 38.36 ± 0.091b | |
| EGFR L858R | 5% | 18.02 ± 0.234 |
| 1% | 17.69 ± 0.127 | |
| 0.1% | 34.95 ± 0.139 | |
| 0.05% | 38.97 ± 0.389b | |
| BRAF V600E | 5% | 20.85 ± 0.057 |
| 1% | 23.94 ± 0.063 | |
| 0.1% | NDa | |
| 0.05% | ND | |
Multi-gene detection and dual mutation discrimination mode
In our MPRP method, RCA was used to pre-amplify the target DNA before the real time PCR reaction. This step not only amplifies and enriches the circularized padlock probe but also increases the volume of input material for the subsequent multi-gene detection, which provides possibilities for targeted mutation screening using 96-well PCR panels.After enrichment of the single-base mutation, multiplex real time PCR is used for the quantification of gene mutations, which further increases the specificity of SNP detection. Dual mutation discrimination mode prevents the false positive results that can be caused by high sensitivity. Furthermore, instead of the traditional fluorescent detection method utilizing a fluorescent reader or microscope that is difficult to calculate and has low sensitivity,17,24,26 the real time PCR method provides automatic detection of signals which can be calculated and presented with visual results using professional analysis software. In our study, we detected the mutation with the mutant ratio as low as 0.1%, which is much better than the sensitivity of traditional real time PCR such as ARMS-PCR.33,34
Clinical evaluation of MPRP assay
Developing a non-invasive diagnostic approach to detect tumor-related mutations is highly desirable for precision cancer treatment. In order to demonstrate its potential in clinical application, a MPRP assay was performed using cell free DNA extracted from the plasma of 8 lung cancer patients to determine the mutation involved in EGFR T790M, EGFR L858R and BRAF V600E, respectively. Meanwhile, for comparison the mutation ratios of these samples were also detected using digital PCR, which provides highly sensitive and absolute quantification of target DNA.33,35,36 Both MPRP assay and digital PCR consistently detected one or two EGFR mutations in 4 patients from a total of 8 patients, and the gene mutation level reflected by the Ct value of the MPRP assay was highly correlated with the mutation ratio determined by digital PCR (Table S1†). Digital PCR only exhibited higher sensitivity in detection of the EGFR L858R mutation in patient P6 compared to the MPRP assay (Table 4, Fig. S4† and 2). There was no BRAF V600E mutation detected by the MPRP assay in any of the patients. Most of the results obtained by MPRP assay were consistent with those from digital PCR, strongly supporting its reliability. Considering the drawbacks of digital PCR, including its complicated operation, low throughput and expensive costs, the MPRP method is a more convenient, efficient and sensitive way to detect cancer-related mutations and can satisfy the clinical needs.| Patients ID | Mutation type | Mutation detected | |
|---|---|---|---|
| MPRP assay (Ct value) | Digital PCR (mutation ratio) | ||
| a ND: not detected.b The Ct value beyond 35 means that the data is unbelievable and can be considered as undetected. | |||
| P6 | EGFR L858R | 36.76 ± 0.783b | 2.38% |
| EGFR T790M | 27.09 ± 0.525 | 7.59% | |
| BRAF V600E | NDa | 0 | |
| P3 | EGFR L858R | 24.37 ± 0.120 | 4.21% |
| EGFR T790M | 20.97 ± 0.182 | 8.25% | |
| BRAF V600E | ND | 0 | |
| P2 | EGFR L858R | ND | 0 |
| EGFR T790M | 12.62 ± 0.091 | 25.98% | |
| BRAF V600E | ND | 0 | |
| P9 | EGFR L858R | ND | 0 |
| EGFR T790M | 38.31 ± 1.046b | 0 | |
| BRAF V600E | ND | 0 | |
| P5 | EGFR L858R | 21.00 ± 0.059 | 2.30% |
| EGFR T790M | 37.58 ± 1.358b | 0 | |
| BRAF V600E | ND | 0 | |
| P1 | EGFR L858R | ND | 0 |
| EGFR T790M | 36.80 ± 0.128b | 0.26% | |
| BRAF V600E | ND | 0 | |
| P7 | EGFR L858R | ND | 0 |
| EGFR T790M | 36.80 ± 0.801b | 0 | |
| BRAF V600E | ND | 0 | |
| P8 | EGFR L858R | 37.48 ± 0.399b | 0 |
| EGFR T790M | ND | 0 | |
| BRAF V600E | ND | 0 | |
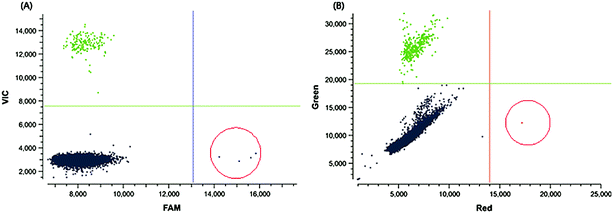 | ||
| Fig. 2 The 2D plot of digital PCR assay. The number of dots in the red ring shows the number of positive droplets. (A) Digital PCR results of the EGFR L858R assay for patient P6. (B) Digital PCR results of the EGFR T790M assay for patient P1. | ||
According to our knowledge, it's an innovative strategy to combine padlock RCA with multiplex real-time PCR for the non-invasive detection of lung cancer-related mutations, which appears to be very promising in clinical application.
