
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceADAM10 modulates SOX9 expression via N1ICD during chondrogenesis at the cranial base†
Runqing Fu a,
Xiaoting Wanga,
Lunguo Xiaa,
Yu Tanb,
Jiaqiang Liuc,
Lingjun Yuana,
Zhi Yang*c and
Bing Fang*a
a,
Xiaoting Wanga,
Lunguo Xiaa,
Yu Tanb,
Jiaqiang Liuc,
Lingjun Yuana,
Zhi Yang*c and
Bing Fang*a
aDepartment of Orthodontics, Shanghai Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University, 500 Quxi Road, Shanghai 200011, China. E-mail: fangbing@sjtu.edu.cn
bThe Second Dental Center, Shanghai Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University, 280 Mohe Road, Shanghai 200011, China
cDepartment of Oral & Cranio-Maxillofacial Science, School of Medicine, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University, 500 Quxi Road, Shanghai 200011, China. E-mail: wcums1981@163.com
First published on 14th November 2018
Abstract
The cranial base is the foundation of the craniofacial structure, and any interruption of the cranial base can lead to facial deformity. The cranial base develops from two synchondroses via endochondral ossification. Chondrogenesis is an important step in endochondral ossification. A disintegrin and metalloprotease (ADAM) 10 participates in the Notch1 signalling pathway, which has been reported to regulate chondrogenesis via a SOX9-dependent mechanism. However, little is known about the function of ADAM10 in chondrogenesis. In this study, adam10-conditional-knockout (cKO) mice exhibited sharper naso-labial angles and flatter skulls than wild-type (WT) mice. In the sagittal plane, SOX9 was more widespread in the cranial base in Adam10-cKO mice than in WT mice. For in vitro experiments, we used the ATDC5 cell line as a model to investigate the role of ADAM10 in chondrogenesis. Plasmid 129 was designed to decrease the expression of Adam10; the resulting downregulation of Adam10 reduced the production of N1ICD. Plasmid 129 increased the expression of SOX9 under chondrogenic induction, and this increase could be inhibited by transfection with exogenous N1ICD. Collectively, these results show that ADAM10 participates in chondrogenesis by negatively regulating SOX9 expression in an N1ICD-dependent manner during cranial base development.
1. Introduction
The cranial base is the foundation of the cranial facial structure. Abnormal cranial bases are related to a variety of diseases that manifest specific facial appearances. In neurofibromatosis type I patients, cranial base shortening, especially in the anterior region, results in a short facial appearance and in mid-face hypoplasia.1 Apert syndrome, a developmental malformation-related syndrome, is also characterized by mid-face hypoplasia caused by early craniosynostosis of the cranial base.2 Moreover, both the length and angle of the cranial base have been proved to be different in Class II and Class III malocclusion patients from in individuals with normal occlusion.3 Specifically, Class III malocclusion patients manifest a reduced cranial base length, while Class II malocclusion patients exhibit a prolonged cranial base.The bone structure of the cranial base develops through endochondral ossification. Over the past few decades, researchers have revealed numerous molecules related to endochondral ossification. For example, Shh initiates chondrogenic differentiation;4,5 Wnt, FGF and TGFβ modulate chondrogenic competence and matrix production;6 and other molecules, such as BMP, PKA, HIF1α, and Notch, regulate endochondral bone formation at different stages.7,8
As a classical member of the ADAM family, ADAM10 acts as an α-secretase enzyme on substrates that are widely distributed in cells, including Notch, N-cadherin, APP and others. Adam10-conditional-knockout (cKO) mice harbouring different Cre enzymes exhibit perinatal fatality, seizures, altered spine morphology and other pathologies,9,10 and Adam10 deletion in endothelial cells impairs blood vessels and the heart.11 In vivo experiments using other models have produced similar results, including dyspoiesis and circulatory system dysfunction.11
In a previous study, we explored the influence of ADAM10 on intramembranous bone formation during maxillary and mandibular development in mice.12 ADAM10 has been found to be highly expressed in the mouse cranial base during the embryonic and youth periods. Furthermore, in the mouse cranial base synchondroses, ADAM10 is mainly expressed in the hypertrophic zones, with decreased expression in the proliferative zones and hardly any expression in the resting zones.13 However, the role of ADAM10 in chondrogenesis remains unclear. In this study, a conditional-knockout mouse model was used to observe the changes caused by the absence of Adam10 expression. The ATDC5 cell line was also used as a model to explore the role of ADAM10 in chondrogenesis.
2. Experimental
Mouse strains and genotyping
Wild-type (WT) mice of different growth stages were purchased from Shanghai Laboratory Animal Company (SLAC). Transgenic mice were raised in the specific pathogen-free (SPF) animal laboratory of Shanghai Ninth People's Hospital. Both Adam10-flox mice (a kind gift from Prof. Zhiqi Xiong) and Wnt1-Cre mice (a kind gift from Prof. Leping Chen) were maintained on a C57 background. All animal operations were approved by the Animal Ethics Committee of Shanghai Ninth People's Hospital. All methods were conducted in accordance with the approved guidelines, and all animals were sacrificed in a humane fashion.In vivo experiments
Embryonic and neonatal mice were sacrificed on ice. Photos of embryos were taken with a stereoscopic microscope (Nikon, Japan).After the bodies had been fixed in 4% paraformaldehyde for over 24 h, they were dehydrated in a series of increasingly concentrated sucrose solutions. Then, frozen sections were generated according to a previously described protocol and stored at −20 °C.14 These sections were brought to room temperature for the immunofluorescence analysis.
Immunofluorescence
Tissue sections and cell slices were permeabilized with 0.3% (v/v) Triton and blocked with 5% (w/v) bovine serum albumin (BSA) for 1 h on a shaker at room temperature. After blocking, phosphate-buffered saline (PBS) was used to wash the samples 3 times, and then primary antibodies against SOX9 (Abcam, USA, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500) and ADAM10 (Abcam, USA, 1:300) were incubated with the samples at 4 °C. A secondary antibody (Invitrogen, USA, 1:1000) was then incubated with the samples for 1 h at room temperature. Hoechst (Invitrogen, US, 1:5000) was used to stain the cell nuclei 10 min at room temperature. After washing with PBS 3 times, the sections and cell slices were mounted with Fluoromount-G (Invitrogen, USA) and stored at −20 °C. Images were obtained with a confocal microscope (Canon, Japan). Each experiment was replicated 3 times.