Experimental
Materials
Human non-small cell lung cancer cell A549 was cultured in DEME medium (ThermoFisher, CA, US) with 10% fetal bovine serum (FBS) at 37 °C in 5% CO2 atmosphere. 5 mL of blood samples from patients with non-small cell lung cancer were collected in a Cell-Free DNA Blood Collection Tube (Streck, La Vista, USA) in the department of thoracic surgery at Hebei Chest Hospital before surgery, according to a protocol approved by the Ethics Committee of these institutions. All patients provided written informed consent. This study was approved by the ethics board of the institute of Hebei Chest Hospital and complied with the Declaration of Helsinki. Within 1 hour, all whole blood samples were centrifuged at 820 g for 10 min. Plasma was collected and subjected to a second centrifugation at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 10 min. The supernatant was then transferred to fresh tubes and stored at −80 °C. Genomic DNA was extracted from the cells using a DNA Extraction Kit (Apexbio, Beijing, China) according to the user manual. Circulating DNA from plasma was extracted with the QIAamp Circulating Nucleic Acid Kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. DNA quantification was performed in a Colibri microvolume spectrophotometer (Titertek-Berthold, Pforzheim, Germany).
000 g for 10 min. The supernatant was then transferred to fresh tubes and stored at −80 °C. Genomic DNA was extracted from the cells using a DNA Extraction Kit (Apexbio, Beijing, China) according to the user manual. Circulating DNA from plasma was extracted with the QIAamp Circulating Nucleic Acid Kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. DNA quantification was performed in a Colibri microvolume spectrophotometer (Titertek-Berthold, Pforzheim, Germany).
MPRP assay
All oligonucleotides in this study were synthesized by Sangon Biotech Co., Ltd (Shanghai, China). The padlock probes and template oligos of EGFR L858R, T790M and BRAF V600E, including both wild-type and mutant, were designed using Primer 3 software (version 4.1.0). The link part of the padlock probe cannot be complementary with the human genomic DNA validated by BLASTN.For the padlock RCA reaction of specificity test, the following were added into three PCR tubes for EGFR L858R, EGFR T790M and BRAF V600E, respectively: 1 μL of each 0.1 pM corresponding padlock probe (both of mutant type and wild type), 1 μL of 0.1 pM corresponding mutant template which was synthesized, 22 μL of ligation solution containing 0.5 μL HiFi Taq DNA ligase (NEB, Ipswich, MA), and 1× HiFi Taq DNA ligase reaction buffer. For the padlock RCA reaction of sensitivity test, the following were added into the PCR tubes for each dilution series of standard DNA: 1 μL of each 0.1 pM padlock probe of mutant types (pEG858C, pEG790A and pBR600A), 1 μL of 0.1 pM standard DNA, 21 μL of ligation solution containing 0.5 μL HiFi Taq DNA ligase (NEB, Ipswich, MA), and 1× HiFi Taq DNA ligase reaction buffer. For the padlock RCA reaction of clinical test, the following were added into the PCR tubes for each sample: 1 μL of each 0.1 pM padlock probe of mutant types (pEG858C, pEG790A and pBR600A), 5 μL of plasma DNA, 17 μL of ligation solution containing 0.5 μL HiFi Taq DNA ligase (NEB, Ipswich, MA), and 1× HiFi Taq DNA ligase reaction buffer. The mixture was incubated at 55 °C for 1 hour after being heated at 95 °C for 3 min. After the circularization of the padlock probe, 10 U exonuclease I (NEB, Ipswich, MA) and 40 U exonuclease III (NEB, Ipswich, MA) were added to digest the ssDNA and dsDNA, respectively. Then, the RCA reaction was immediately performed to elongate the padlock probe circulated for subsequent real time PCR assay in 25 μL of 1× Thermopol buffer (50 mM Tris–HCL, 10 mM MgCL2, 10 mM (NH4)2SO4, 4 mM DTT, PH 7.5@RT), 4 μL 5 M betaine (ThermoFisher, USA), 14 mM dNTPs mix, 1 μL 100 μM uniF, 1 μL 100 μM uniR and 5 μL ligation products. After heating at 95 °C for 3 min and cooling down in ice, 1× BSA, 0.5 μL betaine, 6 mM MgSO4 and 10 U μL−1 of Bst DNA polymerase (NEB, Ipswich, US) were added. The mixture was incubated at 65 °C for 2 h before heating at 85 °C for 10 min to deactivate the polymerase. Then, quantitative real-time PCR testing was performed in a 20 μL reaction containing 1× PerFecTa Multiplex qPCR ToughMix (Quanta Biosciences, Gaithersburg, USA), 400 nM of each uniF and uniR, 200 nM of each multiplex probes, and 1 μL RCA product. For specificity test, the PCR probes included both wild type and mutant type for EGFR L858R, EGFR T790M and BRAF V600E, respectively. For sensitivity test and clinical test, the PCR probes included three probes of mutant type (P858M, P790M and PBM) simultaneously. This was programmed as: 95 °C for 3 min, followed by 40 cycles of 94 °C for 10 s, 60 °C for 20 s on a CFX96 real-time PCR instrument (Bio-Rad, Hercules, CA). All experiments were replicated to ensure reproducibility.
Digital PCR assay
Detection of the EGFR mutation L858R and T790M in cell free DNA was carried out on the Naica digital PCR system (Stilla Technologies, Villejuif, France) with Sapphire chips (Stilla Technologies, Villejuif, France) in a 25 μL reaction mix containing the following components: 1× PerFecTa Multiplex qPCR ToughMix, 40 nM FITC (Saint Louis, MO, USA), 1 μL of primer and probes multiplex mix and 3 μL of DNA template. The chip was loaded into the Naica Geode thermocycler to compartmentalize the droplets and perform the PCR reaction. PCR conditions were 95 °C for 10 min, followed by 45 cycles of 95 °C for 20 s and 60 °C for 30 s. After amplification, the Sapphire chips were imaged using the Naica Prism3 reader and the fluorescent data were analyzed using Crystal Miner software (Stilla Technologies, Villejuif, France). Each patient sample was tested in duplicate. NTC and EGFR Gene-Specific Multiplex Reference Standard gDNA HD802 (Horizon Discovery, Cambridge, UK) were used as negative and positive controls, respectively. Negative and positive droplets were also used to check the fluorescence spill-over compensation.Conclusions
In this study, we developed a MPRP system based on rolling circle amplification and multiplex real time PCR using universal primers for SNP discrimination, which showed high specificity and sensitivity for multiplex detection of tumor-related mutations including EGFR mutations L858R and T790M and BRAF mutation V600E. In order to validate its potential application in clinical diagnosis, we tested the samples from the patients' plasma using both MPRP assay and digital PCR assay. The coincident results between the two methods indicated that the MPRP assay provided a more sensitive, specific and convenient method for the detection of cancer-related mutations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
None declared.Notes and references
- E. Sakai, M. Nakayama, H. Oshima, Y. Kouyama, A. Niida, S. Fujii, A. Ochiai, K. I. Nakayama, K. Mimori, Y. Suzuki, C. P. Hong, C. Y. Ock, S. J. Kim and M. Oshima, Cancer Res., 2018, 78(5), 1334–1336 CrossRef PubMed.
- T. S. Mok, Y. L. Wu, S. Thongprasert, C. H. Yang, D. T. Chu, N. Saijo, P. Sunpaweravong, B. Han, B. Margono, Y. Ichinose, Y. Nishiwaki, Y. Ohe, J. J. Yang, B. Chewaskulyong, H. Jiang, E. L. Duffield, C. L. Watkins, A. A. Armour and M. Fukuoka, N. Engl. J. Med., 2009, 361, 947–957 CrossRef PubMed.
- A. S. Kim, A. N. Bartley, J. A. Bridge, S. Kamel-Reid, A. J. Lazar, N. I. Lindeman, T. A. Long, J. D. Merker, A. J. Rai, D. L. Rimm, P. G. Rothberg, P. Vasalos and J. T. Moncur, JAMA Oncol., 2018, 4(6), 838–841 CrossRef PubMed.
- L. Horn, J. Natl. Compr. Cancer Netw., 2015, 13, 676–678 CrossRef.
- G. P. Kalemkerian, N. Narula, E. B. Kennedy, W. A. Biermann, J. Donington, N. B. Leighl, M. Lew, J. Pantelas, S. S. Ramalingam, M. Reck, A. Saqi, M. Simoff, N. Singh and B. Sundaram, J. Clin. Oncol., 2018, 36, 911–919 CrossRef PubMed.
- L. Odogwu, L. Mathieu, G. Blumenthal, E. Larkins, K. B. Goldberg, N. Griffin, K. Bijwaard, E. Y. Lee, R. Philip, X. Jiang, L. Rodriguez, A. E. McKee, P. Keegan and R. Pazdur, Oncologist, 2018, 23(6), 740–745 CrossRef PubMed.
- F. Fenizia, A. De Luca, R. Pasquale, A. Sacco, L. Forgione, M. Lambiase, A. Iannaccone, N. Chicchinelli, R. Franco, A. Rossi, A. Morabito, G. Rocco, M. C. Piccirillo and N. Normanno, Future Oncol., 2015, 11, 1611–1623 CrossRef PubMed.
- E. Crowley, F. Di Nicolantonio, F. Loupakis and A. Bardelli, Nat. Rev. Clin. Oncol., 2013, 10, 472–484 CrossRef PubMed.
- S. Cui, L. Ye, H. Wang, T. Chu, Y. Zhao, A. Gu, L. Xiong, C. Shi and L. Jiang, Clin. Lung Cancer, 2018, 19(3), e313–e322 CrossRef PubMed.
- L. Zhu, S. Zhang, Y. Xun, Y. Jiang, B. Xia, X. Chen, L. Wang, H. Jiang and S. Ma, Pathol. Oncol. Res., 2017 DOI:10.1007/s12253-017-0286-3.
- H. Nakamura, H. Koizumi, H. Sakai, H. Kimura, T. Miyazawa, H. Marushima, H. Saji and M. Takagi, Clin. Lung Cancer, 2018, 19(2), 170–174 CrossRef PubMed.
- P. Brown, Future Oncol., 2016, 12, 451–452 CrossRef PubMed.
- S. P. Jonstrup, J. Koch and J. Kjems, RNA, 2006, 12, 1747–1752 CrossRef PubMed.
- P. M. Lizardi, X. Huang, Z. Zhu, P. Bray-Ward, D. C. Thomas and D. C. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef PubMed.
- X. Qian and R. V. Lloyd, Diagn. Mol. Pathol., 2003, 12, 1–13 CrossRef PubMed.
- J. Pickering, A. Bamford, V. Godbole, J. Briggs, G. Scozzafava, P. Roe, C. Wheeler, F. Ghouze and S. Cuss, Nucleic Acids Res., 2002, 30, e60 CrossRef PubMed.
- L. Xie, T. Wang, T. Huang, W. Hou, G. Huang and Y. Du, Sci. Rep., 2014, 4, 6300 CrossRef PubMed.