:500) and ADAM10 (Abcam, USA, 1:300) were incubated with the samples at 4 °C. A secondary antibody (Invitrogen, USA, 1:1000) was then incubated with the samples for 1 h at room temperature. Hoechst (Invitrogen, US, 1:5000) was used to stain the cell nuclei 10 min at room temperature. After washing with PBS 3 times, the sections and cell slices were mounted with Fluoromount-G (Invitrogen, USA) and stored at −20 °C. Images were obtained with a confocal microscope (Canon, Japan). Each experiment was replicated 3 times.
Cell culture
The ATDC5 cell line was cultured in F-12 medium/Dulbecco's modified Eagle's medium (DMEM) (HyClone, USA) containing 10% foetal bovine serum (FBS; Gibco, USA) and 1% penicillin–streptomycin (HyClone, US) in a humidified atmosphere with 5% CO2 at 37 °C. Insulin-Transferrin-Selenium (ITS) solution (Gibco, US) was used to induce chondrogenesis in the ATDC5 cell line.Western blot assays
Protein quantification was performed via western blot assays. Radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, China) was used to lyse cells on ice. After adding loading buffer and heating the lysates to 95 °C for 10 min, the lysates were resolved on sodium dodecyl sulfate (SDS)-polyacrylamide gels. Then, the separated proteins were transferred from the gel to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA). After blocking in 5% (w/v) skim milk (Becton, Dickinson and Company, USA) for 1 h, the membrane was incubated with primary antibodies against ADAM10 (Abcam, USA, 1:1000), N1ICD (Cell Signaling Technology, USA, 1:1000), and SOX9 (Abcam, USA, 1:1000) at 4 °C overnight. A horseradish peroxidase (HRP)-conjugated secondary antibody (Cell Signaling Technology, USA, 1:3000) was applied for development. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Invitrogen, USA, 1:3000) was used as a loading control. Each experiment was replicated 3 times.
Plasmid transfection
Plasmids were designed to knock down the expression of Adam10; plasmid 129 was designed to target the sequence coding the ADAM10 metalloproteinase structural domain, and plasmid 132 was designed to target the sequence coding the ADAM10 disintegrin domain. In addition, a full-length ADAM10 plasmid was designed to overexpress ADAM10, and an N1icd plasmid was designed to overexpress N1ICD. A scrambled plasmid was transfected into cells as a control. All plasmids were designed with a green fluorescence tag, and the FuGENE® HD transfection reagent (Progema, USA) was used to transport plasmids into the cells at selected ratios. Each experiment was replicated 3 times.Statistical analysis
The densities of the bands obtained from the western blot assays were analysed with ImageJ (NIH, USA), and the data were processed with SPSS (SPSS, USA). A P value of 0.05 was chosen as the level of significance. Bar graphs were generated with Prism (GraphPad, US).3. Results and discussion
Adam10-conditional-knockout (cKO) mice exhibited a unique morphology
Adam10-flox/flox mice were mated with Wnt1-Cre mice to generate Adam10-cKO mice to study the effects of adam10 on chondrogenesis in the mouse cranial base. None of the cKO mice survived to birth. Adam10-cKO mice were still alive at E15.5, so 3 Adam10-cKO mice and 3 WT mice at E15.5 were used for experiments. The Adam10-cKO mice exhibited unique characteristics. In addition to having a smaller body than WT mice, Adam10-cKO mice had a relatively more acute naso-labial angle, as indicated by the red line in Fig. 1a, b and e. Additionally, the cranium of Adam10-cKO mice was flatter than that of WT mice (Fig. 1a and b). Immunofluorescence staining showed that Adam10-cKO mice possessed shorter resting and proliferative zones than did WT mice (Fig. 1c, d, f and g). SOX9 was significantly expressed in the resting zone and proliferative zone in Adam10-cKO mice, while it was hardly expressed in the same zones in WT mice (Fig. 1c and d).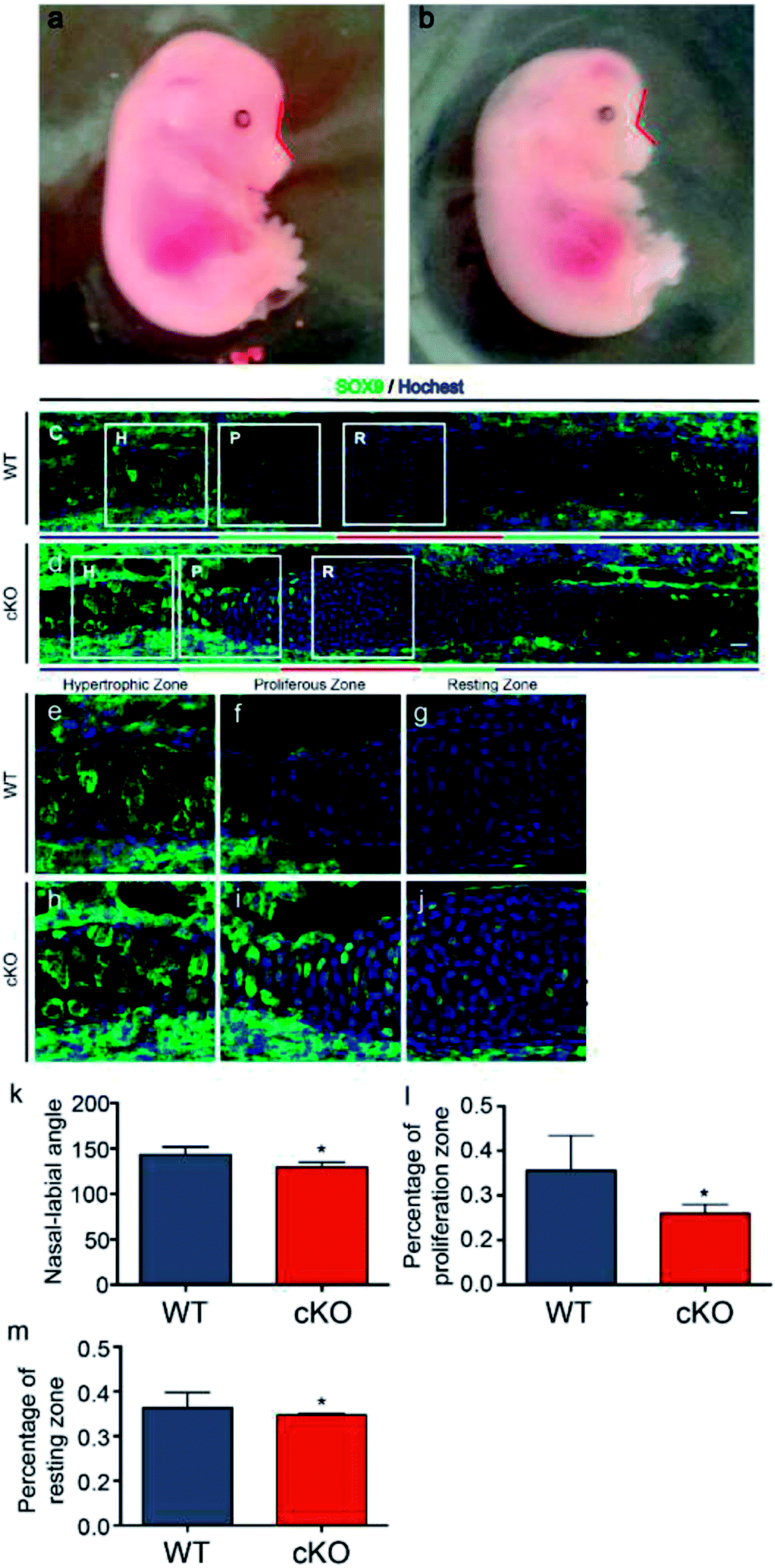 | ||
| Fig. 1 Changes in morphology and SOX9 expression during chondrogenesis in adam10-cKO mice. Images (a) and (b) show the morphology of WT mice and Adam10-cKO mice (cKO). The red lines depict the naso-labial angles of the WT and Adam10-cKO mice. Images c and d depict the changes in SOX9 expression and the structure of the synchondroses. The green fluorescence in images c and d shows the protein expression of SOX9. In images (c) and (d), R represents the resting zone, P represents the proliferative zone and H represents the hypertrophic zone. Beneath images (c) and (d), the red line indicates the length of the resting zone, the green line indicates the length of the proliferative zone, and the blue line indicates the length of the hypertrophic zone. The areas surrounded by white frames in image (c) are amplified in images (e), (f), and (g) by 200×. The areas surrounded by white frames in image (d) are amplified in images (h), (i), and (j) by 200×. Graphs (k), (l), and (m) illustrate the statistical results of the comparisons between WT and Adam10-cKO mice regarding the naso-labial angles, the percentage of the proliferation zone and the percentage of the resting zone, respectively. * represents a significant difference between Adam10-cKO mice and WT mice. P < 0.05. | ||
As the cranial base is the foundation of the hard craniofacial tissue, its length and angle influence facial appearance in mammals.15 Understanding cranial base formation and the involved regulatory mechanisms is thus important, and as shown in previous studies, chondrogenesis is a vital step in cranial base formation.16
Because ADAM10 is highly expressed in the embryonic and infant stages, Wnt1-Cre mice were mated with Adam10-flox/flox mice to knock out Adam10. However, Adam10-cKO mice were unable to survive to birth. Thus, caesarean delivery was performed to obtain live Adam10-cKO mice. E15.5 was the latest time point to which the Adam10-cKO mice survived. The significance of Adam10 in neural and cardiovascular development might underlie the high mortality. Adam10-cKO mice showed a special phenotype; the poorly developed flat cranium might be a consequence of the cranial hypoplasia and obvious oedema. Hypoplasia of the cranial base was an important potential reason for the acute angle between the nose and rostrum. Confocal immunofluorescence microscopy confirmed that the resting zone and proliferating zone in the synchondroses of the Adam10-cKO mice were much shorter than those of the WT mice. Additionally, abundant expression of SOX9 in the Adam10-cKO mice indicated the advancement of chondrogenic differentiation. Surprisingly, SOX9 was expressed in the resting zones and the proliferative zones. Most previous studies have focused on the changes in SOX9 expression in the cranial base during development. In this study, the spatial expression of SOX9 in cranial base synchondroses was revealed by immunofluorescence. B. Balczerski, P. Francis-West et al. reported that in mice, the onset of chondrogenesis occurs much later in the cranial base than in the trunk, and they found that SOX9 mRNA could be detected until E11.5.17 Moreover, Nie X. et al. reported that SOX9 showed a similar spatial expression pattern as that in our WT group by E14.18 In this study, the immunofluorescence images showed the central area of the synchondroses without the extending hypertrophic zones; the expression of SOX9 might be lower in the hypertrophic zones. Moreover, Col2a1Cre;RosaNotch mice have been reported to exhibit decreased cell proliferation and increased SOX9 expression in growth plates, which might help explain the short cranial base in adam10-cKO mice,19 since Notch is an important substrate for the cleavage of ADAM10. Earlier exhaustion of cells and insufficient proliferation in the resting zone could have resulted in insufficient cells for differentiation, causing a relatively shorter cranial base than normal. Furthermore, the length of the cranial base was not adequate to support the development of the brain, which might have resulted in the specific morphological characteristics and high mortality of the Adam10-cKO mice.
Expression of ADAM10 in the ATDC5 cell line
To conveniently investigate the effects of ADAM10 on chondrogenesis, the ATDC5 cell line was used for in vitro research. The ATDC5 cell line, which was first isolated from a differentiating mouse teratocarcinoma, tends to undergo chondrogenic differentiation in a chondrogenic induction environment, and increasing numbers of researchers are using it as a model to investigate the mechanisms of chondrogenesis.20,21 Chondrogenic induction in the ATDC5 cell line was achieved through the application of ITS.Immunofluorescence analysis revealed the changing location of ADAM10 and western blot assays revealed the changing protein expression of ADAM10 during chondrogenic induction in ATDC5 cells. As the immunofluorescence results showed, ADAM10 was initially located in the cytoplasm and cell membrane (Fig. 2a). ADAM10 became concentrated in the nucleus soon after induction; then, it returned to the cytoplasm and was evenly distributed 24 h after induction (Fig. 2a–f). A previous study revealed that a cleaved fragment of ADAM10 might translocate into the nucleus to modulate gene expression,22 which we intend to investigate further in future experiments.