- Y. L. Wu, L. V. Sequist, C. P. Hu, J. Feng, S. Lu, Y. Huang, W. Li, M. Hou, M. Schuler, T. Mok, N. Yamamoto, K. O'Byrne, V. Hirsh, N. Gibson, D. Massey, M. Kim and J. C. Yang, Br. J. Cancer, 2017, 116, 175–185 CrossRef PubMed.
- W. W. Chen, L. Balaj, L. M. Liau, M. L. Samuels, S. K. Kotsopoulos, C. A. Maguire, L. Loguidice, H. Soto, M. Garrett, L. D. Zhu, S. Sivaraman, C. Chen, E. T. Wong, B. S. Carter, F. H. Hochberg, X. O. Breakefield and J. Skog, Mol. Ther.--Nucleic Acids, 2013, 2, e109 CrossRef PubMed.
- R. Kanagal-Shamanna, Methods Mol. Biol., 2016, 1392, 43–50 CrossRef PubMed.
- B. O. Zhang, C. W. Xu, Y. Shao, H. T. Wang, Y. F. Wu, Y. Y. Song, X. B. Li, Z. Zhang, W. J. Wang, L. Q. Li and C. L. Cai, Exp. Ther. Med., 2015, 9, 1383–1388 CrossRef PubMed.
- R. Wang, L. Wang, H. Zhao and W. Jiang, Biosens. Bioelectron., 2016, 86, 834–839 CrossRef PubMed.
- X. Qi, S. Bakht, K. M. Devos, M. D. Gale and A. Osbourn, Nucleic Acids Res., 2001, 29, E116 CrossRef PubMed.
- M. Mignardi, A. Mezger, X. Qian, L. La Fleur, J. Botling, C. Larsson and M. Nilsson, Nucleic Acids Res., 2015, 43, e151 CrossRef PubMed.
- C. Yuan, X. W. Lou, E. Rhoades, H. Chen and L. A. Archer, Nucleic Acids Res., 2007, 35, 5294–5302 CrossRef PubMed.
- R. Li, Y. Wang, P. Wang and J. Lu, Luminescence, 2017, 32, 1574–1581 CrossRef PubMed.
- G. J. Lohman, R. J. Bauer, N. M. Nichols, L. Mazzola, J. Bybee, D. Rivizzigno, E. Cantin and T. C. Evans, Jr, Nucleic Acids Res., 2016, 44, e14 CrossRef PubMed.
- J. Li, T. Deng, X. Chu, R. Yang, J. Jiang, G. Shen and R. Yu, Anal. Chem., 2010, 82, 2811–2816 CrossRef PubMed.
- J. Li, J. H. Jiang, X. M. Xu, X. Chu, C. Jiang, G. Shen and R. Q. Yu, Analyst, 2008, 133, 939–945 RSC.
- L. Liu, Z. Tang, K. Wang, W. Tan, J. Li, Q. Guo, X. Meng and C. Ma, Analyst, 2005, 130, 350–357 RSC.
- M. P. Johnson, L. M. Haupt and L. R. Griffiths, Nucleic Acids Res., 2004, 32, e55 CrossRef PubMed.
- W. Liu, D. Dong, Z. Yang, D. Zou, Z. Chen, J. Yuan and L. Huang, Sci. Rep., 2015, 5, 12723 CrossRef PubMed.
- X. Zhang, N. Chang, G. Yang, Y. Zhang, M. Ye, J. Cao, J. Xiong, Z. Han, S. Wu, L. Shang and J. Zhang, Oncotarget, 2017, 8, 112014–112023 Search PubMed.
- N. Ogasawara, H. Bando, Y. Kawamoto, T. Yoshino, K. Tsuchihara, A. Ohtsu and H. Esumi, Jpn. J. Clin. Oncol., 2011, 41, 52–56 CrossRef PubMed.
- G. Perkins, H. Lu, F. Garlan and V. Taly, Adv. Clin. Chem., 2017, 79, 43–91 Search PubMed.
- W. N. Feng, W. Q. Gu, N. Zhao, Y. M. Pan, W. Luo, H. Zhang, J. M. Liang, J. Yang and Y. M. Deng, Transl. Oncol., 2018, 11, 542–545 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05259j |
| This journal is © The Royal Society of Chemistry 2018 |