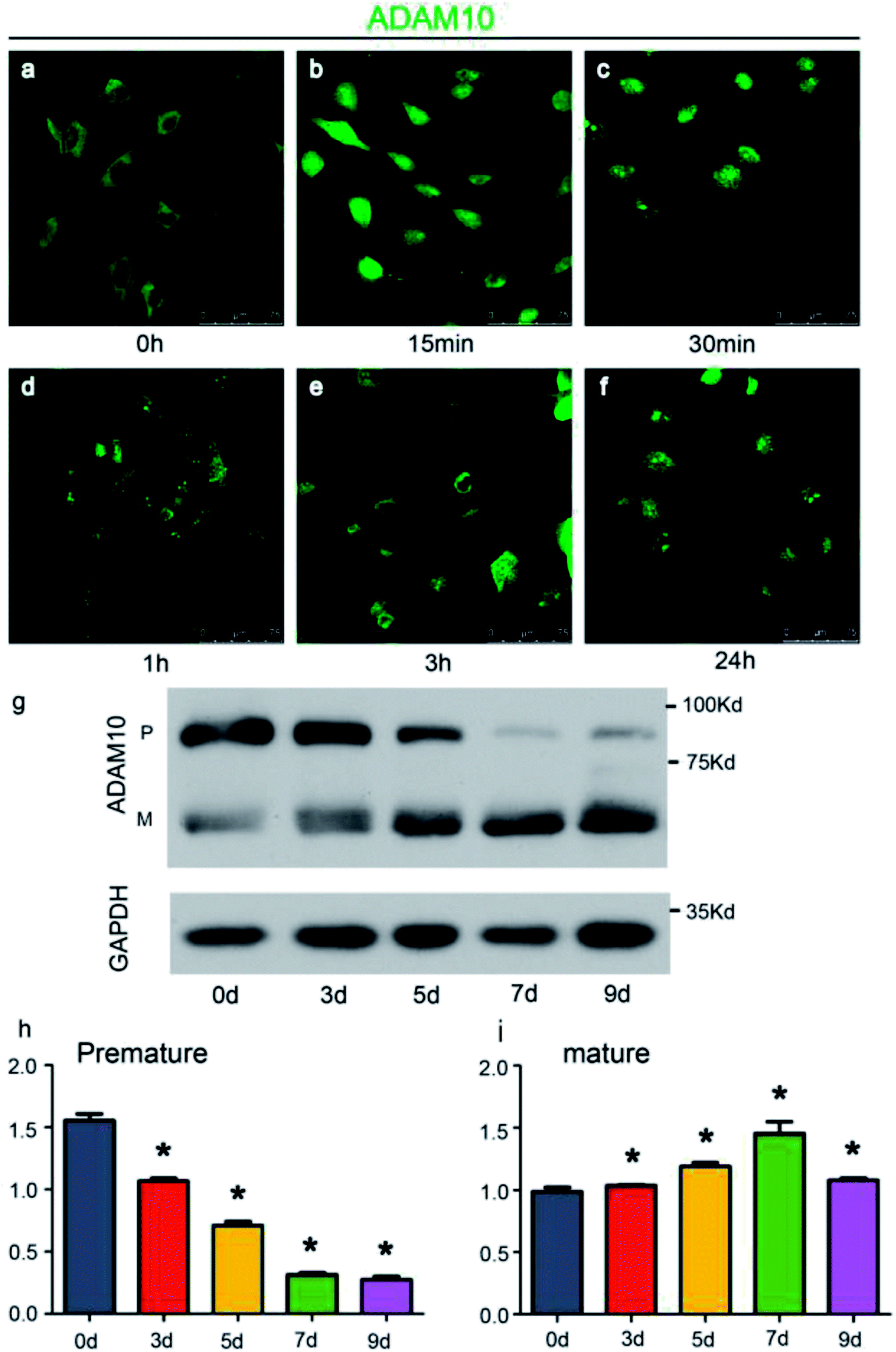 | ||
| Fig. 2 ADAM10 was expressed in the ATDC5 cell line. Panels (a–f) illustrate the changing location of ADAM10 in the ATDC5 cell line immediately after ITS was applied. The ADAM10 molecules are indicated by green fluorescence. Panel (g) shows the protein expression level of ADAM10 after induction by ITS. Graph (h) shows the relative grey value of the premature ADAM10 protein bands, while graph (i) shows the relative grey value of the mature ADAM10 protein bands. The data are presented as the means with standard deviations (SDs). * represents significant differences between 0 d and the other time points. P < 0.05. | ||
Western blot assays showed that under the influence of ITS, the expression of premature ADAM10 increased for the first 7 days and decreased on day 9, while the expression of mature ADAM10 continued to increase until day 9 (Fig. 2g–i). Chubinskaya S. reported that ADAM10 expression is high in infant and adolescent stages and declines beginning in middle age.23 The expression pattern of ADAM10 in the ATDC5 cell line exhibited a similar tendency during chondrogenic induction.
Down-regulation of Adam10 via plasmid transfection
FuGENE® HD transfection reagent was used to transfect ATDC5 cells with plasmids. Reagent:plasmid ratios from 2:1 to 5:1 were tested for the green fluorescent protein (GFP)-tagged plasmid transfection into ATDC5 cells, and a 3:1 was found to be the most appropriate, as this ratio resulted in high transfection efficiency without negative effects on cell morphology (ESI figure†). Plasmids 129 and 132, which were tagged with GFP, were designed to decrease the expression of ADAM10. After these plasmids were transfected into the ATDC5 cells at a ratio of 3:1, the green fluorescence showed that over 90% of the cells were successfully transfected (Fig. 3a and b). Western blot assays showed that both plasmid 129 and plasmid 132 decreased the synthesis of ADAM10 protein in the ATDC5 cell line. Moreover, plasmid 129 decreased ADAM10 expression more efficiently than plasmid 132. Thus, plasmid 129 was selected for further investigation (Fig. 3c–e).
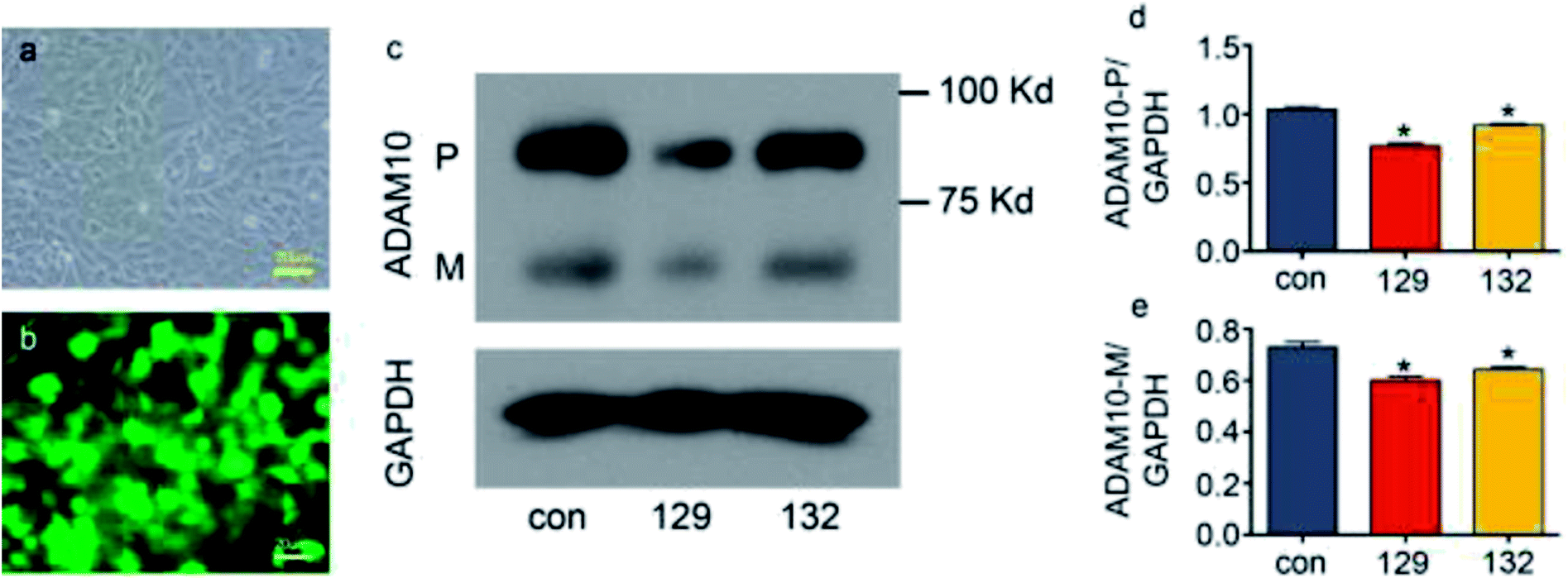 | ||
| Fig. 3 Knockdown of ADAM10 in the ATDC5 cell line via plasmid transfection. Panel (a) shows ATDC5 cells under a light microscope. Panel (b) shows the same visual field as that of panel (a) under a fluorescence microscope. The green fluorescence indicates the cells transfected with plasmids. Panel (c) shows the protein level of ADAM10 after plasmid 129 and plasmid 132 transfection. Panel (d) and panel (e) show the relative grey values of premature ADAM10 and mature ADAM10, respectively, normalized to the grey value of GAPDH. * represents significant differences between the control group and the other groups. P < 0.05. | ||
ADAM10 is a typical proteolytic enzyme that cleaves molecules to produce functional fragments or degrades the extracellular matrix.24–26 ADAM10 has been proved to participate in many biological and pathological processes. In vivo studies have shown that ADAM10 participates in embryonic development; embryonic stem cell migration is the first process in which ADAM10 is involved.27 Moreover, ADAM10 is involved in the development of the nervous system, small intestine, cardiovascular system and renal cortex.11,28,30 In adults, ADAM10 regulates physical functions such as haematopoiesis and secondary lymphoid tissue architecture maintenance.31,32 Abnormal ADAM10 expression may lead to serious consequences. For example, excessive ADAM10 degrades the matrix while synthesis is not sufficient. This imbalance exacerbates inflammation, resulting in damage to cartilage.33,34 Moreover, extracellular matrix loss caused by ADAM10 overexpression facilitates the metastasis of tumour cells.29,35,36 In contrast, low expression of ADAM10 induces the accumulation of amyloid-β (Aβ), which leads to Alzheimer's disease.37 Thus, ADAM10 has been regarded as a meaningful therapeutic target for Alzheimer's disease.38
ADAM10 modulated SOX9 expression via the Notch1 intracellular domain (N1ICD)
Few studies have been performed to determine the role of ADAM10 in chondrogenic differentiation. Eun-Jung Jin reported that ADAM10 inhibits cell proliferation in the mesenchymal bud during endochondral ossification.39 Recently, Zhao R. et al. reported that knocking down Adam10 in endothelial cells causes a lack of osteoclastogenesis in chondro-osseous junctions in a Notch-dependent manner.40 In addition to exploring the direct effects of ADAM10 on endochondral ossification, researchers have also investigated the downstream effects of ADAM10 during hard tissue development. Watanabe N. et al. reported that Notch1 suppresses the proliferation and differentiation of early chondrogenic cells.41 Additionally, Yoko Hosaka et al. reported that Notch signalling modulates the terminal stage of endochondral ossification via an RBPjκ-dependent pathway,42 and Timothy J. Mead and Katherine E. Yutzey reported that conditional loss of function of the Notch signalling pathway in mice leads to deformity of the axial skeletal with overexpression of SOX9 in the limbs.19 To explore the influence of ADAM10 on the Notch1 signalling pathway in the ATDC5 cell line, the expression level of the cleavage product of ADAM10, N1ICD, was detected via western blot assay. After transfection of cells with plasmid 129, the protein expression of N1ICD was effectively reduced (Fig. 4a and b).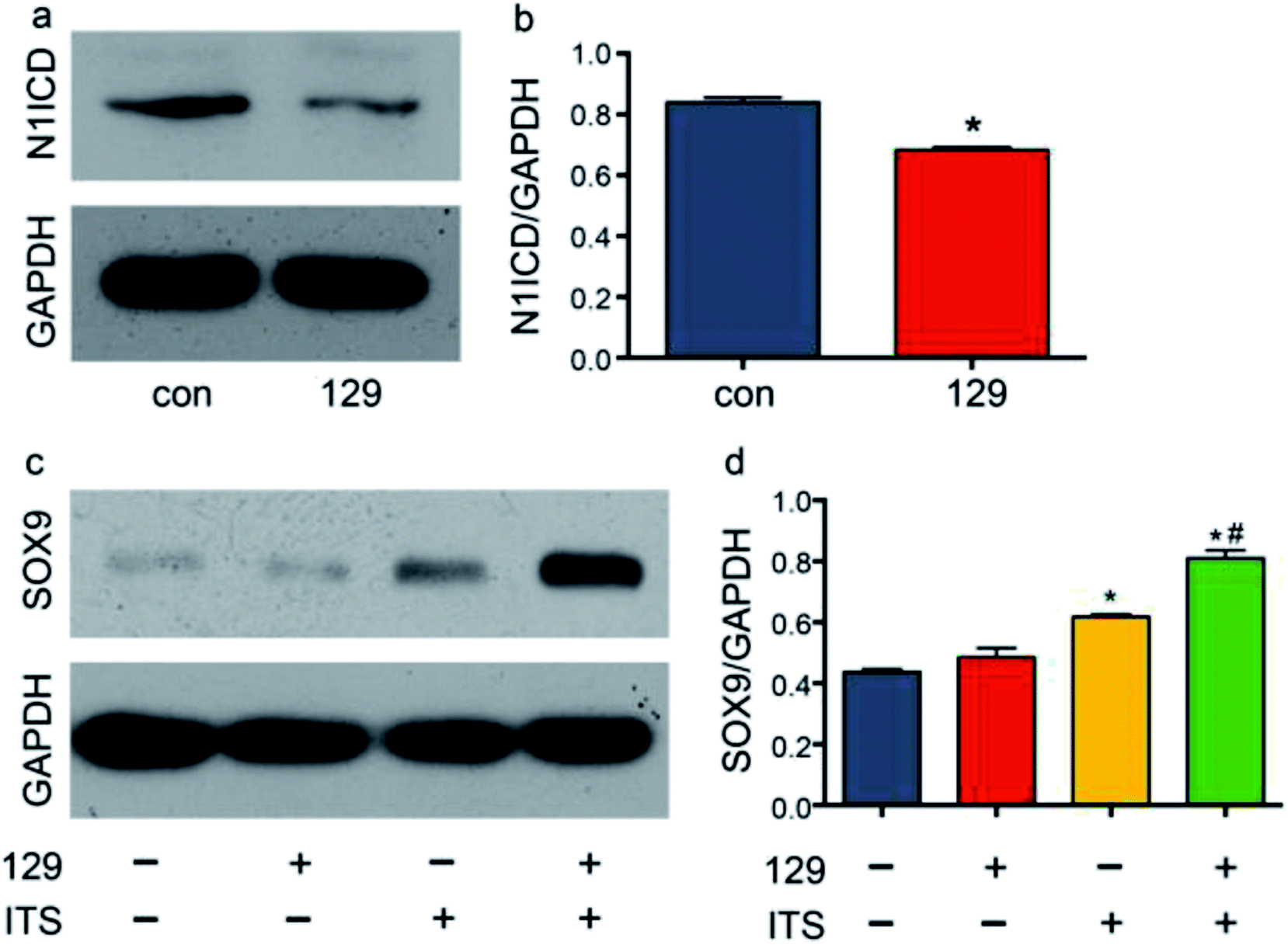 | ||
| Fig. 4 Knockdown of ADAM10 changed the expression of N1ICD and SOX9. Panel (a) shows the protein expression of N1ICD after plasmid 129 transfection. Panel (b) shows the relative grey value of the bands in panel (a). Panel (c) shows the protein expression of SOX9 under different conditions. Panel (d) shows the relative grey value of the bands in panel (c). * represents a significant difference between the control group and the other groups. # in panel (d) represents a significant difference between the ITS group and the 129 + ITS group. P < 0.05. | ||
SOX9 is important in chondrogenesis, as it is required for the aggregation of mesenchymal cells, which initiates chondrogenesis. SOX9 also participates in cell proliferation, migration and differentiation together with SOX5 and SOX6.43 In many studies, SOX9 has been regarded as the most important comprehensive marker for chondrogenesis. To determine the influence of ADAM10 on SOX9 expression, scrambled control plasmid, plasmid 129 and ITS were applied to four groups: the control group, the 129 group, the ITS group, and the 129 + ITS group. SOX9 expression was low in the control group and the 129 group, and there was hardly any difference between the two groups. This finding indicated that ADAM10 has no effect on the ATDC5 cell line under common culture conditions. The ITS and 129 + ITS groups showed high levels of SOX9 expression, with a much higher level in the latter group (Fig. 4c and d). After ITS application, ATDC5 cells appeared to favour chondrogenic differentiation. Elevated SOX9 expression in the 129 + ITS group indicated that silencing ADAM10 expression further enhanced chondrogenic differentiation. In other words, SOX9 expression might have been suppressed by ADAM10. However, it was still unclear whether the modulatory effect of ADAM10 on SOX9 is related to N1ICD.
ADAM10 has been identified as the α-secretase that activates the Notch1 signalling pathway through cleavage.44 The western blot assay revealed that plasmid 129 could inhibit the production of N1ICD by knocking down ADAM10. Additionally, Notch1 has been proven to be a vital modulator of chondrogenesis. To investigate the relationship among ADAM10, Notch1 and SOX9, we examined SOX9 expression in ADAM10-knockdown and N1ICD-overexpressing ATDC5 cells. Transfection with the full-length Adam10 (Adam10-FL) plasmid slightly increased SOX9 expression (Fig. 5), indicating that full-length ADAM10 was able to promote the expression of SOX9. This contradictory result might have been caused by the protein formation of ADAM10. During ADAM10 protein synthesis, some fragments are cleaved from the full-length ADAM10, but few studies have been performed to investigate the function of these fragments. Transfection with the N1icd plasmid slightly decreased the expression of SOX9, and this result agreed with our original hypothesis that N1ICD acts as a negative regulator of SOX9 expression. When Adam10 was knocked down by plasmid 129, SOX9 expression was not affected by treatment with 0.1 μg of the N1icd plasmid. However, 0.2 μg of the N1icd plasmid significantly reduced SOX9 expression after transfection with plasmid 129. Treatment with 0.4 μg of the N1icd plasmid had a similar effect as that of 0.2 μg of N1icd. This result directly showed that the increase in SOX9 expression induced by ADAM10 inhibition could be rescued by N1ICD. However, N1ICD overexpression did not significantly decrease the expression of SOX9, possibly due to some other bypass mechanism through which ADAM10 can modulate SOX9 expression. Based on the above findings, we propose that ADAM10 might modulate chondrogenesis via the N1ICD-SOX9 pathway.
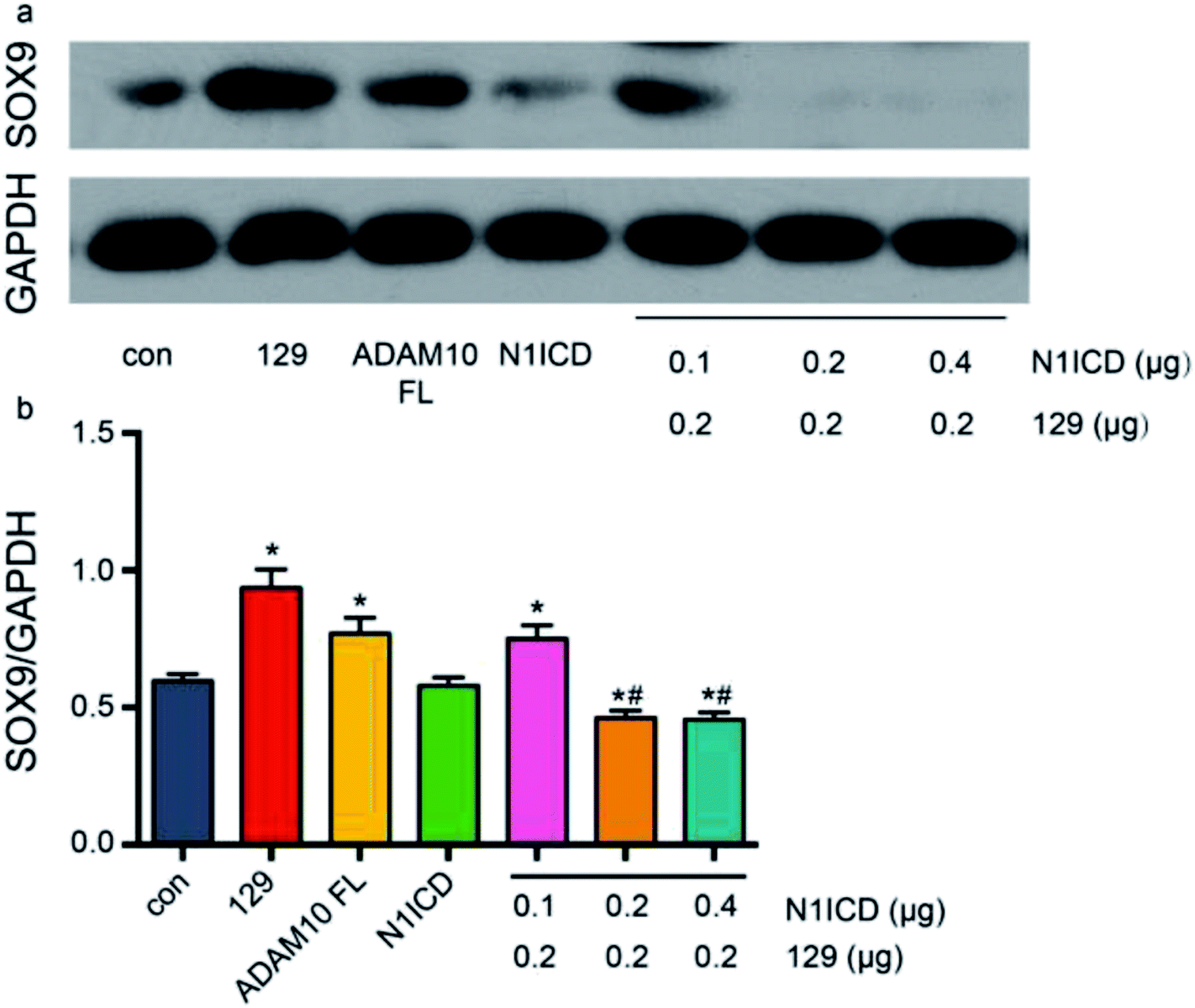 | ||
| Fig. 5 ADAM10 regulated SOX9 expression via N1ICD. All the results were obtained after 4 days of induction. Panel (b) shows the statistical analysis results for the data in image (a). The bars indicate the mean relative grey values with SDs. * indicates significant differences between the scramble group and the other groups. # indicates significant differences between the group transfected with 0.1 μg of N1icd + 0.2 μg of plasmid 129 and the groups transfected with 0.2 or 0.4 μg of N1icd plasmid + 0.2 μg of plasmid 129. | ||
In this study, we focused on the influence of ADAM10 on the ATDC5 cell line during chondrogenic induction. Regarding endochondral bone formation, vessel invasion is an associated event that is closely related to perichondrial development.45 Because ADAM10 is strongly associated with vessel formation,46 whether ADAM10 is involved in perichondrial development warrants investigation. Regarding in vivo experiments, other model mice, such as collagen-II-Cre mice, which specifically express Cre in cartilage, might be more useful than the mice used in this study. In the long term, there is still much more to be explored in this field.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors thank the Laboratory of Orthopaedic Cellular & Molecular Biology and the Shanghai Key Laboratory of Orthopaedic Implant, Department of Orthopaedic Surgery, for providing the cell line and the experimental space. The authors also thank the Laboratory of Neuroscience for sharing the transgenic mice. This study was supported by the National Key Research and Development Programme of China (2016YFC1100200), the National Natural Science Foundation of China (8177115, 81671017, 81400554), the Municipal Human Resources Development Programme for Outstanding Young Talents in Shanghai (2017YQ058), the Excellent Youth Programme of Ninth People's Hospital Affiliated to Shanghai Jiao Tong University School of Medicine (jyyq08201621), and the Interdisciplinary Program of Shanghai Jiao Tong University (YG2017MS01).Notes and references
- W. Cung, L. A. Freedman, N. E. Khan, E. Romberg, P. J. Gardner, C. W. Bassim, A. M. Baldwin, B. C. Widemann and D. R. Stewart, Eur. J. Med. Genet., 2015, 58, 584–590 CrossRef PubMed.
- Premalatha, V. P. Kannan and Madhu, J. Indian Soc. Pedod. Prev. Dent., 2010, 28, 322–325 CrossRef PubMed.
- A. Gong, J. Li, Z. Wang, Y. Li, F. Hu, Q. Li, D. Miao and L. Wang, Angle Orthod., 2016, 86, 668–680 CrossRef PubMed.
- W. Liu, G. Li, J. S. Chien, S. Raft, H. Zhang, C. Chiang and D. A. Frenz, Dev. Biol., 2002, 248, 240–250 CrossRef CAS PubMed.
- M. Enomoto-Iwamoto, T. Nakamura, T. Aikawa, Y. Higuchi, T. Yuasa, A. Yamaguchi, T. Nohno, S. Noji, T. Matsuya, K. Kurisu, E. Koyama, M. Pacifici and M. Iwamoto, J. Bone Miner. Res., 2000, 15, 1659–1668 CrossRef CAS PubMed.
- M. Wuelling and A. Vortkamp, Endocr. Dev., 2011, 21, 1–11 CAS.
- F. Long and D. M. Ornitz, Cold Spring Harbor Perspect. Biol., 2013, 5, a008334 CrossRef PubMed.
- K. Hata, Y. Takahata, T. Murakami and R. Nishimura, J. Bone Metab., 2017, 24, 75–82 CrossRef PubMed.
- J. Zhuang, Q. Wei, Z. Lin and C. Zhou, Gene, 2015, 555, 150–158 CrossRef CAS PubMed.
- E. Jorissen, J. Prox, C. Bernreuther, S. Weber, R. Schwanbeck, L. Serneels, A. Snellinx, K. Craessaerts, A. Thathiah, I. Tesseur, U. Bartsch, G. Weskamp, C. P. Blobel, M. Glatzel, B. De Strooper and P. Saftig, J. Neurosci., 2010, 30, 4833–4844 CrossRef CAS PubMed.
- K. Glomski, S. Monette, K. Manova, B. De Strooper, P. Saftig and C. P. Blobel, Blood, 2011, 118, 1163–1174 CrossRef CAS PubMed.
- Y. Tan, R. Fu, J. Liu, Y. Wu, B. Wang, N. Jiang, P. Nie, H. Cao, Z. Yang and B. Fang, Biochem. Biophys. Res. Commun., 2016, 475, 308–314 CrossRef CAS PubMed.
- R. Q. Fu, Y. Tan, Z. Yang and B. Fang, Shanghai Kouqiang Yixue, 2013, 22, 601–606 CAS.
- Y. Tian, Y. Xu, Q. Fu, M. Chang, Y. Wang, X. Shang, C. Wan, J. V. Marymont and Y. Dong, Mol. Cell. Endocrinol., 2015, 403, 30–38 CrossRef CAS PubMed.
- A. Gong, J. Li, Z. Wang, Y. Li, F. Hu, Q. Li, D. Miao and L. Wang, Angle Orthod., 2015 DOI:10.2319/032315-186.1.
- S. I. Blaser, N. Padfield, D. Chitayat and C. R. Forrest, Pediatr. Radiol., 2015, 45(suppl. 3), S485–S496 CrossRef PubMed.
- B. Balczerski, S. Zakaria, A. S. Tucker, A. G. Borycki, E. Koyama, M. Pacifici and P. Francis-West, Dev. Biol., 2012, 371, 203–214 CrossRef CAS PubMed.
- X. Nie, Acta Odontol. Scand., 2006, 64, 97–103 CrossRef CAS PubMed.
- T. J. Mead and K. E. Yutzey, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14420–14425 CrossRef CAS PubMed.
- Y. Yao and Y. Wang, J. Cell. Biochem., 2013, 114, 1223–1229 CrossRef CAS PubMed.
- W. Chen, X. Zhang, R. K. Siu, F. Chen, J. Shen, J. N. Zara, C. T. Culiat, S. Tetradis, K. Ting and C. Soo, J. Bone Miner. Res., 2011, 26, 1230–1241 CrossRef CAS PubMed.
- T. Arima, H. Enokida, H. Kubo, I. Kagara, R. Matsuda, K. Toki, H. Nishimura, T. Chiyomaru, S. Tatarano, T. Idesako, K. Nishiyama and M. Nakagawa, Cancer Sci., 2007, 98, 1720–1726 CrossRef CAS PubMed.
- S. Chubinskaya, G. Cs-Szabo and K. E. Kuettner, J. Histochem. Cytochem., 1998, 46, 723–729 CrossRef CAS PubMed.
- J. M. White, Curr. Opin. Cell Biol., 2003, 15, 598–606 CrossRef CAS PubMed.
- L. M. Christian, Fly (Austin), 2012, 6, 30–34 CrossRef CAS PubMed.
- S. Wetzel, L. Seipold and P. Saftig, Biochim. Biophys. Acta, 2017, 1864, 2071–2081 CrossRef CAS PubMed.
- J. Lin, X. Yan, A. Markus, C. Redies, A. Rolfs and J. Luo, Dev. Dyn., 2010, 239, 1246–1254 CrossRef CAS PubMed.
- X. Yan, J. Lin, A. Rolfs and J. Luo, Dev., Growth Differ., 2011, 53, 726–739 CrossRef CAS PubMed.
- J. C. Jones, S. Rustagi and P. J. Dempsey, Annu. Rev. Physiol., 2016, 78, 243–276 CrossRef CAS PubMed.
- Z. Yang, P. F. Li, R. C. Chen, J. Wang, S. Wang, Y. Shen, X. Wu, B. Fang, X. Cheng and Z. Q. Xiong, Cereb. Cortex, 2017, 27, 919–932 CrossRef PubMed.
- S. Weber, S. Wetzel, J. Prox, T. Lehmann, J. Schneppenheim, M. Donners and P. Saftig, Biochem. Biophys. Res. Commun., 2013, 442, 234–241 CrossRef CAS PubMed.
- D. R. Gibb, S. J. Saleem, D. J. Kang, M. A. Subler and D. H. Conrad, J. Immunol., 2011, 186, 4244–4252 CrossRef CAS PubMed.
- T. Isozaki, B. J. Rabquer, J. H. Ruth, G. K. Haines, 3rd and A. E. Koch, Arthritis Rheum., 2013, 65, 98–108 CrossRef CAS PubMed.
- T. Kobayakawa, N. Takahashi, Y. Sobue, K. Terabe, N. Ishiguro and T. Kojima, Biochem. Biophys. Res. Commun., 2016, 478, 1230–1235 CrossRef CAS PubMed.
- N. Woods, J. Trevino, D. Coppola, S. Chellappan, S. Yang and J. Padmanabhan, Oncotarget, 2015, 6, 35931–35948 CrossRef PubMed.
- P. Jing, N. Sa, X. Liu, X. Liu and W. Xu, Exp. Mol. Pathol., 2016, 100, 132–138 CrossRef CAS PubMed.
- A. Pliassova, J. P. Lopes, C. Lemos, C. R. Oliveira, R. A. Cunha and P. Agostinho, Mol. Neurobiol., 2015 DOI:10.1007/s12035-015-9491-9.
- O. A. Bianco, P. R. Manzine, C. M. Nascimento, F. A. Vale, S. C. Pavarini and M. R. Cominetti, Int. Psychogeriatr., 2015, 1–6, DOI:10.1017/S1041610215001842.
- E. J. Jin, Y. A. Choi, J. K. Sonn and S. S. Kang, Mol. Cells, 2007, 24, 139–147 CAS.
- R. Zhao, A. Wang, K. C. Hall, M. Otero, G. Weskamp, B. Zhao, D. Hill, M. B. Goldring, K. Glomski and C. P. Blobel, J. Orthop. Res., 2014, 32, 224–230 CrossRef CAS PubMed.
- N. Watanabe, Y. Tezuka, K. Matsuno, S. Miyatani, N. Morimura, M. Yasuda, R. Fujimaki, K. Kuroda, Y. Hiraki, N. Hozumi and K. Tezuka, J. Bone Miner. Metab., 2003, 21, 344–352 CrossRef CAS PubMed.
- Y. Hosaka, T. Saito, S. Sugita, T. Hikata, H. Kobayashi, A. Fukai, Y. Taniguchi, M. Hirata, H. Akiyama, U. I. Chung and H. Kawaguchi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1875–1880 CrossRef CAS PubMed.
- C. F. Liu and V. Lefebvre, Nucleic Acids Res., 2015, 43, 8183–8203 CrossRef CAS PubMed.
- G. van Tetering, P. van Diest, I. Verlaan, E. van der Wall, R. Kopan and M. Vooijs, J. Biol. Chem., 2009, 284, 31018–31027 CrossRef CAS PubMed.
- H. M. Kronenberg, Ann. N. Y. Acad. Sci., 2007, 1116, 59–64 CrossRef CAS PubMed.
- V. Caolo, G. Swennen, A. Chalaris, A. Wagenaar, S. Verbruggen, S. Rose-John, D. G. Molin, M. Vooijs and M. J. Post, Angiogenesis, 2015, 18, 13–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05609a |
| This journal is © The Royal Society of Chemistry 2018 |